

Abstract
Persistent infections and amyloid disorders afflict a significant number of people worldwide. It would appear at first glance that the treatment of these afflictions should be entirely unrelated; however, in both cases components of the adaptive immune system have been harnessed in an attempt to provide some therapeutic relief. Given that the ability of a pathogen to establish persistence often depends in part on a shortcoming of the adaptive immune response, it seems logical to devise immunotherapies with the intention of supplementing (or replacing) the insufficient immunologic element. A case in point is an intervention referred as immunocytotherapy, which relies upon the adoptive transfer of pathogen-specific T lymphocytes into a persistently infected host. Remarkably, the adoptively transferred T lymphocytes not only have the capacity to clear the persistent infection, but can also provide the recipient with protection against subsequent rechallenge (i.e., immunologic memory). Treatment of amyloid disorders (e.g., Alzheimer disease, sporadic inclusion-body myositis) with a similar therapeutic approach is complicated by the fact that the aberrant protein accumulations are self-derived. Focusing the adaptive response on these aberrant self-proteins has the potential to result in autoimmune pathology. This review critically evaluates the importance of immunotherapeutic approaches for the treatment of persistent infections and amyloid disorders, and attempts to delineate the interventions that are most likely to succeed in an exceedingly complex disorder such as sporadic inclusion-body myositis.
Persistent infection and immunocytotherapy
Persistent viral infections profoundly impact the human population by adversely affecting health and by placing an ever-increasing economic burden on society. Consider the societal strains imposed by just two known persistent viral infections: hepatitis B virus (HBV) and HIV-1. Despite the existence of an efficacious vaccine for 20 years,1,2 the impact of HBV on society remains daunting. It is estimated that nearly 2 billion people worldwide have serological evidence (past or present) of an HBV infection. Moreover, 350 million people presently bear the burden of a persistent HBV infection, and approximately 1 million people per year succumb to associated complications, which include cirrhosis and hepatocellular carcinoma (HCC).3-5 HBV is capable of establishing persistence in approximately 5% to 10% of infected adults. However, these percentages are relatively low when compared to those observed during perinatal transmission of HBV. Infants born to HBV-positive women have a 70% to 90% risk of harboring a persistent HBV infection for life6 (presumably due to the induction of immunologic tolerance).
HIV-1, on the other hand, was identified in the early 1980s as the causative agent7 of the deadly AIDS virus, which has had a devastating impact on the global community, especially those residing in developing nations.8 Approximately 40 million people worldwide are infected with HIV-1,8 and the direct cost to treat one patient now exceeds $20,000 per year.9 Over the past two decades, considerable progress has been made in deciphering the life cycle and pathogenesis of HIV-1, which has spawned potent treatment modalities such as the highly active antiretroviral therapy (HAART).10 Nevertheless, HIV-1 still appears to have the upper hand, possessing the ability to persist latently in quiescent cells and render HAART ineffective in its pursuit to achieve complete viral eradication.11-13 To further complicate matters, HIV-1 also appears to infect the CNS to a similar degree observed in the lymphoid tissues (and with similar kinetics),14,15 and through an associated immunosuppression can open the flood gates to a number of opportunistic infections that include Toxoplasma gondii, Crytococcus neoformans, Epstein-Barr virus (EBV), JC virus, and cytomegalovirus. Each of these pathogens has the potential to establish persistence in the CNS and further promote unwanted neurologic complications.16,17
Based on the adverse effects associated with these two human pathogens alone, it could be argued that the hands of society carry a sizeable burden. However, these pathogens represent only two of the known persistent viral infections. The human population is faced with a myriad of other possibilities, which further add to the gravity of the problem. Given the magnitude of this seemingly insurmountable challenge, we as biomedical researchers must devise strategies to relieve patients of infections once they have established persistence. It is important that this statement be considered in its entirety. Relief from an established persistent infection should be considered in a category separate from the prevention of infection. For example, the discovery of vaccination represents a milestone in the maintenance of human health,18 and the administration of vaccines is usually an effective means to prevent a pathogen from establishing persistence. Once a pathogen has established persistence, the immune system often becomes overburdened and in many cases vaccination no longer represents a viable treatment strategy. Consequently, we are forced to consider other options such as pharmacologic inhibitors of a pathogen’s life cycle (e.g., HAART, ribavirin, acyclovir) and cytokines (e.g., type 1 interferons), which are therapies that do not routinely sterilize the host of the invading pathogen and can be associated with unwanted toxic side effects.
