

Abstract
Here, we describe the characterization of new RNA‐cleaving DNAzymes that showed the highest catalytic efficiency at pH 4.0 to 4.5, and were completely inactive at pH values higher than 5.0. Importantly, these DNAzymes did not require any divalent metal ion cofactors for catalysis. This clearly suggests that protonated nucleic bases are involved in the folding of the DNAzymes into catalytically active structures and/or in the cleavage mechanism. The trans‐acting DNAzyme variants were also catalytically active. Mutational analysis revealed a conservative character of the DNAzyme catalytic core that underpins the high structural requirements of the cleavage mechanism. A significant advantage of the described DNAzymes is that they are inactive at pH values close to physiological pH and under a wide range of conditions in the presence of monovalent and divalent metal ions. These pH‐dependent DNAzymes could be used as molecular cassettes in biotechnology or nanotechnology, in molecular processes that consist of several steps. The results expand the repertoire of DNAzymes that are active under nonphysiological conditions and shed new light on the possible mechanisms of catalysis.
Keywords: cleavage reactions, directed evolution, DNA, enzyme catalysis, RNA
1. Introduction
For many years, the term “enzyme” has been understood to mean a protein molecule that accelerates a biochemical process. To date, protein enzymes capable of catalyzing more than 5000 biochemical reaction types have been identified.1 At the beginning of the 1980s, Cech, Altman, and co‐workers discovered RNA molecules that showed an enzyme‐like activity, namely ribozymes.2, 3 Ribozymes occur both in prokaryotic and eukaryotic organisms and mainly catalyze the cleavage of RNA chains. Ten years later, the application of a new method of in vitro selection allowed the isolation of several synthetic ribozymes that were able to catalyze a variety of chemical reactions.4, 5, 6 Subsequently, with the use of the in vitro selection strategy, the range of catalytic nucleic acids was extended to include DNA oligonucleotides with enzymatic properties, termed DNAzymes.
Unlike ribozymes, DNAzymes have not been found in nature as yet. The first DNAzyme was isolated in 1994 by Breaker and Joyce. They used the in vitro selection strategy to identify a DNA catalyst that showed a ribonuclease activity and was able to catalyze the Pb2+‐dependent cleavage of an RNA phosphodiester linkage.7 So far, DNAzymes that catalyze RNA cleavage are the largest group of catalytic DNA molecules. Subsequently, the in vitro selection approach has also allowed the identification of DNA molecules that show other catalytic properties. DNAzymes with ligation,8 porphyrin metalation,9 N‐glycosylase,10 thymidine dimer repair,11 and carbon–carbon bond formation6 activities have been obtained and characterized.
Most of the self‐cleaving, naturally occurring, and man‐made ribozymes and DNAzymes require one or more divalent metal ions and a neutral pH for their activity. The metal cofactors Mg2+, Mn2+, and Ca2+ are usually the most effective. Recently, DNAzymes that require lanthanide and silver ions have also been obtained.12, 13, 14 The ions directly contribute to the chemical catalysis and/or to the folding of DNAzymes or ribozymes into higher‐order structures.15, 16, 17, 18, 19, 20, 21 Usually, a broader specificity towards metal ions is observed. When a particular ribozyme or DNAzyme is considered to be metal‐ion specific, it also performs catalysis in the presence of other ions albeit with a lower efficiency. Presumably, catalytic ions are bound in strong metal‐ion‐binding sites that can accommodate ions with similar properties. However, the ions are bound with diverse affinities or are slightly differently coordinated.22
The cleavage reaction of an RNA phosphodiester bond is the most common reaction catalyzed by known ribozymes and DNAzymes. It is a transesterification reaction that occurs through a general acid–base catalytic mechanism. In the reaction, two products are formed: an oligonucleotide with a 2′,3′‐cyclic phosphate at the 3′ end and an oligonucleotide with a free 5′‐OH group at the 5′ end. For several ribozymes, the catalytic mechanisms have been thoroughly studied. It has been proposed that a divalent metal ion is directly involved in the catalysis and plays the role of a general base. The ionized metal ion hydrate abstracts a proton from the 2′‐hydroxyl group of the ribose residue located in the vicinity of the phosphate group at the cleavage site. The resulting alkoxide anion attacks the phosphorus internucleotide bond with simultaneous breakage of the PO3′−O5 bond. Another metal ion may play a role of an acid that protonates the leaving 5′OH group and facilitates the breakage of the internucleotide chain.23
In subsequent studies of selected ribozymes, convincing data have been accumulated that nucleic acid bases may also be directly involved in a general acid–base catalysis.24 High‐resolution crystallographic data and the results of biochemical and biophysical experiments have been used to propose detailed cleavage mechanisms for a few naturally occurring ribozymes. A similar mechanism of RNA cleavage has also been suggested for DNAzymes, although it has not been investigated in detail. In particular, the possible involvement of nucleic base residues of DNAzymes in catalysis has not been shown.
Most artificial nucleic acid enzymes have been found to function under physiological or near‐physiological conditions and, therefore, it is necessary to look for ribozymes and DNAzymes with different abilities to catalyze the repertoire of chemical reactions under extreme conditions, at non‐physiological pH values or at high temperatures. Such studies are performed to acquire new information regarding the activity of nucleic acid enzymes, the mechanisms of catalysis, and their practical applications.25, 26 In a pioneering experiment, Yayasena and Gold isolated ribozymes that were able to cleave spontaneously at pH 4.0.27 Liu et al. obtained DNAzymes that are highly active under acidic conditions, with an optimum pH of 3.8, by using two steps of in vitro selection.28 Further extensive studies have led to the construction of trans‐acting DNAzymes active at various pH values in the range of 3.0 to 6.0. Note that the selected DNAzymes exhibited broad metal‐ion specificities.28, 29, 30, 31, 32 When considering the possible mechanisms of catalysis performed by ribozymes or DNAzymes at low pH values, it should be taken into account that under such conditions metal ions might be totally redundant. Some nucleic base residues are partly or fully protonated and they may act as positively charged centers during DNAzyme folding or they may play the role of either a general acid or base in the cleavage mechanism.
Here we describe the results of a study aimed at the characterization of unusual RNA‐cleaving DNAzymes that were capable of inducing the cleavage of a single RNA bond under acidic conditions with an optimum pH of around 4.0 to 4.5. Very importantly, these DNAzymes did not require any divalent metal ion cofactors for catalysis. This clearly suggests that protonated nucleic bases are involved in the folding of DNAzymes into catalytically active structures and/or in the reaction mechanism. The trans‐acting DNAzyme variants were also catalytically active. The results of mutagenesis of four selected nucleotides of the enzyme strand of the trans‐acting DNAzyme show that these positions probably contribute to the formation of the DNAzyme catalytic core. The results expand the repertoire of DNAzymes active under nonphysiological conditions and shed new light on the possible mechanisms of catalysis.
2. Results and Discussion
2.1. Isolation of New RNA‐Cleaving DNAzymes
The applied selection procedure was adopted from our earlier studies on the selection of RNA‐cleaving DNAzymes.33 A DNA combinatorial library with a 23‐nucleotide‐long random region corresponded to approximately 7×1013 different sequence variants (Figure 1A and Figure S1 in the Supporting Information). At each selection step, the cleavage products were separated by using polyacrylamide gel electrophoresis and DNA was eluted from the gel by using 0.3 m sodium acetate (pH 5.2). Surprisingly, after elution of the DNA pools at an acidic pH, spontaneous cleavage reactions were observed. After 10 rounds of selection, at which the cleavage extent exceeded 16 %, the DNA library was cloned into pCR2.1‐TOPO plasmids and 46 individual clones were sequenced.
