

Abstract
Purpose
To investigate the role and significance of TP53 mutation in diffusely anaplastic Wilms tumor (DAWT).
Experimental Design
All DAWTs registered on National Wilms Tumor Study-5 (n=118) with available samples were analyzed for TP53 mutations and copy loss. Integrative genomic analysis was performed on 39 selected DAWTs.
Results
Following analysis of a single random sample, 57 DAWT (48%) demonstrated TP53 mutations, 13(11%) copy loss without mutation, and 48(41%) lacked both (defined as TP53-wildtype (wt)). Patients with Stage III/IV TP53-wt DAWTs (but not those with Stage I/II disease) had significantly lower relapse and death rates than those with TP53 abnormalities. In-depth analysis of a subset of 39 DAWT showed 7(18%) to be TP53-wt: these demonstrated gene expression evidence of an active p53 pathway. Retrospective pathology review of TP53-wt DAWT revealed no or very low volume of anaplasia in 6/7 tumors. When samples from TP53-wt tumors known to contain anaplasia histologically were available, abnormal p53 protein accumulation was observed by immunohistochemistry.
Conclusion
These data support the key role of TP53 loss in the development of anaplasia in WT, and support its significant clinical impact in patients with residual anaplastic tumor following surgery. These data also suggest that most DAWTs will show evidence of TP53 mutation when samples selected for the presence of anaplasia are analyzed. This suggests that modifications of the current criteria to also consider volume of anaplasia and documentation of TP53 aberrations may better reflect the risk of relapse and death and enable optimization of therapeutic stratification.
Keywords: p53, Wilms Tumor, anaplasia, pediatric cancers, Tumor staging: correlation of clinical and molecular markers
INTRODUCTION
The most common pediatric renal malignancy is Wilms tumor (WT) (1). Histopathologic categories of WT differing in their prognosis were first reported by Beckwith and Palmer, who identified the presence of anaplasia (defined as nuclear hyperchromasia and enlargement and atypical polyploid mitoses) in 5% of WTs (2). Subsequent studies sub-classified anaplastic WT into those with focal or diffuse anaplasia (DAWT), based on the circumscription and distribution of anaplasia. WTs lacking anaplasia are classified as favorable histology (FHWT) (3). The presence of anaplasia has been consistently associated with a poorer prognosis compared to FHWT, and the current therapeutic protocols continue to rely on these histologic classifications for treatment stratification (4–6). Anaplasia has long been considered to represent a clonal event within a WT that was originally of favorable histology. This concept has largely been based on the examination of anaplastic and non-anaplastic regions of the same tumor, in which TP53 mutation was identified only in the anaplastic region (7–9). Studies of small convenience cohorts have identified TP53 mutations in 50–86% of DAWT with the mutation frequency varying with the methods used and samples analyzed (8–10). The presence of TP53 mutations detected by immunopositivity (due to abnormal accumulation of mutant p53 protein) has also been associated with both anaplasia and with poorer prognosis in WTs (11, 12). Recently, 25/40 (62.5%) selected patients with DAWT were reported to show either TP53 mutation or copy loss; the 15 patients (37.4%) lacking both had a significantly better outcome (20% relapsed) than those with detectable TP53 abnormalities (56% relapsed, p=0.008), with similar differences in overall survival (13). However, correlations between stage, 17p13 abnormality, and outcome could not be analyzed. The current study addresses the prevalence of TP53 mutation and 17p13 copy loss in 118 DAWT registered in the National Wilms Tumor Study (NWTS)-5 cooperative group protocol and discusses the benefits and problems associated with the use of TP53 status to therapeutically stratify patients with DAWT. We accomplish this through participation with the National Cancer Institute’s “Therapeutically Applicable Research to Generate Effective Treatments” (TARGET) initiative which investigates high-risk pediatric tumors through comprehensive integrative genomics (http://ocg.cancer.gov/program/target). We then select 39 of these 118 patients for in-depth investigation by comprehensive integrative genomic analysis focusing on TP53 and relevant pathways.
EXPERIMENTAL PROCEDURES
This study was conducted as a part of the TARGET initiative. The raw sequencing data (BAM files) are deposited in the Sequence Read Archive (SRA) at the National Center for Biotechnology Information, and are accessible through the database of genotypes and phenotypes (dbGAP, http://www.ncbi.nlm.nih.gov/gap) under the accession number phs000471. The gene expression, chromosome copy number, as well as the results of sequence analysis (i.e. MAF and summary files) and clinical information on the cases studied is available through the TARGET Data Matrix (http://target.nci.nih.gov/dataMatrix/TARGET_DataMatrix.html), annotated within MIAME compliant MAGE-TAB files fully describing the methods, the specimen processing details, and the quality control parameters. A summary of the methods used in this study is provided below.
Specimens
All tumors registered on the NWTS-5 protocol that were classified as DAWT based on central pathology review and for which pre-therapy tumor was banked by the Children’s Oncology Group (COG) Biopathology Center (118 DAWTs), were evaluated by target capture sequencing and MLPA. For in-depth analysis, 39 of the 118 patients who also had sufficient normal DNA from peripheral blood or kidney and for which at least 50% of the slides demonstrated anaplasia were evaluated comprehensively. Frozen sections were evaluated and tumors with < 70% viable tumor cellularity were excluded. RNA and DNA were co-extracted from tissue using a modification of the DNA/RNA AllPrep kit (Qiagen). DNA was extracted from blood using the QiaAmp blood midi kit (Qiagen). Studies were performed with the approval of the Lurie Children’s Hospital Institutional Review Board.
Sequencing
For target capture sequencing, TP53 probes were designed using Agilent’s SureDesign online web-based tool (https://earray.chem.agilent.com/suredesign/) using RefSeq IDs limited to coding exons (chromosomal location 7571645-7590899). Each region was padded with an additional 10 bases on both the 5′ and 3′ ends. Probe density was specified at 2x and 98.7% of the target region was covered by a probe. Genomic DNA libraries were constructed according to BCCA-GSC plate-based and paired-end library protocols on a Biomek FX liquid handling robot (Beckman-Coulter, USA). The pooled libraries were hybridized to the RNA probes. Post-capture material was purified and enriched with 10 cycles of PCR. Paired-end 100 base reads were sequenced per pool in a single lane of an Illumina HiSeq2500 instrument. The predicted impact of the variants were annotated with the following algorithms: SIFT using Ensembl Variant Effect Predictor Version 75 (14), PolyPhen Version 2 (15), MutationTaster (16) or Provean Version 1.1.3 (17, 18).