Another important problem in our pursuits to relieve patients of a persistent infection relates to the issue of how to proceed once a pathogen has gained access to the CNS. The CNS participates in nearly every aspect of our existence, from complex cognition to the metronome-like beating of our heart. Because the CNS factors so heavily into the equation that maintains our livelihood, it is important that this vital tissue compartment be equipped with a collection of protective mechanisms that limit potential sources of damage. Consequently, the CNS is fitted with an array of immune-dampening mechanisms that limit the toxicity (and in some cases the effectiveness) of a pathogen-specific immune response.19 This chink in the armor of our immunologic defenses enables a broad selection of pathogens to utilize the CNS as a safe haven.20 Unfortunately, the establishment of persistence within this safe haven can result in severe neurologic disturbances and impair nearly every aspect of our well-being.
Given the aforementioned challenges, it would appear that our plight to cleanse humans of a broad spectrum of pathogens that establish persistence in the CNS as well as the periphery has risen to the level of the unattainable. However, a series of seminal studies conducted in the lymphocytic choriomen-ingitis virus (LCMV) model system has revealed that it is possible to achieve the seemingly unattainable— total body clearance of a pathogen once it has established persistence.21 LCMV is a noncytopathic murine as well as human pathogen that has been instrumental in elucidating a number of fundamental concepts in virology and immunology.22 For example, a remarkable therapeutic approach (referred to as immunocytotherapy) was pioneered over four decades ago in the LCMV model system by Mogens Volkert and colleagues.23 Immunocytotherapy can be employed to completely cleanse a host of a persistent viral infection by reconstituting the cellular immune response. If mice are infected at birth or in utero with LCMV (referred to as LCMV carrier mice), the virus establishes lifelong persistence in every tissue compartment (e.g., spleen, thymus, lymph nodes, liver, lung, heart, kidney, CNS, etc.) (figure A).24 This route of transmission is analogous to the mode by which many viruses are passed from mother to offspring in the human population (e.g., HBV). Because LCMV establishes persistence in the thymus, T lymphocytes become tolerant to the virus, and thus, attempts to eliminate the pathogen through vaccination have been entirely unsuccessful.25 To further complicate matters, neurons are the predominant LCMV-infected cell population residing in the CNS parenchyma (figure B).26,27 Unless placed under extenuating circumstances (e.g., electrical inactivation),28 neurons do not readily express antigen-presenting machinery.29 In fact, one might question whether T lymphocytes are even capable of purging virus from neurons through direct interactions. Nevertheless, the immunocytotherapeutic administration of LCMV-specific memory T lymphocytes (both CD8+ and CD4+) obtained from syngeneic immune mice can completely eliminate virus from all tissue compartments (including the CNS neurons) of LCMV carrier mice following adoptive transfer.23,26 In this model system, it was determined that as few as 350,000 CD8+ and 7,000 CD4+ memory T lymphocytes are the minimum cellular requirements for total body clearance.30 It is important to emphasize that the success of this therapeutic intervention depends on the cooperative behavior of both T cell subsets—a point that will become pertinent later in this review. Moreover, clearance also depends on the ability of the transferred T lymphocyte population to produce the cytokine IFN-"),31,32 which might facilitate noncytopathic clearance of the pathogen.33
Figure.