Figure 1.
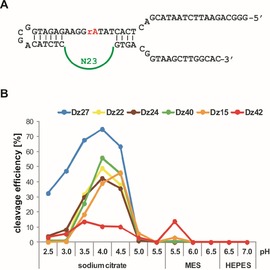
A) The sequence of the DNA pool used for selection, which contained a single cleavage site (rA) and a 23‐nucleotide random region (N23). B) The pH profiles of the six DNAzymes that represent the selected groups of sequences: Dz27 and Dz22 (group I), Dz24 (group II), Dz40 (group III), Dz15 (group IV), and the unclassified variant Dz42. The pH profiles were determined by conducting cleavage reactions in three different buffers that spanned pH 2.5–7.0. The cleavage efficiencies were measured after 60 min incubation at 25 °C.
The variants were categorized into four major groups based on similarities in the nucleotide composition of the initially randomized region (Figure S2). In the first group, which consisted of 22 clones, 19 clones had an identical sequence in the initially randomized region, whereas 17 clones had an A/G mutation at the same position in the constant region. The second family consisted of five clones and the sequence that corresponded to the random region was more differentiated. In the third family, 8 out of 9 clones had the same sequence. The fourth family consisted of 8 clones and the sequences that corresponded to the random region were the same in 7 clones. Single variant no. 42 was not been assigned to any of the families. The sequence alignments of all obtained variants revealed specific sequence motifs that were composed of two, three, and four nucleotides of the same type. Both the polypyrimidine and polypurine tracks, CCC, AA, TT (for group I), CCC, AA, CCCC, AAA, CCC, TT (for group II), CCC, AA, CCCC, AAA, CC, TT, CC (for group III), and TT, AAA, CCCC, AAA, CC, TT, TT (for group IV), can be seen.
The combinatorial library used in this study corresponded to approximately 7×1013 different sequence variants. In fact, all the theoretically possible sequences were present in the initial library. The 23‐random region is basically the longest nucleotide stretch that allows a fully represented library to be obtained in a standard experiment.34, 35 Velez et al. have suggested that short combinatorial libraries of catalytic DNA molecules can form relatively simple secondary and tertiary structural motifs.36 Although longer randomized regions have been used in several in vitro selections of DNAzymes, relatively simple structural motifs were usually identified to show catalytic properties.37, 38, 39, 40, 41, 42
2.2. Selected DNAzymes are Activated Under Low‐pH Conditions
Our observations on the selection progress and results of the initial screening of the catalytic activity of selected DNAzymes suggested that their cleavage occurred at an acidic pH. Six variants were chosen for more detailed study: Dz27 and Dz22 represented the first group of sequences, Dz24 and Dz40 were from the second and third group, respectively; moreover, a member of the fourth group (Dz15) and the unclassified variant (Dz42) were used in further experiments. The pH profile of these DNAzymes was determined under three buffer conditions that spanned a pH range from 2.5 to 7.0. The cleavage extent was measured after 60 min incubation at 25 °C (Figures 1B and S3).
DNAzyme Dz27 reached the highest cleavage efficiency of 75 % during incubation at pH 4.0. A decrease in the pH value to 2.5 resulted in cleavage efficiency that was approximately twofold lower, and at pH 6.0 to 7.0 this variant showed a negligibly low activity or was completely inactive (Figure 1B and Figure S3). Another member of group I, Dz22, showed a catalytic activity at pH values of 3.0 to 5.0, whereas the highest cleavage efficiency of 38 to 49 % was determined at pH 4.0 to 4.5. It should be noted that Dz22 was active at a narrower pH range than Dz27, despite the fact that both DNAzymes represented the same family of sequences (group I). Furthermore, the cleavage efficiency of Dz22 at pH 4.0 was approximately 25 % lower than that of Dz27. The differences in catalytic activity between these two DNAzymes might be connected with the different nucleotide composition of the catalytic region 3′ end (Figure S2). We suggest that the differences in the nucleotide sequences may have an impact on the thermodynamic stability of both variants and on their folding into correct secondary structures, which are essential for the achievement of high catalytic performance. At pH values of 2.5 to 7.0, DNAzymes from the second and third groups were slightly less active than variant Dz27. At pH 4.0, Dz24 and Dz40 were cleaved to 42 and 56 %, respectively. DNAzyme Dz15, from group IV, reached maximum catalytic activity at pH 4.5 instead of pH 4.0, like the other variants. The catalytic efficiency of Dz15 at pH 4.5 was approx. 50 %. It turned out that Dz42, which was not classified into any selection group, did not exhibit a high catalytic activity at any of the examined pH values. In summary, all the tested DNAzymes showed the highest cleavage activity at pH 4.0 to 4.5. Interestingly, the DNAzymes were completely inactive or showed a negligibly low activity at pH values higher than 5.0 (Figure 1B and Figure S3).
2.3. pH‐Dependent DNAzymes do not Require Divalent Metal Ions for Catalysis
To establish whether the selected DNAzymes required metal‐ion cofactors for catalysis, we determined the dependence on metal ions of the Dz15 and Dz27 variants at pH 4.5 and 4.0, respectively. The catalytic activity was assayed in the presence of 1 and 0.1 mm Cd2+, Mg2+, Zn2+, Sr2+, Ca2+, Mn2+, Co2+, Ni2+, and also in the presence of monovalent ions Na+ and K+ (100 mm; Figure S4). Note that the cleavage reactions were carried out in 50 mm sodium citrate buffer, however, a very similar catalytic activity and pH profile for DNAzyme Dz27 were observed when the buffer was replaced by 50 mm ammonium acetate with no sodium ions present (data not shown). The cleavage extent of Dz15 at pH 4.5 with no metal ions added was approximately 50 % (Figure S4 A, C(+) lane). The presence of Mg2+, Zn2+, Sr2+, Ca2+, Mn2+, Co2+, Ni2+, or Na+ ions did not significantly affect the catalytic activity of the Dz15 variant, whereas Cd2+ (1 mm) and K+ (100 mm) inhibited the cleavage reaction by approximately 10 %. Interestingly, divalent Cd2+ ions showed an inhibitory effect similar to monovalent K+ ions, but at a much lower concentration. It is known that divalent cations are much more effective in stabilizing the nucleic acid structure than monovalent ones, which is essential for achieving high catalytic efficiency in DNAzymes. The same observation has been made by Jayasena and Gold, who selected self‐cleaving RNAs under low pH conditions.27 The impact of metal cofactors on the catalytic activity of Dz27 at pH 4.0 was similar to that observed for Dz15 at pH 4.5. Most of the tested metal ions did not influence the cleavage reaction of this variant (Figure S4 B). Exceptionally, a strong inhibitory effect was observed for 1 mm Cd2+ ions and the cleavage extent of Dz27 decreased by over twofold.
Importantly, the DNAzymes obtained in our study were not dependent on divalent metal ion cofactors and were able to function without any metal ions. In contrast, DNA enzymes isolated previously by Li and co‐workers, which were active under acidic conditions from pH 3 to 6, required metal ions for catalysis. DNAzyme pH4DZ1 required both Mn2+ and Cd2+ for optimal activity, however, Ni2+ ions inhibited the cleavage reaction. Conversely, DNAzyme pH5DZ1 was metal nonselective, with a slight preference for Mn2+ions.28
Note that most DNAzymes require divalent metal ions for their activity. The first RNA‐cleaving DNAzymes able to function in the absence of divalent metal cofactors were obtained in 1997 by the group of Geyer and Sen. These DNAzymes required only monovalent ions for their activity.43 Moreover, some organic ion cofactors have also been used for in vitro selection of DNAzymes. For example, Roth and Breaker identified the histidine‐dependent HD2 DNAzyme.44 The observation that our simple DNAzymes are functional with no divalent metal ions present made them very attractive objects for further study.
