

Abstract
Chromosomal translocations are a genomic hallmark of many hematologic malignancies. Often as initiating events, these structural abnormalities result in fusion proteins involving transcription factors important for hematopoietic differentiation and/or signaling molecules regulating cell proliferation and cell cycle. In contrast, epigenetic regulator genes are more frequently targeted by somatic sequence mutations, possibly as secondary events to further potentiate leukemogenesis. Through comprehensive whole-transcriptome sequencing of 231 children with acute lymphoblastic leukemia (ALL), we identified 58 putative functional and predominant fusion genes in 54.1% of patients (n = 125), 31 of which have not been reported previously. In particular, we described a distinct ALL subtype with a characteristic gene expression signature predominantly driven by chromosomal rearrangements of the ZNF384 gene with histone acetyltransferases EP300 and CREBBP. ZNF384-rearranged ALL showed significant up-regulation of CLCF1 and BTLA expression, and ZNF384 fusion proteins consistently showed higher activity to promote transcription of these target genes relative to wild-type ZNF384 in vitro. Ectopic expression of EP300-ZNF384 and CREBBP-ZNF384 fusion altered differentiation of mouse hematopoietic stem and progenitor cells and also potentiated oncogenic transformation in vitro. EP300- and CREBBP-ZNF384 fusions resulted in loss of histone lysine acetyltransferase activity in a dominant-negative fashion, with concomitant global reduction of histone acetylation and increased sensitivity of leukemia cells to histone deacetylase inhibitors. In conclusion, our results indicate that gene fusion is a common class of genomic abnormalities in childhood ALL and that recurrent translocations involving EP300 and CREBBP may cause epigenetic deregulation with potential for therapeutic targeting.
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children and a leading cause of disease-related death in childhood (Inaba et al. 2013; Pui et al. 2015). While it is a prototype of cancer that can be cured by chemotherapy alone, current ALL therapy relies primarily on conventional cytotoxic agents with significant acute and long-term side effects (Robison 2011). Therefore, a better understanding of genomic landscape of ALL is critical for developing molecularly targeted therapy and implementing genomics-based precision medicine in this cancer. One of genomic hallmarks of ALL is chromosomal translocation that gives rise to tumor-specific fusion proteins with novel and often oncogenic functions (Wiemels 2008; Hunger and Mullighan 2015). For example, reciprocal translocation t(12;21) results in chimeric transcripts of two critical hematopoiesis transcription factors ETV6 and RUNX1, and this fusion protein specifically promotes B-cell precursor expansion, enhances self-renewal capacity, and impairs B-lymphocyte differentiation (Hong et al. 2008; Schindler et al. 2009; Tsuzuki and Seto 2013). As initiating events, some of these large structural abnormalities arise early in life in children with ALL (Greaves and Wiemels 2003; Bateman et al. 2015) and frequently involve genes related to hematopoiesis and/or cell proliferation/cycling. Recent advents in genomic sequencing enabled the discovery of a variety of novel gene rearrangements in ALL, many of which are confined to these two pathways (Atak et al. 2013; Roberts and Mullighan 2015; Iacobucci et al. 2016; Yasuda et al. 2016).
Epigenetic modification is highly regulated during hematopoiesis and is important for proper maintenance of hierarchical differentiation (Ji et al. 2010; Abraham et al. 2013; Stergachis et al. 2013; Lara-Astiaso et al. 2014). However, chromosomal rearrangements in epigenetic regulator genes are relatively rare in hematologic malignancies. Instead, most somatic alterations in these genes are sequence mutations, possibly as a subsequent cooperating event. For example, CREBBP is recurrently mutated in ALL (Mullighan et al. 2011) and B-cell lymphoma (Pasqualucci et al. 2011), resulting in the loss of histone acetyltransferase (HAT) activity. While the exact biological consequences of these events remain unknown in the context of disease pathogenesis, these data point to the importance of the EP300/CREBBP signaling axis in normal and malignant hematopoiesis (Chalmers et al. 1990; Blobel 2000; Kimbrel et al. 2009). Mutations in histone genes themselves (e.g., histone gene cluster on 6p22) (Mullighan et al. 2007), histone modifying enzymes (e.g., SETD2, ASXL2, KMT2D, KDM6A) (Huether et al. 2014; Mar et al. 2014; Zhu et al. 2014), and chromatin remodeling genes (e.g., ASXL1, EZH2) (Nikoloski et al. 2010; Balasubramani et al. 2015) have also been reported, albeit with low frequencies. More recently, EP300 fusion with the transcription factor ZNF384 has been reported in ALL (Gocho et al. 2015; Ping et al. 2015; Yasuda et al. 2016), and ZNF384 is also implicated in chromosomal translocation with a number of partner genes (Martini et al. 2002; La Starza et al. 2005; Zhong et al. 2008; Nyquist et al. 2011; Yasuda et al. 2016), although the biological functions of these fusions are largely unknown.
In the current study, we systematically described gene fusion events in childhood ALL, by whole-transcriptome sequencing, and identified a distinct ALL subtype characterized by ZNF384 rearrangements and potential sensitivity to histone deacetylase inhibition.
Results
We performed whole-transcriptome sequencing of 231 children with newly diagnosed ALL who were consecutively enrolled in the Ma-Spore frontline ALL trials (Supplemental Table S1). Primarily of Asian descent (e.g., Chinese, Filipino, Indian, Malay), patients had a median age of 4.6 yr at diagnosis. Unsupervised hierarchical clustering and principal component analysis of global gene expression profile identified seven major ALL subgroups (Fig. 1A; Supplemental Fig. S1). Consistent with previous reports, cases with T-cell immunophenotype, ETV6-RUNX1, TCF3-PBX1, and hyperdiploid karyotype were readily identified by transcription signatures (Fig. 1A). Interestingly, ALL blasts from 12 patients did not carry these known cytogenetic abnormalities and formed a distinctive cluster. Further analyses revealed that 11 cases (91.6%) within this subgroup had gene fusion events involving the transcription factor ZNF384 (Fig. 1A,B), all of which were confirmed by reverse transcription polymerase chain reaction (RT-PCR) amplification and Sanger sequencing (Supplemental Fig. S2). Overall, we have identified 130 fusion genes in 144 patients, 100 of which to our knowledge have not been reported previously (Supplemental Table S2). In 125 patients, a chimeric gene either was the only fusion event detected or was the clearly predominant fusion (58 unique fusions in total) (Supplemental Fig. S3). Thirty-seven predominant fusion genes (24 known and 13 novel) directly fused coding sequences of two genes with a predicted in-frame chimeric protein (Supplemental Fig. S3D). In contrast, 21 predominant fusion transcripts (three known and 18 novel) involved noncoding sequences from one or both partner genes, leading to aberrant expression of important signaling genes in some instances (e.g., P2RY8-CRLF2). Notably, ETV6, PAX5, ZNF384, KMT2A, and TCF3 were repeatedly targeted by chromosomal translocations with diverse partner genes (Fig. 1C), highlighting their importance in ALL leukemogenesis.
Figure 1.

Whole-transcriptome sequencing of childhood ALL. (A) Unsupervised hierarchical clustering of global gene expression profile from 231 children with newly diagnosed ALL. Columns indicate ALL patients, and rows are genes; gene overexpression and underexpression are shown in red and green, respectively. ALL immunophenotype, cytogenetics, and selected chromosomal translocations are indicated for each sample above the heatmap, and the text below denotes seven major ALL subgroups identified on the basis on gene expression profiles. “Predominant fusion” refers to likely functional and driver fusion event, as defined in the Methods and Supplemental Figure S3A. (B) Pie chart of major gene fusions and cytogenetic abnormalities identified in B-ALL and T-ALL. Percentage indicates prevalence in the entire cohort; B-ALL and T-ALL are discriminated by solid fill and dot pattern, respectively. (C) Circos plot of the predominant fusions involving frequently translocated genes (i.e., genes with three or more different translocation partners identified). The line links the two partner genes in a fusion, with line width indicative of the prevalence of the specific fusion.