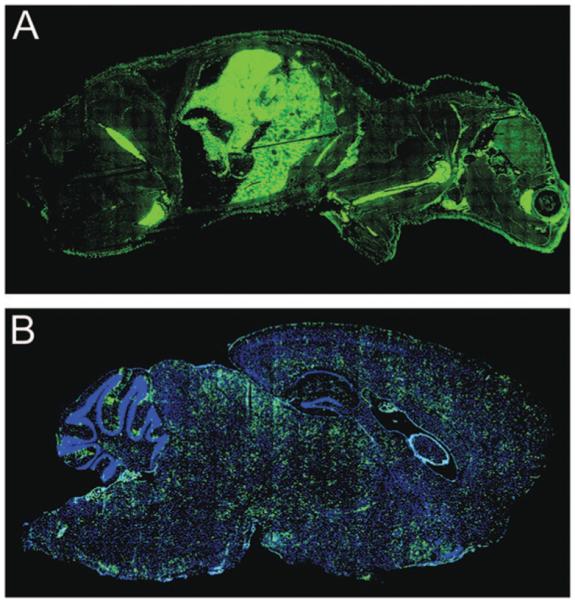
Systemic localization of LCMV in a carrier mouse persistently infected from birth. (A) Whole body distribution of LCMV (green) in a carrier mouse derived from persistently infected parents. The head of the mouse appears on the right side of the image. The virus was labeled using a polyclonal anti-LCMV antibody. Note the abundance of LCMV in nearly every tissue compartment. (B) An enlarged sagittal view of an LCMV carrier brain. Note the even distribution of LCMV (green) throughout the parenchyma. Neurons represent the primary infected cell population in the parenchyma. The meninges, choriod plexus, and ependyma are also infected. Nuclei are shown in blue.
At present our understanding of the precise mechanisms that underlie total body clearance in LCMV carrier mice following the administration of immunocytotherapy is at an elementary level. Although it might appear at first glance that the mode of clearance reflects nothing more than a potent antiviral immune response, consider the magnitude of the antigenic burden that the transferred memory T lymphocytes must face following transfer into LCMV carrier mice (figure). Nearly every tissue compartment is inundated with LCMV, and from this fact emerges a fundamental question: how does the transferred T lymphocyte population retain functionality in the presence of such high antigenic loads and at the same time clear virus without inducing a lethal immunopathology? Studies have convincingly demonstrated that a persistent viral infection can result in functional exhaustion of a primary pathogen-specific T cell response,34 thus rendering the response ineffective in its pursuits to clear the invading infectious agent. It is postulated that exposure to elevated/sustained antigenic loads achieved during a persistent viral infection is a major factor that contributes to this exhaustion, yet the transferred population of memory T lymphocytes fail to show signs of functional fatigue upon injection into the heavily burdened LCMV carrier mice. The transferred cells not only retain function, but also cleanse the infected mice without inducing a lethal immuno-pathology. It is well known that T lymphocytes (especially cytotoxic lymphocytes) possess the means to ward off an invading pathogen through the induction of target cell death.35 In fact, the fatal meningitis observed following an intracerebral injection of adult mice with LCMV occurs as a direct consequence of T cell-mediated pathology in the CNS.36 Thus, one of the most intriguing aspects of immunocytotherapy in LCMV carrier mice is that total body clearance can be attained without fatal immunopathologic consequences. This process becomes even more impressive when one considers that in addition to persistently infected neurons, the transferred population of memory T lymphocytes must also cleanse virus from the meninges, ependymal cells, and choriod plexus (figureB)—the very same structures targeted in adult mice that succumb to the aforementioned LCMV-induced meningitis.36
Given that an assemblage of cellular constituents can accomplish a monumental task—relief from a systemic viral infection to which the host is largely tolerant—it is important that we as biomedical researchers fully dissect (and ultimately improve upon) this remarkable process. A comprehensive understanding of the factors that underlie successful immunocytotherapy in the LCMV carrier model system may translate into more efficacious treatments for humans bearing the burden of a persistent infection. It is important to note that the success of this therapeutic intervention is not unique to the LCMV model system. Immunocytotherapy has been used to successfully cleanse patients of HBV,37 cytomegalovirus (CMV),38,39 EBV,40 and EBV-induced Hodgkin lymphoma.41 However, attempts to purge patients of HIV-1 were unsuccessful.42 HIV-1 patients received a monoclonal population of virus-specific cytotoxic lymphocytes (CTL) rather than a clonally diverse repertoire of CD8+ and CD4+ T cells—a strategy commonly employed in immunocytotherapeutic treatment trials. The administration of monoclonal CTL, which failed to cleanse patients of HIV-1, was also unsuccessful in the treatment of mice persistently infected with LCMV.43 When faced with elevated antigenic loads and a highly mutable pathogen, clonal diversity and T-cell cooperation become necessary elements of a successful therapy. It is known that CD4+ T cells are required to sustain the activities of CTL during the clearance of a persistent viral infection44; however, the mechanistic link between these two T cell subsets remains elusive. Immunocytotherapeutic treatment of LCMV carrier mice represents an ideal model system to uncover the nature of the cooperative behavior between these two subsets.