2.4. Secondary Structures of DNAzymes Dz15 and Dz27, Active at Low pH
To characterize the secondary structure of pH‐dependent DNAzymes, two variants, Dz15 and Dz27, were chosen. These DNAzymes represented different selection groups and were cleaved at optimum pH values of 4.0 and 4.5. In experiments to probe the secondary structure, these variants contained deoxyadenosine instead of adenosine at the cleavage site to avoid spontaneous catalysis. Because the methods used for RNA mapping are of limited use for DNA mapping, S1 nuclease digestion and modification with dimethyl sulfate (DMS) were used, which have been applied previously for mapping Cd2+‐dependent DNAzymes.33 The secondary structures of DNAzymes Dz15 and Dz27, shown in Figures 2A, B, were predicted in silico by using the RNAstructure 5.6 program,45 in which the data from DMS probing and constraints from S1 nuclease mapping were included (Figure S5). It is worth noting that computer algorithms are built on thermodynamic data for nucleotide interactions at pH 7.0, which creates typical Watson–Crick base pairs. Similar conditions were used for experimental mapping of the DNAzyme structures. Neither computer nor experimental structural data were able to take into account the possible protonation of nucleotides and formation of nonstandard base pairs. Thus, the proposed structures of DNAzymes reflect base‐pair schemes that exist at pH 7.0. Possibly some additional nonstandard interactions are formed at pH 4.0 to 4.5, at which the DNAzymes indicate optimal activity.
Figure 2.
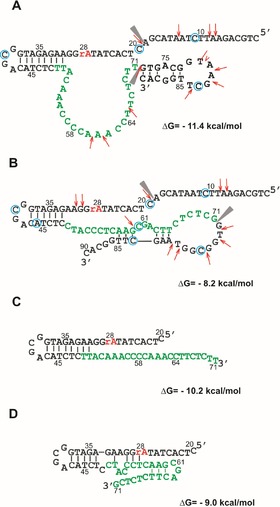
Secondary‐structure arrangements of full‐length DNAzymes: A) Dz15 and B) Dz27. In the structures, the DMS‐modified cytosine residues are marked by blue circles and S1 nuclease digestion sites are denoted by red arrows. The sites at which the sequences were truncated are denoted by black triangles. Secondary‐structure models of the shortened DNAzymes: C) Dz15WS and D) Dz27WS were predicted by using the RNAstructure 5.6 program. Green letters mark the initially randomized region of DNAzymes and the catalytic cleavage site is marked as rA in red.
The proposed structures of both DNAzymes revealed a characteristic hairpin (A31–T49) with a four‐nucleotide stable loop of the GNRA type (N=any nucleotide, R=purine), which was present in the initial DNA library. Additionally, in the proposed structure of Dz15 a hairpin motif with a seven‐nucleotide loop was present at the 3′ terminus. The initially randomized region (T49–T71) had a single‐stranded form with no defined structural motifs present (Figure 2A). Interestingly, the Dz15 variant resembled a known RNA‐cleaving DNAzyme 10‐23, which has a catalytic core composed of 15 unpaired nucleotides.38 However, in the catalytic region of the Dz27 variant the hairpin motif (C57–G86) and the three‐nucleotide‐membered internal loop were present (Figure 2B). In an additional in silico analysis, the Mfold,46 RNAfold,47 and KineFold48 programs were applied. The first two programs are used to generate DNA secondary structures based on the algorithm of the minimum free energy, whereas the third program is capable of folding DNA by taking into account pseudoknot structures. The secondary‐structure arrangements proposed by these programs were, however, similar to those predicted by the RNAstructure 5.6 program and the experimental data.
Note that the proposed secondary structures of Dz15 and Dz27are not fully consistent with the experimental data (Figure 2). For example, the presumably catalytic region of Dz15 is a single‐stranded stretch and this correlates well with the S1 nuclease mapping data because this region was digested by the enzyme. However, the cytosine residues present within this region are not modified by DMS, which suggests that they may participate in bond formation, for example, long‐range interactions. Importantly, in the case of DNAzymes that operate at low pH, it is likely that noncanonical base pairs are formed in their structures with the involvement of protonated cytosine residues, for example, C+−C or C+−C−G or protonated adenines A+−A+.49 However, the presence of such interactions cannot be detected by using the available biochemical probing methods; this requires high‐resolution techniques, such as NMR spectroscopy or crystallography.
2.5. DNAzymes Dz15 and Dz27 Truncated at the 5′and 3′ Ends Retain Their Catalytic Properties
Based on the results of the structural probing of DNAzymes Dz15 and Dz27, their truncated variants were designed, in which 19‐nucleotide fragments from the 5′ and 3′ ends were deleted (Figures 2C, D). The pH profiles of their catalytic activities were determined at pH 2.5 to 7.0. The cleavage extent was measured after incubation for 60 min at 25 °C. Truncated Dz15WS reached the highest cleavage efficiency of approximately 50 % at pH 4.0 to 4.5, the same as its full‐length version. A decrease in the pH value to 3.5 or an increase to 5.0 resulted in a 1.5‐fold lower cleavage extent, whereas at pH 2.5 to 3.0 and 5.5 to 7.0 this variant showed a negligibly low activity (Figure 3A). The other truncated variant, Dz27WS, was active at pH 3.0 to 4.5. The highest cleavage extent was determined at pH 4.0 and 4.5. Dz27WS was 65 % cleaved at pH 4.0 and 4.5, whereas the cleavage extent of the full‐length variant was 10 % higher at pH 4.0 but remained unchanged at pH 4.5. Note that the Dz27WS variant was able to cleave at a narrower pH range compared with full‐length Dz27, which was active at pH values between 2.5 and 4.5 (Figures 1 and 3A).
Figure 3.
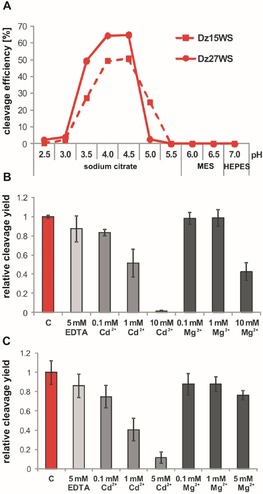
The impact of pH and metal ions on the cleavage of the cis‐acting DNAzymes Dz15WS and Dz27WS. (A) pH profiles of DNAzymes Dz15WS and Dz27WS. The cleavage efficiencies were determined in three different buffers spanning a pH range from 2.5 to 7.0. Dependence of the relative cleavage yield of B) Dz15WS at pH 4.5 and C) Dz27WS at pH 4.0 in the presence of selected metal ions and EDTA. The relative cleavage yields were determined after incubation for 60 min at 25 °C.
It has been shown that distal sequence elements could enhance the activity and selectivity of Co2+‐dependent DNAzyme.50 However, our earlier studies of Cd2+‐dependent DNAzyme have proven that its 19‐nucleotide‐long terminal fragments are not required for catalytic activity.33 It has also been suggested that if DNAzymes do not adopt Watson–Crick interactions in acidic conditions, as in pH3DZ1, pH4DZ1, and pH5DZ1, they will need a large number of nucleotides for the formation of the correct tertiary structures.32
Metal ions were shown not to be necessary for the catalytic activity of Dz15WS and Dz27WS, similar to previous observations for their full‐length variants. The cleavage activity was tested at pH 4.5 in the presence of Cd2+, Mg2+, and EDTA (Figure 3B, C). Importantly, the presence of 5 mm EDTA had essentially no impact on the catalytic properties of Dz15WS and Dz27WS. At a higher concentration of divalent metal ions, 1 mm Cd2+ and 10 mm Mg2+, the cleavage extent of Dz15WS decreased by twofold, and at 10 mm Cd2+ the reaction was almost completely inhibited. The Dz27WS variant behaved similarly.