In nine of 11 ALL cases with ZNF384 rearrangements, the breakpoints in this gene were invariably between exon 2 and exon 3, resulting in deletion of only the 5′ UTR and then in-frame fusion of the entire coding sequence with the other genes (Fig. 2A; Supplemental Fig. S4). EP300 was the predominant fusion partner (n = 6), with its N terminus domains (amino acid residues 1 to 510) juxtaposed with the full-length ZNF384 protein (Fig. 2A). As a result, the EP300-ZNF384 fusion protein lacked the critical bromodomain and the HAT domain. A similar fusion was also observed between ZNF384 and CREBBP (n = 1) (Fig. 2A), another member of the p300/CBP coactivator family. The CREBBP-ZNF384 fusion lost most of the CREBBP domains related to HAT activity but retained the KIX domain that is responsible for CREB and MYB interaction (Dai et al. 1996; Radhakrishnan et al. 1997). Other ZNF384 rearrangements involved TCF3 and TAF15 (n = 3 and 1, respectively) (Fig. 2A). We next performed whole-exome sequencing of ZNF384-rearranged cases (n = 10) with available tumor, and matched germline samples to identify additional genomic abnormalities. An average of 4.3 nonsilent point mutations or insertions/deletions were observed per case (range from one to 11) (Fig. 2B; Supplemental Table S3), with no significant differences in frequency by ZNF384 fusion partners. Somatic mutations in epigenetic genes were common in this group of ALL, e.g., CREBBP (n = 2), KDM6A (n = 2), CHD4 (n = 1), and CHD8 (n = 1). In particular, CREBBP mutations were only observed in cases with TCF3- and TAF15-ZNF384 fusions, mutually exclusive with EP300 or CREBBP translocations (Fig. 2B). Taken together, genomic lesions (mutation or translocation) of epigenetic regulator genes were present in 81.8% of ZNF384-rearranged ALL. Mutations in RAS-PI3K pathway were also noted, e.g., PIK3CD, PTPN11, RIT1, and FLT3 (Supplemental Table S3). In an independent validation cohort of 602 Chinese children with newly diagnosed ALL (including 239 cases with BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, TCF3-HLF, or MLL [also called KMT2A] rearrangements), we observed comparable frequencies of EP300-ZNF384 (2.0%, n = 12) and CREBBP-ZNF384 fusions (1.8%, n = 11) as detected by RT-PCR and Sanger sequencing. Our subsequent functional studies primarily focused on EP300-ZNF384 and CREBBP-ZNF384 fusions because of their predominance in ZNF384-rearranged ALL.
Figure 2.

Genomic abnormalities in ZNF384-rearranged ALL. (A) Schematic representation of ZNF384 rearrangement in ALL. (Left) The wild-type proteins and their domain structures. Arrows indicate breakpoints observed in ALL samples, and colored blocks represent protein functional domains. (Right) The predicted ZNF384 fusion proteins. Dashed vertical lines mark the junction between fusion partners and are color-coded according to the breakpoint (arrows) in the left panel. (B) ZNF384-rearranged cases were also subjected to whole-exome sequencing, and putative functional genomic lesions (translocation, frameshift, and missense mutations) in selected genes are indicated for each case (left). (Right) Bar graph denotes the cumulative frequency of genomic lesion in each gene in ZNF384-rearranged ALL. (C) Heatmap shows the top 50 differentially expressed genes. Columns indicate genes, and rows are patients or patient groups. Expression value is indicated for individual patient with ZNF384 rearrangement; for cases without ZNF384 rearrangement (wild-type), expression is shown as the mean expression value for each ALL subgroup. The normalized expression level for each gene (z-score) is indicated by a color (red and green for over- and underexpression, respectively).
In spite of the variety of fusion partners, ZNF384-rearranged ALL was characterized by a common gene expression signature (Fig. 2C). Of 353 genes differentially expressed in this ALL subtype, there was a significant enrichment of ZNF384 binding sites as determined by ChIP-seq in B-lymphoblastoid cells (Rosenbloom et al. 2013), particularly in the promoter and/or enhancer regions (P = 6.8 × 10−14, Fisher's exact test) (Supplemental Table S4). Two of the most significantly up-regulated genes in this group were CLCF1 and BTLA, expressed at an average of 15.5- to 15.0-fold higher than cases with wild-type ZNF384, respectively (Fig. 3A,B). CLCF1 encodes cardiotrophin-like-cytokine factor 1, which binds to CRLF1 to form a compound cytokine, eventually leading to activation of the JAK-STAT signaling and B-cell proliferation in vivo (Senaldi et al. 2002; Crabe et al. 2009; Savin et al. 2015; Sims 2015). Important for lymphocyte development and activation, BTLA expression is highly regulated during B-cell differentiation and is also directly linked to B-cell receptor signaling cascade (Vendel et al. 2009; Llinas et al. 2011; Kannan et al. 2015). Because ZNF384 binding sites were identified within the promoter and enhancer regions of both BTLA and CLCF1 (Supplemental Fig. S5), we hypothesized that ZNF384 may function as a direct transcription activator of these two target genes. Thus, we evaluated the expression of a luciferase reporter gene placed downstream from the promoter and/or enhancer elements of BTLA or CLCF1. In HEK293T cells, ectopic expression of wild-type ZNF384 readily promoted luciferase gene transcription with the BTLA promoter alone or both the promoter and enhancer of BTLA (Fig. 3C), but not with promoters/enhancers void of ZNF384 binding sites (Supplemental Fig. S6). Overexpression of ZNF384 fusions further increased BTLA promoter and enhancer activity relative to cells transfected with wild-type ZNF384, pointing to plausible gain of function by fusion gene. Consistent results were observed when we tested the CLCF1 promoter and enhancer (Fig. 3C; Supplemental Fig. S7). Similarly, in human leukemia cells, BTLA and CLCF1 promoter and enhancer activity was consistently and significantly higher in the ZNF384-rearranged ALL cell line JIH-5 compared with the ZNF384 wild-type ALL cell line Nalm6 or with reporter gene construct without ZNF384 binding sites (Fig. 3D; Supplemental Fig. S6).
Figure 3.