Of the 39 cases analyzed in-depth, 21 underwent whole genome sequencing (WGS) and 18 whole exome sequencing (WES). WGS libraries were constructed and sequenced using Complete Genomics Inc. (CGI) technology (http://www.completegenomics.com/customer-support/documentation/162097975.html) (19). Variants were identified through a pipeline that is described on the TARGET DataMatrix]. The variants were filtered retaining all somatic, non-synonymous variants in exons with a somatic score >−10, somatic rank ≥ 0.1, and FET p <0.05. WES was performed on the Illumina HiSeq platform. Variant calling from the aligned BAM files was performed using both the ATLAS and SAMtools and annotation and filtering was performed using the SACBE annotation pipeline (20, 21). In addition, the WES BAM files were also analyzed using Bambino (22) and then combined. High-quality somatic, non-synonymous exonic variants from both platforms were identified and annotated using the Oncotator algorithm (http://www.broadinstitute.org/oncotator/).
For mRNA sequencing, following first and second strand cDNA synthesis, quantification, and quality verification, plate-based libraries were prepared following the BCCA-GSC paired-end protocol. The libraries were sequenced on the Illumina HiSeq 2000/2500 platform using v3 chemistry and HiSeq Control Software version 2.0.10, aligned to GRCh37-lite genome-plus-junctions (23) using BWA Version 0.5.7 (24). Variants were then detected on positive- and negative-split BAMs separately and annotated with SnpEff (25) (Ensembl 66) and SnpSift (26) (dbSNP137 and COSMIC64).
Copy Number Analysis
Within the 39 samples analyzed in-depth, nucleic acid labeling, hybridization, and array scanning were performed on tumor and corresponding normal samples according to the manufacturer’s protocol for the Affymetrix 6.0 SNP array. The resulting .cel files were processed using Affymetrix Genotyping Console (GTC) 4.0 software. Reference normalization utilized a diploid chromosome for each sample, as previously described (27). Circular binary segmentation (CBS) was performed in R using the DNAcopy BioConductor package. When Affy SNP 6.0 data was not available, copy number data were calculated based on the CGI relative coverage smoothed in 100-kb windows, corrected for the GC content, and normalized using composite baseline coverage from multiple healthy samples. The determination of areas of gain and loss was accomplished by importing segmented (Level 3) data into IGV (http://www.broadinstitute.org/igv/). Segmented regions containing at least 8 markers in which the log2 value was ≥ +0.5 or ≤ −0.5 were considered regions of gain or loss, respectively.
Within the 118 initial cases, analysis for 17p13 copy number changes was performed with Multiplex Ligation Probe Amplification (MLPA) using the SALSA MLPA Kit (MRC Holland), using methods previously described (28). The probes used are provided in Supplemental Table S4. The raw data peaks were visualized by using Genemarker to ensure the presence of distinct peaks; samples that passed these criteria were then normalized by using population normalization. Samples with TP53 peak values < 0.75 for two out of three TP53 probes were classified as having copy number loss.
Gene Expression Analysis
Total RNA was analyzed using the Affymetrix U133+2 chip, according to the manufacturer’s protocol. The arrays were scanned and analyzed using the Gene-Chip Operating Software (GCOS). Robust Multichip Average (RMA) normalization was performed using the ExpressionFileCreator module of the Broad Institute’s GenePattern server (http://www.broadinstitute.org/cancer/software/genepattern/); the data were collapsed by using the CollapseDataset module.
Statistical evaluation
Unsupervised analysis was performed using Non-negative Matrix Factorization Consensus Version 5 (NMF) (29). Comparisons of gene expression between two groups of samples were performed using Significance Analysis of Microarray 4.0 (30) following removal of probe-sets classified as absent in >95% of the samples analyzed; genes with SAM q-values of < 0.1 and Student’s t-test BH-corrected p values <0.05 were considered significant. Supervised hierarchical clustering was performed using the Hierarchical Clustering module of the Broad Institute’s Gene Pattern server (http://www.broadinstitute.org/cancer/software/genepattern/). Gene Set Enrichment Analyses, version 2.0.14, (http://www.broadinstitute.org/gsea) (31) was used to perform a comparison between tumors with or without TP53 abnormalities by using the c2 Biocarta module (v5). Analyses were run using 1000 permutations, phenotype permutation, and default basic and advanced fields. We used the default GSEA settings (excluding genes sets with <15 genes or >500 genes). Significant enrichment was defined as those lists with FDR <20%, and a p-value <5%. Time to relapse or progression was characterized by the Kaplan-Meier curve of Disease Free Survival (DFS). The log-rank test was used to compare the difference in DFS between DAWTs with TP53 mutation and/or copy loss and DAWTs without either. Statistical significance was assessed at the 0.05 level.
RESULTS
TP53 mutations and copy number changes in 118 DAWTs
To determine the overall frequency of TP53 mutation in unselected DAWTs, we identified all patients with DAWT registered on the NWTS-5 protocol for which tumor samples were available (n=118), and analyzed these for TP53 copy number alterations using multiplex ligation probe amplification (MLPA) and for variants in TP53 using target capture sequencing that covered the coding region (chromosomal location 7571645-7590899). Variants were retained that had a total tumor read count >10, alternative base count in the tumor compared to reference genome >3 with an allelic fraction ≥ 0.05, and a minor allele frequency ≤0.005 in the relevant population in dbSNP142. TP53 variants were identified in 57/118 DAWTs (48%), with four tumors showing two different TP53 variants. The details of each variant are provided in Supplemental Table S1. All single nucleotide variants (SNVs) were present in COSMIC version 76 and were evaluated as damaging by at least one algorithm (see methods). Of note, 21/61 (34%) of the TP53 mutations involved the exon 10 tetramerization motif, with 13 involving p.R342 (five c.1024C>T nonsense mutations and eight c.1025G>C missense mutations); one nonsense mutation removed the tetramerization domain entirely (p.R306*). Four TP53 variants were splice site mutations and the remaining mutations were distributed over or just outside the DNA binding domain (Figure 1). Given the prior association between Wilms tumor and constitutional TP53 splice site mutations in patients with LiFraumeni syndrome (32–34), we sequenced available DNA samples from blood or normal kidney of 2 out of the 4 patients with somatic splice site mutations and did not find the variants in the germline. All TP53 variants were verified by comprehensive sequencing of tumor DNA or mRNA (see below).