Earlier studies, which used Raman spectroscopy to investigate the interactions of metal ions with DNA, have revealed that transition‐metal ions show higher affinities for DNA bases, whereas alkaline‐earth‐metal ions show higher affinities for phosphate residues. The Cd2+ or Zn2+ ions interact preferentially with N3 of cytosine or N7 of guanine bases.51, 52 As we mentioned earlier, cytosine residues seem to play an important role in the folding of pH‐dependent DNAzymes. Thus, the significant inhibitory effect of Cd2+ ions on the cleavage reaction of Dz15WS and Dz27WS might result from the interactions of these ions with cytosines and then incorrect folding of the DNAzymes into catalytically active structures.
The dependence on temperature of the cleavage reaction rate constants (k obs) for Dz15WS and Dz27WS was also determined. At a temperature of 37 °C, the k obs value of the Dz15WS variant (0.012 min−1) was almost two times higher than that at 25 °C (0.007 min−1; Figure 4A). Interestingly, the k obs values for Dz27WS at 25 and 37 °C (0.035 and 0.036 min−1, respectively) were almost identical (Figure 4B). It is worth noting that both DNAzymes catalyzed the cleavage reaction at different rates. In comparison with the Dz27WS variant, which is a representative of group I of the selected DNAzymes, the group IV member, Dz15WS, was threefold less active at 25 °C and fivefold less active at 37 °C (Figure 4). Based on the uncatalyzed RNA cleavage rate, calculated to be 10−7 min−1,53 DNAzymes Dz15WS and Dz27WS accelerated RNA cleavage by a factor of 7 to 35×103 fold, respectively.
Figure 4.
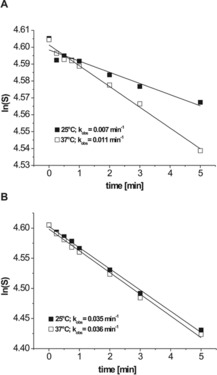
The cleavage rate constant (k obs) of pH‐dependent DNAzymes A) Dz15WS and B) Dz27WS. The assays were carried out at pH 4.0. The reactions were performed at 25 (▪) and at 37 °C (□).
We suggest that the differences observed in the cleavage efficiency of Dz15WS and Dz27WS might be connected, in particular, with the differences in the nucleotide composition of their catalytic regions, which has an impact on the folding of these DNAzymes into correct structures. The catalytic region of the Dz15WS variant is comprised of stretches of two, three, and four nucleotides of the same type (5′TTacAAACCCCAAACCTTctcTT3′) and this may result in a molecule that folds into a structure with a lower catalytic activity at an acidic pH.
2.6. Structural Changes in DNAzymes Dz15WS and Dz27WS at Low pH
We used UV and CD spectroscopy to test whether any structural changes occurred in DNAzymes Dz15WS and Dz27WS under different pH values (Figure S6). In these experiments, we used noncleavable variants of these DNAzymes, in which adenosine at the cleavage site was replaced by deoxyadenosine. The CD spectra indicated the presence of B‐form DNA, revealed by a positive Cotton effect at λ≈277 nm and a negative Cotton effect at λ≈250 nm.54 In addition, the decrease in pH from 7.0 to 2.5 was accompanied by a significant decrease in the positive signal at around λ=280 nm (Figure S6, upper panel), which might be related to the transition from B‐ to Z‐form DNA.55 However, no negative signals at λ=294 nm and at short wavelengths (λ≈205 nm) were detected, and this observation did not fully support the B‐to‐Z transition of pH‐dependent DNAzymes.
Interestingly, the UV spectra of DNAzymes showed substantial changes depending on the pH conditions (Figure S6, bottom panel). A significant decrease in the absorption peak at λ≈265 nm as the pH value was decreased to 4.0 might be explained by changes in the hydrogen bonds between the paired bases in the double helix, which limits the resonance behavior of the aromatic rings of the bases. Additionally, on changing the pH from 3.5 to 5.5, a shift in the absorption maximum towards longer wavelengths was observed. Moreover, at pH 2.5 to 3.0, an increase in the absorption was noted. This might be connected to the lack of specific interactions between unusual base pairs that involve protonated residues (non‐Watson–Crick interactions).
It has been shown that DNA strands rich in cytosines could form a quadruplex structure (termed intercalated) or an i‐tetraplex/i‐motif.56 Elbaz et al. have designed a pH‐induced, switchable, activated and deactivated E6‐Mg2+‐dependent DNAzyme that was based on the i‐tetraplex structure.57 The CD spectra of cytosine quadruplexes have dominant positive bands at λ=290 nm and negative bands at λ=260 nm. The formation of such structures is promoted by acidic pH conditions, which are needed for the formation of hemiprotonated C−C+‐type base pairs. However, in the CD spectra of pH‐dependent DNAzymes Dz15WS and Dz27WS, dominant positive Cotton effects at λ=290 nm were not observed, which could indicate the generation of i‐tetraplex structures (Figure S6).
As mentioned earlier, all the obtained DNAzymes revealed specific sequence motifs within their initially randomized regions, which were composed of two, three, and four nucleotides of the same type (Figure S2). It has been shown that single‐stranded nucleic acids, composed of regular homopyrimidine or homopurine repeats, might be subjected to significant structural rearrangements in a low pH environment due to the formation of unusual base pairs that involve protonated residues.58 This process is accompanied by marked changes in the CD spectra.59 The CD and UV data for DNAzymes Dz15WS and Dz27WS showed that substantial changes in the absorption spectra occurred at pH 3.5 to 5.0 (Figure 5 and Figure S6). The largest decrease in the intensity of the absorption bands was observed at pH 4.0 to 4.5. This correlates with the conditions under which these DNAzymes show the highest catalytic activity. Thus, the CD and UV spectroscopic data at pH 4.0 to 4.5 characterize the catalytically active forms of DNAzymes Dz15WS and Dz27WS.
Figure 5.
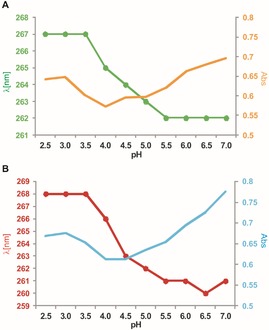
Dependence of the changes in UV absorption intensity (plain lines) and shift in the absorption maximum from λ=261 to 268 nm (filled circles), which were determined for A) Dz15WS and B) Dz27WS at different pH values. A noncleavable version of the DNAzymes was used, in which adenosine at the cleavage site was replaced by its deoxy analogue. The following reaction conditions were applied: 50 mm sodium citrate, 25 °C; total concentration of DNAzymes 10 μm.
2.7. trans‐Acting Variants of DNAzymes Dz15WS and Dz27WS are Catalytically Active
trans‐Acting DNAzymes Dz15WS_E and Dz27WS_E were synthetized. They are derivatives of Dz15WS and Dz27WS in which the oligonucleotide substrate and deoxyribozyme strands were separated in the four‐nucleotide loop between C40 and G41 (Figure 6). The catalytic activity of these variants was determined at 37 °C at pH 2.5 to 5.0. The DNAzyme components were used in 100‐fold excess relative to the oligonucleotide substrate with adenosine at the cleavage position (wild‐type DNAzymes).
