

Abstract
The molecular mechanism by which the homodimeric enzyme Cu/Zn superoxide dismutase (SOD) causes neural damage in amytrophic lateral sclerosis is yet poorly understood. A striking, as well as an unusual, feature of SOD is that it maintains intrasubunit disulfide bonds in the reducing environment of the cytosol. Here, we investigate the role of these disulfide bonds in folding and assembly of the SOD apo protein (apoSOD) homodimer through extensive protein engineering. The results show that apoSOD folds in a simple three-state process by means of two kinetic barriers: 2D⇌2M⇌M2. The early predominant barrier represents folding of the monomers (M), and the late barrier the assembly of the dimer (M2). Unique for this mechanism is a dependence of protein concentration on the unfolding rate constant under physiological conditions, which disappears above 6 M Urea where the transition state for unfolding shifts to first-order dissociation of the dimer in accordance with Hammond-postulate behavior. Although reduction of the intrasubunit disulfide bond C57–C146 is not critical for folding of the apoSOD monomer, it has a pronounced effect on its stability and abolishes subsequent dimerization. Thus, impaired ability to form, or retain, the C57–C146 bond in vivo is predicted to increase the cellular load of marginally stable apoSOD monomers, which may have implications for the amytrophic lateral sclerosis neuropathology.
Keywords: protein folding, protein stability, disulfide bond, transition-state shifts, protein engineering
The mechanism by which mutant superoxide dismutase (SOD) leads to neural damage in the familial form of amyotrophic lateral sclerosis (ALS) is yet unknown (1–3). In analogy with other neurodegenerative disorders (4), however, an increasing body of observations (5–16) suggest that the ALS disease mechanism is coupled to destabilization or misfolding of the SOD structure (17), manifested ultimately by cellular inclusions of SOD aggregates (3, 18). From a strictly energetic perspective, the native SOD structure is distinctive by containing one oxidized disulfide bond per monomer (C57–C146) in the reducing environment of the cytosol (19) (Fig. 1). Normally, disulfide bonds are maintained only under oxidizing conditions in the extracellular space (20, 21) where they increase protein stability by confining the configurational entropy of the denatured ensemble (22). Indications that the integrity of the C57–C146 bond may have bearing on the molecular events in ALS were recently provided by Tiwari and Hayward (15), who demonstrated that ALS-associated SOD mutants are more susceptible to chemical disulfide reduction than the wild-type protein. More detailed elucidation of the structural and energetic effects accompanying mutational perturbations and disulfide reduction has so far been prevented by scarce knowledge about how the SOD homodimer folds (23).
Fig. 1.

Structural comparison of the SOD homodimer and monomer. (Left) Structure of the human SOD homodimer (1SPD), showing the positions of the cysteines, the loop containing residues 49–84, and the Cu and Zn ions. (Right) Loop displacements upon dissociation of the SOD homodimer (illustrated on the right-hand-side monomer), obtained by structural superposition of holoSODwt (blue) (51) and holoSOD50/51/E133Q (orange) (41).
In this study, we shed further light on this issue by mapping out the folding and assembly reaction of metal-depleted SOD through kinetic and thermodynamic analysis of mutant protein, with particular focus on the role of the intrasubunit disulfide bond C57–C146. After elimination of artifactual cross-linking by substitution of the solvent-accessible cysteines 6 and 111, the oxidized SOD apoenzyme (apoSOD) homodimer reveals a conspicuous chevron plot indicative of a three-state process in which the rate-limiting step shifts from the first-order nucleation of the monomers at low-urea concentrations, to the transition state for monomer dissociation at high-urea concentrations. A benefit of this transition-state change is that information about monomer folding and dimer formation can be derived from kinetic data alone. The results show that the C57–C146 bond only marginally affects the folding process of the monomeric protein, but is crucial for its stability and dimerization in the absence of Cu and Zn.
Materials
Proteins. The single and multiple SOD mutations C6A, C111A, C6A/C111A, and C6A/C111A/C57A/C146A and F50E/G51E (monomeric) variants thereof were constructed as described in ref. 12. Samples of apoSOD (molecular mass of 32 kDa for dimer) were prepared in two ways: (i) on a chelate column under mildly acidic conditions (pH 3.5) according to the procedures in ref. 24 and dialyzed against 10 mM EDTA at pH 7; and (ii) by unfolding in 4 M guanidinium chloride in the presence of 50 mM EDTA and then refolding by dialysis against 10 mM Mes (pH 6.3) with 10 mM EDTA (standard buffer). Reducing conditions were obtained by the addition of 2 mM Tris-(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) in the unfolding buffer and 0.5 mM TCEP·HCl in all other steps.
Measurements. Equilibrium titrations were done on a J-810 CD spectrometer (Jasco, Easton, MD) or a π*-180 instrument (Applied Photophysics, Surrey, U.K.), and the kinetic measurements were carried out on an SX-17MV stopped-flow fluorimeter (Applied Photophysics) at 25°C. Protein concentration was 4 μM or 70 μM monomer.
Data Analysis. In the kinetic analysis, monomeric apoSOD was assumed to display a minimal two-state transition between the denatured state (D) and the fully folded monomer (M), KD–M = [D]/[M] = ku/kf:
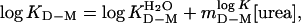 |
[1] |
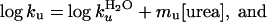 |
[2] |
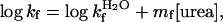 |
[3] |
where the slopes 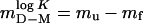 are commonly taken as a measure of solvent exposure in the equilibrium unfolding process and the activation process of unfolding and refolding, respectively (25). The parameters were derived from equilibrium data by using the equation
are commonly taken as a measure of solvent exposure in the equilibrium unfolding process and the activation process of unfolding and refolding, respectively (25). The parameters were derived from equilibrium data by using the equation
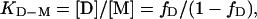 |
[4] |
where fD is the fraction of D, and protein stability was derived as 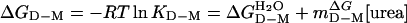 . The plots of log kf and log ku vs. [urea] were fitted by
. The plots of log kf and log ku vs. [urea] were fitted by
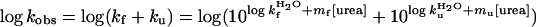 |
[5] |
and the position of the transition-state ensemble was obtained from
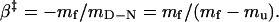 |
[6] |
The φ-values were calculated from wild-type and mutant data by the standard relation (25)
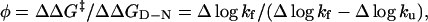 |
[7] |
where ΔΔG‡ and ΔΔGD–N are the mutant-induced destabilization of the transition-state ensemble and the folded structure, respectively. A φ-value of 1 suggests that the structure around the mutated residue is native-like in the transition state ensemble, whereas a φ-value of 0 suggests that it is unfolded. Fractional φ-values indicate that the targeted region is partly structured in the transition state ensemble.