Meticulous examination of the immunotherapeutic process will undoubtedly elevate our basic understanding of memory T lymphocytes. T lymphocytes have the capacity to purge a pathogen from a cell in a noncytopathic manner,33 and it is worthwhile to theorize that memory T lymphocytes have evolved to rely more so on this mechanism of clearance to minimize tissue injury upon pathogen rechallenge. This could explain the total body clearance achieved in the LCMV model system without fatal immunopathologic consequences. Studies have also shown that successful clearance in LCMV carrier mice depends on the presence of an intact bone marrow compartment,45 which signifies that the transferred T lymphocyte population alone is incapable of mediating clearance. We have recently observed that immunocytotherapy induces a marked recruitment of professional antigen-presenting cells (APCs) into all tissue compartments (including the immunologically specialized CNS) of the recipient mice (unpublished observations). Therefore, the distinct possibility exists that interactions between tissue-infiltrating APCs and memory T lymphocytes result in the release of cytokines that noncytopathically purge virus from cells. An indirect, bystander mechanism of clearance would minimize the amount of damage that might occur as a consequence of direct cellular engagement. If this mechanism does indeed govern the success of immunocytotherapy, then it is conceivable that the administration of a potent antiviral factor could supplement an existing immunocytotherapeutic regimen and provide relief from a hard-to-treat pathogen. It is also possible that therapeutic manipulation of host APCs could improve upon clearance following immunocytotherapy. Only further experimentation will enable us to determine whether these possibilities can ultimately be realized.
Immunotherapy to treat amyloid disorders
Because the adaptive component of the immune system has an irreducible role in the clearance of an invading pathogen, it seems logical (and perhaps intuitive) to devise immunocytotherapy, an approach that relies upon the transfer of fully functional immune cells into a persistently infected host that it is lacking such cells. The transferred immune cells perform their natural functions in a timely manner and with a remarkable degree of efficiency. In fact, because memory cells possess the desired quality of self-renewal, they are maintained in the transfer recipient following the clearance phase and can provide protection against pathogen rechallenge or resurgence from a latent reservoir. Researchers are presently attempting to harness these advantageous qualities of the adaptive immune system in an effort to relieve patients of degenerative disorders that result from aberrant protein accumulation.46 This is best exemplified in the field of Alzheimer disease (AD) research.47 Patients with AD harbor CNS plaques comprised of β-amyloid (Aβ). Based on the working hypothesis that Aβ is the causative agent of AD, Schenk and colleagues immunized PDAPP transgenic mice (a mouse strain that develops amyloid plaques over time) with Aβ1-42 plus adjuvant.48 Remarkably, the therapeutic regimen significantly lowered plaque loads in aged mice. Based on the success of this intervention in mice, a clinical trial was initiated in humans with AD.49 Similar to the murine study, patients were immunized with Aβ1-42. Unfortunately, despite showing some promise, the trial was discontinued because 18 patients (6%) developed severe meningoencephalitis.50,51
In contrast to the immunocytotherapeutic protocols designed to treat persistent viral infections, reduction of plaque loads in AD models depends primarily on the humoral component of the adaptive immune response (rather than T lymphocytes). In fact, the T-lymphocyte response is thought to be responsible for the meningoencephalitis observed in the adversely impacted subset of patients.47 Consequently, researchers are now attempting to devise Aβ immunization strategies that promote antibody rather than T-lymphocyte responses. However, it should be noted that the immune system is a double-edged sword, and the redirection of an immune response toward a self-protein (in this case, Aβ) might always be fraught with undesired pathologic consequences in a subset of patients given the genetic diversity in the human population. In this subset of individuals, the very qualities of the immune system deemed beneficial for the treatment of a persistent viral infection become disadvantageous. For example, replication of a pathogen depends on genetic material that is separable from our own. In other words, the immune system has the capacity to eliminate proteinaceous as well as genetic material associated with a pathogen, and once this is accomplished, the pathogen-specific response abates and target cell engagement is no longer required. In the case of an amyloid disorder, the situation is quite different. Aβ is not derived from foreign genetic material but rather from a source that cannot be eliminated by the immune system. Consequently, the redirection of an adaptive immune response toward a self- (or altered self-) protein has the potential to become detrimental and uncontrollable if the source of the aberrant protein cannot be regulated. Thus, it seems that passive administration of an antibody directed against a self-protein is a much safer approach for the treatment of amyloid disorders. The antibody dose can be modified in each individual, and most importantly, the treatment can be discontinued immediately if adverse side effects become apparent.
Treatment of an amyloid disorder such as sporadic inclusion-body myositis (s-IBM) is even more complicated. Similar to AD, abnormal accumulation of Aβ can be found in the muscle of s-IBM patients, which suggests that passive administration of Aβ-specific antibodies might represent a viable treatment option. However, the efficacy of this therapeutic intervention will depend primarily on the modus operandi of Aβ-specific antibodies. Abnormal Aβ accumulation is an intracellular rather than extracellular phenomenon in s-IBM. In AD, it is thought Aβ-specific antibodies can directly bind to extracellular plaques and facilitate removal through an Fc receptor-mediated scavenging mechanism.52 If this is the predominant mode of action, then it is unlikely that Aβ-specific antibodies will be able to purge s-IBM patients of intracellular inclusions. However, it was also proposed in AD that Aβ-specific antibodies function by peripherally sequestering Aβ, thus limiting the formation of new plaques in the brain. Sequestration of Aβ with antibodies might represent an excellent means to thwart the progression of s-IBM if Aβ is an essential and causative element of disease pathogenesis.
Another mode of treatment in s-IBM patients emerges from the fact that a clonal and fully functional population of CTL can be found interacting with cells comprising the muscle.53 The presence of a clonal T-cell population in a degenerative disorder immediately gives rise to the assumption that the process is autoimmune in nature; however, it is equally probable that the CTL are reactive against a persistent pathogen that has sought refuge in the muscle. Indeed, there is precedence in the literature to support that individuals harboring a persistent viral infection (e.g., HIV-1 and HTLV-1) can present with s-IBM,54 yet it remains to be determined whether an infectious agent represents a causative element in the disease process. Given the established link between inflammatory metabolites and amyloidogenesis,55 it is plausible that chronic CTL engagement of muscles fibers triggers intracellular Aβ accumulation in s-IBM patients. If this is the case, then it becomes of the utmost importance to impede the inflammatory response. However, one must approach this quest with caution. It is not sensible to interfere with the inflammatory response if s-IBM patients harbor a persistent infection in the muscle. To do so would undoubtedly favor the pathogen. However, if s-IBM is an autoimmune disease, then blockade of immune function might provide some relief (assuming that the associated amyloidosis is not a self-perpetuating pathogenic process). The key to treatment may rest in defining the specificity of CTL clones isolated from s-IBM patients. Through the use of peptide libraries and protein databases, it should be possible to determine whether the CTL found in s-IBM patients are specific to a pathogen or a self-protein. Because treatment of persistent infection and autoimmune disease lie at the opposite ends of the spectrum, determination of CTL specificity could provide a much needed clue to unlock the etiology of the disease and direct clinicians toward the most appropriate therapeutic interventions.
Footnotes
Disclosure: The author reports no conflicts of interest.
References
- 1.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307:178–180. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 2.Michel ML, Pontisso P, Sobczak E, Malpiece Y, Streeck RE, Tiollais P. Synthesis in animal cells of hepatitis B surface antigen particles carrying a receptor for polymerized human serum albumin. Proc Natl Acad Sci U S A. 1984;81:7708–7712. doi: 10.1073/pnas.81.24.7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer. 1988;61:1942–1956. doi: 10.1002/1097-0142(19880515)61:10<1942::aid-cncr2820611003>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 4.Kane M. Global programme for control of hepatitis B infection. Vaccine. 1995;13(Suppl 1):S47–49. doi: 10.1016/0264-410x(95)80050-n. [DOI] [PubMed] [Google Scholar]
- 5.Alter MJ. Epidemiology and prevention of hepatitis B. Semin Liver Dis. 2003;23:39–46. doi: 10.1055/s-2003-37583. [DOI] [PubMed] [Google Scholar]
- 6.Xu ZY, Liu CB, Francis DP, et al. Prevention of perinatal acquisition of hepatitis B virus carriage using vaccine: preliminary report of a randomized, double-blind placebo-controlled and comparative trial. Pediatrics. 1985;76:713–718. [PubMed] [Google Scholar]
- 7.Barre-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 8.Valdiserri RO, Ogden LL, McCray E. Accomplishments in HIV prevention science: implications for stemming the epidemic. Nat Med. 2003;9:881–886. doi: 10.1038/nm0703-881. [DOI] [PubMed] [Google Scholar]
- 9.Bozzette SA, Berry SH, Duan N, et al. The care of HIV-infected adults in the United States. HIV Cost and Services Utilization Study Consortium. N Engl J Med. 1998;339:1897–1904. doi: 10.1056/NEJM199812243392606. [DOI] [PubMed] [Google Scholar]
- 10.Pomerantz RJ, Horn DL. Twenty years of therapy for HIV-1 infection. Nat Med. 2003;9:867–873. doi: 10.1038/nm0703-867. [DOI] [PubMed] [Google Scholar]
- 11.Chun TW, Carruth L, Finzi D, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 12.Finzi D, Hermankova M, Pierson T, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 13.Wong JK, Hezareh M, Gunthard HF, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 14.Resnick L, DiMarzo-Veronese F, Schupbach J, et al. Intra-blood-brain-barrier synthesis of HTLV-III-specific IgG in patients with neurologic symptoms associated with AIDS or AIDS-related complex. N Engl J Med. 1985;313:1498–1504. doi: 10.1056/NEJM198512123132402. [DOI] [PubMed] [Google Scholar]
- 15.Ho DD, Rota TR, Schooley RT, et al. Isolation of HTLV-III from cerebro-spinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Engl J Med. 1985;313:1493–1497. doi: 10.1056/NEJM198512123132401. [DOI] [PubMed] [Google Scholar]
- 16.McArthur JC. NeuroAIDS: diagnosis and management. Hosp Pract. 1997;32:73–84. doi: 10.1080/21548331.1997.11443542. [DOI] [PubMed] [Google Scholar]
- 17.Mamidi A, DeSimone JA, Pomerantz RJ. Central nervous system infections in individuals with HIV-1 infection. J Neurovirol. 2002;8:158–167. doi: 10.1080/13550280290049723. [DOI] [PubMed] [Google Scholar]
- 18.Plotkin SA. Vaccination against the major infectious diseases. C R Acad Sci III. 1999;322:943–951. doi: 10.1016/s0764-4469(00)87191-7. [DOI] [PubMed] [Google Scholar]
- 19.Wekerle H. Immune protection of the brain– efficient and delicate. J Infect Dis. 2002;186:S140–S144. doi: 10.1086/344937. [DOI] [PubMed] [Google Scholar]
- 20.Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol. 2003;3:493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homann D. Immunocytotherapy. Curr Top Microbiol Immunol. 2002;263:43–65. doi: 10.1007/978-3-642-56055-2_4. [DOI] [PubMed] [Google Scholar]
- 22.Borrow P, Oldstone MBA. Lymphocytic choriomeningitis virus. Lippincott-Raven; Philadelphia: 1997. [Google Scholar]
- 23.Volkert M. Studies on immunological tolerance to LCM virus. A preliminary report on adoptive immunization of virus carrier mice. Acta Pathol Microbiol Scand. 1962;56:305–310. [PubMed] [Google Scholar]
- 24.Fazakerley JK, Southern P, Bloom F, Buchmeier MJ. High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: relevance to arenavirus disease and mechanisms of viral persistence. J Gen Virol. 1991;72:1611–1625. doi: 10.1099/0022-1317-72-7-1611. [DOI] [PubMed] [Google Scholar]
- 25.vonHerrath MG, Berger DP, Homann D, Tishon T, Sette A, Oldstone MB. Vaccination to treat persistent viral infection. Virology. 2000;268:411–419. doi: 10.1006/viro.1999.0130. [DOI] [PubMed] [Google Scholar]
- 26.Oldstone MB, Blount P, Southern PJ, Lampert PW. Cytoimmunotherapy for persistent virus infection reveals a unique clearance pattern from the central nervous system. Nature. 1986;321:239–243. doi: 10.1038/321239a0. [DOI] [PubMed] [Google Scholar]
- 27.Oldstone MB. Immunotherapy for virus infection. Curr Top Microbiol Immunol. 1987;134:211–229. doi: 10.1007/978-3-642-71726-0_9. [DOI] [PubMed] [Google Scholar]
- 28.Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995;269:549–552. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- 29.Joly E, Mucke L, Oldstone MB. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 1991;253:1283–1285. doi: 10.1126/science.1891717. [DOI] [PubMed] [Google Scholar]
- 30.Berger DP, Homann D, Oldstone MB. Defining parameters for successful immunocytotherapy of persistent viral infection. Virology. 2000;266:257–263. doi: 10.1006/viro.1999.0074. [DOI] [PubMed] [Google Scholar]
- 31.Tishon A, Lewicki H, Rall G, VonHerrath M, Oldstone MB. An essential role for type 1 interferon-gamma in terminating persistent viral infection. Virology. 1995;212:244–250. doi: 10.1006/viro.1995.1477. [DOI] [PubMed] [Google Scholar]
- 32.Planz O, Ehl S, Furrer E, et al. A critical role for neutralizing-antibody-producing B cells, CD4(+) T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. Proc Natl Acad Sci USA. 1997;94:6874–6879. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 34.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 35.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Ann Rev Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 36.McGavern DB, Homann D, Oldstone MB. T cells in the central nervous system: the delicate balance between viral clearance and disease. J Infect Dis. 2002;186:S145–S151. doi: 10.1086/344264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lau GK, Suri D, Liang R, et al. Resolution of chronic hepatitis B and anti-HBs seroconversion in humans by adoptive transfer of immunity to hepatitis B core antigen. Gastroenterology. 2002;122:614–624. doi: 10.1053/gast.2002.31887. [DOI] [PubMed] [Google Scholar]
- 38.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 39.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 40.Heslop HE, Ng CY, Li C, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 41.Smith CA, Ng CY, Loftin SK, et al. Adoptive immunotherapy for Epstein-Barr virus-related lymphoma. Leuk Lymphoma. 1996;23:213–220. doi: 10.3109/10428199609054823. [DOI] [PubMed] [Google Scholar]
- 42.Brodie SJ, Lewinsohn DA, Patterson BK, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med. 1999;5:34–41. doi: 10.1038/4716. [DOI] [PubMed] [Google Scholar]
- 43.Moskophidis D, Laine E, Zinkernagel RM. Peripheral clonal deletion of antiviral memory CD8+ T cells. Eur J Immunol. 1993;23:3306–3311. doi: 10.1002/eji.1830231237. [DOI] [PubMed] [Google Scholar]
- 44.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jamieson BD, Butler LD, Ahmed R. Effective clearance of a persistent viral infection requires cooperation between virus-specific Lyt2+ T cells and nonspecific bone marrow-derived cells. J Virol. 1987;61:3930–3937. doi: 10.1128/jvi.61.12.3930-3937.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White AR, Hawke SH. Immunotherapy as a therapeutic treatment for neurodegenerative disorders. J Neurochem. 2003;87:801–808. doi: 10.1046/j.1471-4159.2003.02064.x. [DOI] [PubMed] [Google Scholar]
- 47.Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Curr Opin Immunol. 2004;16:599–606. doi: 10.1016/j.coi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 48.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 49.Bayer AJ, Bullock R, Jones RW, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 50.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 51.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 52.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 53.Dalakas MC. The future prospects in the classification, diagnosis and therapies of inflammatory myopathies: a view to the future from the “bench-to-bedside”. J Neurol. 2004;251:651–657. doi: 10.1007/s00415-004-0353-z. [DOI] [PubMed] [Google Scholar]
- 54.Cupler EJ, Leon-Monzon M, Miller J, Semino-Mora C, Anderson TL, Dalakas MC. Inclusion body myositis in HIV-1 and HTLV-1 infected patients. Brain. 1996;119:1887–1893. doi: 10.1093/brain/119.6.1887. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Q, Powers ET, Nieva J, et al. Metabolite-initiated protein misfolding may trigger Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:4752–4757. doi: 10.1073/pnas.0400924101. [DOI] [PMC free article] [PubMed] [Google Scholar]