Figure 6.
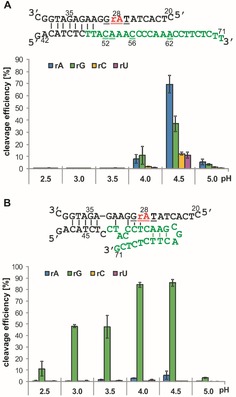
Effect of RNA base identity at the cleavage position on the reaction efficiency of trans‐acting DNAzymes: A) Dz15WS_E and B) Dz27WS_E. The oligonucleotide substrates were synthesized with adenosine (rA), guanosine (rG), cytidine (rC), or uridine (rU) at the cleavage site. All mutated nucleotides are denoted by a double underline. Cleavage reactions were performed in sodium citrate buffer pH 2.5–5.0 at 37 °C for 3 h.
The pH profile of the cleavage efficiency of Dz15WS E was similar to that of the cis‐acting variant (Figures 3A and 6A).The highest activity was observed at pH 4.5, with approximately 70 % cleavage, whereas the cis‐acting variant was cleaved to 65 %. However, these comparable cleavage yields were determined for the cis‐acting variant after 60 min at 25 °C, whereas for the trans‐acting DNAzyme the reaction was performed for 3 h at 37 °C. Thus, the trans‐acting derivative was substantially less active than its parental cis‐acting variant. Variant Dz15WS_E was only 8 and 5 % cleaved during incubation at pH 4.0 and 5.0, respectively, in contrast with the corresponding cis‐acting variant, which was 65 and 2 % cleaved. Conversely, cis‐acting Dz27WS was cleaved at pH 4.0 to 4.5 with a 50 % yield, whereas its trans‐acting derivative Dz27WS_E was marginally active. Only 2 and 5 % cleavage yields were detected at pH 4.0 and 4.5, respectively (Figures 3A and 6B). Thus, a decrease in activity of over tenfold occurred compared with the cis‐acting DNAzyme. This might be a result of the removal of the four‐nucleotide stable loop of GNRA type (N=any nucleotide, R=purine) present in cis‐acting Dz27WS, which seems to be essential for DNAzyme folding and its catalytic activity.
We also tested the effect of nucleoside substitution at the cleavage site on the reaction efficiency. In the initial DNAzyme library, a single adenosine (rA) was present in this position and this nucleoside was present in the oligonucleotide substrate, which was used in trans‐acting DNAzymes Dz15WS_E and Dz27WS_E. Thus, three additional oligonucleotide substrates were synthesized with guanosine (rG), cytidine (rC), and uridine (rU) residues in the corresponding position. The replacement of adenosine by other nucleosides had a diverse effect on the cleavage efficiency. For Dz15WS_E, the largest effect, a sevenfold decrease in cleavage efficiency, was observed at pH 4.5 in the presence of pyrimidine residues at the cleavage site, and a smaller twofold decrease was observed in the presence of guanosine residue (Figure 6A). The cleavage extent was significantly reduced at pH 4.0, to give 10 and 2 % cleavage for substrates with purine and pyrimidine bases, respectively. At pH values lower than 4.0, essentially no catalytic activity was observed (Figure 6A). Unexpectedly, Dz27WS_E was active only with a guanosine residue at the cleavage position. The DNAzyme was cleaved to 80 % at pH 4.0 to 4.5 and to 50 % at pH 3.0 to 3.5. A lower activity was observed at pH 2.5 and 5.0, to give approximately 10 and 5 % cleavage efficiency, respectively. In the presence of pyrimidine residues at the cleavage site, the Dz27WS_E variant was almost completely inactive (Figure 6B).
2.8. Mutational Analysis Shows Strong Conservation of the DNAzyme Core
We also determined the cleavage activity of trans‐acting DNAzymes Dz15WS_E and Dz27WS_E when additional ribonucleotides immediately adjacent to the catalytic cleavage site were present in the oligonucleotide substrate (Figure 6A).
Santoro and Joyce selected the DNAzyme that was able to cleave anywhere along the 12nt length of the RNA sequence.38 Moreover, the cleavage efficiency of another DNAzyme, the 17E variant, was only twofold lower in the presence of the all‐RNA substrate than in the presence of the DNA/RNA chimeric substrate.60
For variant Dz15WS E, the replacement by uridine of 5′ thymidine immediately adjacent to the catalytic cleavage site resulted in a substantial reduction in the cleavage efficiency at pH 4.5 (Figure 6A, Figure S7, and Table 1A). When the 3′ deoxyguanosine was replaced with guanosine, catalysis decreased by approximately fourfold. However, the largest effect, an eightfold lower catalytic activity, was observed at pH 4.5 if both these deoxynucleosides, G29 and T27, were simultaneously replaced with guanosine and uridine. For variant Dz27WS E, mutations at position G29 or T27 caused a dramatic 800‐fold decrease in the cleavage efficiency at pH 4.5 (Figure 6B, Figure S7, and Table 1B). These data might suggest that residues of the oligonucleotide substrate immediately adjacent to the cleavage site are involved in interactions with some other part of the DNAzyme–substrate complex. Alternatively, a specific conformation of the polynucleotide chain around the cleavage site is required for efficient catalysis to occur. Subsequently, we examined how changes in individual nucleotides within the DNAzyme strand could impact its catalytic performance. A statistical analysis of the sequences of all clones from the in vitro selection experiment was conducted. The variations observed at each position of the sequence that corresponded to the 23‐nucleotide random region of the initial library are summarized in Figure S8. A detailed examination of the prevalence of particular bases revealed that nucleotides at positions 52, 53, 56, and 62 were highly conserved. Positions 52 and 53 were occupied by cytidines with 98 and 83 % frequency, respectively. Additionally, adenosine also appeared at position 53 with approximately 17 % frequency. More than 93 % of the selected variants had cytidine at position 56 and adenosine at position 62. In other positions in the in vitro selected sequences, at least two or more nucleotides occurred with comparable frequencies.
Table 1.
The cleavage efficiency of trans‐acting DNAzymes Dz15WS_E (A) and Dz27WS_E (B) with oligonucleotide substrates Sub1, Sub2, Sub3, and Sub4, which contained additional ribonucleotides immediately adjacent to the cleavage site.
| Substrate | pH | Cleavage efficiency (A) [%] | Cleavage efficiency (B) [%] | ||||
|---|---|---|---|---|---|---|---|
| rU | rA | rG | rU | rA | rG | ||
| Sub1 27‐T rA G‐29 | 3.5 | – | 0.10 | – | – | 0.19 | – |
| 4.0 | – | 16.8 | – | – | 1.95 | – | |
| 4.5 | – | 62.4 | – | – | 15.44 | – | |
| Sub2 27‐rU rA G‐29 | 3.5 | 0.09 | 0.07 | – | 0.20 | 0.04 | – |
| 4.0 | 0.72 | 10.3 | – | 1.28 | 0.26 | – | |
| 4.5 | 1.52 | 50.1 | – | 0.0006 | 0.23 | – | |
| Sub3 27‐T rA rG‐29 | 3.5 | – | 0.02 | 0.05 | – | 0.04 | 0.06 |
| 4.0 | – | 0.37 | 0.05 | – | 0.02 | 0.03 | |
| 4.5 | – | 4.28 | 0.07 | – | 0.01 | 0.02 | |
| Sub4 27‐rU rA rG‐29 | 3.5 | 0.10 | 0.02 | 0.16 | 0.21 | 0.04 | 0.40 |
| 4.0 | 0.14 | 0.33 | 0.26 | 0.52 | 0.02 | 0.18 | |
| 4.5 | 0.17 | 2.01 | 0.22 | 0.11 | 0.01 | 0.14 | |
The cleavage assays were carried out in sodium citrate buffer (pH 3.5, 4.0, and 4.5). The reactions were performed at 37 °C for 3 h. trans‐Acting Dz15WS_E was chosen for directed mutagenesis studies. We analyzed the effect of individual mutations in position 52, 53, 56, and 62. The cleavage efficiency of the examined mutants at pH 4.5 is presented in Figure S9. It turned out that nearly every substitution reduced catalysis by more than 100‐fold (A53G, C56T, and A62G). Only the C52T and A53C mutations seemed to be tolerated to some extent, and only lowered the catalytic efficiency by seven‐ and ninefold, respectively. Thus, the mutagenesis assays revealed that nucleotides in the four examined positions probably contribute to the formation of the DNAzyme catalytic core and/or contribute directly to chemical catalysis. The cytosine residue in position 56 and adenine residues in positions 53 and 62 seem to play a particularly important role in the functioning of DNAzyme Dz15WS_E.