The stability of dimeric apoSOD, ΔG2D–M2 =–RT ln K2D–M2, was derived from equilibrium denaturation data according to
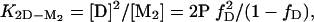 |
[8] |
assuming linear free-energy dependence on [urea]. Data analysis was done by using the kaleidagraph software (Synergy Software, Reading, PA).
Results
Disulfide Reduction Leads to Dissociation of the apoSOD Dimer. The dimer integrity of oxidized and reduced SOD was analyzed by gel chromatography on a Sephadex S-75 column (Pharmacia). To minimize the effects of local charge differences between the holo- and apo-species, 150 mM NaCl was added to the standard buffers. The concentration of the eluted protein was ≈20 μM monomer. Oxidized apoSODwt elutes at 10.5 ml, corresponding to the dimeric species (Fig. 2a). This peak is well separated from that of the monomeric double mutant F50E/G51E (apoSOD50/51) at 12 ml (Fig. 2a). Upon reduction of the cysteines, apoSODwt elutes mainly as a folded monomer with only a small residual of dimers. For comparison, holoSODwt remains dimeric in both its oxidized and disulfide-reduced state (data not shown). The results suggest that assembly of the SOD homodimer requires either the oxidized disulfide linkage between Cys-57 and Cys-146, or coordinated metal ions; the absence of both promotes dissociation. Consistently, the fully cystein-depleted mutant C6A/C111A/C57A/C146A (SODCallA) elutes as a folded monomer in its apo state and as a dimer with coordinated metals, although the dimeric state of this radical construct travels somewhat slower than the wild-type ditto (Fig. 2b). The apo state of the double mutant C6A/C111A (apoSOD6/111), in which only the solvent-accessible cysteines have been replaced, monomerizes upon disulfide reduction in a manner identical to that of the wild-type protein (Fig. 2c).
Fig. 2.

Size-exclusion chromatograms showing the dimer/monomer composition of wild-type and mutant SOD with and without the C57–C146 disulfide linkage. Dimers elute at 10–11 ml and folded monomers at 12 ml. (a) Oxidized apoSODwt (black), reduced apoSODwt (red), and oxidized apoSOD50/51 (blue). (b) Oxidized apoSODwt (black), fully cysteine-depleted apoSOD (red), and fully cysteine-depleted holoSOD (blue). (c) Oxidized apoSOD6/111 (black) and reduced apoSOD6/111 (red). AU, absorbance unit.
Monomeric SOD Shows Classical Two-State Folding Behavior. Equilibrium denaturation of the reduced apoSODwt monomer by urea reveals a sigmoidal unfolding transition consistent with a two-state process (see Scheme 1), where D is the denatured ensemble and M is the folded monomer (data not shown). Fitting of Eq. 4 yields a midpoint of [urea] = 1.51 M and mD–M = 1.42 M–1, corresponding to log 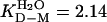 or a stability of
or a stability of 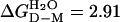 kcal/mol in the absence of urea (Eq. 1 and Table 1). Compared with other single-domain proteins, this stability is notably low (Fig. 4). Additional evidence for two-state folding is provided by the kinetics. The chevron plot of apoSODwt, i.e., a plot of log kf and log ku vs. urea, is characteristically v-shaped with slopes mf =–1.05 M–1and mu = 0.48 M–1 (Eqs. 2, 3, and 5) that sum up to
kcal/mol in the absence of urea (Eq. 1 and Table 1). Compared with other single-domain proteins, this stability is notably low (Fig. 4). Additional evidence for two-state folding is provided by the kinetics. The chevron plot of apoSODwt, i.e., a plot of log kf and log ku vs. urea, is characteristically v-shaped with slopes mf =–1.05 M–1and mu = 0.48 M–1 (Eqs. 2, 3, and 5) that sum up to 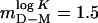 M–1 from equilibrium data (Fig. 3 and Table 1). Moreover, the sum of log
M–1 from equilibrium data (Fig. 3 and Table 1). Moreover, the sum of log 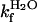 and log
and log 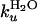 matches log
matches log 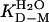 , yielding the relation KD–M = ku/kf. By these criteria, the monomeric state of apoSODwt classifies as a “classical” two-state folder (26). That is, monomeric apoSODwt describes a highly concerted transition over a pointed free-energy barrier, in accord with the majority of other single-domain proteins (27). The position of the transition-state ensemble on this barrier profile is β‡ = 0.69 (Eq. 6), in good correspondence with the values observed for other two-state folders (27). The reduced state of the monomeric variant apoSOD50/51 displays overall an identical chevron plot but with a slightly decreased value of log
, yielding the relation KD–M = ku/kf. By these criteria, the monomeric state of apoSODwt classifies as a “classical” two-state folder (26). That is, monomeric apoSODwt describes a highly concerted transition over a pointed free-energy barrier, in accord with the majority of other single-domain proteins (27). The position of the transition-state ensemble on this barrier profile is β‡ = 0.69 (Eq. 6), in good correspondence with the values observed for other two-state folders (27). The reduced state of the monomeric variant apoSOD50/51 displays overall an identical chevron plot but with a slightly decreased value of log 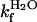 (Fig. 3 and Table 1). Although this small decrease of the refolding rate constant (
(Fig. 3 and Table 1). Although this small decrease of the refolding rate constant (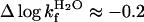 ) is a notable and consistent feature of the F50E/G51E mutation, it is nevertheless too small to permit structural interpretation by φ-value analysis (28).
) is a notable and consistent feature of the F50E/G51E mutation, it is nevertheless too small to permit structural interpretation by φ-value analysis (28).
Scheme 1.