Figure 1. TP53 Mutations in DAWTs.

Illustrated is the location of the TP53 mutations (n=61) within the known p53 protein domains. The number of variants detected for each is provided in the parenthesis. TAD = Transcription-Activation Domain; DNABD = DNA binding domain; TM = Tetramerization Domain.
Loss of TP53 (17p13) was analyzed by MLPA in 116/118 tumors with available tumor DNA. Of the 53 tumors with a single TP53 mutation, 42 (79%) also showed loss of one copy of 17p13, 9 (17%) lacked 17p13 loss, and 2 lacked available DNA. Seven of the nine tumors with TP53 mutations lacking 17p13 loss demonstrated a mutant TP53 allelic fraction of >75%, providing evidence for loss of the chromosome segment harboring the wild type TP53 and duplication of the chromosome segment containing the mutant allele. Thus, of the 55 fully evaluable tumors carrying TP53 mutations (including four tumors with two different TP53 mutations (compound heterozygotes)), only two are predicted to have retained one wildtype allele. Thirteen of the 61 TP53 mutation-negative DAWTs had loss of 17p13. Overall 48/118 DAWT (41%) lacked TP53 mutation and/or copy loss (defined as TP53-wt). The mean age at diagnosis of all 118 DAWTs was 58 months (range 8.4–184.8 months). This is higher than the reported mean age at diagnosis for FHWTs (42 months) (35). There was no significant difference in gender, predominant histologic pattern (blastemal, epithelial, stromal, or mixed), or nephrogenic rest status between FHWTs and DAWTs. Clinical and pathologic features and the presence or absence of copy number changes and TP53 mutations are provided in Supplemental Table S2.
Relapse occurred in 34/118 patients (29%; median time to relapse 313 days). All patients who relapsed died, 33 from disease and 1 from toxicity of therapy. Eight additional patients showed disease progression despite therapy and all died of disease. The remaining patients (n=76) did not relapse and are alive at a mean of 2378 days follow-up (range 301–4520 days, Supplemental Table S2). When analyzed by stage, there was no overt difference in prevalence of TP53 abnormalities by stage (Table 1). Those patients with stage I–II disease showed no difference in outcome based on TP53 status; however those patients with stage III or IV disease and TP53 abnormalities in their tumors showed a significantly worse disease free and overall survival compared with those that were TP53-wt (p=0.0006 and p=0.0007, respectively) (Table 1, Figure 2).
Table 1.
Clinical Outcome by Stage and TP53 status
| Disease Stage | N (% of stage group) | relapse or progression | Death from disease |
|---|---|---|---|
|
| |||
| Stage I (n=14) | |||
|
|
|||
| TP53 abnormality | 8 (57%) | 3 | 2 |
|
| |||
| No TP53 abnormality | 6 (43%) | 2 | 1 |
|
| |||
| Stage II (n=37) | |||
|
|
|||
| TP53 abnormality | 18 (48.6%) | 4 | 4 |
|
| |||
| No TP53 abnormality | 19 (51.4%) | 3 | 3 |
|
| |||
| Stage III (n=57) | |||
|
|
|||
| TP53 abnormality | 39 (68.4%) | 24 | 24 |
|
| |||
| No TP53 abnormality | 18 (31.6%) | 3 | 3 |
|
| |||
| Stage IV (n=10) | |||
|
|
|||
| TP53 abnormality | 5 (50%) | 3 | 3 |
|
| |||
| No TP53 abnormality | 5 (50%) | 0 | 0 |
Figure 2. Association between stage, TP53 status and Disease Free Survival.

Kaplan-Meier curve of Disease Free Survival (DFS) (in years), comparing DAWTs with TP53 mutation and/or copy loss and DAWTs without either. Numbers at risk are listed at the bottom of the graphs. All patients who relapsed or progressed also died.
Integrative In-Depth Analysis of 39 DAWT
Comprehensive genomic analysis of 39 of the above 118 DAWT was performed (see Supplemental Tables S1 and S2 for specific samples examined). Included were DAWTs with sufficient case-matched peripheral blood or normal kidney samples and whose tumor demonstrated anaplasia in at least 50% of reviewed slides (regardless of the volume of anaplasia in each slide), as recorded at the time of central pathology review. Characterization included either whole genome sequencing (WGS; n=21) or whole exome sequencing (WES n=18) as well as global chromosome segment copy number analysis (Affymetrix 6.0), gene expression analysis (Affymetrix U133+2), and mRNA sequencing (mRNA seq).
Recurrent mutations previously reported in FHWT were also identified in a minority of DAWT including those involving WT1 (1), WTX (1), CTNNB1(1), DROSHA (1), DGCR8 (1), and SIX1(1) (Supplemental Table S3). TP53 was the only gene with somatic mutations more highly represented in DAWTs than in FHWTs (sequencing and verification of all TARGET high risk Wilms tumors will be reported in full when the analysis is completed). Twenty seven high-quality, somatic, non-synonymous variants in TP53 (transcript NM_001126112) were identified in 25(64%) of these 39 DAWT by either WGS or WES (Supplemental Table S1). Variants in other genes within the p53 pathway were not identified. Of the 39 cases, 37 had sufficient RNA to perform mRNAseq. Two additional cases with TP53 mutations were identified by mRNAseq, but not by WGS (PAJMRL) or WES (PAKXXF). Unfiltered variants detected by WGS and WES were also evaluated for TP53 mutations that failed filtering; one additional TP53 mutation was identified (PALLCK), for a total of 30 TP53 mutations in 28 of the 39 DAWT. These three additional TP53 variants were verified by target capture (above).