3. Conclusions
We present a comprehensive characterization of highly efficient RNA‐cleaving DNAzymes that work at low pH, which were obtained by using the in vitro selection approach. All the studied DNAzymes showed the highest catalytic efficiency at pH 4.0 to 4.5 and were completely inactive at pH values higher than 5.0. Unlike most of the catalytic nucleic acids identified so far, which require divalent metal ions for catalytic activity,28, 37, 38, 39, 40, 41, 42 the full‐length DNAzymes and their truncated variants did not require any divalent metal cofactors for catalysis.
At a low pH, at which the selected DNAzymes were active, extensive protonation of nucleic bases occurs, mainly at N1 of adenosine and N3 of cytidine residues. Under such conditions, positively charged bases may directly contribute to the chemical catalysis rather than metal ions, by playing the role of a base or an acid in a general acid–base cleavage mechanism. Positively charged bases could also enable the folding of DNAzymes into the correct secondary structures that are essential for achieving high catalytic efficiency. It is worth noting that the UV and CD spectroscopy data of the selected DNAzymes showed major changes in their structures at pH 4.0 to 4.5, which coincides with conditions conducive to their highest activity.
The trans‐acting versions of the selected DNAzymes reached maximum activity at a pH value of around 4.5, similar to their cis‐acting parent variants. Mutagenesis studies of the cleavage site showed that cleavage efficiency was significantly higher for purine than for pyrimidine residues in this position. Interestingly, a dramatic decrease in the cleavage extent occurred in the presence of an oligonucleotide substrate with additional ribonucleotides immediately adjacent to the cleavage site. Thus, these residues may be involved in interactions with some other part of the DNAzyme–substrate complex. Alternatively, the presence of additional ribonucleotides may change the conformation of oligonucleotide substrate and the spatial arrangement of bases located in the vicinity of the cleavage site. This may influence the possible contribution of bases to the DNAzyme folding or their direct involvement in the cleavage mechanism. This assumption was supported by the results of mutagenesis for four selected nucleotides of the enzyme strand of the trans‐acting DNAzyme. The changes at these positions resulted in barely active DNAzymes. Thus, the pH‐dependent DNAzymes require the formation of highly specific spatial folds and they are catalytically active only at strictly specified pH conditions.
Novel DNAzymes able to function under extreme conditions, such as high temperatures, high or low pH, and in the presence of heavy metal ions, are constantly being discovered and developed. A large advantage of the DNAzymes described herein is that they are inactive at pH values close to the physiological value and under a wide range of conditions, in the presence of monovalent and divalent metal ions, or at different temperatures. The only factor that triggers their catalytic activity is the lowering of the pH to 4.0 to 4.5. These pH‐dependent DNAzymes could be used as molecular cassettes in biotechnology or nanotechnology, in molecular processes that consists of several steps, or as specific molecular tools, such as biosensors. Such cassettes allow the environmental conditions to be changed to a large extent, but during one step only the cassette can be cut off by lowering the pH. The trans‐acting variants of the obtained DNAzymes are relatively small, only approximately 31 nucleotides in length, which greatly simplifies their usage. Finally, these DNAzymes might be applied to monitor pH changes in cells during metabolic processes. The results described herein widen the repertoire of DNAzymes that work under non‐physiological conditions and shed new light on the possible mechanisms of catalyzed reactions.
Experimental Section
Oligonucleotides and Reagents
All oligonucleotides used in this study (see the Supporting Information, Figure S1) were prepared by automated DNA–RNA synthesis by using the standard phosphoramidite chemistry (FutureSynthesis, Poznan, Poland or Integrated DNA Technologies, Leuven, Belgium). The random‐sequence DNA library was synthesized by using an equimolar mixture of the four standard phosphoramidities. The DNA oligonucleotides were purified by using 8 or 12 % denaturing polyacrylamide gel electrophoresis prior to usage. DNA bands were excised, eluted with 0.3 m sodium acetate (pH 5.2) and 1 mm EDTA, and precipitated with 3 volumes of ethanol. The DNA concentration was measured by using a NanoDrop spectrometer. Radioactive nucleotides [γ‐32P]ATP and [α‐32P]dCTP with specific activity of 5000 Ci mmol−1 were purchased from Hartmann Analytic. T4 polynucleotide kinase, S1 nuclease, Taq polymerase, and dNTPs were from MBI Fermentas. All other chemicals and salts were from Sigma–Aldrich and Serva.
Selection Protocol
The selection procedure followed a protocol described previously.33 Briefly, the starting DNA pool was generated by extending primer P3 in the presence of primer M1 in a 300 μL reaction volume for seven thermal cycles. Then primers P3 and P4 were added to the reaction mixture for seven additional cycles of PCR amplification. Adenosine (rA) was introduced through the usage of P3 primer. The PCR products were precipitated with ethanol and purified by using electrophoresis in a 12 % denaturing polyacrylamide gel. The band corresponding to the random DNA pool was excised from the gel, then the DNA was eluted and precipitated in ethanol. The DNA pool containing a single rA was incubated in HEPES (50 mm, pH 7.0), NaCl (100 mm), and CdCl2 at 25 °C for a designated time period. The concentration of CdCl2 in the mixture was reduced from 1 mm for the first selection round to 100 μm for round eleven. Subsequently, the DNA cleavage product was purified in 12 % denaturing polyacrylamide gel, eluted from the gel by using 0.3 m sodium acetate (pH 5.2) and 1 mm EDTA, then precipitated with ethanol. Two rounds of PCR were used to amplify the recovered 62‐nt cleavage product. PCR products were purified by electrophoresis in a 12 % denaturing polyacrylamide gel, and eluted from the gel with 0.3 m sodium acetate (pH 5.2) and 1 mm EDTA, precipitated in ethanol, and finally used to initiate the next round of selection.
The tenth generation of selectively amplified DNA was cloned by using the TA‐TOPO Cloning Kit (Invitrogen). Plasmid DNA was extracted from Escherichia coli cells that contained the vector by using a BIO BASIC INC MiniPreps Kit. The 46 randomly selected clones were submitted to the Oligo.pl service, IBB PAN Warsaw, for sequencing.