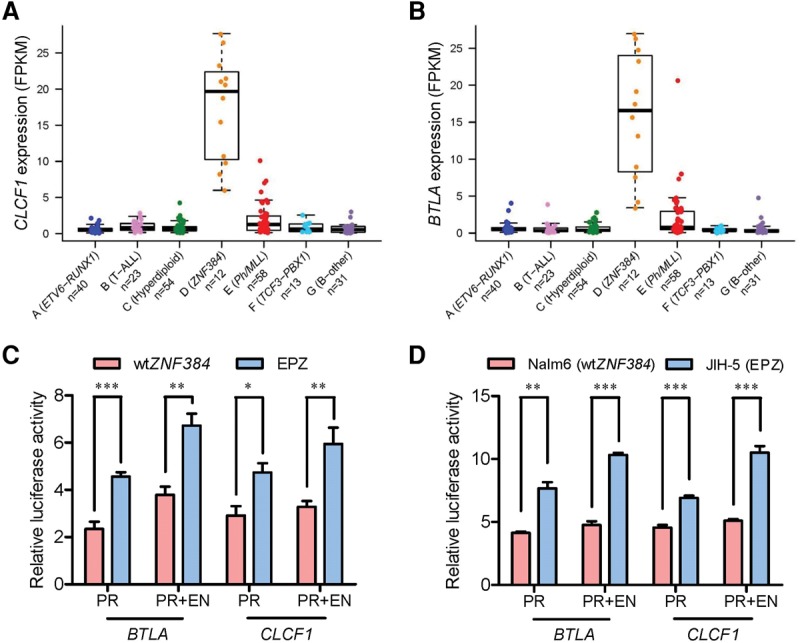
ZNF384 fusions transactivated CLCF1 and BTLA expression. (A,B) Expression of the CLCF1 (A) and BTLA (B) genes in seven ALL subgroups identified from hierarchical clustering. Each sample is represented by a dot and is color-coded according to the subgroups it belongs to. (C,D) Luciferase reporter gene assay of BTLA and CLCF1 promoter/enhancer activity. HEK293T cells were transiently transfected with pGL3 construct (luciferase gene with BTLA or CLCF1 promoter [PR] and/or enhancer [EN]), pcDNA construct (wild-type ZNF384 [wtZNF384] or EP300-ZNF384 [EPZ]), and pGL-TK (Renilla luciferase) (C). Similar sets of reporter gene assay were performed in the ALL cell lines Nalm6 (wtZNF384) and JIH-5 (EPZ) (D). Firefly luciferase activity was measured 24 h post-transfection and normalized to Renilla luciferase activity. Relative luciferase activity indicates the ratio over the value from pGL3 basic vector alone. All experiments were performed in triplicate and repeated at least three times. (PR) Promoter; (EN) enhancer; (wtZNF384) wild-type ZNF384; (EPZ) EP300-ZNF384 fusion. Statistical significance was evaluated by using two-sided Student's t-test: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
To directly examine the effects of ZNF384 fusion on hematopoietic phenotypes, we next evaluated colony forming potential of hematopoietic stem and progenitor cells (HSPCs) expressing different fusion genes in vitro. In response to cytokine stimulation for myelopoiesis (SCF, IL3, IL6, and EPO), purified mouse HSPCs gave rise to colonies characteristic of common myeloid progenitor cells (CFU-GEMM), granulocyte-macrophage progenitor cells (CFU-GM), and also erythroid progenitor cells (BFU-E and CFU-E). Upon overexpression of EP300-ZNF384 or CREBBP-ZNF384 fusion, the number of colonies giving rise to those four myeolopoietic progenitor populations was reduced dramatically (60.0% to 29.2% of that from HSPCs transduced with mock vectors) (Fig. 4A). In contrast, with cytokine condition for B-cell differentiation (IL7), HSPCs with forced expression of ZNF384 fusions were significantly more likely to develop into pre-B progenitor cells relative to the control (Fig. 4B). These data suggest that the EP300-ZNF384 and CREBBP-ZNF384 fusions altered the potential of HSPCs for lymphoid versus myeloid differentiation. However, unlike other leukemia lesions (e.g., BCR-ABL1), ZNF384 fusion did not improve the sustained growth of HSPCs in a serial replating experiment (Supplemental Fig. S8), indicating that they may not be capable of inducing overt oncogenic transformation. Similarly, when ZNF384 fusions were introduced into the mouse hematopoietic progenitor cell line Ba/f3, there was a significant increase in proliferation with a modestly higher proportion of cells in S phase compared with the empty vector control (Fig. 4C,D). With IL3-independent growth as a measurement of oncogenic transformation, EP300- and CREBBP-ZNF384 fusions did not transform Ba/f3 cells in vitro, although with modest anti-apoptotic effects (Fig. 4E). We hypothesized that ZNF384 fusions are not oncogenic by themselves but instead may increase the transforming potential of other oncogenic mutations. In fact, Ba/f3 cells became IL3 independent 7 d upon cotransduction of ZNF384 fusion and NRASG12D, significantly more quickly than cells expressing NRASG12D alone (Fig. 4F). Similar effects were observed when EP300-ZNF384 or CREBBP-ZNF384 fusions were cotransduced with leukemogenic gene CRLF2 (Supplemental Fig. S9).
Figure 4.
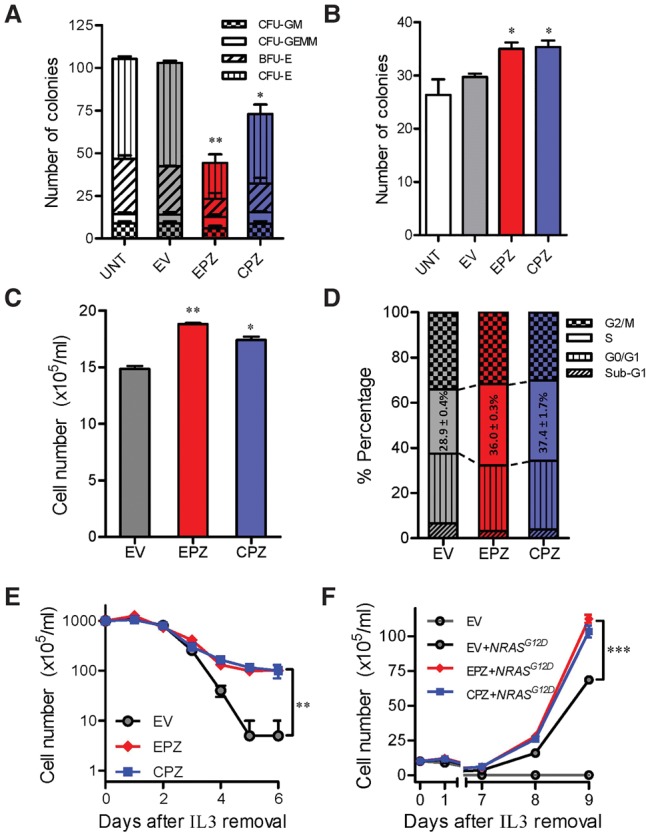
EP300- and CREBBP-ZNF384 fusions influence hematopoiesis and hematopoietic progenitor cell transformation in vitro. (A,B) Effects of fusion genes on mouse hematopoiesis in vitro. Mouse Lin−Sca1+c-Kit+ cells were lentivirally transduced with empty vector (gray), EP300-ZNF384 (red), or CREBBP-ZNF384 (blue). Cells were then cultured in methylcellulose medium supplemented with cytokines for myelopoisis (A) or pre-B-cell differentiation (B), and colony formation was assessed 12–14 d later. (C,D) ZNF384 fusion genes and proliferation of mouse hematopoietic progenitor cell Ba/f3. Ba/f3 cells were lentivirally transduced with empty vector (grey), EP300-ZNF384 (red), or CREBBP-ZNF384 (blue) and then cultured in the presence of IL3 (10 ng/mL). After 48 h, cell density was examined using Trypan blue (C), and cell cycle distribution was evaluated using a standard PI staining protocol (D). (E,F) Effects of fusion genes on Ba/f3 transformation. Following transduction of empty vector or ZNF384 fusion genes, Ba/f3 cells were cultured in IL3-depleted medium with cytokine-independent cell growth as a measure of oncogenic transformation. The number of viable cells was evaluated daily (E). A similar set of experiments was performed with cotransduction of oncogenic NRASG12D (F). All experiments were performed in triplicate and repeated twice. (EV) Empty vector; (EPZ) EP300-ZNF384 fusion; (CPZ) CREBBP-ZNF384 fusion; (CFU-GM) colony-forming unit–granulocyte, macrophage; (CFU-GEMM) colony-forming unit–granulocyte, erythrocyte, macrophage, megakaryocyte; (BFU-E) burst-forming unit–erythroid; (CFU-E) colony-forming unit–erythroid. Statistical significance of the differences between EPZ versus EV or between CPZ versus EV was estimated by using two-sided Student's t-test (A–C) or two-way ANOVA (E,F): (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Finally, we tested the effects of EP300-ZNF384 and CREBBP-ZNF384 fusions on histone modification. Wild-type (full-length), truncated, and fusion EP300 and CREBBP proteins were ectopically expressed in Sf9 insect cells, purified to homogeneity, and then subjected to HAT activity assay. EP300- and CREBBP-ZNF384 fusion proteins showed a 75% and 90% reduction of HAT activity of wild-type proteins, respectively, comparable to truncated EP300 and CREBBP proteins alone (Fig. 5A). This suggested that the loss of enzymatic activity was primarily due to the truncation of the HAT domain. The addition of fusion proteins (but not truncated EP300) also significantly repressed HAT activity of wild-type EP300 (Fig. 5B; Supplemental Fig. S10), indicating a dominant-negative effect that was dependent on fusion with ZNF384. Along the same line, expression of EP300- or CREBBP-ZNF384 fusion proteins significantly decreased global H3 acetylation (at both K9 and K27 residues) in Ba/f3 cells compared with the vector control (Fig. 5C; Supplemental Fig. S11). To test the feasibility of restoring histone acetylation as a therapeutic strategy for ALL cases with these ZNF384 fusions, we sought to evaluate the cytotoxicity of the histone deacetylase inhibitor vorinostat in Ba/f3 cells. NRASG12D-transformed Ba/f3 cells underwent apoptosis upon vorinostat treatment in a dose-dependent fashion, but cotransduction of the EP300- or CREBBP-ZNF384 fusion significantly potentiated cytotoxic effects of histone deacetylase inhibition (Fig. 5D), with a concomitant increase in overall histone 3 acetylation (Supplemental Fig. S12). In parallel, in a panel of human ALL cell lines representative of common molecular subtypes (i.e., ETV6-RUNX1, TCF3-PBX1, BCR-ABL1, DUX4-IGH, MLL-rearranged, and ZNF384-rearranged), EP300-ZNF384–positive JIH-5 cells showed the lowest level of acetyl H3 at both the K9 and K27 residues and also the highest sensitivity to vorinostat compared with ALL cells with wild-type ZNF384 (Supplemental Fig. S13). Taken together, these results point to the possibility that ZNF384-rearranged ALL may benefit from therapeutic agents targeting histone acetylation regulation.