DNA chromosomal segment analysis
Segmental loss of 17p13 including the TP53 locus was identified in 25/28 DAWTs containing mutant TP53. In 21/25 of these DAWTs the complete chromosome 17 short arm was lost. The remaining three tumors with TP53 mutations, but lacking copy loss, either had two different TP53 mutations (PAKFYV) or demonstrated a mutant allelic fraction >95% and segmental uniparental disomy of 17p (PAJLKC, PADXAY), consistent with loss of the normal allele and duplication of the mutant allele. Of the 11 DAWTs lacking TP53 sequence variants, loss of one allele was identified in four tumors (three showing loss restricted to the short arm and one showing loss of the entire chromosome 17). One of these tumors (PAKUIT) demonstrated an additional small segment of loss (defined by 23 SNPs) involving the 3′ end of TP53, resulting in homozygous loss of this region. Normal 17p13 copy number was present in seven cases, and they lacked evidence of uniparental disomy of 17p13. Therefore, 7/39 DAWT were identified as TP53-wildtype (TP53-wt). Of note, neither 17p loss nor TP53 mutation was identified in any of the 77 TARGET FHWT comprehensively analyzed (36).
Segmental loss at 11p13 (the location of WT1) was identified in six DAWTs, and segmental loss at Xq11.2 (the location of WTX) was identified in eight cases (Supplemental Table S3); these proportions are similar to what was seen in FHWTs (37). The seven TP53-wt DAWT showed fewer total copy number alterations in their genomes (defined as segments with ≥ 8 markers and log2 ratio ≥ +0.5 or ≤ −0.5) when compared to the DAWTs carrying TP53 mutations and/or copy loss. In fact, TP53-wt DAWT showed a distribution of segmental gains and losses similar to the 77 TARGET FHWTs previously reported (36). DAWTs with TP53 abnormalities had 169 ± 226 segments of gain or loss, compared with TP53-wt DAWTs which had 55 ± 51 segments of gain or loss (p=0.016), and with FHWTs which had 47 ± 52, (p=0.006) segments of gain or loss. In addition to loss of 17p, the most common copy number changes in DAWTs with TP53 abnormalities included loss of 4q (identified in 69% (22/32) DAWT with TP53 abnormalities, 14% (1/7) TP53-wt DAWT, and 8% (2/77) FHWT), loss of 14q (66% (21/32) DAWT with TP53 abnormalities, 14% (1/7) TP53-wt DAWTs, and 11.7% (9/77) FHWT), loss of 16q (62.5% (20/32) DAWT with TP53 abnormalities, 28.6% (2/7) TP53-wt DAWTs, and 26% (20/77) FHWT), and loss of chromosome 22 (56% (18/32) DAWT with TP53 abnormalities, 14% (1/7) TP53-wt DAWTs, and 12% (9/77) FHWT) (Figure 3A, Supplemental Table S3). These findings are similar to previous reports of DAWT (37, 38). Of note, gain of 1q was identified in 50% (16/32) of DAWT with TP53 abnormalities, comparable with 51% (39/77) identified in relapsed FHWT (18), similar to previous reports (31). There was a lower rate of 1q gain in TP53-wt DAWT (2/7, 28.6%), likewise consistent with the previously reported data from FHWT (31) considering that this group was not selected for relapse status. Of note, none of the DAWTs demonstrated amplification of 12q15, a region encompassing the MDM2 gene which encodes an E3 ubiquitin ligase that targets p53 for degradation (39).
Figure 3. Comparison of Copy number variations and gene expression in DAWTs with and without TP53 abnormalities.

A. Copy number variations (CNVs; defined as segments with ≥ 8 markers and log2 ratio ≥ +0.5 or ≤ −0.5) identified in DAWTs with TP53 abnormalities (top panel) or without TP53 abnormalities (bottom panel). Each row represents one DAWT. Segments in red represent CN loss and those in blue CN gain. The seven TP53-wt samples showed fewer copy number alterations in their genome when compared to the DAWTs carrying TP53 mutations and/or copy loss, and are similar to those previously reported in FHWTs. The tumors with TP53 abnormalities had recurrent loss of chromosomes 4q, 14q, 16q, 17p, and 22.
B. Hierarchical analysis of genes differentially expressed in TP53 mutant DAWT:
Statistical Analysis for Microarray (SAM) comparing the seven TP53-wt tumors with the remaining 31 tumors identified 35 genes differentially expressed (SAM q<0.10 and Student’s t-test BH corrected p value < 0.05). Hierarchical analysis of these 35 genes shows all to be upregulated in the tumors lacking TP53 mutation or 17p13 copy number loss (red is upregulation and blue indicates downregulation). These include several genes located on 17p, and other genes associated within apoptotic pathways known to be regulated by p53 (such as BAX and CDKN1A).
Gene Expression Analysis
Unsupervised analysis of the 38 tumors for which gene expression data was available did not show clustering of the TP53-wt DAWT. Supervised analysis comparing the seven TP53-wt DAWT with the remaining 31 tumors showing TP3 mutation and/or copy loss using Statistical Analysis for Microarray (SAM) identified 35 differentially expressed genes (SAM q<0.10 and Student’s t-test BH corrected p value < 0.05). Hierarchical analysis of these 35 genes (Figure 3B) shows all to be upregulated in tumors lacking TP53 abnormalities. Nineteen are located on 17p11-13. In addition, genes within apoptotic pathways known to be positively regulated by p53 (such as BAX and CDKN1A) were upregulated. Notably, TP53 itself was not significantly transcriptionally upregulated in TP53-wt tumors compared to the remaining 31 tumors, because some TP53-mutant tumors express relatively high levels of the mutant mRNA. To identify canonical pathways and biologic functions associated with TP53-wt tumors, we performed Gene Set Enrichment Analysis Version 2.0.14 (GSEA) (31) using the same comparison groups. Two gene sets were significantly enriched (FDR< 20%), including genes involved in the Biocarta p53 (p=0.012, FDR=0.190) and FAS apoptosis (p=0.004, FD=0.19) pathways. In summary, analysis of gene expression patterns supports an active p53 pathway within the TP53-wt tumors.
Histology and p53 Immunohistochemistry
It has long been recognized that tumors with TP53 mutations often (but not always) show nuclear accumulation of the abnormal protein (40, 41). Immunohistochemistry is widely used clinically to document the presence of TP53 mutation in many tumor types due to its speed, low cost, and ability to assess localized areas showing features suspicious for anaplasia. We therefore examined the available TP53-wt tumors for abnormal protein accumulation by p53 immunohistochemistry using the pre-diluted DO7 antibody (Ventana catalogue # 790-2912, Tucson, AZ). It is important to note that for the COG biology studies, frozen and formalin-fixed tissue samples are randomly taken by the local pathologist prior to histologic evaluation. The sample that is frozen is commonly taken from a different area than the sample placed in formalin. For six of the seven TP53-wt tumors that underwent in-depth molecular analyses, a randomly submitted formalin-fixed sample was submitted to the COG Biopathology Center (BPC). Unequivocal anaplasia was present histologically in 3/6 of these samples and each of these three tumors demonstrated unequivocal evidence of abnormal nuclear p53 protein accumulation in the regions of anaplasia (Figure 4). The individual blocks available from the BPC for the remaining three cases did not show histologic evidence of anaplasia, and did not show abnormal protein accumulation. This suggests that for the three evaluable TP53-wt DAWTs, the failure to detect TP53 abnormalities in the frozen samples results from failure to sample tissue containing the anaplastic clone. Lastly, retrospective analysis of the full set of slides from the seven TP53-wt tumors demonstrated five to show a very low volume of anaplastic cells (<1%). This varied highly in pattern, with some showing small foci of anaplastic cells in a small number of slides and others showing individual quite rare individual anaplastic cells in most slides. One of these six cases was evaluated by three pathologists, who were not able to confidently make the diagnosis of anaplasia. The only TP53-wt DAWT to show appreciable volumes of anaplasia (PAJMLZ) had stage II disease, relapsed and died.
Figure 4. Abnormal protein accumulation by p53 immunohistochemistry.
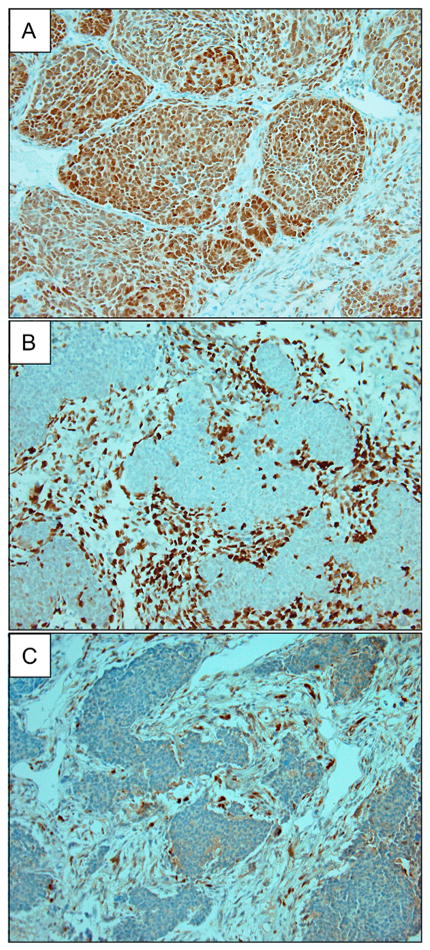
Archival tissue blocks containing histologic evidence of anaplasia were available for three of the seven DAWTs determined to be TP53-wt by the absence of TP53 mutation and copy number loss in a single randomly selected frozen tumor sample (the remaining four cases did not have archival tissue within the biopathology center containing clear histologic evidence of anaplasia). (Original magnification 40X for all.)
A) PAJMLZ: The block containing anaplasia shows diffuse abnormal p53 protein accumulation.
B) PALEZT: The block contains anaplastic cells that were stromal that surrounded non-anaplastic blastemal cells. Immunohistochemistry shows nodules of blastemal cells that were largely negative for p53 protein, with anaplastic stromal cells containing marked accumulation of p53 protein.
C) PAJNRH: The block had a microscopic focus of anaplasia, which shows intense abnormal nuclear p53 staining.
DISCUSSION
TP53, the most commonly mutated gene in human cancer, encodes a transcription factor that regulates the expression of genes involved in cell cycle, senescence and apoptosis. Until recently, sequencing of the TP53 gene has largely relied on assessment of the highly conserved DNA binding domain (exons 5–8), explaining why most mutations (>80%) in Cosmic have been reported in these domains. Such studies underestimate TP53 mutations identified in WTs, a third of which we demonstrate to occur in exon 10, the tetramerization domain. Many mutant p53 proteins evade degradation and accumulate to high levels that are therefore able to be detected by immunohistochemistry. As a result, p53 immunohistochemistry is often used as an alternative simple and low cost surrogate for detecting TP53 mutations in tumors. However, not all TP53 mutations result in abnormal protein accumulation (such as many nonsense mutations). It is therefore not surprising that there is a considerable lack of correlation between p53 immunopositivity and TP53 mutation detected historically by sequencing (41, 42). This lack of consistency has also been demonstrated in anaplastic WTs (8, 11).
We report TP53 mutations detected by sequencing alone to occur with an overall frequency of 48% in 118 unselected patients with DAWT registered on a single cooperative group protocol, and TP53 segmental copy loss without mutation was identified in another 11%. While the majority of mutations involved the TP53 DNA binding domain, a high proportion (34%) occurred in the tetramerization motif, also known as the oligomerization domain. Mutations in this domain have previously been shown to result in defective transcription, ubiquitylation,and protesome mediated degradation (43–45). WTs are infrequently associated with Li-Fraumeni syndrome (LFS), and 5 out of 6 reported patients with both LFS and WT had TP53 splice site mutations (46). While we identified four DAWTs with splice site mutations, no evidence of constitutional TP53 mutations was identified the two with available normal samples.
To better understand the nature of TP53-wt DAWTs, we performed an in-depth genomic analysis of 39 DAWT. We identified mutations in WT1, WTX, CTNNB1, DROSHA, DGCR8, and SIX1/2 in patients with DAWTs, supporting the concept that TP53 mutation occurs as a second event within WTs of multiple underlying genetic causes. While the majority (82%) of these 39 tumors demonstrated TP53 abnormalities (mutation and/or loss), seven DAWT were TP53-wt. We did not identify any mutations in other genes within the p53 pathway in TP53-wt DAWT, or in any other genes with known biologic relevance. Gene expression analysis documented an intact p53 pathway and did not provide evidence of disruption of another pathway in TP53-wt DAWTs. By gene expression and segmental copy number analysis, TP53-wt DAWTs were not distinguishable from a previously reported larger group of FHWTs that were analyzed in the same manner (36). Immunohistochemical analysis of all three evaluable DAWTs that were TP53-wt by sequencing (performed on a randomly selected frozen sample) displayed unequivocally abnormal p53 protein accumulation in an archival sample from a different area of the tumor (again randomly selected) that contained histologic evidence of anapasia. Lastly, retrospective histologic analysis of the original set of H&E slides from all 7 TP53-wt DAWT (by sequencing and copy number alone) demonstrated all but one to have no or a low volume of anaplasia. Of note, in 1/39 tumors the diagnosis of anaplasia was not definitive upon retrospective review by 3 pathologists. Establishing a confident diagnosis of anaplasia (based on nuclear enlargement, nuclear hyperchromasia, and atypical mitoses) by histology alone is difficult, due to the fact that neither nuclear enlargement nor atypical mitoses are able to be evaluated in a dichotomous fashion with full confidence. Tumors borderline for these criteria have been classified as “nuclear unrest”, and have been shown to be most appropriately grouped with FHWT based on clinical and pathologic features (47).
The data acquired during this in-depth, integrative analysis supports the hypothesis that for TP53-wt DAWTs the frozen tissue that underwent comprehensive genomic analysis contained no or low volume of anaplastic cells. By gene expression and copy number analysis this sample was comparable with FHWTs. The diagnosis of DAWT is established by a pathologist after analyzing multiple representative slides taken from all regions of the tumor. In contrast, tissue samples for banking or biologic studies are taken without knowledge of the histology and are often limited to a small number of samples. This results in a variety of possible outcomes, as illustrated in Figure 5. Conceptually, clonal development within a FHWT resulting from TP53 mutation may occur within an undifferentiated cell that may then proliferate in a discohesive manner throughout much of the WT, resulting in scattered individual anaplastic cells that may be quite difficult to detect both histologically and molecularly (Figure 5A). Anaplasia in such tumors is by current definition classified as diffuse, despite the very small percentage of anaplastic cells. In some tumors these cells may differentiate and grow cohesively resulting in scattered nodules of anaplastic cells (Figure 5B). Well-defined nodules of anaplastic cells are easier to identify, depending on their size.
Figure 5. Tumor heterogeneity and sample selection in detecting TP53 abnormalities.

The predominant underlying cause of failure to detect TP53 abnormalities is heterogeneity in tumors with a low volume of anaplasia. The diagnosis of DAWT is established by a pathologist after analyzing multiple representative slides taken from all regions of the tumor. In contrast, single randomly selected tissue samples are taken for banking or biologic studies without knowledge of the histology. In some cases anaplasia may be diffuse, but may still be due to a very small percentage of cells, making it difficult to detect TP53 abnormalities. If there are scattered nodules of anaplasia, area(s) of anaplasia may not be selected; or there might be only a very small focus of anaplasia within the sample selected, insufficient for the detection of TP53 abnormalities.
The most intriguing finding of this study is that stage III and IV DAWTs containing TP53 abnormalities within a randomly selected sample had a relapse and a death rate of 61%, compared to a 13% relapse and death rate in DAWT that were determined to be TP53-wt in a randomly selected sample. Stage I and II DAWTs did not show a significant impact of TP53 abnormality on outcome. These data together with the finding that 6/7 TP53-wt tumors had a very low volume of anaplasia suggests that the absence or presence of TP53 abnormalities is a surrogate for the overall extent of TP53 abnormalities within the tumor, in turn reflecting the burden of anaplasia. The burden of anaplasia may have less impact on outcome for stage I and II tumors, which are completely resected, than for stage III and IV tumors, which by definition have a high risk of residual tumor following nephrectomy. Our observations therefore raise the important question of whether or not the diagnostic criteria of anaplasia need to be re-evaluated, or alternatively if stratification based on p53 status is appropriate. Unfortunately, analysis of the copy number and sequencing data within the 70 patients containing TP53 abnormalities indicates that neither method alone is sufficiently sensitive to be clinically useful. Sequencing alone missed 13/70 (19%, those DAWTs in which only 17p13 copy loss was detected), and copy number analysis alone missed 13/70 (19%, those DAWT with TP53 mutations involving both alleles). This demonstrates that both modalities (ie sequencing and copy number analysis of TP53) would need to be performed in order to rely on TP53 status to guide therapy. Platforms utilizing next-generation sequencing that enable the analysis of both are rapidly becoming available. Due to the retrospective nature of this study, and to our reliance on material available from the BPC, we were unable to perform immunohistochemistry on anaplasia-containing tumor for all cases and therefore were unable to document its sensitivity.
In conclusion, there is an opportunity for refining our diagnostic and/or therapeutic strategy for DAWTs based on TP53 status. However, two issues need to be addressed. First, the analysis of TP53 abnormalities performed on random sections of DAWT is insufficient. A better alternative would be for the institutional pathologist to identify a block (or a microdissected region) that optimally demonstrates anaplasia, and to submit this block or unstained slides for evaluation by p53 immunohistochemistry, 17p13 copy loss, and/or TP53 sequencing. Second, a new classification system for anaplasia needs to be developed that better takes into account the location and burden of anaplasia; this must show acceptable intra- and inter-reviewer reliability testing and must correlate with outcome better than the current classification. Given these two changes, an algorithm could then be evaluated that allows for the identification of the most cost-effective method of stratification that is also able to be performed within an optimal time frame.
Lastly, the available wealth of information regarding TP53 mutation in WTs may provide important lessons regarding the implementation of precision medicine overall. First, despite the sophisticated tools now available, there are limitations to our ability to detect mutations that likely exist outside of the protein coding region (for example, it is possible if not likely that at least some of the DAWTs with 17p13 copy number loss alone have undetected variants impacting TP53 directly or indirectly). More importantly, our ability to recognize the mutant anaplastic clone by histology enabled the full evaluation of the data. This ability is not known to exist for the vast majority of other mutations that may be associated with differences in prognosis. Therefore, in the absence of histologic clues, when attempting to implement precision medicine by analyzing pre-therapy diagnostic tumor samples, we are in many ways limited to pathogenetic clonal events that exist in the vast majority of cells analyzed, rather than progression-related events such as TP53 mutation, which may also be important determinants of prognosis. Relapse samples, on the other hand, have undergone selection, and similar analyses using these samples warrant prioritization as they will provide important information regarding both pathogenetic genetic events as well as subsequent progression-related events.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Anaplasia in Wilms Tumor (WT) has long been associated with TP53 mutation, although determining its significance has been limited by both methodology and the patient populations analyzed. We show 41% of 118 prospectively identified DAWT to lack TP53 abnormalities by sequencing or copy number analysis performed on a random sample. This lack was associated with 1) no or a very low volume of anaplastic cells in the entire tumor, 2) evidence of abnormal TP53 protein accumulation (consistent with mutation) in other samples of the same tumor selected for the presence of anaplasia, and 3) a significantly lower relapse rate compared to those with TP53 abnormalities in in patients with stage III or IV disease. Future studies that evaluate volume and location of anaplasia and confirm TP53 abnormalities may enable optimized therapeutic stratification and thereby avoid toxic therapy in some children.
Acknowledgments
The TARGET initiative is supported by NCI Grant U10 CA98543. Work performed under contracts from the NCI, US NIH within HHSN261200800001E includes specimen processing (the COG Biopathology Center), WGS (CGI, Inc.), WES (Baylor College of Medicine), miRNAseq, RNAseq, and target capture sequencing (BCCA Genome Sciences Center). The content of this publication is solely the responsibility of the authors and does not necessarily reflect the views or policies of the National Institutes of Health or the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The authors thank Patee Gesuwan and Leandro Hermida and the Data Coordinating Center for their support. We also thank the Clinical Applications of Core Technology Laboratory of the Hartwell Center for Bioinformatics and Biotechnology of St. Jude Children’s Research Hospital for performing the copy number analysis, and the Northwestern University Genomic Core facility for performing the methylation analysis. The authors are grateful for the expertise of Karen Novik and Laura Monovich, Patricia Beezhold, Donna Kersey, Debbie-Payne Turner, Mary McNulty, and Yvonne Moyer. This work would not be possible without the dedication of all the experts within the many clinical disciplines at the local institutions and centrally within the COG and NWTSG.
Financial support:
TARGET U10 CA98543 contract HHSN261200800001E (see acknowledgements); The Children’s Oncology Group award numbers NIH U10CA180886, NIH U10CA180899; NIH U10CA098413; NIH U10CA42326 (EJP); U10CA98543 (JSD, EJP); U24 CA114766; UO1CA88131 (EJP); NCI T32 CA079447, (ALW); Dutch Cancer Society (AO); American and Lebanese Syrian Associated Charities of St. Jude (CGM, JM).
Footnotes
The authors disclose no potential conflicts of interest
References
- 1.Howlader NNA, Krapcho M, Garshell J, Miller D, Altekruse SF, et al. In: SEER Cancer Statistics Review, 1975–2012. Cronin KA, editor. National Cancer Institute; 2014. posted to the SEER website, April 2015. [Google Scholar]
- 2.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41(5):1937–48. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. Epub 1978/05/01. [DOI] [PubMed] [Google Scholar]
- 3.Faria P, Beckwith JB, Mishra K, Zuppan C, Weeks DA, Breslow N, et al. Focal versus diffuse anaplasia in Wilms tumor--new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. The American journal of surgical pathology. 1996;20(8):909–20. doi: 10.1097/00000478-199608000-00001. Epub 1996/08/01. [DOI] [PubMed] [Google Scholar]
- 4.Dome JS, Cotton CA, Perlman EJ, Breslow NE, Kalapurakal JA, Ritchey ML, et al. Treatment of anaplastic histology Wilms’ tumor: results from the fifth National Wilms’ Tumor Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(15):2352–8. doi: 10.1200/jco.2005.04.7852. Epub 2006/05/20. [DOI] [PubMed] [Google Scholar]
- 5.Pritchard-Jones K, Moroz V, Vujanic G, Powis M, Walker J, Messahel B, et al. Treatment and outcome of Wilms’ tumour patients: an analysis of all cases registered in the UKW3 trial. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2012;23(9):2457–63. doi: 10.1093/annonc/mds025. Epub 2012/03/15. [DOI] [PubMed] [Google Scholar]
- 6.Dome JSPE, Graf N. Risk Stratification for Wilms Tumor: Current Approach and Future Directions ASCO. 2014. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Soffer SZ, Kim ES, Yokoi A, Moore JT, McCrudden KW, et al. p53 accumulation in favorable-histology Wilms tumor is associated with angiogenesis and clinically aggressive disease. Journal of pediatric surgery. 2002;37(3):523–7. doi: 10.1053/jpsu.2002.30858. Epub 2002/03/06. [DOI] [PubMed] [Google Scholar]
- 8.Bardeesy N, Beckwith JB, Pelletier J. Clonal expansion and attenuated apoptosis in Wilms’ tumors are associated with p53 gene mutations. Cancer research. 1995;55(2):215–9. Epub 1995/01/15. [PubMed] [Google Scholar]
- 9.Bardeesy N, Falkoff D, Petruzzi MJ, Nowak N, Zabel B, Adam M, et al. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nature genetics. 1994;7(1):91–7. doi: 10.1038/ng0594-91. Epub 1994/05/01. [DOI] [PubMed] [Google Scholar]
- 10.Malkin D, Sexsmith E, Yeger H, Williams BR, Coppes MJ. Mutations of the p53 tumor suppressor gene occur infrequently in Wilms’ tumor. Cancer research. 1994;54(8):2077–9. Epub 1994/04/15. [PubMed] [Google Scholar]
- 11.Lahoti C, Thorner P, Malkin D, Yeger H. Immunohistochemical detection of p53 in Wilms’ tumors correlates with unfavorable outcome. The American journal of pathology. 1996;148(5):1577–89. Epub 1996/05/01. [PMC free article] [PubMed] [Google Scholar]
- 12.Franken J, Lerut E, Van Poppel H, Bogaert G. p53 Immunohistochemistry expression in Wilms tumor: a prognostic tool in the detection of tumor aggressiveness. The Journal of urology. 2013;189(2):664–70. doi: 10.1016/j.juro.2012.09.115. Epub 2012/10/06. [DOI] [PubMed] [Google Scholar]
- 13.Maschietto M, Williams RD, Chagtai T, Popov SD, Sebire NJ, Vujanic G, et al. TP53 mutational status is a potential marker for risk stratification in Wilms tumour with diffuse anaplasia. PloS one. 2014;9(10):e109924. doi: 10.1371/journal.pone.0109924. Epub 2014/10/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics (Oxford, England) 2010;26(16):2069–70. doi: 10.1093/bioinformatics/btq330. Epub 2010/06/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. Epub 2010/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods. 2014;11(4):361–2. doi: 10.1038/nmeth.2890. Epub 2014/04/01. [DOI] [PubMed] [Google Scholar]
- 17.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS one. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. Epub 2012/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choy Y, editor. A Fast Computation of Pairwise Sequence Alignment Scores Between a Protein and a Set of Single-Locus Variants of Another Protein. ACM Conference on Bioinformatics, Computational Biology and Biomedicine; 2012; Orlando (FL), USA. New York (NY), USA. 2012. [Google Scholar]
- 19.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science (New York, NY) 2010;327(5961):78–81. doi: 10.1126/science.1181498. Epub 2009/11/07. [DOI] [PubMed] [Google Scholar]
- 20.Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome medicine. 2013;5(2):11. doi: 10.1186/gm415. Epub 2013/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome medicine. 2013;5(6):57. doi: 10.1186/gm461. Epub 2013/06/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics (Oxford, England) 2011;27(6):865–6. doi: 10.1093/bioinformatics/btr032. Epub 2011/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morin R, Bainbridge M, Fejes A, Hirst M, Krzywinski M, Pugh T, et al. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTechniques. 2008;45(1):81–94. doi: 10.2144/000112900. Epub 2008/07/10. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. Epub 2009/05/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. doi: 10.4161/fly.19695. Epub 2012/06/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Frontiers in genetics. 2012;3:35. doi: 10.3389/fgene.2012.00035. Epub 2012/03/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pounds S, Cheng C, Mullighan C, Raimondi SC, Shurtleff S, Downing JR. Reference alignment of SNP microarray signals for copy number analysis of tumors. Bioinformatics (Oxford, England) 2009;25(3):315–21. doi: 10.1093/bioinformatics/btn624. Epub 2008/12/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gratias EJ, Jennings LJ, Anderson JR, Dome JS, Grundy P, Perlman EJ. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children’s Oncology Group. Cancer. 2013;119(21):3887–94. doi: 10.1002/cncr.28239. Epub 2013/08/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(12):4164–9. doi: 10.1073/pnas.0308531101. Epub 2004/03/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. Epub 2001/04/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. Epub 2005/10/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlegelberger B, Kreipe H, Lehmann U, Steinemann D, Ripperger T, Gohring G, et al. A child with Li-Fraumeni syndrome: Modes to inactivate the second allele of TP53 in three different malignancies. Pediatric blood & cancer. 2015;62(8):1481–4. doi: 10.1002/pbc.25486. Epub 2015/03/20. [DOI] [PubMed] [Google Scholar]
- 33.Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20(34):4621–8. doi: 10.1038/sj.onc.1204621. Epub 2001/08/11. [DOI] [PubMed] [Google Scholar]
- 34.Varley JM, Chapman P, McGown G, Thorncroft M, White GR, Greaves MJ, et al. Genetic and functional studies of a germline TP53 splicing mutation in a Li-Fraumeni-like family. Oncogene. 1998;16(25):3291–8. doi: 10.1038/sj.onc.1201878. Epub 1998/07/29. [DOI] [PubMed] [Google Scholar]
- 35.Gadd S, Huff V, Huang CC, Ruteshouser EC, Dome JS, Grundy PE, et al. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children’s Oncology Group Study. Neoplasia (New York, NY) 2012;14(8):742–56. doi: 10.1593/neo.12714. Epub 2012/09/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer cell. 2015;27(2):286–97. doi: 10.1016/j.ccell.2015.01.003. Epub 2015/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams RD, Al-Saadi R, Natrajan R, Mackay A, Chagtai T, Little S, et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes, chromosomes & cancer. 2011;50(12):982–95. doi: 10.1002/gcc.20907. Epub 2011/09/02. [DOI] [PubMed] [Google Scholar]
- 38.Wittmann S, Zirn B, Alkassar M, Ambros P, Graf N, Gessler M. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes, chromosomes & cancer. 2007;46(2):163–70. doi: 10.1002/gcc.20397. Epub 2006/11/14. [DOI] [PubMed] [Google Scholar]
- 39.Xu C, Fan CD, Wang X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene. 2015;34(3):281–9. doi: 10.1038/onc.2013.557. Epub 2014/01/15. [DOI] [PubMed] [Google Scholar]
- 40.Beniers AJ, Efferth T, Fuzesi L, Granzen B, Mertens R, Jakse G. p53 expression in Wilms’ tumor: a possible role as prognostic factor. International journal of oncology. 2001;18(1):133–9. Epub 2000/12/15. [PubMed] [Google Scholar]
- 41.Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih Ie M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24(9):1248–53. doi: 10.1038/modpathol.2011.85. Epub 2011/05/10. [DOI] [PubMed] [Google Scholar]
- 42.Watanabe G, Ishida T, Furuta A, Takahashi S, Watanabe M, Nakata H, et al. Combined Immunohistochemistry of PLK1, p21, and p53 for Predicting TP53 Status: An Independent Prognostic Factor of Breast Cancer. The American journal of surgical pathology. 2015;39(8):1026–34. doi: 10.1097/pas.0000000000000386. Epub 2015/07/15. [DOI] [PubMed] [Google Scholar]
- 43.Chene P. The role of tetramerization in p53 function. Oncogene. 2001;20(21):2611–7. doi: 10.1038/sj.onc.1204373. Epub 2001/06/23. [DOI] [PubMed] [Google Scholar]
- 44.Lang V, Pallara C, Zabala A, Lobato-Gil S, Lopitz-Otsoa F, Farras R, et al. Tetramerization-defects of p53 result in aberrant ubiquitylation and transcriptional activity. Molecular oncology. 2014;8(5):1026–42. doi: 10.1016/j.molonc.2014.04.002. Epub 2014/05/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lubin DJ, Butler JS, Loh SN. Folding of tetrameric p53: oligomerization and tumorigenic mutations induce misfolding and loss of function. Journal of molecular biology. 2010;395(4):705–16. doi: 10.1016/j.jmb.2009.11.013. Epub 2009/11/17. [DOI] [PubMed] [Google Scholar]
- 46.Scott RH, Stiller CA, Walker L, Rahman N. Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. Journal of medical genetics. 2006;43(9):705–15. doi: 10.1136/jmg.2006.041723. Epub 2006/05/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hill DA, Shear TD, Liu T, Billups CA, Singh PK, Dome JS. Clinical and biologic significance of nuclear unrest in Wilms tumor. Cancer. 2003;97(9):2318–26. doi: 10.1002/cncr.11325. Epub 2003/04/25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.