Synthetic DNAzyme Variants
The cis‐acting DNAzymes for structural probing and determination of catalytic activity were synthesized by using a previously described procedure.33 Briefly, equimolar amounts of two DNA oligomers P3 (or P3.1) and P4 and plasmid that contained individual DNAzyme template (60 ng) were annealed. The reaction mixture contained both oligomers (0.5 μm each), MgCl2 (2 mm), (NH4)2SO4 (20 mm), Tris‐HCl (75 mm, pH 8.8 at 25 °C), Tween 20 (0.01 %), [α‐32P]dCTP (10 μL), dNTP (0.2 mm each), and DNA Taq polymerase (0.05 U μL−1). The reaction was performed for 25 thermal cycles (92 °C/30 s, 60 °C/30 s, and 72 °C/1 min). The dsDNA template was precipitated with ethanol and purified by using electrophoresis in a 12 % denaturing polyacrylamide gel. The band corresponding to the cis‐acting DNAzyme (90 nt) was cut out, eluted, and precipitated with ethanol.
Shortened DNAzyme Variants
Variants Dz15WS, Dz27WS, from which 19 nucleotides from both the 5′ and 3′ ends of the full‐length DNAzymes Dz15 and Dz27 were deleted, trans‐acting DNAzymes Dz15WS_E, Dz27WS_E, Dz15WS_E C52T, Dz15WS_E A53C, Dz15WS_E A53G, Dz15WS_E C56T, and Dz15WS_E A62G, and their oligonucleotide substrates (Sub1, Sub2, Sub3, Sub4, Sub5, Sub6, and Sub7) were prepared by using automated DNA synthesis (FutureSynthesis, Poznan).
Structural Probing
5′‐32P‐End‐labelled noncleavable DNAzymes (with single rA replaced by dA at the cleavage site) were used to probe the structures. DNAzymes were supplemented with tRNA carrier to a final concentration of 2 A260 mL−1 and renatured in HEPES (80 mm, pH 7.0), NaCl (40 mm), and MgCl2 (10 mm) by heating at 65 °C for 5 min and cooling to RT. Subsequently, a 1/20 volume of DMS in ethanol (1:12 v/v) was added and the samples were incubated at 25 °C for 10 or 30 minutes. The reaction was terminated by standard ethanol precipitation (twice). The pellet was dissolved in a ice‐cold solution of hydrazine in water (10 μL; 1:1, v/v) and incubated for 10 min at 0 °C. After precipitation with ethanol (twice), the pellet was dissolved in dilute piperidine (20 μL, 1/10) and incubated at 60 °C for 15 min. After precipitation with ethanol (twice), the DNA was dissolved in urea (8 m)/dyes/EDTA (20 mm) and loaded on a denaturing polyacrylamide gel.
Limited digestions with nuclease S1 were carried out as follows: 5′‐32P‐end‐labeled noncleavable DNAzyme variants in Tris‐HCl (10 mm; pH 7.5), NaCl (40 mm), and MgCl2 (10 mm) were renatured by heating at 100 °C for 2 min and cooling at 0 °C for 5 min. Reactions were performed at 25 °C for different time intervals with the enzyme (275 U m−). The reactions were terminated by adding a solution of urea (8 m)/dyes/EDTA (20 mm) and immediately freezing the samples in dry ice. The reaction products were analyzed by using polyacrylamide gel electrophoresis.
Catalytic Cleavage Reaction
Prior to self‐cleavage, the full‐length cis‐acting DNAzymes and their shortened variants, internally 32P‐labeled or 5′‐32P‐end‐labeled, were subjected to a denaturation–renaturation procedure by incubating at 65 °C for 1 min, slowly cooling to 35 °C, and then incubating at 25 °C (or 37 °C) for 10 min. The cleavage reactions were carried out in sodium citrate (50 mm; pH 2.5–5.5) or MES (pH 5.5–6.5) or HEPES (pH 6.5–7.0) buffers at 25 or 37 °C and were quenched at various time points with a stop‐buffer that contained urea (8 m), EDTA (20 mm), and dyes for electrophoresis. In some experiments (see Figure 3 and Figure S4), mono‐ or divalent metal ions or EDTA were added before the cleavage reactions. The products of the catalytic cleavage were analyzed by using denaturing 12 % polyacrylamide gels.
The trans‐acting DNAzymes were prepared by mixing of 5′‐32P‐end‐labeled DNA substrates (0.5 nm) with DNA enzymes (500 nm) to obtain a 100:1 DNAzyme–substrate ratio. The mixtures were heated for 10 min at 37 °C and under these conditions all the substrate molecules were bound in substrate–DNAzyme complexes. Subsequently, the sodium citrate buffer (pH 2.5–5.0) was added and the reactions proceeded at 37 °C. The cleavage reactions were quenched at specified time points with equal volumes of a mixture of urea (8 m), EDTA (20 mm), and dyes for electrophoresis. Products of the catalytic cleavage were analyzed by using denaturing 20 % polyacrylamide gels.
For quantitative analysis, a FLA 5100 image analyzer (Fuji) and MultiGauge software (Fuji) were used. The rate constant for the cleavage reaction (k obs) was determined by plotting the natural logarithm of the fraction of DNA that remained unreacted versus the reaction time.
Spectroscopic Measurements
Spectroscopic studies (CD, UV) were carried out with noncleavable versions of cis‐acting DNAzymes Dz15WS and Dz27WS, in which the single rA at the cleavage site was replaced by dA. The DNAzymes were subjected to a denaturation–renaturation procedure by incubating them at 65 °C for 1 min, slowly cooling to 35 °C, and then incubating at 25 °C for 10 min. The final concentration of DNA was 10 μm in all the experiments. Appropriate volumes of sodium citrate buffer (50 mm, pH 2.5–7.0) were titrated, and absorption spectra were recorded by using an Agilent Cary 60 UV/Vis spectrophotometer, whereas CD spectra were recorded by using a Jasco J‐715 spectropolarimeter over the range of λ=400–220 nm, with 0.1 cm cuvettes and a sample volume of 250 μL. The spectra are the average of five sequential accumulations for each sample and were recorded in 50 mm sodium citrate.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by Wroclaw Research Center EIT+ under the project “Biotechnologies and advanced medical technologies–BioMed” (POIG 01.01.02‐02‐003/08‐00) financed by the European Regional Development Fund (Operational Programme Innovative Economy, 1.1.2). This publication was also supported by the Polish Ministry of Science and Higher Education, under the KNOW program.