Figure 5.
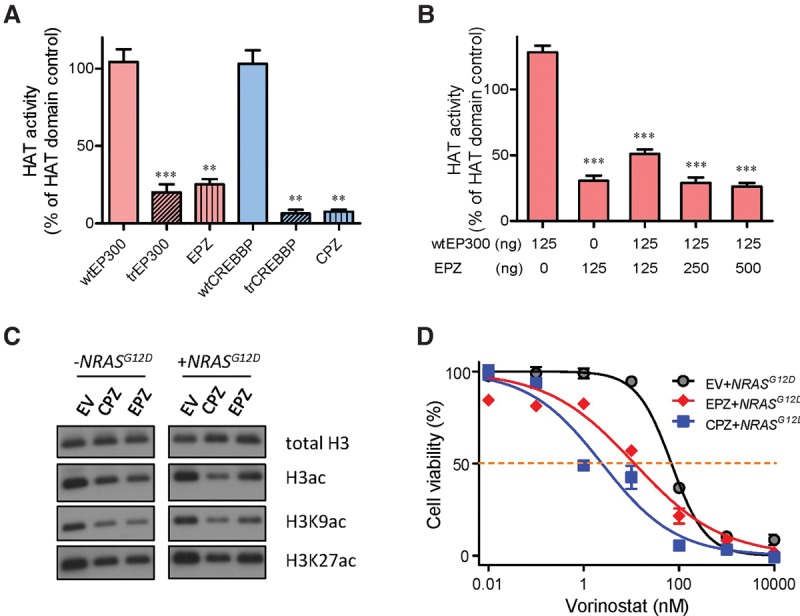
EP300- and CREBBP-ZNF384 fusions resulted in loss of HAT activity, global histone acetylation deregulation, and sensitivity to HDAC inhibition. (A,B) HAT enzymatic activity was measured for various EP300 and CREBBP proteins: wild-type, truncated, and ZNF384 fusions. Proteins were expressed in Sf9 insect cells and purified to homogeneity. (A) HAT activity was determined for each protein individually. (B) In parallel, wild-type EP300 was mixed with increasing amount of EP300-ZNF384 fusion protein to examine dominant-negative effects of the latter on HAT activity. (C) Global H3, H3K9, and H3K27 acetylation were evaluated by Western blot in Ba/f3 cells overexpressing EP300-/CREBBP-ZNF384 fusion, with total H3 as a loading control. (D) Cytotoxicity of HDAC inhibitor vorinostat was examined in NRASG12D-transformed Ba/f3 cells expressing EP300-ZNF384 (red) or CREBBP-ZNF384 (blue) or transduced with empty vector (gray). Cells were incubated with vorinostat for 48 h, and viability was then measured using a MTT assay. Experiments were performed in triplicate and repeated at least three times. (wtEP300) Wild-type EP300; (trEP300) truncated EP300; (wtCREBBP) wild-type CREBBP; (trCREBBP) truncated CREBBP; (EPZ) EP300-ZNF384 fusion; (CPZ) CREBBP-ZNF384 fusion; (EV) empty vector. EPZ- or CPZ-transduced cells were significantly more sensitive to vorinostat than EV, as determined by two-way ANOVA (P < 0.001). (A,B) Statistical significance was evaluated by using a two-sided Student's t-test: (**) P < 0.01; (***) P < 0.001.
Discussion
Chromosomal translocation in leukemia is arguably one of the first recognized genetic abnormalities in cancer (Rowley 2013). These nonrandom rearrangements are likely the founding events during leukemogenesis (Greaves 1997; Wiemels 2008). While focal genomic lesions (e.g., sequence mutations) can evolve during ALL progression and relapse (Mullighan et al. 2008; Yang et al. 2008; Ma et al. 2015), gene fusions arising from translocations usually remain unchanged across subclones over time, suggesting its fundamental role in the pathogenesis of this type of cancer (Anderson et al. 2011). By applying whole-transcriptome sequencing, we have identified a large number of fusion genes (Supplemental Table S2; Supplemental Fig. S3), and a few observations are particularly noteworthy. First, in this largely unselected cohort of childhood ALL, a readily expressed and predominant fusion gene was identified in 54.1% of patients, suggesting that gene fusion is a common form of genomic abnormality in ALL. Second, while most fusions were rare, a handful of genes (e.g., PAX5, ETV6, TCF3, ZNF384, and KMT2A) were repeatedly targeted by rearrangements with substantial diversity in their fusion partners; therefore, it is possible that a variety of mechanisms exist to deregulate these key genes during leukemogenesis. Third, of the 37 fusions (24 known and 13 novel) predicted to encode an in-frame chimeric protein and thus are most likely functional, many involved gene defects that may be therapeutically targeted (e.g., ABL1, CREBBP, PDGFR), highlighting opportunities for rationally designed novel ALL therapy. While we primarily focused on in-frame fusions, it is formally possible that “out-of-frame” fusions can lead to loss of function and are thus consequential in leukemogenesis. It should also be noted that our base-resolution whole-transcriptome sequencing enables a variety of genomic discoveries in addition to gene fusion detection and their associated gene expression signature. For example, future analyses of this data set would focus on novel transcript isoforms, allele-specific or -biased expression, etc. Finally, unlike most prior genomic profiling studies of ALL, our patient population is exclusively Asian and thus affords a unique opportunity to identify ancestry-related difference in the transcriptomic structure of ALL, which should be considered in the future development of targeted therapy for this cancer.
The fact that ZNF384 rearrangements are largely specific to ALL strongly argues that this gene plays a potentially important role in leukemogenesis even though little is known about the exact biological function of this zinc finger transcription factor. First identified for its direct interaction with focal adhesion protein BCAR1 (Janssen and Marynen 2006), ZNF384 shuttles between cytoplasm and nucleus and can alter DNA structure in response to changes in cell shape and cytoskeleton architecture (Feister et al. 2000). Loss of ZNF384 in vivo impairs bone formation (Morinobu et al. 2005) and leads to defects in spermatogenesis (Nakamoto et al. 2004), although the effects on hematopoiesis have not been comprehensively examined in these animal models. Across different hematopoietic compartments, ZNF384 expression is fairly ubiquitous, with only modest down-regulation in hematopoietic stem cells (Novershtern et al. 2011), and also remains largely constant during B-cell differentiation (Hardy fractions A–F) (Supplemental Fig. S14) in contrast with other known transcription factors involved in hematopoiesis (Novershtern et al. 2011). ZNF384-rearranged ALLs exhibit immunophenotypes that are highly characteristic of Pro-B cells, suggesting that these fusion genes may specifically stall B-lymphocyte differentiation at this stage. Forced expression of EP300-ZNF384 indeed inhibited mouse Pro-B cell differentiation in vivo (Yasuda et al. 2016), but in the more primitive HSPCs, this fusion protein blocked the differentiation along the myeloid and granulocyte lineages (Fig. 4A). Surprisingly, ZNF384-rearranged ALL often also expresses cell surface markers of myeloid lineage (CD13 and CD33) (Supplemental Table S1B), suggesting that EP300-ZNF384 might function differently depending upon the exact precursor cell population in which it occurs. We postulate that the juxtaposition of the partner gene with ZNF384 in the fusion protein confers novel functions that may interfere with normal hematopoiesis at multiple stages. So far, five partner genes (EP300, CREBBP, TAF15, TCF3, and ESWR1) (Martini et al. 2002; Zhong et al. 2008; Nyquist et al. 2011; Gocho et al. 2015) have been identified in ZNF384-rearranged ALL, and it remains unclear whether there is a common mechanism for leukemogenesis in these cases. Our preliminary data thus far indicated that at least the TAF15- and TCF3-ZNF384 fusions also positively regulated BTLA and CLCF1 transcription and potentiated oncogenic transforming effects in Ba/f3 cells (especially with CRLF2), although with distinctive effects on HSPC function in vitro compared with EP300- and CREBBP-ZNF384 fusions (Supplemental Figs. S7, S15). In fact, expression of ZNF384 was also significantly higher in cases with ZNF384 fusions than those without (P < 0.01, two-sided Student's t-test) (Supplemental Fig. S16). Taken together, these findings suggest the gain of ZNF384 transcription factor activity as a common feature in ZNF384 fusion functions but also point to differential effects possibly driven by the specific fusion partner genes involved. The contribution of ZNF384 fusions to ALL leukemogenesis is likely to be multifactorial, and future mechanistic studies are warranted to determine the exact requirement of each functionality of these fusions.
While ZNF384 rearrangement is predominantly observed in B-ALL, these leukemia blast cells also often express myeloid markers (La Starza et al. 2005; Gocho et al. 2015; Ping et al. 2015), pointing to its possible link to biphenotypic leukemia. Clinically, ZNF384-rearranged ALLs do not exhibit other unique presenting features and tend to be associated with a favorable prognosis in retrospective studies. In the context of the multi-institutional prospective Ma-Spore frontline ALL trial, ZNF384 fusions had no significant effects on prednisone treatment in vivo, leukemia burden at the end of induction therapy, or event-free survival (Supplemental Table S1A), although this could have been influenced by our relatively limited sample size.
EP300 and CREBBP are two highly related proteins best known for their lysine acetyltransferase activity especially in the context of transcription coactivation (Ait-Si-Ali et al. 1998; Iyer et al. 2004; Liu et al. 2008). Canonically, EP300 and CREBBP can directly influence chromatin conformation by targeted histone lysine acetylation (Liu et al. 2008), and concomitant EP300 binding and H3K27 acetylation is a hallmark of promoter/enhancer activation (Vo and Goodman 2001). During hematopoiesis, EP300 and CREBBP are involved in a complex series of transcriptional events mainly through interactions with other hematopoietic transcription factors (e.g., GATA1, TCF3, and RUNX1) (Blobel 2000). Leukemia-associated translations involving CREBBP have been identified in acute myeloid leukemia (KAT6A-CREBBP and KMT2A-CREBBP) (Yang 2004), whereas EP300 translocation is exceedingly rare, and in these cases, the fusion proteins retain most of the EP300/CREBBP functional domains. In contrast, in lymphoid malignancies, CREBBP was most frequently targeted by sequence mutation or focal copy number changes that mainly affect the HAT catalytic domain (Mullighan et al. 2011; Pasqualucci et al. 2011). Although uncommon in leukemia, EP300 mutations are also somewhat enriched in the HAT domain (Shigeno et al. 2004). In ZNF384-rearranged ALL, nearly all cases harbored genomic lesions in EP300 and/or CREBBP that would lead to loss of HAT activity, suggesting that the selective truncation of this domain is critical. The global decrease of H3 acetylation that possibly resulted from dominant-negative EP300-ZNF384 fusion also presented a potential therapeutic opportunity for small molecules such as histone deacetylase inhibitors (Fig. 5; Supplemental Fig. S13). Plausibly, histone deacetylation is required for leukemia maintenance in these ALL cases, thus restoring acetyl marks by vorinostat is anti-leukemic. While our data indicated a correlation between EP300-ZNF384 fusion, aberrant histone acetylation, and sensitivity to vorinostat, future studies are needed to establish the causal relationship and delineate the exact mechanistic details. The function of EP300/CREBBP is incredibly complex, and therefore, deregulation of histone acetylation might be one of many mechanisms that link these fusions to leukemogenesis and sensitivity to this class of drugs.
In conclusion, we have comprehensively described gene fusions in childhood ALL and identified ZNF384-rearranged ALL as a distinct subtype with characteristic genomic lesions targeting epigenetic modulator genes. These findings shed lights on genomic complexity of ALL and also potential genomics-guided therapeutic options.
Methods
Patients and samples
Included in this study were 231 children with newly diagnosed ALL treated in Ma-Spore frontline ALL clinical trials (Yeoh et al. 2012). Gross cytogenetics and common translocations were determined at the time of diagnosis (i.e., BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, STIL-TAL1, MLL rearrangement, and hyperdiploid karyotype) by karyotyping and reverse transcription PCR in the reference laboratory at National University Hospital, Singapore (Yeoh et al. 2012). Subjects were selected based on sample availability. To test the frequency of EP300-ZNF384 and CREBBP-ZNF384 fusions, 602 children with newly diagnosed ALL from Beijing Children's Hospital, China (Gao et al. 2012), were included as the validation cohort (239 cases with BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, TCF3-HLF, or MLL rearrangements). Total RNA and genomic DNA were extracted from diagnostic bone marrow. Peripheral blood or bone marrow specimens collected during clinical remission were used to prepare germline genomic DNA whenever applicable. This study was approved by the institutional review boards at St. Jude Children's Research Hospital, USA; National University of Singapore, Singapore; and Beijing Children's Hospital, China.
Whole-transcriptome sequencing
TruSeq stranded mRNA library prep kit (Illumina) was used for whole-transcriptome library preparation, and paired-end sequencing was performed using the Illumina HiSeq 2000/2500 platform with a 101-bp read length at Hartwell Center of Biotechnology at St. Jude Children's Research Hospital. Sequencing reads were aligned to the human genome (hg19) reference sequence using TopHat2 (v2.0.12) (Trapnell et al. 2012). Samples were excluded if the percentage of exonic regions with 10× or more coverage was <30%, read duplication rate >55%, or a strong 3′-bias of transcript coverage. Relative transcript expression levels (fragments per kilobase of transcript per million mapped reads [FPKM]) were estimated based on supporting reads. For each gene, expression was summarized as the sum of FPKMs for all transcript isoforms and normalized across all qualified samples by Cuffnorm (v2.2.1, with the default geometric algorithm) (Trapnell et al. 2012) and by loess function of R stats package (version 3.1.3) (R Core Team 2015). The FPKM values were log2 transformed for all subsequent analyses. Unsupervised hierarchical clustering (Ward.D2 method for clustering with Euclidean distance for samples and correlation for genes) (Murtagh and Legendre 2014) was performed after excluding genes with invariable expression (coefficient of variation <1) or extremely low or high expression (mean log2[FPKM + 1] < 0.585 or maximal log2[FPKM + 1] > 8, respectively). As a result, 490 most variably expressed genes were included in the final clustering and principal component analyses. Genes differentially expressed by ALL subgroup were identified using the SAM algorithm with fold change of more than two and false-discovery rate (q-value) <0.1% (Tusher et al. 2001). By using ZNF384 ChIP-seq data in the lymphoblastoid GM12878 cell line from ENCODE Project (Rosenbloom et al. 2013), we identified putative ZNF384 binding sites in genes differentially expressed in the ZNF384-rearranged ALL following previously established procedures (Qian et al. 2013).
Gene fusion was identified by two independent algorithms, namely, TopHat-Fusion (Kim and Salzberg 2011) and CICERO (Roberts et al. 2014). The initial screen for fusion transcript was performed using TopHat-Fusion with default parameters and predicted fusion genes involving mitochondria chromosome or with breakpoint-spanning read count of less than three in all suspected samples were excluded. In parallel, sequence reads were realigned by using the STAR (v2.4.0d) (Dobin et al. 2013) program and then analyzed by using the CICERO algorithm. Fusions called by CICERO with a high-confidence score (more than 400) were also added to candidate fusions identified by TopHat-Fusion. Manual review was applied to generate the final fusion gene list.
ETV6-RUNX1, TCF3-PBX1, BCR-ABL1, STIL-TAL1, and MLL fusions were screened during ALL diagnostic work-up, and we observed 100% concordance with the whole-transcriptome sequencing results. Fusions involving ZNF384, ETV6, PAX5, or RUNX1 were prioritized for validation by PCR amplification of breakpoint region of the chimeric transcript in the corresponding patient cDNA, followed by Sanger sequencing for final confirmation (PCR and sequencing primers are listed in Supplemental Table S5). The remainders of the fusions were validated by PCR on the basis of gene function (i.e., SPI1, MYB, LMO1, RAG1, and ABL1) and sample availability. In total, 74 fusions in 125 samples were validated. We subjectively classified fusions into “predominant” versus “minor” to indicate the likelihood of a functional event versus a random and passenger event, following a multistep process considering predicted open reading frame of the chimeric transcript, adequate expression of the fusion, and also involvement of gene paralogs (Supplemental Fig. S3A). Wherever applicable, chromosomal translocations were visualized using Circos (Krzywinski et al. 2009).
Whole-exome sequencing
Matched germline and tumor DNA from ALL cases with ZNF384 rearrangements were subjected to whole-exome sequencing. The Nextera rapid capture expanded exome kit was used for capture and library preparation with insert size ∼200 bp predominantly (ranges from 100–800 bp). Paired-end sequencing (2 × 101 bp read length) was performed using the Illumina HiSeq 2000/2500 platform at St. Jude Children's Research Hospital. Reads were aligned to the human genome (GRCh37) reference sequence (human_g1k_v37.fasta) using Burrows-Wheeler aligner MEM algorithm (version 0.7.12) (Li and Durbin 2009). Across 10 tumor–germline pairs (20 samples), we observed a median of 88% of exonic regions covered by at least 10× and 69% exome by at least 20×. All steps of the data processing prior to variant calling were performed according to the GATK best practices using GATK (version 3.5) (McKenna et al. 2010; DePristo et al. 2011) and Picard tools (version 1.141; http://broadinstitute.github.io/picard). Somatic mutations (single-nucleotide variants and short insertion/deletions) were called by using MuTect2 (Cibulskis et al. 2013). Mutations with one of the following features in the tumor data were excluded: (1) The read depth was less than 20; (2) the mutant allele frequency was <20%; (3) all reads supporting the mutation call came from the same mapping direction; (4) two and more mutations called in the same sample clustered together (within a 30-bp window). A total of 56 mutations remained, all of which were reviewed manually using the Integrative Genomics Viewer (Robinson et al. 2011). Twenty-one mutations were deemed false positive because of read mapping error due to genomic sequence duplication and thus removed from subsequent analyses. To address the possibility of variants missed by whole-exome sequencing due to low coverage in some cases, we also curated variant calls from the matched whole-transcriptome sequencing data. In fact, eight mutations originally excluded because of low mutant allele frequency or low read depth in whole-exome sequencing were validated in whole-transcriptome sequencing and thus confirmed as somatic mutation. The final list thus consisted of 43 high-confidence somatic mutations.
ZNF384 target gene promoter and enhancer activity by luciferase assays
Full-length chimeric transcripts (EP300-, CREBBP-, TCF3-, and TAF15-ZNF384) were amplified from patient cDNA and cloned into pcDNA3.1+/Hydro(+) vector (Thermo Fisher Scientific) with a C terminus Flag tag. Putative promoter and enhancers of BTLA and CLCF1 were identified from ENCODE ChIP-seq data in GM12878 cell line and were cloned into pGL3 luciferase vector (Promega): BTLA promoter, Chr 3: 112217785–112218842; BTLA enhancer, Chr 3: 112208658–112209560; CLCF1 promoter, Chr 11: 67140939–67141906; and CLCF1 enhancer, Chr 11: 67135923–67136631. To ensure specificity of these assays, we also tested pGL3 constructs void of predicted ZNF384-binding sites at the BTLA and CLCF1 loci (deletion of sequence Chr 3: 112218257–112218467 within BTLA promoter and Chr 3: 112208991–112209202 within BTLA enhancer; deletion of sequence Chr 11: 67141234–67141499 within CLCF1 promoter and Chr 11: 67136225–67136404 within in CLCF1 enhancer). All chromosomal coordinates were according to human genome hg19.
For the luciferase assay, 2.5 × 104 HEK293T cells cultured in a 96-well plate were transiently transfected with 50 ng of pGL3 construct (luciferase gene with promoter and/or enhancer of interest), 50 ng of pcDNA construct (wild-type ZNF384 or ZNF384 fusion), and 5 ng of pGL-TK (Renilla luciferase) using Lipofectamine 2000 (Invitrogen). Similar luciferase assays were performed in leukemia cell lines Nalm6 and JIH-5. Nalm6 was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ), and the JIH-5 cells (Ping et al. 2015) were a gift from Dr. Suning Chen at Soochow Children's Hospital and Dr. Hans G. Drexler at DSMZ. Nalm6 or JIH-5 cells (1 × 106) were transiently transfected using Amaxa cell line nucleofector kit T (Lonza). Firefly luciferase activity was measured after 24 h post-transfection using the dual luciferase assay (Promega). The results were normalized to Renilla luciferase activity. These experiments were repeated at least three times, and each sample was assayed in triplicate.
In vitro differentiation assay of mouse HSPCs
EP300-, CREBBP-, TCF3-, and TAF15-ZNF384 transcripts were cloned into the pcL20c-IRES-GFP lentiviral vector, and lentiviral supernatants were produced by transient transfection of HEK293T cells using calcium phosphate (Xu et al. 2015). Lin−Sca1+c-Kit+ HSPCs were sorted from bone marrow of C57/BL6 mice (8–10 wk old) by using flow cytometry (Taggart et al. 2016) and lentivirally transduced with ZNF384 fusions for 72 h followed by GFP selection (Ye et al. 2015). For in vitro colony formation assays, 500 GFP+ HSPCs were plated in 35-mm Petri dishes in Methocult M3434 or M3630 methylcellulose medium (Stemcell Technologies) to examine differentiation potential for myelopoiesis or into pre-B cells, respectively. Methocult M3434 medium consists of 2% methylcellulose, SCF (50 ng/mL), IL3 (10 ng/mL), IL6 (10 ng/mL), and EPO (3 U/mL), and M3630 is supplemented with IL7 (10 ng/mL). Serial replating was done at 500 cells per 35-mm Petri dish in the same medium above. The number of colonies was counted 12–14 d after plating.
Mouse hematopoietic progenitor cell Ba/f3 proliferation and transformation assay
Flag-tagged ZNF384 fusions were introduced into Ba/f3 cells by lentiviral transduction. GFP-positive cells were sorted 48 h later and plated at 1 × 105 cells/mL density in the presence of 10 ng/mL IL3. Cell growth was monitored daily using Trypan blue, and cell cycle distribution was determined 48 h after plating using a standard propidium iodide (PI) staining protocol.
To examine transformation potential, Ba/f3 cells expressing different ZNF384 fusion genes were further transduced with NRASG12D (retroviral, pMSCV) or CRLF2 (lentiviral, pcL20C). IL3 was removed 48 h later, and the number of viable cells was monitored daily by Trypan blue. The NRASG12D and CRLF2 constructs were gifts from Dr. Charles Mullighan at St. Jude Children's Research Hospital. Retroviral particles were produced using ecotropic HEK293T cells (American Type Culture Collection) (Xu et al. 2015). Each experiment was performed three times.
HAT activity assay and global histone acetylation
Wild-type (full-length) and truncated EP300 and CREBBP, EP300-ZNF384 fusion, and CREBBP-ZNF384 fusion were cloned into the pFastBac/HBM-TOPO vector with a C terminus His-tag. Each protein was ectopically expressed in Sf9 cells and purified to homogeneity by affinity chromatography (Invitrogen) (Supplemental Fig. S17; Delvecchio et al. 2013). His-tag was removed by AcTEV protease (Invitrogen) for all recombinant proteins, and HAT activity was then measured using HAT assay kit (Active Motif). The recombinant wild-type EP300 HAT domain (Active Motif) was included as a positive control.
In transformed Ba/f3 cells, ZNF384 fusion gene expression and histone H3 acetylation were evaluated by Western blot, using anti-acetyl-histone H3 antibody (Millipore, 06-599, 1:1000 dilution), anti-histone H3 antibody (Cell Signaling, 4499S, 1:1,000 dilution), anti-histone H3 (acetyl K9) antibody (Abcam, ab4441, 1:1,000 dilution), and anti-histone H3 (acetyl K27) antibody (Abcam, ab4729, 1:1,000 dilution). Histone H3 was used as a loading control. The relative quantification of immunoblots were analyzed using Image Studio lite version 4.0.21 (LI-COR Biosciences). Following the same procedures, six human ALL cell lines (JIH-5, Nalm6, SEM, 697, SUP-B15, and REH) representative of the major molecular subtypes were also assayed for histone acetylation by Western blotting. ALL cell lines were obtained from the American Type Culture Collection or DSMZ unless otherwise indicated.
Vorinostat cytotoxicity
NRASG12D-transformed Ba/f3 cells overexpressing EP300-ZNF384 or CREBBP-ZNF384 and the six human ALL cells described above were incubated with increasing concentrations of vorinostat for 48 h, after which cell viability was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Experiments were performed in triplicate and repeated at least three times, and results were plotted using Graph Prism 6.
Statistical analysis
All statistical analyses were performed using Graphpad Prism and/or R (R Core Team 2015); all tests were two-sided, and the level of statistical significance was P < 0.05.
Data access
Sequencing data have been submitted to the European Genome-Phenome Archive (EGA; http://www.ebi.ac.uk/ega), which is hosted by the European Bioinformatics Institute (EBI), under accession number EGAS00001001858.
Supplementary Material
Acknowledgements
We thank the patients and parents who participated in the clinical trials included in this study and Dr. Yongjin Li at St. Jude Children's Research Hospital for his advice on CICERO algorithm. This work was supported by the National Institutes of Health (CA021765), the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital, and the National Medical Research Council in Singapore (NMRC/CSA/0053/2013). T.M. is supported by the Mie Prefecture Study-Abroad Scholarship in Japan. C.G. is supported by National Cancer Institute Diversity Supplement (CA176063-S1). S.L. is supported by China Scholarship Council and National Natural Science Foundation of China (81300434). S.H. is supported by Jiangsu Province Key Project (BL2013014). S.C. is supported by the National Science Foundation of China (81600114). H.Z. is a St. Baldrick's International Scholar (318318) and is supported by the National Science Foundation of China (81300401).
Author contributions: J.J.Y. and A.E.J.Y. are principal investigators of this study and take responsibility for the integrity of the data and the accuracy of the data analysis. M.Q. analyzed the genomic sequencing data; H.Z., S.L., C.J., X.Z., Y.L., C.G., TN.L., R.Z., T.M., and Z.Y. performed the experiments; J.J.Y., M.Q., and H.Z. wrote the manuscript; Z.L., T.C.Q., H.A., A.M.T., S.S., D.B., S.H., S.C., HY.Z., and C-H.P. provided advice on study design and participated in data collection; and J.J.Y., M.Q., and H.Z. interpreted the data and the research findings. All authors reviewed the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.209163.116.
References
- Abraham BJ, Cui K, Tang Q, Zhao K. 2013. Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics 14: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi F, Magnaghi-Jaulin L, Girault JA, Robin P, Knibiehler M, Pritchard LL, Ducommun B, et al. 1998. Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature 396: 184–186. [DOI] [PubMed] [Google Scholar]
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. 2011. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469: 356–361. [DOI] [PubMed] [Google Scholar]
- Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A, et al. 2013. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet 9: e1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM, Lahdesmaki H, Rao A. 2015. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1–BAP1 complex. Nature Commun 6: 7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman CM, Alpar D, Ford AM, Colman SM, Wren D, Morgan M, Kearney L, Greaves M. 2015. Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia 29: 58–65. [DOI] [PubMed] [Google Scholar]
- Blobel GA. 2000. CREB–binding protein and p300: molecular integrators of hematopoietic transcription. Blood 95: 745–755. [PubMed] [Google Scholar]
- Chalmers JJ, Kim E, Telford JN, Wong EY, Tacon WC, Shuler ML, Wilson DB. 1990. Effects of temperature on Escherichia coli overproducing β-lactamase or human epidermal growth factor. Appl Environ Microbiol 56: 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabe S, Guay-Giroux A, Tormo AJ, Duluc D, Lissilaa R, Guilhot F, Mavoungou-Bigouagou U, Lefouili F, Cognet I, Ferlin W, et al. 2009. The IL-27 p28 subunit binds cytokine-like factor 1 to form a cytokine regulating NK and T cell activities requiring IL-6R for signaling. J Immunol 183: 7692–7702. [DOI] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka Y, Hou DX, Yasukawa T, Kanei-Ishii C, Takahashi T, Ishii S. 1996. CBP as a transcriptional coactivator of c-Myb. Genes Dev 10: 528–540. [DOI] [PubMed] [Google Scholar]
- Delvecchio M, Gaucher J, Aguilar-Gurrieri C, Ortega E, Panne D. 2013. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat Struct Mol Biol 20: 1040–1046. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feister HA, Torrungruang K, Thunyakitpisal P, Parker GE, Rhodes SJ, Bidwell JP. 2000. NP/NMP4 transcription factors have distinct osteoblast nuclear matrix subdomains. J Cell Biochem 79: 506–517. [PubMed] [Google Scholar]
- Gao C, Zhao XX, Li WJ, Cui L, Zhao W, Liu SG, Yue ZX, Jiao Y, Wu MY, Li ZG. 2012. Clinical features, early treatment responses, and outcomes of pediatric acute lymphoblastic leukemia in China with or without specific fusion transcripts: a single institutional study of 1,004 patients. Am J Hematol 87: 1022–1027. [DOI] [PubMed] [Google Scholar]
- Gocho Y, Kiyokawa N, Ichikawa H, Nakabayashi K, Osumi T, Ishibashi T, Ueno H, Terada K, Oboki K, Sakamoto H, et al. 2015. A novel recurrent EP300–ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia 29: 2445–2448. [DOI] [PubMed] [Google Scholar]
- Greaves MF. 1997. Aetiology of acute leukaemia. Lancet 349: 344–349. [DOI] [PubMed] [Google Scholar]
- Greaves MF, Wiemels J. 2003. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 3: 639–649. [DOI] [PubMed] [Google Scholar]
- Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, et al. 2008. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science 319: 336–339. [DOI] [PubMed] [Google Scholar]
- Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. 2014. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun 5: 3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunger SP, Mullighan CG. 2015. Acute lymphoblastic leukemia in children. N Engl J Med 373: 1541–1552. [DOI] [PubMed] [Google Scholar]
- Iacobucci I, Li Y, Roberts KG, Dobson SM, Kim JC, Payne-Turner D, Harvey RC, Valentine M, McCastlain K, Easton J, et al. 2016. Truncating erythropoietin receptor rearrangements in acute lymphoblastic leukemia. Cancer Cell 29: 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Greaves M, Mullighan CG. 2013. Acute lymphoblastic leukaemia. Lancet 381: 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, Caldas C. 2004. p300/CBP and cancer. Oncogene 23: 4225–4231. [DOI] [PubMed] [Google Scholar]
- Janssen H, Marynen P. 2006. Interaction partners for human ZNF384/CIZ/NMP4—zyxin as a mediator for p130CAS signaling? Exp Cell Res 312: 1194–1204. [DOI] [PubMed] [Google Scholar]
- Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. 2010. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467: 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S, Kurupati RK, Doyle SA, Freeman GJ, Schmader KE, Ertl HC. 2015. BTLA expression declines on B cells of the aged and is associated with low responsiveness to the trivalent influenza vaccine. Oncotarget 6: 19445–19455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Salzberg SL. 2011. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 12: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrel EA, Lemieux ME, Xia X, Davis TN, Rebel VI, Kung AL. 2009. Systematic in vivo structure-function analysis of p300 in hematopoiesis. Blood 114: 4804–4812. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Starza R, Aventin A, Crescenzi B, Gorello P, Specchia G, Cuneo A, Angioni A, Bilhou-Nabera C, Boque C, Foa R, et al. 2005. CIZ gene rearrangements in acute leukemia: report of a diagnostic FISH assay and clinical features of nine patients. Leukemia 19: 1696–1699. [DOI] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. 2014. Immunogenetics. Chromatin state dynamics during blood formation. Science 345: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. 2008. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451: 846–850. [DOI] [PubMed] [Google Scholar]
- Llinas L, Lazaro A, de Salort J, Matesanz-Isabel J, Sintes J, Engel P. 2011. Expression profiles of novel cell surface molecules on B-cell subsets and plasma cells as analyzed by flow cytometry. Immunol Lett 134: 113–121. [DOI] [PubMed] [Google Scholar]
- Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, Song G, Easton J, Harvey RC, Wheeler DA, et al. 2015. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE, Silverman LB, et al. 2014. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun 5: 3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini A, La Starza R, Janssen H, Bilhou-Nabera C, Corveleyn A, Somers R, Aventin A, Foa R, Hagemeijer A, Mecucci C, et al. 2002. Recurrent rearrangement of the Ewing's sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res 62: 5408–5412. [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinobu M, Nakamoto T, Hino K, Tsuji K, Shen ZJ, Nakashima K, Nifuji A, Yamamoto H, Hirai H, Noda M. 2005. The nucleocytoplasmic shuttling protein CIZ reduces adult bone mass by inhibiting bone morphogenetic protein–induced bone formation. J Exp Med 201: 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. 2007. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446: 758–764. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. 2008. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322: 1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, et al. 2011. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471: 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtagh F, Legendre P. 2014. Ward's hierarchical agglomerative clustering method: Which algorithms implement Ward's criterion? J Classif 31: 274–295. [Google Scholar]
- Nakamoto T, Shiratsuchi A, Oda H, Inoue K, Matsumura T, Ichikawa M, Saito T, Seo S, Maki K, Asai T, et al. 2004. Impaired spermatogenesis and male fertility defects in CIZ/Nmp4-disrupted mice. Genes Cells 9: 575–589. [DOI] [PubMed] [Google Scholar]
- Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, et al. 2010. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 42: 665–667. [DOI] [PubMed] [Google Scholar]
- Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, et al. 2011. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144: 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyquist KB, Thorsen J, Zeller B, Haaland A, Troen G, Heim S, Micci F. 2011. Identification of the TAF15–ZNF384 fusion gene in two new cases of acute lymphoblastic leukemia with a t(12;17)(p13;q12). Cancer Genet 204: 147–152. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. 2011. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471: 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping N, Qiu H, Wang Q, Dai H, Ruan C, Ehrentraut S, Drexler HG, MacLeod RA, Chen S. 2015. Establishment and genetic characterization of a novel mixed-phenotype acute leukemia cell line with EP300-ZNF384 fusion. J Hematol Oncol 8: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, Vora A, Baruchel A, Silverman LB, Schmiegelow K, et al. 2015. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol 33: 2938–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian M, Jin W, Zhu X, Jia X, Yang X, Du Y, Wang K, Zhang J. 2013. Structurally differentiated cis-elements that interact with PU.1 are functionally distinguishable in acute promyelocytic leukemia. J Hematol Oncol 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. 1997. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 91: 741–752. [DOI] [PubMed] [Google Scholar]
- Roberts KG, Mullighan CG. 2015. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol 12: 344–357. [DOI] [PubMed] [Google Scholar]
- Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, et al. 2014. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison LL. 2011. Late effects of acute lymphoblastic leukemia therapy in patients diagnosed at 0–20 years of age. Hematology 2011: 238–242. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41: D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley JD. 2013. Genetics. A story of swapped ends. Science 340: 1412–1413. [DOI] [PubMed] [Google Scholar]
- Savin VJ, Sharma M, Zhou J, Gennochi D, Fields T, Sharma R, McCarthy ET, Srivastava T, Domen J, Tormo A, et al. 2015. Renal and hematological effects of CLCF-1, a B-cell-stimulating cytokine of the IL-6 family. J Immunol Res 2015: 714964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, Hock H. 2009. TEL-AML1 corrupts hematopoietic stem cells to persist in the bone marrow and initiate leukemia. Cell Stem Cell 5: 43–53. [DOI] [PubMed] [Google Scholar]
- Senaldi G, Stolina M, Guo J, Faggioni R, McCabe S, Kaufman SA, Van G, Xu W, Fletcher FA, Boone T, et al. 2002. Regulatory effects of novel neurotrophin-1/B cell-stimulating factor-3 (cardiotrophin-like cytokine) on B cell function. J Immunol 168: 5690–5698. [DOI] [PubMed] [Google Scholar]
- Shigeno K, Yoshida H, Pan L, Luo JM, Fujisawa S, Naito K, Nakamura S, Shinjo K, Takeshita A, Ohno R, et al. 2004. Disease-related potential of mutations in transcriptional cofactors CREB-binding protein and p300 in leukemias. Cancer Lett 213: 11–20. [DOI] [PubMed] [Google Scholar]
- Sims NA. 2015. Cardiotrophin-like cytokine factor 1 (CLCF1) and neuropoietin (NP) signalling and their roles in development, adulthood, cancer and degenerative disorders. Cytokine Growth Factor Rev 26: 517–522. [DOI] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. 2013. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 154: 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart J, Ho TC, Amin E, Xu H, Barlowe TS, Perez AR, Durham BH, Tivnan P, Okabe R, Chow A, et al. 2016. MSI2 is required for maintaining activated myelodysplastic syndrome stem cells. Nat Commun 7: 10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki S, Seto M. 2013. TEL (ETV6)-AML1 (RUNX1) initiates self-renewing fetal pro-B cells in association with a transcriptional program shared with embryonic stem cells in mice. Stem Cells 31: 236–247. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendel AC, Calemine-Fenaux J, Izrael-Tomasevic A, Chauhan V, Arnott D, Eaton DL. 2009. B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol 182: 1509–1517. [DOI] [PubMed] [Google Scholar]
- Vo N, Goodman RH. 2001. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem 276: 13505–13508. [DOI] [PubMed] [Google Scholar]
- Wiemels J. 2008. Chromosomal translocations in childhood leukemia: natural history, mechanisms, and epidemiology. J Natl Cancer Inst Monogr 2008(39): 87–90. [DOI] [PubMed] [Google Scholar]
- Xu H, Zhang H, Yang W, Yadav R, Morrison AC, Qian M, Devidas M, Liu Y, Perez-Andreu V, Zhao X, et al. 2015. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat Commun 6: 7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ. 2004. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 32: 959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, Wang J, Morrison D, Devidas M, Hunger SP, et al. 2008. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood 112: 4178–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, Imoto N, Kohsaka S, Kunita A, Doi K, et al. 2016. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet 48: 569–574. [DOI] [PubMed] [Google Scholar]
- Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M, Levantini E, Welner RS, Bach CS, Zhang J, et al. 2015. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 17: 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh AE, Ariffin H, Chai EL, Kwok CS, Chan YH, Ponnudurai K, Campana D, Tan PL, Chan MY, Kham SK, et al. 2012. Minimal residual disease–guided treatment deintensification for children with acute lymphoblastic leukemia: results from the Malaysia-Singapore acute lymphoblastic leukemia 2003 study. J Clin Oncol 30: 2384–2392. [DOI] [PubMed] [Google Scholar]
- Zhong CH, Prima V, Liang X, Frye C, McGavran L, Meltesen L, Wei Q, Boomer T, Varella-Garcia M, Gump J, et al. 2008. E2A-ZNF384 and NOL1-E2A fusion created by a cryptic t(12;19)(p13.3; p13.3) in acute leukemia. Leukemia 22: 723–729. [DOI] [PubMed] [Google Scholar]
- Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, Yan X, Wei W, Pang Y, Cheng H, et al. 2014. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet 46: 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.