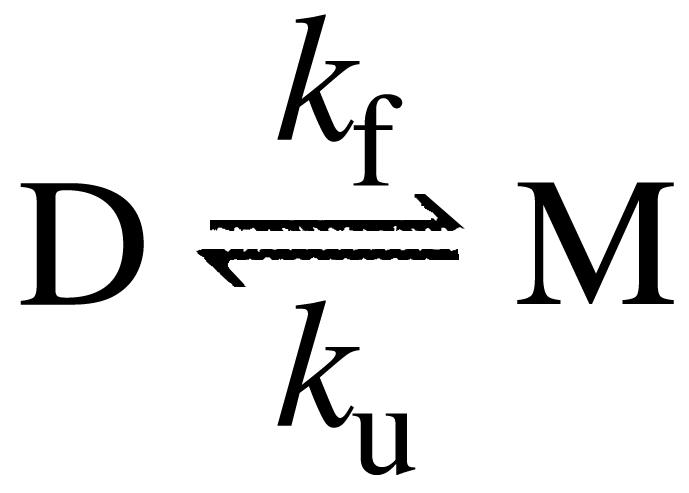
Table 1. Kinetic and thermodynamic parameters for wild-type and mutant apoSOD.
| apoSODwt reduced | apoSODwt oxidized* | apoSOD50/51 reduced | apoSODC6A reduced | apoSODC111A reduced | apoSOD6/111 reduced | apoSOD6/57/111/146 reduced | apoSOD50/51/6/111 reduced† | apoSOD50/51/6/11 oxidized | |
|---|---|---|---|---|---|---|---|---|---|
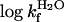 |
-1.00 ± 0.02 | -0.51 ± 0.05 | -1.22 ± 0.02 | -1.35 ± 0.03 | -1.14 ± 0.02 | -1.64 ± 0.04 | -1.73 ± 0.03 | -1.85 ± 0.03 | -1.36 ± 0.04 |
| mf (M-1) | -1.05 ± 0.03 | — | -1.04 ± 0.02 | -1.22 ± 0.05 | -1.18 ± 0.05 | -1.25 ± 0.11 | -1.29 ± 0.09 | -1.15 ± 0.1 | -0.95 ± 0.05 |
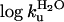 |
-3.15 ± 0.03 | -3.81 ± 0.08 | -3.16 ± 0.02 | -3.15 ± 0.03 | -2.71 ± 0.02 | -2.80 ± 0.03 | -2.90 ± 0.02 | -2.93 ± 0.03 | -3.59 ± 0.07 |
| mu (M-1) | 0.48 ± 0.01 | — | 0.48 ± 0.01 | 0.44 ± 0.01 | 0.47 ± 0.01 | 0.45 ± 0.01 | 0.44 ± 0.01 | 0.45 ± 0.01 | 0.46 ± 0.02 |
| mu — mf (M-1) | 1.53 ± 0.03 | — | 1.52 ± 0.02 | 1.66 ± 0.05 | 1.65 ± 0.05 | 1.70 ± 0.11 | 1.73 ± 0.09 | 1.67 ± 0.10 | 1.41 ± 0.05 |
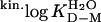 |
-2.15 ± 0.04 | -3.30 ± 0.1 | -1.94 ± 0.02 | -1.80 ± 0.04 | -1.57 ± 0.03 | -1.16 ± 0.05 | -1.17 ± 0.04 | -1.08 ± 0.04 | -2.23 ± 0.08 |
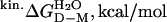 |
2.92 ± 0.05 | 4.48 ± 0.13 | 2.64 ± 0.03 | 2.45 ± 0.05 | 2.14 ± 0.04 | 1.58 ± 0.07 | 1.59 ± 0.05 | 1.47 ± 0.05 | 3.03 ± 0.11 |
| eq·MP,§ M | 1.51 ± 0.02 | — | 1.30 ± 0.02 | ‡ | ‡ | ‡ | ‡ | ‡ | 1.92 ± 0.03 |
 ,§ kcal/mol·M ,§ kcal/mol·M |
-1.93 ± 0.19 | — | -1.93 ± 0.19 | ‡ | ‡ | ‡ | ‡ | ‡ | -1.93 ± 0.19 |
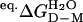 ,§ kcal/mol ,§ kcal/mol |
2.91 ± 0.30 | — | 2.52 ± 0.25 | ‡ | ‡ | ‡ | ‡ | ‡ | 3.71 ± 0.37 |
Superscript “kin.” and “eq.” refer to parameters derived from kinetic and equilibrium data, respectively (Eqs. 1-5).
Estimated from reduced apoSODwt and the differences between reduced and oxidized apoSOD50/51/6/111.
mf locked to 〈mf〉 in Eq. 5 due to low midpoint.
Midpoints too low for determination of native base line.
From global fit with shared 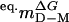 .
.
Fig. 4.

Stability histogram for 27 single-domain proteins adapted from ref. 27. The apparent stabilities of the reduced apoSODwt monomer and the native holoSODwt dimer are found on the extreme sides of the distribution.
Fig. 3.

Chevron plots of wild-type and mutant SOD monomers display simple two-state folding behavior. Units are in s–1. (a) Reduced apoSODwt (•) and reduced apoSOD50/51 (○). (b) Reduced apoSODwt (•), reduced apoSODC6A (▵), reduced apoSODC111A (▴), and reduced apoSOD6/111 (▾). (c) Reduced apoSOD50/51/6/111 (□) and oxidized apoSOD50/51/6/111 (▪).
The Role of Cys-6 and Cys-111 in Folding and Stability. Because SODwt tends to aggregate under conditions promoting disulfide bond formation, we have used the much more controllable variant apoSOD6/111, lacking free cysteine residues, as pseudo wild-type in the kinetic analysis of the oxidized protein (29). The procedure is validated as follows. Neither of the single mutations C6A or C111A nor the double mutant C6A/C111A shows any appreciable change of the folding m-values, indicating that their influence on the folding trajectory is negligible (25) (Fig. 3 and Table 1). With respect to the energetics, reduced C6A affects mainly the refolding limb of the chevron plot. Truncation of the thiol group destabilizes the transition-state ensemble (ΔΔG‡ = 2.3RTΔlog kf = 0.47 kcal/mol) and the folded monomer (ΔΔGD–N = 0.47 kcal/mol) to a similar extent, yielding a φ-value of 1.0 (Eq. 7). Thus, the structure around this position seems to form early in folding and constitutes part of the critical nucleus in the transition-state ensemble (25). The small decrease of ku accompanying reduced C6A is rarely seen for truncations of methylene groups but could, in this case, stem from the desolvation penalty of the thiol moiety that is larger in the folded protein than in the more expanded transition-state ensemble. In contrast, the mutation C111A affects mainly the unfolding kinetics, indicative of a more selective destabilization of the folded monomer (Fig. 3 and Table 1). The values of ΔΔG‡ = 0.19 kcal/mol, ΔΔGD–N = 0.8 kcal/mol, and φ = 0.24 suggest that the structural environment of C111 is largely unfolded in the transition state. Finally, the result of reduced apoSOD6/111 is simply the sum of the two single mutants, providing further (site-specific) evidence that the substitutions C6A and C111A do not significantly influence the folding trajectory of the apoSOD monomer (Fig. 3 and Table 1). As an additional control, the fully cysteine-depleted quadruple mutant mimics precisely the behavior of reduced apoSOD6/111 (Table 1).
The Intrasubunit Disulfide Linkage Has Minor Influence on Folding but Is Important for Monomer Stability. To investigate the role of the C57–C146 disulfide bond in the early folding events, we used apoSOD50/51/6/111, which is the only construct in this study that allows selective detection of monomer unfolding and stability under oxidizing conditions. From the chevron plots in Fig. 3, it is apparent that the main contribution of the C57–C146 disulfide bond is to promote monomer stability by decreasing the unfolding rate constant (ΔΔGD–N = 1.56 kcal/mol, Table 1 and Fig. 3). The corresponding effect on the transition-state ensemble is notably smaller at ΔΔG‡ = 0.67 kcal/mol, yielding an apparent φ-value of 0.43 (Table 2). At a molecular level, the interpretation of this φ-value is somewhat different from that of side-chain truncations (22). In contrast to the enthalpic contribution of side-chain contacts, disulfide linkages stabilize folded structures by reducing the configurational entropy of disordered states. The fractional effect of the C57–C146 disulfide bond on the apoSOD transition-state ensemble, suggests then that the part of the polypeptide chain that is restricted by this linkage is largely disordered in the rate-limiting step for folding. That is, the contacts anchoring loop around position 57 are not required for the early folding events and consolidate mainly on the downhill side the folding barrier. The result is not surprising because it is expected that the correct topological context needs to be firmly established before disulfide-bond formation in the reducing environment of the cytosol.
Table 2. Data for the apoSOD6/111 dimer.
| apoSOD6/111 4 μM oxidized | apoSOD6/111 70 μM oxidized | apoSODWT 4 μM oxidized* | |
|---|---|---|---|
| MP†, M | 2.72 ± 0.03 | 3.06 ± 0.02 | 3.55 ± 0.04 |
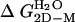 ,† kcal/mol ,† kcal/mol |
-15.80 ± 0.57 | -15.6 ± 0.47 | -19.20 ± 0.54 |
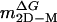 ,† kcal/mol·M ,† kcal/mol·M |
3.11 ± 0.25 | 3.26 ± 0.15 | — |
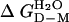 ,‡ kcal/mol ,‡ kcal/mol |
-3.32 ± 0.12 | -3.32 ± 0.12 | -4.49 ± 0.13 |
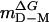 ,‡ kcal/mol·M ,‡ kcal/mol·M |
1.93 ± 0.08 | 1.93 ± 0.08 | — |
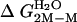 ,§ kcal/mol ,§ kcal/mol |
-12.48 ± 0.58 | -12.28 ± 0.48 | -14.71 ± 0.56 |
| m2M-M2§ | 1.18 ± 0.26 | 1.33 ± 0.17 | — |
| log kf¶ | -1.1 ± 0.07 | -1.1 ± 0.01 | — |
| mf¶ M-1 | -0.83 ± 0.06 | -0.83 ± 0.02 | — |
| log kd¶ | -2.79 ± 0.02 | -2.80 ± 0.03 | — |
| md,¶ M-1 | 0.09 ± 0.01 | 0.09 ± 0.01 | — |
| log ka∥ | 6.39 ± 0.58 | 6.23 ± 0.48 | — |
Estimated from apoSOD6/111 dimer and difference between reduced apoSOD50/51 and apoSOD50/51/6/111 (compare Table 1).
Derived from equilibrium data (Eq. 8).
Estimated from oxidized apoSOD50/51/6/111 and differences between apoSODwt and apoSOD50/51 (kinetic data).
Calculated from 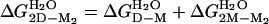 .
.
Derived from fits with global md in Fig. 5.
Calculated from 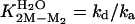 .
.
Folding of the apoSOD Homodimer. As expected, the apparent stability of the oxidized apoSOD6/111 homodimer increases at increased protein concentration. The transition midpoint shifts from 2.7 M urea at 4 μM apoSOD6/111 to 3.1 M urea at 70 μM (Table 2), and the hidden surface area of the dimer interface contributes to an apparent m-value that is ≈0.2 units larger than for the monomer. The values of 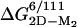 , as derived from equilibrium unfolding data at 4 and 70 μM protein (Eq. 8), are –15.8 ± 0.6 kcal/mol and –15.6 ± 0.5 kcal/mol, respectively, in good correspondence with earlier reports on the apo protein (30) (Table 2). The refolding kinetics of dimeric apoSOD6/111 resembles closely that of the oxidized monomer apoSOD50/51/6/111 (Fig. 5 and Table 1): the values of mf are within experimental errors, whereas the small offset in logkf of 0.26 matches that observed for the dimer-splitting substitutions F50E/G51E. Moreover, the refolding rate constant for dimeric apoSOD6/111 shows no dependence on protein concentration: the refolding limbs obtained at 4 μM and 70 μM apoSOD6/111 are indistinguishable (Fig. 5 and Table 1). On this basis, we conclude that refolding of the SOD homodimer is rate limited by the first-order formation of the monomer. We note, however, that an additional faster phase emerges at high protein concentrations, whose amplitude constitutes <20% of the total fluorescence change. This minor phase is not resolved with the monomeric protein, and its origin is yet unclear. One possibility is that it stems from transient aggregation of denatured or partly folded protein in the absence of the hydrophilic substitutions F50E/G51E (31). Another possibility is that a subfraction of the SOD molecules assemble before the global refolding transition, or form monomeric intermediates. The major difference between the monomeric and homodimeric protein is seen in the unfolding kinetics: the values of logku are substantially decreased, and the chevron plot shows a pronounced downward kink at around 5 M urea (Fig. 5). Similar kinks have been described for other proteins and are the hallmarks for changes of the rate-limiting step (32, 33); i.e., the transition state shifts closer to the native state along the experimental progress coordinate in accordance with the Hammond postulate (34). In the case of SOD, it is evident that the unfolding reaction proceeds over the transition state for monomer formation at low [urea], to become rate limited by the first-order dissociation of the dimer above 5 M urea (see Scheme 2). In direct support of this folding model, increased protein concentration affects solely the steeper region of the unfolding limb. A qualitative description of the effect is provided by the free-energy profiles in Fig. 5. Below 5 M urea, the unfolding time course is modulated by both the activation barrier for unfolding of M and the second-order equilibrium between M2 and M. Under these conditions, elevated protein concentrations slow down the unfolding process by shifting the equilibrium toward M2, i.e., the initial free-energy difference between M2 and M is increased. Above 5 M urea, the dependence on protein concentration vanishes because the rate-limiting step moves to become the first-order dissociation of M2.Tovisualizethe dimer folding in a single chevron plot, we have tentatively treated the unfolding kinetics as first-order at all concentrations of denaturant. The second-order component in the unfolding reaction below 5M urea does not cause any detectable deviation from exponential time courses. The [M]2/[M2] equilibrium constant at 0 M urea was estimated from the stability difference between dimeric and monomeric protein,
, as derived from equilibrium unfolding data at 4 and 70 μM protein (Eq. 8), are –15.8 ± 0.6 kcal/mol and –15.6 ± 0.5 kcal/mol, respectively, in good correspondence with earlier reports on the apo protein (30) (Table 2). The refolding kinetics of dimeric apoSOD6/111 resembles closely that of the oxidized monomer apoSOD50/51/6/111 (Fig. 5 and Table 1): the values of mf are within experimental errors, whereas the small offset in logkf of 0.26 matches that observed for the dimer-splitting substitutions F50E/G51E. Moreover, the refolding rate constant for dimeric apoSOD6/111 shows no dependence on protein concentration: the refolding limbs obtained at 4 μM and 70 μM apoSOD6/111 are indistinguishable (Fig. 5 and Table 1). On this basis, we conclude that refolding of the SOD homodimer is rate limited by the first-order formation of the monomer. We note, however, that an additional faster phase emerges at high protein concentrations, whose amplitude constitutes <20% of the total fluorescence change. This minor phase is not resolved with the monomeric protein, and its origin is yet unclear. One possibility is that it stems from transient aggregation of denatured or partly folded protein in the absence of the hydrophilic substitutions F50E/G51E (31). Another possibility is that a subfraction of the SOD molecules assemble before the global refolding transition, or form monomeric intermediates. The major difference between the monomeric and homodimeric protein is seen in the unfolding kinetics: the values of logku are substantially decreased, and the chevron plot shows a pronounced downward kink at around 5 M urea (Fig. 5). Similar kinks have been described for other proteins and are the hallmarks for changes of the rate-limiting step (32, 33); i.e., the transition state shifts closer to the native state along the experimental progress coordinate in accordance with the Hammond postulate (34). In the case of SOD, it is evident that the unfolding reaction proceeds over the transition state for monomer formation at low [urea], to become rate limited by the first-order dissociation of the dimer above 5 M urea (see Scheme 2). In direct support of this folding model, increased protein concentration affects solely the steeper region of the unfolding limb. A qualitative description of the effect is provided by the free-energy profiles in Fig. 5. Below 5 M urea, the unfolding time course is modulated by both the activation barrier for unfolding of M and the second-order equilibrium between M2 and M. Under these conditions, elevated protein concentrations slow down the unfolding process by shifting the equilibrium toward M2, i.e., the initial free-energy difference between M2 and M is increased. Above 5 M urea, the dependence on protein concentration vanishes because the rate-limiting step moves to become the first-order dissociation of M2.Tovisualizethe dimer folding in a single chevron plot, we have tentatively treated the unfolding kinetics as first-order at all concentrations of denaturant. The second-order component in the unfolding reaction below 5M urea does not cause any detectable deviation from exponential time courses. The [M]2/[M2] equilibrium constant at 0 M urea was estimated from the stability difference between dimeric and monomeric protein, 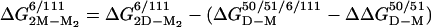 , where
, where 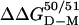 is the adjustment for the mutation F50E/G51E (Table 2). The value of
is the adjustment for the mutation F50E/G51E (Table 2). The value of 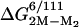 ≈ –12 kcal/mol yields a dissociation constant of 1.5 nM, which is in precise agreement with previous estimates (30).
≈ –12 kcal/mol yields a dissociation constant of 1.5 nM, which is in precise agreement with previous estimates (30).
Fig. 5.

Chevron plots for the apoSOD6/111 dimer at protein concentrations of 4 μM (blue) and 70 μM (red). For comparison, shown are the data for the monomer apoSOD50/51/6/111 (black). Units are in s–1. The fits are from a three-state model according to Scheme 2 (52). Below are the corresponding folding free-energy profiles at 0, 2.7, and 6.5 M urea, as estimated from transition-state theory with prefactor of 106 s–1 (53), and the apparent stabilities fD/(1 – fD) of M and M2 by using chevron and equilibrium data, respectively. At low [urea], the dimeric and monomeric proteins seem to fold over the same transition state (‡′), where the offset in logkf is due to the F50E/G51E mutation. Above the transition midpoint, logku decreases with increasing protein concentrations as the free-energy difference between ‡′ and M2 goes up. This concentration dependence vanishes at high [urea] where the rate-limiting step shifts to the first-order dissociation of M2→‡′′ according to Hammond-postulate behavior (33).
Scheme 2.

Discussion
Disulfide Reduction Promotes Dimer Dissociation. Compared with other homodimeric proteins, the ratio between interfacial and intramonomer contacts in SOD is relatively small (35). Characteristic for such proteins is that monomer folding precedes dimerization, producing interfaces that are overall less optimized than those of obligatory dimers where folding and association occur in one highly concerted step (35, 36). Consistently, the crystal structures of the ALS mutants A4V and I113T are reported to display marked distortions of the dimer interface although the structural effect on the individual monomers is small (37). Hough et al. (37) suggest that the decreased stability of the dimer interface expected to result from this distortion could be an integral part of the ALS disease mechanism. Along the same line, neurodegeneration in Parkinson's disease has been linked to an interface mutation in the homodimeric protein DJ-1 (38), and familial amyloid polyneuropathy has been linked to dissociation of the transthyretin tetramer into alternatively folded monomers (39). In this study, we reveal an additional aspect of the SOD homodimer by showing that its association equilibrium is also critically dependent on the intrasubunit disulfide bond C57–C146. Upon reduction of this disulfide bond, the apo protein completely fails to dimerize at physiological concentrations [see also the recent work by Furukawa et al. (40), arriving at the same conclusion]. That is, SOD needs to maintain the C57–C146 bond oxidized under the reducing pressure in the cytosol to ensure full dimer stability. This redox sensitivity is a most unusual property of intracellular proteins that could constitute an Achilles' heel of the SOD molecule. From this perspective, it is interesting to note that ALS-associated mutants exhibit an increased susceptibility to disulfide reduction (15), implicating a scenario where the effect of otherwise benign mutations is augmented by loss of the C57–C146 bond (15). Consistently, we have evidence for the susceptibility of mutant SOD proteins to thioredoxin and thioredoxin reductase, the major disulfide reduction system of the cell responsible for keeping the cytosol reduced (M.O., M.J.L., and A.H., unpublished results).
Loop Dynamics Can Modulate Interface Stability The structural connection between the dimer interface and the disulfide link is by means of the large loop between strands 4 and 5 (Fig. 1). The loop forms part of the interface (residues 50–54) and is anchored to the β barrel by the C57–C146 bond. It encompasses also the Cu and Zn ions through the coordinating residues H46, H48, H63, H71, H80, and D83 (41). The importance of this loop in modulating the docking of the two SOD monomers is clearly emphasized by superposition of the crystal structures of the dimeric and monomeric proteins: the loop displays a significant displacement on either side of the disulfide anchoring point in the monomer (41). It is thus plausible that cleavage of the C57–C146 bond favors the monomeric species entropically by simply increasing the loop dynamics. Coordination of Zn and Cu is then expected to have an opposing effect by constraining the loop, supported by the observation that the completely cysteine-depleted mutant elutes as a dimer in the holo state but monomerizes upon removal of the metals (Fig. 2).
The SOD Monomer Shows Low Stability and Slow Folding. The monomeric species produced by disulfide reduction of the apoSOD homodimer displays all of the kinetic and thermodynamic characteristics of fully structured subunits (Fig. 3). It folds cooperatively in a classical two-state reaction (27) that is overall indistinguishable from that of the structured monomer obtained by the interface mutations F50E/G51E (41). We note, however, that the stability and refolding rate constant of the reduced apoSODwt monomer are unusually low in relation to other single-domain proteins (Fig. 4). The equilibrium constant [D]/[M] at 0 M urea is 7/103 (Table 1) compared, e.g., with 1/103 to 1/107 for the structurally analogous Ig-like β-sandwich proteins (42). With respect to the dynamics, reduced apoSODwt unfolds on average every 23 min and then refolds in 10 s. The corresponding lifetimes for the Ig-like β-proteins are 1–7 min and 0.004–0.7 s, respectively (42). For the oxidized apoSODwt monomer, [D]/[M] decreases to 5/104, whereas the lifetimes of D and M change to 3 s and 108 min, respectively (Table 1). Accordingly, the refolding rate constant of the apoSOD monomer (0.1–0.3 s–1) is among the lowest observed for single-domain proteins, in good agreement with results from theory (23). It is also much lower than expected from its value of the topological parameter “relative contact order” (0.15) that empirically correlates with the refolding rate constants across two-state proteins (43).
φ-Value Analysis. The φ-value of 1.0 for the substitution C6A implies that the structural environment of the thiol moiety consolidates early in the folding process and is close to native-like in the transition-state ensemble. The interactions involved are those that close up strands 1 and 8 in the β-barrel. The result agrees well with observations from simulations where this region is identified as important for two-state folding kinetics (23). Correspondingly, the low φ-value of 0.24 for the C111A mutant indicates that the loop between strands 6 and 7 is largely unstructured in the transition state. However, these results need to be verified by additional, more conservative mutations because the replacement C to A involves the truncation of a buried group with polar character that could be suspicious in φ-value analysis due to mismatched desolvation terms (25). A more reliable probe for the folding process is the C57–C146 disulfide linkage that yields a φ-value of 0.43. This result suggests that the large loop containing residues 49–84 is mainly disordered around position 57 in the transition state and attains its native structure after the rate-limiting step for folding. That is, the folding process of the SOD monomer is only marginally affected by the disulfide link. This finding is not surprising because the SOD monomer most likely needs to efficiently adopt its correct topological context to allow disulfide bond formation and metal incorporation in the reducing environment of the cytosol. The first folding step of SOD in vivo is thus likely to be the slow, cooperative formation of the relatively unstable disulfide-reduced monomer. In support of this conclusion, it was recently reported (40) that disulfide formation in vivo takes place in connection with copper loading of the folded SOD monomer through interactions with the copper chaperone (CCS).
Folding of the SOD Homodimer. Folding of the apoSOD homodimer reveals a unique kinetic fingerprint, consistent with a simple three-state process (Scheme 2), where M is a high-energy intermediate at experimentally accessible protein concentrations (Fig. 5). A characteristic feature of the dimer chevron plot is that the folding process is rate-limited by the monomer formation under physiological conditions and that the transition state shifts to dimer dissociation at severely destabilizing conditions at high [urea], allowing full description of the parameters in Scheme 2. The apparent stability of the oxidized apoSODwt dimer, 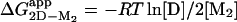 , is ≈7 kcal/mol at 4 μM protein and 0 M urea, as estimated from equilibrium unfolding. For the holo protein, the corresponding value of
, is ≈7 kcal/mol at 4 μM protein and 0 M urea, as estimated from equilibrium unfolding. For the holo protein, the corresponding value of 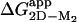 is ≈11 kcal/mol, as derived from data in ref. 30. The native SOD homodimer has thus a stability that falls in the typical range of other proteins (Fig. 4), possibly controlled by the in vivo requirements for efficient steady-state processing and degradation. It is then conceivable that the unusually low ΔGD–M of the apoSOD monomer is an adaptation to maintain the native dimer at a biologically suitable stability.
is ≈11 kcal/mol, as derived from data in ref. 30. The native SOD homodimer has thus a stability that falls in the typical range of other proteins (Fig. 4), possibly controlled by the in vivo requirements for efficient steady-state processing and degradation. It is then conceivable that the unusually low ΔGD–M of the apoSOD monomer is an adaptation to maintain the native dimer at a biologically suitable stability.
Implications for Gain of Function. Taken together, the results in this study show that reduction of the C57–C146 disulfide bond is critical for the integrity of the dimer interface and shifts the equilibrium toward marginally stable monomers. For several of the ALS-associated mutations, these monomers even reach the critical point where they are fractionally unfolded at physiological conditions (M.J.L. and M.O., unpublished results). On this basis, the disulfide-reduced monomer stands out as an interesting candidate for noxious gain of function, either directly through excessive sampling of folding intermediates or unfolded conformations that are structurally promiscuous or, indirectly, through misfolding and aggregation (14, 17, 18, 44–46). Consistently, the monomeric species has been implicated as a common precursor in SOD aggregation after oxidative damage of His and Phe side chains (47). Moreover, trapping of the dimeric state by the introduction of an intersubunit disulfide bond is reported to completely abolish aggregation of the A4V mutant that otherwise forms pore-like assemblies similar to those of other disease-associated proteins (44, 48). The most compelling evidence that SOD molecules lacking the C57–C146 linkage are powerful inducers of cytotoxic function is found in the list of ALS-associated mutants. C146R and seven different C-terminal truncations (2) provoke disease although they miss one of the key thiol groups. The presence of disulfide-reduced SOD is also found to be the least common denominator in spinal-cord extracts from transgenic mice carrying the ALS-linked mutations G85R, D90A, G93A and the truncation variant G127insTGGG (S. Marklund, personal communication). An attractive feature of disease models linked to an increased load of monomeric protein is that they reconcile the structurally diverse set of mutations associated with ALS. Elevated levels of noxious monomers could arise from mutations that promote disulfide reduction (15), weaken the dimer interface (37), decrease the global stability (11–13, 47) or folding cooperativity (49) of either the monomeric or dimeric species, or adversely affect metal loading through the copper chaperone (CCS) (50). The low stability of the reduced monomer provides also an explanation for sporadic ALS. In these cases, impaired function of the cellular “housekeeping” system, i.e., chaperones, quality control, and degradation mechanisms, might cause the reduced form of wild-type SOD to enter the same cytotoxic pathway as the ALS-associated mutants, consistent with observations of cytosolic SOD-containing aggregates in both familial and sporadic ALS (18). Understanding the roles of the physiological disulfide-reduction systems in the pathology of SOD mutations and ALS is an obvious future goal.
Acknowledgments
We thank the Swedish Research Council and Hjärnfonden for financial support.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SOD, superoxide dismutase; apoSOD, SOD apoenzyme; ALS, amyotrophic lateral sclerosis.
References
- 1.Liochev, S. I. & Fridovich, I. (2003) Free Radical Biol. Med. 34, 1383–1389. [DOI] [PubMed] [Google Scholar]
- 2.Andersen, P. M., Sims, K. B., Xin, W. W., Kiely, R., O'Neill, G., Ravits, J., Pioro, E., Harati, Y., Brower, R. D., Levine, J. S., et al. (2003) Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 62–73. [DOI] [PubMed] [Google Scholar]
- 3.Wood, J. D., Beaujeux, T. P. & Shaw, P. J. (2003) Neuropathol. Appl. Neurobiol. 29, 529–545. [DOI] [PubMed] [Google Scholar]
- 4.Dobson, C. M. (2003) Nature 426, 884–890. [DOI] [PubMed] [Google Scholar]
- 5.Kunst, C. B., Mezey, E., Brownstein, M. J. & Patterson, D. (1997) Nat. Genet. 15, 91–94. [DOI] [PubMed] [Google Scholar]
- 6.Shinder, G. A., Lacourse, M. C., Minotti, S. & Durham, H. D. (2001) J. Biol. Chem. 276, 12791–12796. [DOI] [PubMed] [Google Scholar]
- 7.Okado-Matsumoto, A. & Fridovich, I. (2002) Proc. Natl. Acad. Sci. USA 99, 9010–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durham, H. D., Roy, J., Dong, L. & Figlewicz, D. A. (1997) J. Neuropathol. Exp. Neurol. 56, 523–530. [DOI] [PubMed] [Google Scholar]
- 9.Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., Reaume, A. G., Scott, R. W. & Cleveland, D. W. (1998) Science 281, 1851–1854. [DOI] [PubMed] [Google Scholar]
- 10.Johnston, J. A., Dalton, M. J., Gurney, M. E. & Kopito, R. R. (2000) Proc. Natl. Acad. Sci. USA 97, 12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez, J. A., Valentine, J. S., Eggers, D. K., Roe, J. A., Tiwari, A., Brown, R. H., Jr., & Hayward, L. J. (2002) J. Biol. Chem. 277, 15932–15937. [DOI] [PubMed] [Google Scholar]
- 12.Lindberg, M. J., Tibell, L. & Oliveberg, M. (2002) Proc. Natl. Acad. Sci. USA 99, 16607–16612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiDonato, M., Craig, L., Huff, M. E., Thayer, M. M., Cardoso, R. M., Kassmann, C. J., Lo, T. P., Bruns, C. K., Powers, E. T., Kelly, J. W., et al. (2003) J. Mol. Biol. 332, 601–615. [DOI] [PubMed] [Google Scholar]
- 14.Stathopulos, P. B., Rumfeldt, J. A., Scholz, G. A., Irani, R. A., Frey, H. E., Hallewell, R. A., Lepock, J. R. & Meiering, E. M. (2003) Proc. Natl. Acad. Sci. USA 100, 7021–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiwari, A. & Hayward, L. J. (2003) J. Biol. Chem. 278, 5984–5992. [DOI] [PubMed] [Google Scholar]
- 16.Jonsson, P. A., Ernhill, K., Andersen, P. M., Bergemalm, D., Brannstrom, T., Gredal, O., Nilsson, P. & Marklund, S. L. (2004) Brain 127, 73–88. [DOI] [PubMed] [Google Scholar]
- 17.Valentine, J. S. & Hart, P. J. (2003) Proc. Natl. Acad. Sci. USA 100, 3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe, M., Dykes-Hoberg, M., Culotta, V. C., Price, D. L., Wong, P. C. & Rothstein, J. D. (2001) Neurobiol. Dis. 8, 933–941. [DOI] [PubMed] [Google Scholar]
- 19.Parge, H. E., Getzoff, E. D., Scandella, C. S., Hallewell, R. A. & Tainer, J. A. (1986) J. Biol. Chem. 261, 16215–16218. [PubMed] [Google Scholar]
- 20.Derman, A. I., Prinz, W. A., Belin, D. & Beckwith, J. (1993) Science 262, 1744–1747. [DOI] [PubMed] [Google Scholar]
- 21.Battistoni, A., Mazzetti, A. P. & Rotilio, G. (1999) FEBS Lett. 443, 313–316. [DOI] [PubMed] [Google Scholar]
- 22.Clarke, J. & Fersht, A. R. (1993) Biochemistry 32, 4322–4329. [DOI] [PubMed] [Google Scholar]
- 23.Khare, S. D., Ding, F. & Dokholyan, N. V. (2003) J. Mol. Biol. 334, 515–525. [DOI] [PubMed] [Google Scholar]
- 24.Sutter, B., Bounds, P. L. & Koppenol, W. H. (2000) Protein Expression Purif. 19, 53–56. [DOI] [PubMed] [Google Scholar]
- 25.Fersht, A. R. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 26.Fersht, A. R. (1995) Proc. Natl. Acad. Sci. USA 92, 10869–10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson, S. E. (1998) Folding Des. 3, R81–R91. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez, I. E. & Kiefhaber, T. (2003) J. Mol. Biol. 334, 1077–1085. [DOI] [PubMed] [Google Scholar]
- 29.Lepock, J. R., Frey, H. E. & Hallewell, R. A. (1990) J. Biol. Chem. 265, 21612–21618. [PubMed] [Google Scholar]
- 30.Stroppolo, M. E., Malvezzi-Campeggi, F., Mei, G., Rosato, N. & Desideri, A. (2000) Arch. Biochem. Biophys. 377, 215–218. [DOI] [PubMed] [Google Scholar]
- 31.Silow, M. & Oliveberg, M. (1997) Proc. Natl. Acad. Sci. USA 94, 6084–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliveberg, M. (1998) Acc. Chem. Res. 31, 765–772. [Google Scholar]
- 33.Otzen, D. E. & Oliveberg, M. (2002) J. Mol. Biol. 317, 639–653. [DOI] [PubMed] [Google Scholar]
- 34.Hammond, G. S. (1955) J. Am. Chem. Soc. 77, 334–338. [Google Scholar]
- 35.Levy, Y., Wolynes, P. G. & Onuchic, J. N. (2004) Proc. Natl. Acad. Sci. USA 101, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shakhnovich, E. I. (1999) Nat. Struct. Biol 6, 99–102. [DOI] [PubMed] [Google Scholar]
- 37.Hough, M. A., Grossmann, J. G., Antonyuk, S. V., Strange, R. W., Doucette, P. A., Rodriguez, J. A., Whitson, L. J., Hart, P. J., Hayward, L. J., Valentine, J. S. & Hasnain, S. S. (2004) Proc. Natl. Acad. Sci. USA 101, 5976–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson, M. A., Collins, J. L., Hod, Y., Ringe, D. & Petsko, G. A. (2003) Proc. Natl. Acad. Sci. USA 100, 9256–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelly, J. W. (1998) Curr. Opin. Struct. Biol. 8, 101–106. [DOI] [PubMed] [Google Scholar]
- 40.Furukawa, Y., Torres, A. S. & O'Halloran, T. V. (2004) EMBO J. 23, 2872–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferraroni, M., Rypniewski, W., Wilson, K. S., Viezzoli, M. S., Banci, L., Bertini, I. & Mangani, S. (1999) J. Mol. Biol. 288, 413–426. [DOI] [PubMed] [Google Scholar]
- 42.Clarke, J., Cota, E., Fowler, S. B. & Hamill, S. J. (1999) Structure Fold. Des. 7, 1145–1153. [DOI] [PubMed] [Google Scholar]
- 43.Plaxco, K. W., Simons, K. T. & Baker, D. (1998) J. Mol. Biol. 277, 985–994. [DOI] [PubMed] [Google Scholar]
- 44.Chung, J., Yang, H., de Beus, M. D., Ryu, C. Y., Cho, K. & Colon, W. (2003) Biochem. Biophys. Res. Commun. 312, 873–876. [DOI] [PubMed] [Google Scholar]
- 45.Elam, J. S., Taylor, A. B., Strange, R., Antonyuk, S., Doucette, P. A., Rodriguez, J. A., Hasnain, S. S., Hayward, L. J., Valentine, J. S., Yeates, T. O. & Hart, P. J. (2003) Nat. Struct. Biol. 10, 461–467. [DOI] [PubMed] [Google Scholar]
- 46.Julien, J. P. (2001) Cell 104, 581–591. [DOI] [PubMed] [Google Scholar]
- 47.Rakhit, R., Crow, J. P., Lepock, J. R., Kondejewski, L. H., Cashman, N. R. & Chakrabartty, A. (2004) J. Biol. Chem. 279, 15499–15504. [DOI] [PubMed] [Google Scholar]
- 48.Ray, S. S., Nowak, R. J., Strokovich, K., Brown, R. H., Jr., Walz, T. & Lansbury, P. T., Jr. (2004) Biochemistry 43, 4899–4905. [DOI] [PubMed] [Google Scholar]
- 49.Lindberg, M., Tangrot, J. & Oliveberg, M. (2002) Nat. Struct. Biol. 9, 818–822. [DOI] [PubMed] [Google Scholar]
- 50.Falconi, M., Iovino, M. & Desideri, A. (1999) Structure Fold. Des. 7, 903–908. [DOI] [PubMed] [Google Scholar]
- 51.Parge, H. E., Hallewell, R. A. & Tainer, J. A. (1992) Proc. Natl. Acad. Sci. USA 89, 6109–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanchez, I. E. & Kiefhaber, T. (2003) J. Mol. Biol. 325, 367–376. [DOI] [PubMed] [Google Scholar]
- 53.Hagen, S. J., Hofrichter, J., Szabo, A. & Eaton, W. A. (1996) Proc. Natl. Acad. Sci. USA 93, 11615–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]