A. Kasprowicz, K. Stokowa-Sołtys, M. Jeżowska-Bojczuk, J. Wrzesiński, J. Ciesiołka, ChemistryOpen 2017, 6, 46.
References
- 1. Chang A., Schomburg I., Placzek S., Jeske L., Ulbrich M., Xiao M., Sensen Ch. W., Schomburg D., Nucleic Acids Res. 2015, 43, D439–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kruger K., Grabowski P. J., Zaug A. J., Sands J., Gottschling E., Cech T. R., Cell 1982, 31, 147–157. [DOI] [PubMed] [Google Scholar]
- 3. Guerrier-Takada C., Gardiner K., Marsh T., Pace N., Altman S., Cell 1983, 35, 849–857. [DOI] [PubMed] [Google Scholar]
- 4. Bartel D. P., Szostak J. W., Science 1993, 261, 1411–1418. [DOI] [PubMed] [Google Scholar]
- 5. Williams K. P., Ciafre S., Tocchini-Valentini G. P., EMBO J. 1995, 14, 4551–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chandra M., Silverman S. K., J. Am. Chem. Soc. 2008, 130, 2936–2937. [DOI] [PubMed] [Google Scholar]
- 7. Breaker R. R., Joyce G. F., Chem. Biol. 1994, 1, 223–229. [DOI] [PubMed] [Google Scholar]
- 8. Sreedhara A., Li Y., Breaker R. R., J. Am. Chem. Soc. 2004, 126, 3454–3460. [DOI] [PubMed] [Google Scholar]
- 9. Li Y., Sen D., Nat. Struct. Biol. 1996, 3, 743–747. [DOI] [PubMed] [Google Scholar]
- 10. Sheppard T. L., Ordoukhanian P., Joyce G. F., Proc. Natl. Acad. Sci. USA 2000, 97, 7802–7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chinnapen D. J., Sen D., Proc. Natl. Acad. Sci. USA 2004, 101, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang P-J. J., Vazin M., Matuszek Ż., Liu J., Nucleic Acids Res. 2015, 43, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou W., Zhang Y., Huang P-J. J., Ding J., Liu J., Nucleic Acids Res. 2016, 44, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saran R., Liu J., Anal. Chem. 2016, 88, 4014–4020. [DOI] [PubMed] [Google Scholar]
- 15. Schlosser K., Li Y., Chem. Biol. 2009, 16, 311–322. [DOI] [PubMed] [Google Scholar]
- 16. Kim H. K., Rasnik I., Liu J., Ha T., Lu Y., Nat. Chem. Biol. 2007, 3, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim H. K., Liu J., Li J., Nagraj N., Li M., Pavot C. M., Lu Y., J. Am. Chem. Soc. 2007, 129, 6896–6902. [DOI] [PubMed] [Google Scholar]
- 18. Pyle A. M., Science 1993, 261, 709–714. [DOI] [PubMed] [Google Scholar]
- 19. Yarus M., FASEB J. 1993, 7, 31–39. [DOI] [PubMed] [Google Scholar]
- 20. Torabis S. F., Wu P., McGhee C. E., Chen L., Hwang K., Zheng N., Cheng J., Lu Y., Proc. Natl. Acad. Sci. USA 2015, 112, 5903–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou W., Saran R., Chen Q., Ding J., Liu J., ChemBioChem 2016, 17, 159–163. [DOI] [PubMed] [Google Scholar]
- 22. Feig A. L., Uhlenbeck O. C., The RNA World, 2nd ed. (Eds.: J. Atkins, R. Gesteland, T. Cech), Cold Spring Laboratory Press, Cold Spring Harbor, NY, 1999, pp. 287–319. [Google Scholar]
- 23. Ke A., Zhou K., Ding F., Cate J. H., Doudna J. A., Nature 2004, 429, 201–205. [DOI] [PubMed] [Google Scholar]
- 24. Jimenez R. M., Polanco J. A., Lupták A., Trends Biochem. Sci. 2015, 40, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silverman S. K., Trends Biochem. Sci. 2016, 41, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hollenstein M., Molecules 2015, 20, 20777–20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jayasena V. K., Gold L., Proc. Natl. Acad. Sci. USA 1997, 94, 10612–10617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Z., Mei S. H., Brennan J. D., Li Y., J. Am. Chem. Soc. 2003, 125, 7539–7545. [DOI] [PubMed] [Google Scholar]
- 29. Ali M. M., Kandadai S. A., Li Y., Can. J. Chem. 2007, 85, 261–273. [Google Scholar]
- 30. Kandadai S. A., Li Y., Nucleic Acids Res. 2005, 33, 7164–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen Y., Brennan J. D., Li Y., Biochemistry 2005, 44, 12066–12076. [DOI] [PubMed] [Google Scholar]
- 32. Kandadai S. A., Mok W. W., Ali M. M., Li Y., Biochemistry 2009, 48, 7383–7391. [DOI] [PubMed] [Google Scholar]
- 33. Kasprowicz A., Stokowa-Sołtys K., Jeżowska-Bojczuk M., Wrzesiński J., Ciesiołka J., Dalton Trans. 2015, 44, 8138–8149. [DOI] [PubMed] [Google Scholar]
- 34. Ciesiołka J., Illangasekare M., Majerfeld I., Nickles T., Welch M., Yarus M., Zinnen S., Methods Enzymol. 1996, 267, 315–335. [DOI] [PubMed] [Google Scholar]
- 35. Łęgiewicz M., Wichłacz A., Brzezicha B., Ciesiołka J., Nucleic Acids Res. 2006, 34, 1270–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Velez T. E., Singh J., Xiao Y., Allen E. C., Wong O. Y., Chandra M., Kwon S. C., Silverman S. K., ACS Comb. Sci. 2012, 14, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Breaker R. R., Joyce G. F., Chem. Biol. 1995, 2, 655–660. [DOI] [PubMed] [Google Scholar]
- 38. Santoro S. W., Joyce G. F., Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bruesehoff P. J., Li J., Augustine A. J., Y, Lu , Comb. Chem. High Throughput Screening 2002, 5, 327–335. [DOI] [PubMed] [Google Scholar]
- 40. Lu Y., Liu J., Li J., Bruesehoff P. J., Pavot C. M. B., Brown A. K., Biosens. Bioelectron. 2003, 18, 529–540. [DOI] [PubMed] [Google Scholar]
- 41. Liu J., Brown A. K., Meng X., Cropek D. M., Istok J. D., Watson B. D., Lu Y., Proc. Natl. Acad. Sci. USA 2007, 104, 2056–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hollenstein M., Hipolito C., Lam C., Dietrich D., Perrin D. M., Angew. Chem. Int. Ed. 2008, 47, 4346–4350; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4418–4422. [Google Scholar]
- 43. Geyer C. R., Sen D., Chem. Biol. 1997, 4, 579–593. [DOI] [PubMed] [Google Scholar]
- 44. Roth A., Breaker R. R., Proc. Natl. Acad. Sci. USA 1998, 95, 6027–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reuter J. S., Mathews D. H., BMC Bioinformatics 2010, 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zuker M., Nucleic Acids Res. 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofacker I. L., Fontana W., Stadler P. F., Bonhoeffer S., Tacker M., Schuster P., Monatsh. Chem. 1994, 125, 167–188. [Google Scholar]
- 48. Xayaphoummine A., Bucher T., Isambert H., Nucleic Acids Res. 2005, 33, W605–W610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McManus S. A., Li Y., Molecules 2010, 15, 6269–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nelson K. E., Ihms H. E., Mazumdar D., Bruesehoff P. J., Lu Y., ChemBioChem 2012, 13, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duguid J., Bloomfield V. A., Benevides J., Thomas G. J., Biophys. J. 1993, 65, 1916–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Langlais M., Tajmir-Riahi H. A., Savoie R., Biopolymers 1990, 30, 743–752. [DOI] [PubMed] [Google Scholar]
- 53. Li Y., Breaker R. R., J. Am. Chem. Soc. 1999, 121, 5364–5372. [Google Scholar]
- 54. Kypr J., Kejnovská I., Renciuk D., Vorlícková M., Nucleic Acids Res. 2009, 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mazumdar D., Nagraj N., Kim H. K., Meng X., Brown A. K., Sun Q., Li W., Lu Y., J. Am. Chem. Soc. 2009, 131, 5506–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guéron M., Leroy J.-L., Curr. Opin. Struct. Biol. 2000, 10, 326–331. [DOI] [PubMed] [Google Scholar]
- 57. Elbaz J., Lioubashevski O., Wang F., Remacle F., Levine R. D., Willner I., Nat. Nanotechnol. 2010, 5, 417–422. [DOI] [PubMed] [Google Scholar]
- 58. Gray D. M., Liu J. J., Ratliff R. L., Antao V. P., Gray C. W., Struct. Express. 1987, 2, 147–166. [Google Scholar]
- 59. Manzini G., Yathindra N., Xodo L. E., Nucleic Acids Res. 1994, 22, 4634–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li J., Zheng W., Kwon A. H., Lu Y., Nucleic Acids Res. 2000, 28, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary