

Abstract
In prostate cancer, the development of castration resistance is pivotal in progression to aggressive disease. However, understanding of the pathways involved remains incomplete. In this study, we performed a high-throughput genetic screen to identify kinases that enable tumor formation by androgen-dependent prostate epithelial (LHSR-AR) cells under androgen-deprived conditions. In addition to the identification of known mediators of castration resistance, which served to validate the screen, we identified a mitotic-related serine/threonine kinase, NEK6, as a mediator of androgen-independent tumor growth. NEK6 was overexpressed in a subset of human prostate cancers. Silencing NEK6 in castration-resistant cancer cells was sufficient to restore sensitivity to castration in a mouse xenograft model system. Tumors in which castration resistance was conferred by NEK6 were predominantly squamous in histology with no evidence of AR signaling. Gene expression profiling suggested that NEK6 overexpression stimulated cytoskeletal, differentiation and immune signaling pathways and maintained gene expression patterns normal decreased by castration. Phosphoproteome profiling revealed the transcription factor FOXJ2 as a novel NEK6 substrate, with FOXJ2 phosphorylation associated with increased expression of newly identified NEK6 transcriptional targets. Overall, our studies establish NEK6 signaling as a central mechanism mediating castration-resistant prostate cancer.
Keywords: NEK6, prostate cancer, castration resistant, hormone refractory, androgen independent
INTRODUCTION
Prostate cancer is the second most common cause of cancer death in men, and the majority of these deaths occur in patients with metastatic castration-resistant prostate cancer (CRPC). Studies of metastatic tissue from these patients reveal that tumors can become resistant either through persistent activation of the AR pathway (1), or by progression to a state described as androgen pathway independent prostate cancer (APIPC) (2) with or without evidence for neuroendocrine transdifferentiation.
Constitutive activation of kinases such as ERBB2, MAPK, PI3K/Akt, and Src has been implicated in mediating resistance to hormonal therapies through both AR-dependent and AR-independent mechanisms (3). However, inhibitors of many of these kinases have failed to demonstrate significant clinical benefit in unselected patient populations, such as in trials of the ERBB2 inhibitor lapatinib (4), the MTOR inhibitor everolimus (5), and the SRC inhibitor dasatinib (6). Cabozantinib, a small molecule inhibitor of MET and VEGFR2, exhibited clinical activity in metastatic CRPC (7) but a Phase III trial failed to demonstrate improvement in overall survival (8).
Tumor samples from patients with CRPC show increased levels of tyrosine phosphorylation as compared to treatment naïve prostate cancer (9), and the analysis of serine/threonine and tyrosine phosphorylation events in metastatic CRPC demonstrated a number of kinases that are differentially expressed or activated in prostate cancer metastases (10). However, there is limited evidence for activating kinase point mutations in CRPC (11), suggesting that kinase pathways are activated by other (structural genetic, epigenetic, microenvironmental) mechanisms. Kinase signaling has previously been implicated in treatment resistance in other cancer types through re-activation of the targeted signaling pathway, activation of parallel signaling pathways and through inducing transdifferentiation phenotypes (12); kinases conferring resistance are obvious druggable targets for potential therapeutic intervention. Since kinase signaling pathways whose activation could lead to castration resistance in patients with prostate cancer have not been comprehensively catalogued, we have performed an in vivo functional genomic screen to identify novel pathways that may be involved and identified NEK6 as a kinase that mediates androgen independent tumor growth.
MATERIALS AND METHODS
Cell Lines and Antibodies
LH, LHSR-AR, and LHMK-AR cells (13) were cultured in Prostate Epithelial Basal Medium (PrEBM) with Prostate Epithelial Growth Medium (PrEGM) SingleQuot Kit Supplements & Growth Factors (Lonza). LNCaP, C4-2, CL-1 and DU145 were cultured in phenol red free RPMI (Gibco) supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate and 10 mM HEPES. LAPC4 cells were cultured in IMDM supplemented with 10% FBS and 1nM R1881. VCaP and PC-3 cells were cultured in DMEM:F12 (1:1) with L-glutamine (Hyclone) + 10% FBS. RWPE-1 cells were cultured in Keratinocyte-SFM + EGF and bovine pituitary extract (Gibco).
Antibodies used for immunoblotting were: NEK6 (AbCam ab76071 for Figure 1C, Santa Cruz sc-134833 for Supplemental Figure S1A, AbCam ab133494 otherwise), AR (Santa Cruz, sc-7305), Hsp90 (Cell Signaling 4875), beta-actin (Santa Cruz sc-47778 HRP), V5 (Invitrogen P/N 46-0708), FOXJ2 (LifeSpan LS-C31294), AKT1 (Cell Signaling 9272), RAF1 (BD Biosciences 610151), p-T412-RPS6KB1 (Cell Signaling 9234), p-S727-STAT3 (GeneTex 61050), cleaved PARP (Cell Signaling 9541), p53 (Cell Signaling 9282), p21 (Santa Cruz sc-397).
Figure 1.
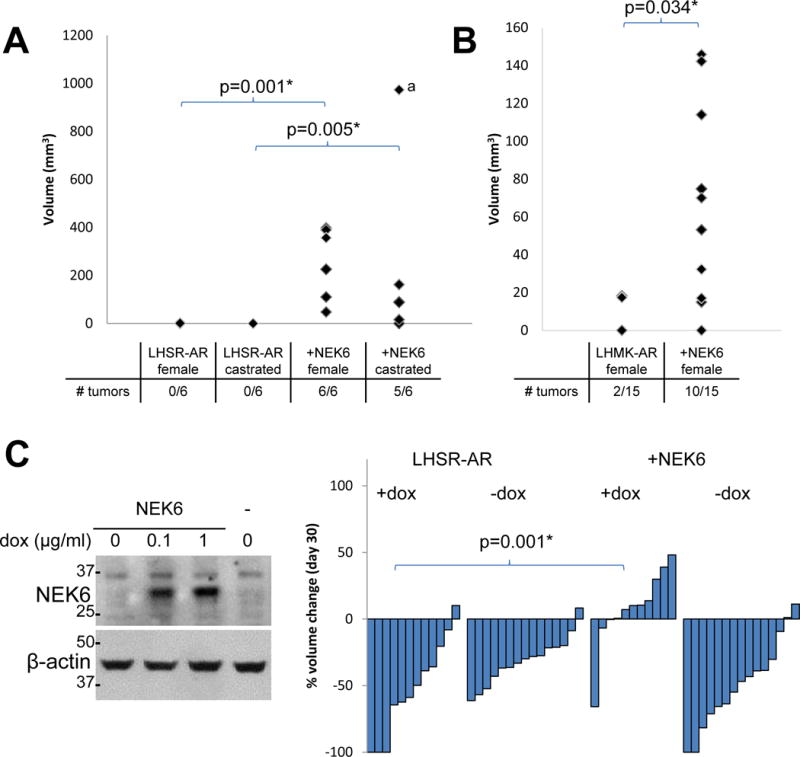
NEK6 confers androgen-independent tumor formation in a xenograft model of androgen-dependent prostate cancer. A. Effects of NEK6 expression on the rate of tumor formation at 60 d in the indicated LHSR-AR cells in female and castrated mice. Of note, if the tumor denoted with a is excluded, the difference in rate of tumor formation between parental LHSR-AR cells and cells expressing NEK6 in castrated mice remains statistically significant (p=0.009). B. The effects of expressing NEK6 on the rate of tumor formation at 60 d for parental LHMK-AR cells and cells expressing NEK6 in female mice. C. Left: Inducible expression of NEK6 in vitro at 48 hrs after addition of doxycycline Right: Waterfall plot of change in tumor volume at 30 d after castration (compared to prior to castration) of xenograft tumors derived from parental LHSR-AR cells and cells with inducible NEK6 expression formed in male mice.
Kinase Library and Androgen Independence Screen
LHSR-AR cells were infected with each virus from the previously described human kinase library (14) in 96-well format; cells from 9–10 of these wells were pooled together to generate 61 total pools. 2×106 cells from each pool in 200 μl 1:1 PBS:matrigel (Corning 354234) were implanted into each of 3 subcutaneous sites of a female BALB/C nude mouse (Charles River, Boston, MA). DNA was isolated from tumors using the Qiagen Blood Spin Mini Kit. For tumor formation in male mice, mice were implanted with testosterone pellet (Innovative Research NA-151) into the lateral neck; pellets were removed at the time of castration. For castration, testes were ligated and removed through a lower abdominal incision. Doxycycline diet at 625 ppm (Test Diet) was fed to mice for target dose of 0.6 – 2 mg/kg.
Tumor Processing and Gene Expression analysis
Tumors were harvested fresh and fragments were immediately fixed in 10% neutral buffered formalin solution (Sigma) for paraffin embedding or flash frozen in liquid nitrogen and stored in −80° C. Protein lysates were generated by mechanical disruption in RIPA buffer (Cell Signaling) and passage through a QiaShredder column; RNA was isolated using the Qiagen RNeasy purification kit with passage through a QiaShredder column and on-column DNase digestion per manufacturer’s protocol. RNA-Seq, comparative marker selection using GENE-E software, and GSEA pre-ranked were performed as described in the Supplemental Methods.
Immunohistochemistry
Prostate cancer microarray slides immunostained for NEK6 were scanned and scored by automated spectral imaging analysis per the Supplemental Methods. Paraffin embedded subcutaneous tumor sections were immunostained using anti-AR (Dako # M3562, dilution 1:200, 1 hr incubation, citrate and microwave retrieval, EnVision detection. BioGenex platform); anti beta-catenin (Santa Cruz sc-1496, dilution 1:100, T3 H1(30), Leica platform), anti-KRT13 (GeneTex EPR3671), anti-NEK6 (as for TMAs but at 1:100 dilution).
Phosphoproteomic analysis
LHSR-AR cells transduced with constructs for doxycycline-inducible expression of wild-type (wt) NEK6 and kinase-dead (kd) NEK6 were cultured in “light,” “medium,” or “heavy” SILAC media in two different permutations of the three conditions of wt +dox, wt −dox (uninduced), and kd +dox. Cells were growth factor starved and then growth factor stimulated for 5 min; protein lysates were subjected to SCX/IMAC and mass spectrometry as previously described (15). Detailed procedures are described in the Supplemental Methods.
RNA interference, Proliferation Assessment, and Cell Cycle Profiling
Inducible suppression of NEK6 was accomplished by using the pLKO-TetOn-puro lentiviral vector with the shRNA sequences listed in the Supplemental Methods. Proliferation assays were performed by plating 100,000 cells in 3 × 60 mm plates at day 0; at the designated time points cells were harvested and counted by Coulter counter (3 measurements from each of 3 plates), and 100,000 cells were replated in 3 new 60 mm plates. For cell cycle profiling after growth factor withdrawal, LHSR-AR cells were growth factor starved for 28 hours, followed by media change to complete PrEGM, and cells were harvested at the designated times points for propidium iodide staining and flow cytometry using a previously described protocol (http://fccf.salk.edu/protocols/cellcycle.php).
ORF constructs and in vitro kinase assays
Details on the ORF constructs and site-directed mutagenesis protocols used in these experiments are found in the Supplemental Methods. For in vitro kinase assays, expression constructs of putative substrates with a C-terminal V5 tag were transfected into 293T cells using TransIT®-LT1 Transfection Reagent (Mirus); cell pellets were lysed and proteins immunoprecipitated with anti-V5 agarose affinity gel (Sigma A7345) in RIPA buffer for on-bead kinase assay using active recombinant GST-NEK6 (Sigma N9788) per manufacturer’s protocol using 0.1 μl GST-NEK6 per reaction.
Study Approval
All patients with tumors included in the tissue microarrays provided written informed consent to allow the collection of tissue and blood and the analysis of clinical and genetic data for research purposes (DFCI Protocol #01–045). All xenograft studies were conducted under the guidelines of the Dana-Farber Cancer Institute (DFCI) Animal Research Facility, and were approved by the DFCI Institutional Care and Use Committee (IACUC) under Protocol #03–013.
RESULTS
Identification of NEK6 as a mediator of castration-resistance in vivo
Previous work in our laboratory (13) demonstrated that primary prostate epithelial cells (PrECs) that are rendered tumorigenic by the expression of the SV40 large T and small t antigens, the catalytic subunit of telomerase (hTERT), activated H-Ras, and the androgen receptor (LHSR-AR cells) form well-differentiated prostate epithelial tumors in mice. These tumors are androgen-dependent and are thus unable to grow in female or castrated mice, unlike xenografts derived from the prostate cancer cell lines VCaP and LNCaP, which were derived from patients with CRPC and thus do not demonstrate robust differential tumor formation capability in non-castrate vs. castrate mice in vivo. Here we performed a high throughput, in vivo genetic screen to identify kinases that permitted LHSR-AR cells to form tumors in female animals. A lentivirally delivered kinase ORF library (14) encompassing 601 kinases and other oncogenes was introduced into these cells in pools of 9–10, and we identified fourteen ORF integrants that allowed for the androgen-independent development of subcutaneous tumors by PCR using vector specific primers (Supplemental Table S1).
These 14 candidates were then introduced into LHSR-AR cells individually and implanted subcutaneously in female BALB/c nude mice for validation of androgen-independent tumor formation. Among the candidates that reproducibly yielded androgen-independent tumors were mutated KRAS; RAF1, a known prostate cancer oncogene (16) that was recently described to confer metastatic potential in an in vivo model (10); ERBB2, AKT1, and constitutively active MEK1, which have been implicated in androgen independence (3); and PIM1 and PIM2, which have previously been demonstrated to be important oncogenes in prostate cancer (17, 18). In addition, we found that a wild-type form (NP_055212.2) of the Never In Mitosis A (NIMA) related kinase 6 (NEK6) scored strongly in this assay.
In addition to conferring tumor formation in female mice, overexpression of NEK6 in LHSR-AR cells also led to tumor formation in castrated mice (Figure 1A), which lack circulating androgens since mice do not synthesize androgens from their adrenal glands (19). In addition, we tested whether NEK6 overexpression can lead to androgen-independent tumor formation in a different genetic context, that is in immortalized PrECs transformed by overexpression of the MYC oncogene and expression of an active form of the p110α subunit of PI3 kinase (rather than with small t antigen and active HRAS), termed LHMK-AR cells (13). Indeed, NEK6 overexpression led to tumor formation in this other genetic context as well (Figure 1B). The kinase activity of NEK6 was essential for conferring castration resistance, as a kinase-dead formof NEK6, K74M/K75M (20), failed to lead to tumor formation of LHSR-AR cells in female or castrated mice (Supplemental Figure S1A).
In addition to demonstrating the role of NEK6 in androgen-independent tumor formation, we determined whether NEK6 confers castration resistance to established tumors by generating LHSR-AR cells in which NEK6 expression is induced with doxycycline (Figure 1C, left). After xenograft tumor formation in male mice in the presence of doxycycline for 35 days, the mice were castrated and the tumors were monitored for response. We found that NEK6 expressing tumors continued to grow even in castrated mice as compared to control tumors derived from parental LHSR-AR cells (p=0.001). However, when NEK6 overexpression was abrogated by doxycycline withdrawal at the time of castration, the resulting tumors were as sensitive to castration as the control tumors. These observations demonstrate that NEK6 can lead to castration resistance in established tumors, and suggest that therapeutic targeting of NEK6 can restore sensitivity to castration in tumors where its activity is increased.
The tumors formed due to NEK6 overexpression mostly lacked AR expression, except for in small regions of the tumors (Figure 2A). Hematoxylin and eosin (H&E) staining demonstrated squamous differentiation in these tumors, with the more mature differentiated regions demonstrating keratin deposition and AR loss. Androgen-independent tumors derived from expression of the other kinases identified in the screen also have regions of AR-positivity and negativity that vary in proportion and intensity (Supplemental Table S2). NEK6 overexpressing tumors established in male mice demonstrate strong nuclear AR staining with no squamous differentiation (Figure 2B, left panels). However, after castration, these tumors lose nuclear AR over time and develop more keratinization between nests of tumor cells (Figure 2B, bottom panels). This observation suggests that NEK6-mediated castration resistance does not require AR activity, and that transdifferentiation to a squamous phenotype is a result of or response to castration.
Figure 2.
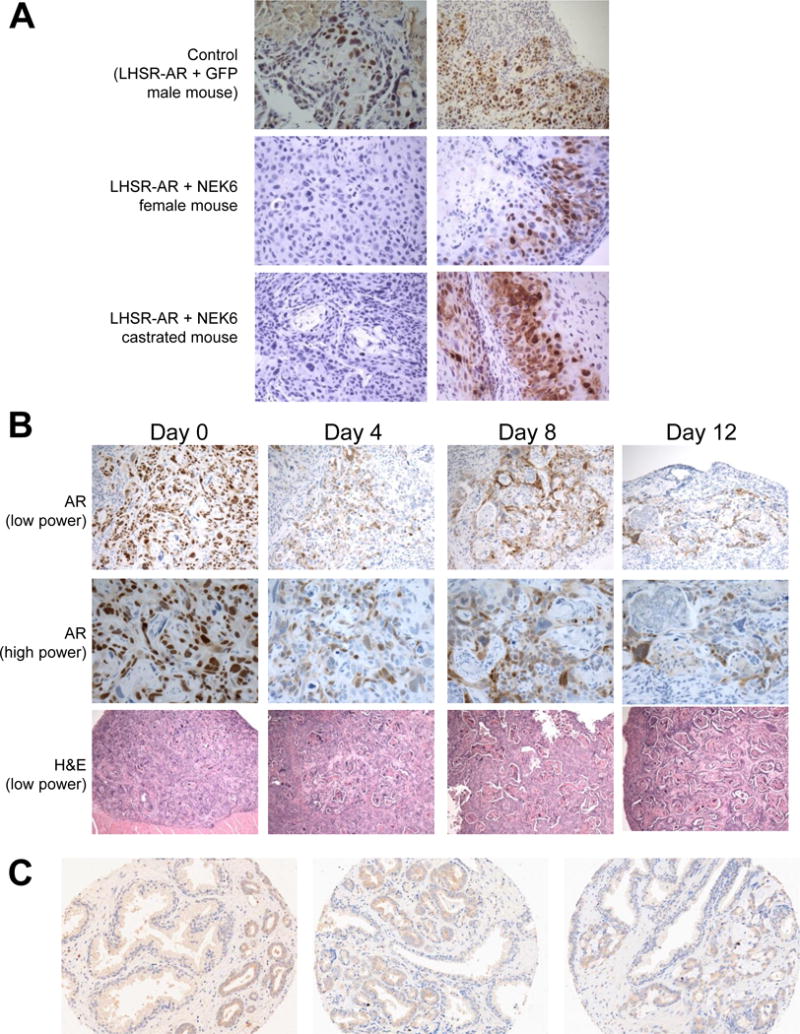
A. NEK6-mediated androgen-independent tumors are primarily squamous in histology and AR negative. Sections of tumors derived from parental LHSR-AR cells expressing GFP in male mice, and cells expressing NEK6 in female and castrated mice were stained with AR antibody (brown). B. Sections of tumors derived from LHSR-AR cells overexpressing NEK6 in male mice with implanted testosterone pellet harvested prior to castration (Day 0) and at 4, 8, and 12 d after castration. C. Immunohistochemical staining at 20× magnification for NEK6 in prostate cancer tissue microarrays. Low (left and middle) and high grade (right) cases are represented, each core showing tumor infiltrating in between benign glands with higher expression seen in the tumor.
NEK6 is aberrantly expressed in human prostate cancer
NEK6 is overexpressed in several malignant tissues and cell lines and has been previously been implicated in cell transformation in vitro (21,22) and in vivo (23). Cancer types previously described to have high frequency of NEK6 overexpression include cancers of the liver (21), stomach (22), breast (21,22), uterus, colon/rectum and ovary (22). In human prostate cancer, the NEK6 locus at chromosome 9q33.3 is present in an amplicon without a known validated prostate cancer oncogene: a statistically significant amplicon at 9q33.2-q34.3 (q=0.0066) was reported by Taylor, et al. (16), while updated analysis of 492 tumors from The Cancer Genome Atlas consortium (TCGA) revealed the peak of this amplicon to be at 9q33.3 (q=0.059)(24). Genomic characterization of human prostate cancers demonstrated amplification or mRNA overexpression (z-score>2) of NEK6 in 7% of primary tumors (provisional results from The Cancer Genome Atlas consortium, www.cbioportal.org) and 6% of metastases (11).
We performed immunohistochemistry (IHC) on primary prostate cancers compiled in seven tumor microarrays (TMA) generated from radical prostatectomy (RP) specimens from 347 patients using an antibody we optimized for IHC (Supplemental Figure S2A and B), and quantified NEK6 expression by spectral imaging. We failed to detect NEK6 protein in most normal prostate tissues, but found that NEK6 is aberrantly expressed in ~16% of tumors (Supplemental Figure S2C). NEK6 expression was greater in tumor samples than in benign glandular epithelium from these same RP specimens (p=0.0024), suggesting that NEK6 expression is associated with tumorigenesis (Figure 2C). The level of NEK6 expression in human tumors is somewhat less than the degree of overexpression achieved in xenografts (Supplemental Figure S2B), as assessed by the fact that the NEK6 antibody was used at a 1:100 dilution in xenografts and at a 1:50 dilution in clinical samples (though differences in sample preparation and the fact that the TMA slides were older could also play a role). We sought to assess whether NEK6 overexpression by IHC correlated with progression to castration resistance in this cohort, but given that these specimens were from patients with primary prostate cancer, there were not enough events in clinical follow-up to assess this outcome.
We analyzed the levels of NEK6 expression in several patient-derived prostate cell lines by immunoblotting (Figure 3A) and found that most prostate cancer cell lines expressed higher levels of NEK6 than immortalized (RWPE, PrEC-LH) and transformed (LHSR-AR) prostate epithelial cells. Interestingly, NEK6 levels were relatively high in VCaP and LNCaP cells, suggesting that high expression of NEK6 was not sufficient to overcome the in vitro androgen dependence of these cells; however, it is important to note that these cells were derived from patients with castration-resistant disease in vivo. The level of exogenous overexpression of NEK6 in LHSR-AR cells leading to the castration-resistance phenotype seen in Figure 1 is somewhat greater than endogenous levels in VCaP and LNCaP cells (Figure 3A, right). This finding, along with the fact that the LHSR-AR xenografts express only somewhat greater levels of NEK6 than what is seen in human primary tumors, would suggest that the phenotype conferred by NEK6 is not due to artifact of supraphysiologic overexpression.
Figure 3.
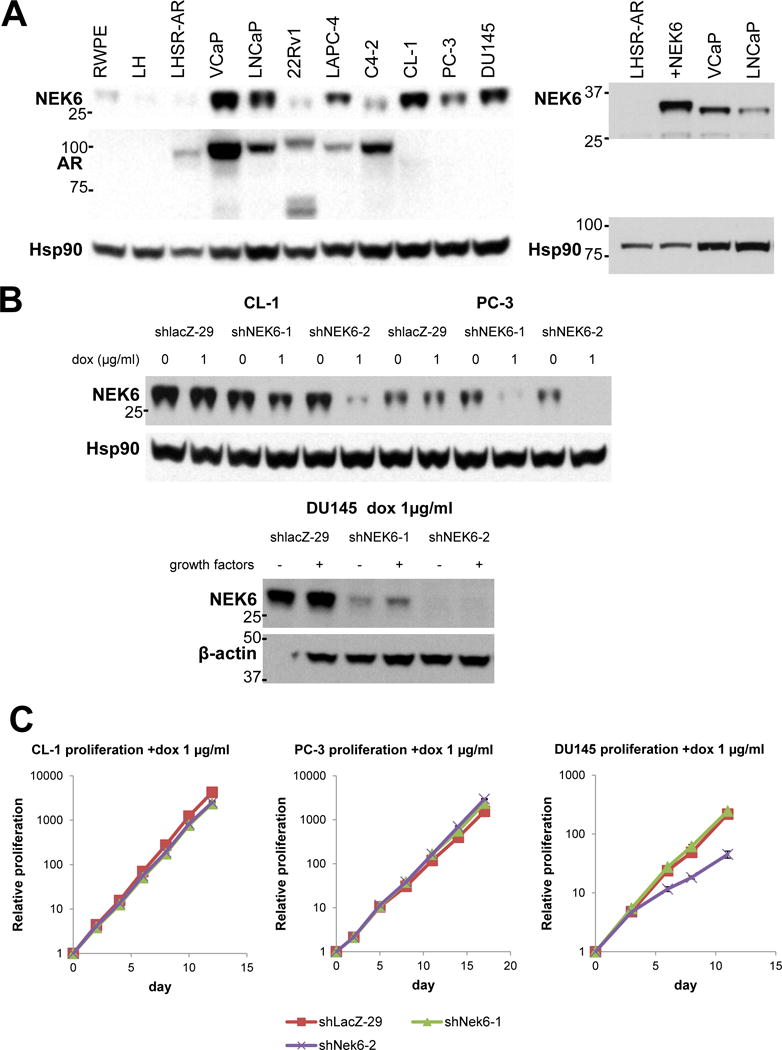
NEK6 is overexpressed in several prostate cancer cell lines compared to immortalized (RWPE, LH) and transformed (LHSR-AR) prostate epithelial cells. A. Left: Expression of NEK6 and AR in prostate cell lines with Hsp90 as loading control; Right: NEK6 expression in LHSR-AR cells (lane 1), LHSR-AR cells overexpressing NEK6 (lane 2), and in VCaP and LNCaP cells. B. Expression of NEK6 in CL-1, PC-3, and DU145 cells with doxycycline inducible expression of 2 shRNAs targeting NEK6 or a control targeting lacZ in the presence or absence of doxycycline (CL-1 and PC-3) or with doxycycline in the presence or absence of growth factor stimulation (DU145). C. Proliferation curves of cells cultured in the presence of doxycycline with cells in 60 mm plates, split and replated every 2–3 d as indicated. The average of 3 replicates with error bars is shown.
To test whether NEK6 was essential for in vitro proliferation of androgen-independent AR-low cell lines (reminiscent of the AR-low xenograft tumors), we introduced doxycycline-inducible shRNAs targeting NEK6 to CL-1, PC-3 and DU145 cells (Figure 3B). Despite near complete suppression of NEK6 expression, there was no effect on proliferation of the cell lines except a modest suppression of DU145 proliferation with shRNA#2. These observations are in consonance with a prior report suggesting that NEK6 is not essential for proliferation of many mammalian cell lines (22).
NEK6 does not activate AR signaling or other known bypass pathways
Since persistence of androgen receptor signaling has been demonstrated to be an important mechanism of castration resistance (1), we determined whether NEK6 influences AR signaling. NEK6-mediated castration resistant tumors mostly lack expression of the AR, but such tumors sometimes harbor areas that express AR. NEK6 overexpression did not lead to an increase in activity of an AR reporter based on the PSA enhancer in LNCaP cells (Supplemental Figure S3A), and inducible overexpression of NEK6 in LHSR-AR cells (Supplemental Figure S3B) failed to increase expression of the AR targets PSA and TMPRSS2 (Supplemental Figure S3C). When we performed Gene set enrichment analysis (GSEA) (25) of an mRNA expression signature of NEK6 activity generated from cells inducibly expressing wild-type vs. a kinase dead form of NEK6 K74M/K75M (20), we found that gene expression changes associated with NEK6 kinase activity failed to correlate (Supplemental Figure S3D) with three previously published signatures of AR activity (26–28).
We explored the possibility that the Wnt/β-catenin pathway mediated AR bypass in these tumors since a prior report demonstrated that the transformation of patient-derived prostate basal cells induced xenograft tumors exhibiting squamous histology with evidence of active β-catenin (29). However, we failed to find evidence of nuclear β-catenin staining in our NEK6-mediated castration resistant tumors (Supplemental Figure S4A).
Previously, NEK6 has been reported to be the major protein kinase that is active on the RPS6KB1 hydrophobic regulatory site, threonine 412 (30) in vitro. In addition, STAT3 phosphorylation at serine 727 has been implicated in the transformation activity of NEK6 previously (21). When we assessed phosphorylation of these proteins in LHSR-AR cells expressing NEK6, we failed to find evidence of RPS6KB1 (Thr412) or STAT3 (Ser727) phosphorylation in cultured cells (Supplemental Figure S4B), or in castration resistant tumors (Supplemental Figure S4C) as compared to controls.
Given that NEK6 has also been described to play a role in the G2/M transition in hepatocellular carcinoma through modulation of cyclin B (31) as well as inhibition of p53-mediated senescence (32), we assessed whether NEK6 overexpression influenced cell cycle progression. We failed to find any difference in cell cycle profiles of cells overexpressing NEK6 at steady state or upon release from growth factor starvation (Supplemental Figure S5A). NEK6 did not increase proliferation rate of LHSR-AR cells in vitro compared to a lacZ control (Supplemental Figure S5B). Thus, NEK6 overexpression in these cells does not appear to be related to cell cycle progression. Consistent with p53 being inactivated in LHSR-AR cells due the expression of SV40 large-T antigen, exposure of LHSR-AR cells to etoposide failed to cause p21 induction, but PARP was induced (Supplemental Figure S5C). Thus NEK6 does not act by modulating the p53 pathway in our cell context. Furthermore, the pro-survival effect of NEK6 appears to be specific to the in vivo context of castration resistance.
Gene Expression changes mediated by NEK6
Since the observed castration resistance mediated by NEK6 does not appear to involve these previously described pathways, we sought to elucidate downstream signaling mediated by NEK6 overexpression in an unbiased manner by examining gene expression changes in NEK6 driven tumors under castrate conditions. We established subcutaneous tumors in male mice with LHSR-AR cells expressing NEK6 under a doxycycline-inducible promoter; after tumors formed, 7 days before planned tumor harvest we split the mice into two groups, with half continuing on doxycycline diet and half discontinuing doxycycline. We then castrated the animals and harvested the tumors at days 0, 2 and 5 after castration. We found that doxycycline withdrawal reduced NEK6 levels to baseline (Figure 4A). We then analyzed gene expression by RNA-Seq on mRNA isolated from tumors harvested from mice with and without doxycycline in their diet at days 2 and day 5 after castration (3 tumors under each condition, 12 samples total).
Figure 4.
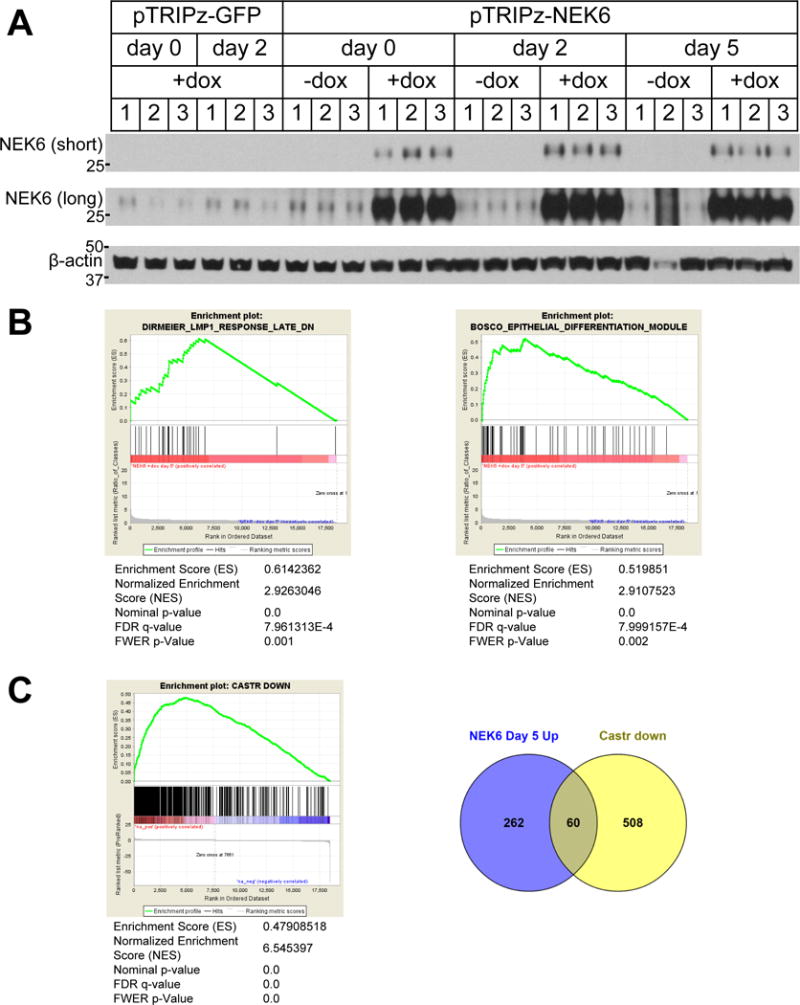
A. Immunoblots of protein lysates of xenograft tumors derived from LHSR-AR cells transduced with doxycycline-inducible constructs (in the pTRIPz vector) for the expression of GFP or NEK6 in male mice, with doxycycline maintained in the diet at time of harvest (+dox) or with doxycycline diet removed 7 d prior to harvest (−dox). Tumors were harvested from non-castrated mice (day 0) or 2 or 5 d after castration. Short and long exposures of the NEK6 immunoblot are shown. B. Gene Set Enrichment Analysis performed on the gene expression signature mediated by NEK6 overexpression at d 5 after castration (GSEA pre-ranked based on ratio of classes to tumors without NEK6 overexpression). The top two curated gene sets (C2) from the molecular signatures database (MSigDB) correlated with the NEK6 signature are shown. C. Left: Genes downregulated in the control (−dox) tumors after castration (fold change <−1.5, signal-to-noise <−1 in the comparison of tumors harvested at d 5 and day 2) were plotted against the NEK6 signature at d 5 using GSEA pre-ranked. Right: Venn diagram of genes downregulated in control tumors after castration intersected with genes upregulated by NEK6 at day 5 (fold change >1.5, signal-to-noise >1 in the comparison of +dox to −dox tumors). The GO terms most highly enriched in this overlap are cytokine-mediated and type I interferon signaling pathways (p=2×10−5).
To characterize gene expression changes associated with NEK6 expression in response to castration, we performed GSEA comparing tumors overexpressing NEK6 (+dox) at day 5 after castration with −dox controls (Figure 4B). The top two curated gene sets from the molecular signatures database (MSigDB collection C2, representing 4725 gene sets) correlated with the NEK6 signature are (i) genes down-regulated in primary B lymphocytes within 60–180 min after activation of LMP1, an oncogene encoded by Epstein-Barr virus (33) and (ii) genes representing the epithelial differentiation module in sputum during asthma exacerbations (34). The GO biological process most significantly enriched among the NEK6-upregulated genes as assessed using the Gene Ontology for Functional Analysis (GOFFA) software (35) was “response to biotic stimulus” (p=0.0005) (Supplemental Table S3); other relevant terms include “apoptotic signaling pathway” (p=0.0056), “cytoskeleton organization” (p=0.0084), cytokine-mediated signaling pathway (p=0.0085) and “epithelial cell differentiation” (p=0.0183). One of these differentiation genes is cytokeratin 13 (KRT13), a known marker of squamous differentiation in the literature (36). KRT13 expression in xenograft tumors expressing NEK6 in the presence of circulating androgens was modest (Supplemental Figure S6, left panels), but increased with castration preferentially in tumors that continued to express NEK6 under a doxycycline inducible promoter as compared to tumors where NEK6 expression was abrogated by doxycycline withdrawal (Supplemental Figure S6, right panels).
We hypothesized that NEK6 may mediate resistance to castration in tumors by maintaining survival signaling that is lost when AR is no longer activated by circulating androgens. To understand components of this survival signaling, we compared gene expression of control tumors (i.e. without NEK6 overexpression) at the day 2 and day 5 time points. We found that the top gene set and six of the top 12 gene sets by GSEA associated with gene expression lost following castration reflected interferon signaling (Supplemental Table S4A). In addition, two prostate cancer related gene sets were also highly correlated with genes downregulated by castration (ranked 9 and 12): genes up-regulated in prostate tumors developed by transgenic mice overexpressing Vav3 (37) and genes up-regulated in prostate cancer samples from African-American patients compared to those from the European-American patients (38). These latter signatures were reported to incorporate elements of NFκB signaling and interferon signaling respectively, suggesting that the loss of circulating androgens leads to a decrease in immune-type signaling in these tumors.
The set of genes upregulated by NEK6 overexpression in castrate conditions correlated strongly with genes downregulated after castration in control tumors (Figure 4C), suggesting that NEK6 maintains gene expression lost with castration. The 60 overlapping genes in these two comparisons are listed in Supplemental Table S4B. The most statistically significant GO biological processes associated with the overlap (Supplemental Table S4C) are “cytokine-mediated signaling pathway” (p=2×10−5) and “type I interferon signaling pathway” (p=2×10−5), suggesting that maintenance of interferon and other immune-type signaling normally lost with castration may be involved in NEK6-mediated castration resistance.
Identification of FOXJ2 and NCOA5 as NEK6 substrates
To gain a more comprehensive understanding of the immediate signaling mediated by NEK6 expression and discover novel in vivo substrates, we performed a phosphoproteomic analysis as a screen to identify candidate substrates with the intent to validate individual substrates. In global phosphorylation analysis from cell lysates, a total of 9418 phosphopeptides (8432 phosphoserine, 952 phosphothreonine, 34 phosphotyrosine) from 3401 proteins were detected. Of these, 59 phosphopeptides from 50 proteins were upregulated by wild-type NEK6 expression as compared to control and kinase-dead NEK6 (Supplemental Table S5). A previously described NEK6 motif with polypeptide sequence [L/F/W/Y-X-X-pS/pT-F/W/Y/M/L/I/V/R/K] (39) was significantly enriched among these phosphopeptides as compared to all phosphopeptides detected in the experiment (8/59 vs. 186/9362, p=1.95×10−5), whereas other common phosphorylation motifs were not enriched (Figure 5A). This observation suggested that this peptide sequence is a true description of the NEK6 phosphorylation motif, and that the 8 proteins with phosphopeptides of this form detected here (FOXJ2, HUWE1, NCOA5, KRT18, TRA2B, HNRNPM, HNRNPA2B1, ZNF326) are likely bona fide in vivo NEK6 substrates.
Figure 5.
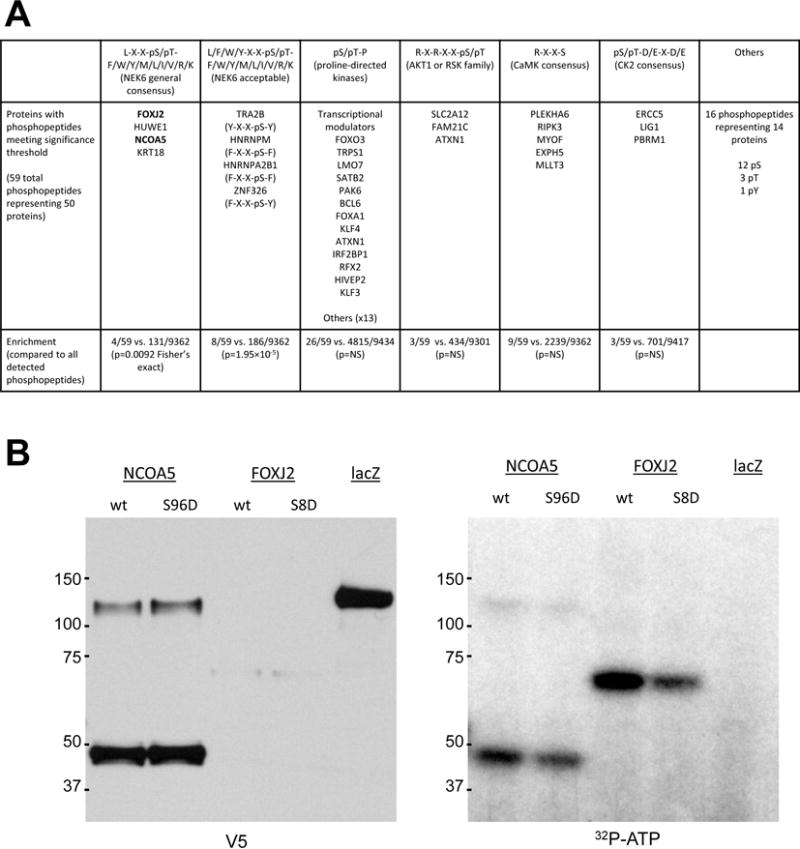
A. Summary of phosphopeptides (categorized by phosphorylation motif) found to be enriched in cells expressing wild-type NEK6 under a doxycycline-inducible promoter as compared to kinase-dead NEK6 or cells not treated with doxycycline. B. NEK6 can phosphorylate NCOA5 and FOXJ2 in vitro at the sites discovered in the phosphoproteomic screen. 293T cells were transfected with expression constructs for wild-type and mutant (S-to-D) versions of NCOA5 or FOXJ2 with a C-terminal V5 tag and immunoprecipitated with anti-V5 antibody. Eluates from 1/5 of the beads were assayed by V5 immunoblot; the remaining 4/5 was subjected to on-bead in vitro kinase assay with recombinant active GST-NEK6 (Sigma).
Two potential substrates identified in this analysisFOXJ2 and NCOA5, are related to families of transcription factors already described to be important in prostate cancer (40,41), and we sought to determine the phosphorylation sites of NEK6 on these proteins. Substitution of the phosphorylation sites of FOXJ2 and NCOA5 identified via mass spectrometry, Ser8 and Ser96 respectively, decreased the in vitro phosphorylation of these substrates (Figure 5B). Additionally, we found that NEK6 phosphorylated FOXJ2 at Thr23 and Ser254 (Figure 6A, lanes 3 and 5), and NCOA5 at Ser21 and Ser151 (Figure 6B, lanes 3 and 4). The phosphorylation of FOXJ2 and NCOA5 in vitro is indeed due to the active GST-NEK6 added to the assay, as excluding it led to no detectable phosphorylation of these substrates (Figure 6C lanes 2 and 6).
Figure 6.
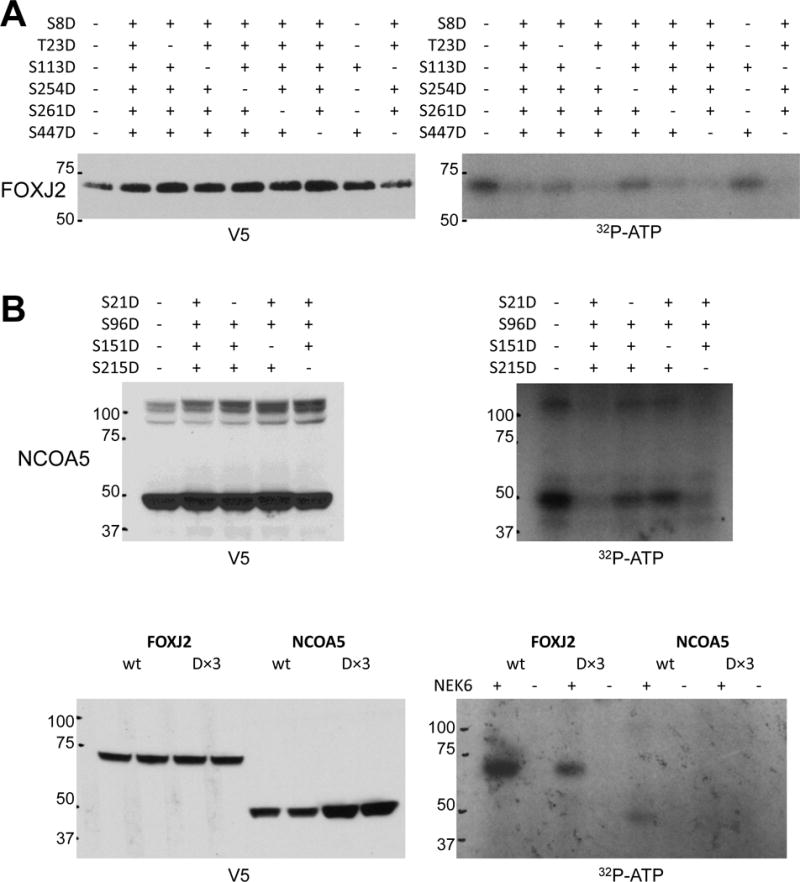
A+B. Mapping NEK6 phosphorylation sites on FOXJ2 and NCOA5. 293T cells were transfected with expression constructs for wild-type and mutant (S-to-D) versions of NCOA5 or FOXJ2 with a C-terminal V5 tag and immunoprecipitated with anti-V5 antibody; + indicates a mutation is present at that residue, − indicates that the residue is wild-type (lane 1 of each blot represents the wild-type protein). 5% of the input assayed by V5 immunoblot is shown in the left panels; the kinase assay is shown in the right panels. FOXJ2 has 10 S/T residues of the previously reported NEK6 motif L/F/W/Y-X-X-pS/pT-F/W/Y/M/L/I/V/R/K; the six residues in FOXJ2 (or homologous residues in FOXJ3) that had been previously demonstrated to be phosphorylated in phosphosite.org were assayed here. NCOA5 has 5 S/T residues in this NEK6 phosphorylation motif; however, mutating Ser201 to aspartic acid dramatically decreased exogenous NCOA5 expression, so the remaining four sites were tested in this assay. C. Kinase assay performed for wild-type and (D×3) forms of FOXJ2 and NCOA5 in the presence (+) or absence (−) of active recombinant GST-NEK6 in the assay.
Gene expression changes mediated by the NEK6 substrate FOXJ2
To determine the role of the identified substrates in mediating signaling downstream of NEK6, we assessed signaling in tumors mediated by phosphomimetic forms of these substrates with serine to aspartic acid substitutions of the phosphorylation sites identified in vitro, which we called FOXJ2(D×3) and NCOA5(D×3). We expressed them under a doxycycline-inducible promoter in tumors formed in male mice, which were harvested for RNA-Seq five days after castration from mice with continued doxycycline treatment or 7 day doxycycline withdrawal as for NEK6 previously. GSEA revealed that the gene expression changes mediated by a phosphomimetic form of FOXJ2 (FDR q-value=0.0044), but not NCOA5 (q=0.477) was statistically significantly correlated with the NEK6 signature (Figure 7A). Since NCOA5 failed to correlate to NEK6 signature, these observations suggest that the gene expression signatures associated with NEK6 and FOXJ2(D×3) expression are not the result of artifact or technical bias related to doxycycline treatment.
Figure 7.
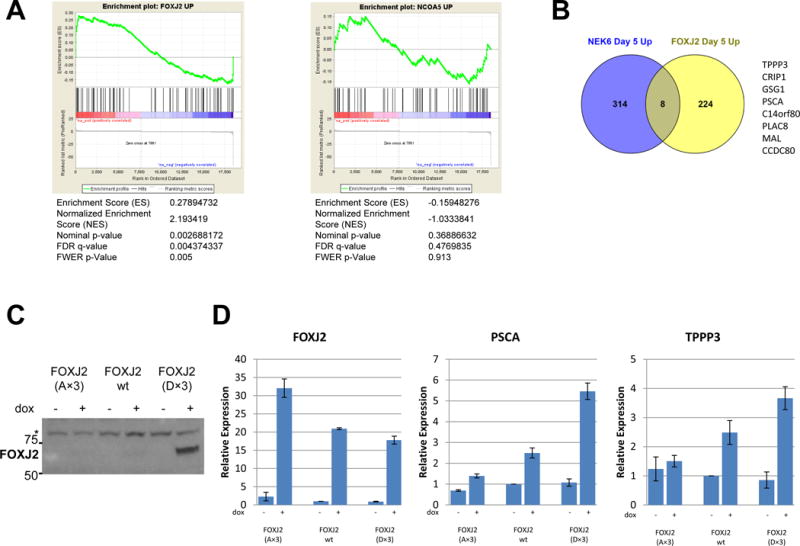
A. RNA-Seq was performed from 3 tumors inducibly expressing phosphomimetic forms of FOXJ2 or NCOA5 with continued doxycycline expression, and 3 matched tumors seven days post doxycycline withdrawal, 5 days after castration. Shown are plots demonstrating correlation of genes upregulated by these phosphomimetic forms with the NEK6 signature at day 5 by GSEA pre-ranked. B. Venn diagram of genes upregulated by NEK6 with genes upregulated by FOXJ2(D×3) at day 5 after castration (fold change >1.5, signal-to-noise >1 in the comparison of +dox to −dox tumors). C. Immunoblot of protein lysates of LHSR-AR cells transduced with doxycycline-inducible constructs (in the pTRIPz vector) for the expression of phosphorylation-deficient (A×3), wild-type, and phosphomimetic (D×3) forms of FOXJ2 treated with or without doxycycline for 48 hours. * indicates a background band used as a loading control. D. Gene expression of FOXJ2, TPPP3 and PSCA by quantitative RT-PCR (as fold-change from control) in cells expressing (A×3), wild-type and (D×3) forms of FOXJ2 under a doxycycline-inducible promoter treated with or without doxycycline for 48 hours. The average of 3 technical replicates (with error bars representing the standard deviation) is shown.
Eight genes overlapped between the gene expression signature mediated by FOXJ2(D×3) and NEK6 at the significance threshold used here (Figure 7B). Three of these eight genes, TPPP3 (Tubulin Polymerization-Promoting Protein Family Member 3), PSCA (Prostate Stem Cell Antigen), and PLAC8 (Placenta-Specific 8), had previously been found to be upregulated by NEK6 expression in vitro in the experiment described in Supplemental Figure S3D, suggesting these three genes as bona fide transcriptional targets of NEK6 signaling with FOXJ2 as the relevant transcription factor. TPPP3 has been previously implicated in proliferation, invasion and migration of cancer cells (42), PSCA has been reported to promote growth and metastasis of prostate cancer (43), and PLAC8 overexpression has been linked to EMT features and increased invasiveness (44), so these transcriptional targets may play a role in the oncogenic functions of NEK6.
Phosphorylation of FOXJ2 by NEK6 appears to increase stability of the FOXJ2 protein, as inducible expression of the phophomimetic form (D×3) led to a significantly higher FOXJ2 protein expression as compared to wild-type and phosphorylation-deficient(A×3) forms (Figure 7C) despite similar mRNA levels (Figure 7D). This higher level of protein expression led to increased mRNA expression of TPPP3 and PSCA in vitro (Figure 7D).
DISCUSSION
We performed an unbiased in vivo functional genomic screen to identify kinases that can confer androgen independence in a model of androgen-dependent prostate tumor formation, and discovered NEK6 as a novel mediator of castration resistance. Microenvironment potentially plays an important role in the development of resistance phenotypes, and performing a genomic screen in vivo allowed us to assess candidates requiring a physiologic microenvironment for their functional effects. We found that NEK6 does not confer any growth or survival advantage to transformed prostate epithelial cells in vitro in terms of proliferation rate or sensitivity to chemotherapy. Thus, the in vivo setting allowed us to discover NEK6 as a gene that confers castration resistance.
The NEK6 gene on chromosome 9q33.3 is located on a region of recurrent copy number gain in prostate cancer, and NEK6 protein is overexpressed in most patient-derived prostate cancer cell lines tested as well as a subset of primary human prostate cancers. NEK6 plays a mechanistic role in the development of castration resistance in our model, and suppressing NEK6 expression in xenograft tumors restores sensitivity to castration, suggesting that its continued activity is required for tumor maintenance in this context. NEK6 is not essential for cell proliferation, and mice homozygous for a targeted null Nek6 allele (Nek6tm1Dgen - DELTAGEN_T518) demonstrate no apparent phenotype (www.infrafrontier.eu), suggesting that NEK6 could serve as a therapeutic target without significant on-target toxicity.
Prior work has implicated NEK6 in cell cycle progression/cyclin B modulation (31), inhibition of p53-mediated senescence (32), the phosphorylation of STAT3 (21), and the abrogation of nuclear translocation of SMAD4 to antagonize TGFβ signaling (45). Here we failed to find evidence to implicate any of these previously described mechanisms in the role of NEK6-mediated castration resistance. In this experimental context, NEK6 overexpression did not promote cell cycle progression in vitro, as we failed to find any difference in cell cycle profiles of cells overexpressing NEK6 at steady state or upon release from growth factor starvation. We also found no evidence that NEK6 was required for cell cycle progression of androgen-independent prostate cancer cells, as there was no effect of NEK6 suppression on proliferation of the tested cell lines (except for a modest suppression of DU145 proliferation with a single shRNA). However, we cannot eliminate the possibility that the expression of NEK6 may affect cell cycle progression, contributing to the castration resistance phenotype in vivo. Similarly, while there was no evidence that NEK6 modulates p53 activity in these experiments, it remains possible that the expression of NEK6 inhibits a senescence phenotype that may occur with castration in a p53-independent manner.
We did not find gene sets annotated as involving STAT3, Wnt/β-catenin or TGFβ/SMAD4 signaling within the top 200 gene sets correlated with the NEK6 signature by GSEA in tumors after castration; rather, cytoskeletal, differentiation, and immune processes are implicated. In particular, NEK6 overexpression maintained the expression of a set of genes involved in interferon signaling that was lost with castration in this model. Relevance of interferon-related gene sets in prostate cancer has been demonstrated previously in signaling differences between cancers in patients with African and European ancestry (38) and in treatment resistance (46). Recent reports also implicated immune signaling (47) and cytoskeletal signaling (by induction of RhoA, Cdc42, and Rac1) (48) as mechanisms of resistance to the anti-androgen enzalutamide. Here, analysis of gene expression changes induced by a phosphomimetic form of a newly identified substrate FOXJ2 revealed that this is the relevant transcription factor for certain important transcriptional targets downstream of NEK6. Of note, expression of a phosphomimetic form of FOXJ2 failed to confer androgen-independent tumor formation, suggesting that a combination of events downstream of NEK6 is required for this phenotype.
Molecular mechanisms underlying transition to androgen pathway independence are not fully characterized: a subset of these cancers express markers of neuroendocrine differentiation, and transdifferentiation to a squamous phenotype has also been described in resistance to hormonal therapy (49). We hypothesize that APIPC represents a diversity of phenotypes and dependencies; a comprehensive understanding of these mechanisms would identify potential therapeutic targets for intervention, and would also allow for biomarker discovery to stratify patients for likelihood of response to targeted therapies. Thus, the signaling pathways described here warrant further study in this patient population.
Supplementary Material
Acknowledgments
We acknowledge Mari Nakabayashi, Matthew Chabot and the Arthur and Linda Gelb Center for Translational Research for the acquisition and annotation of clinical samples; Ted Pei for training on animal husbandry and procedures; Victor Zhivich for technical assistance with mouse husbandry; Jenn Abelin, Christina Hartigan, and Gaelen Guzman for technical assistance with experiments performed at the Broad Proteomics platform; Mukta Bagul and the Broad Genetics Perturbation Platform for providing ORF reagents; Kexin Xu and Myles Brown for providing reagents for the AR reporter assay; Kevin Buczkowski for training on generating cDNA libraries for RNA-Seq; Aaron Thorner and the DFCI Center for Cancer Genome Discovery for performing RNA-Seq; Edward Fox and Yue Shao from the DFCI Microarray Core; Massimo Loda, Edward Stack and the Center for Molecular Oncologic Pathology; and Christine Unitt and the DF/HCC Histopathology Core.
Financial support: This work was supported in part by NCI grant U01CA176058 and the Prostate Cancer Foundation. A.D.C. and I.G. have been supported by the Prostate Cancer Foundation Young Investigator Award and by the Department of Defense Prostate Cancer Research Program through the Physician Scientist Training Award and Postdoctoral Trainee Award, respectively.
Footnotes
CONFLICT OF INTEREST STATEMENT
W.C.H. is a consultant for and receives research support from Novartis.
References
- 1.Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154(11):4010–7. doi: 10.1210/en.2013-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson PS. Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30(6):644–6. doi: 10.1200/JCO.2011.39.1300. [DOI] [PubMed] [Google Scholar]
- 3.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12(6):1665–71. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 4.Whang YE, Armstrong AJ, Rathmell WK, Godley PA, Kim WY, Pruthi RS, et al. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urologic oncology. 2013;31(1):82–6. doi: 10.1016/j.urolonc.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 5.Nakabayashi M, Werner L, Courtney KD, Buckle G, Oh WK, Bubley GJ, et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU international. 2012;110(11):1729–35. doi: 10.1111/j.1464-410X.2012.11456.x. [DOI] [PubMed] [Google Scholar]
- 6.Oh WK, Hayes J, Evan C, Manola J, George DJ, Waldron H, et al. Development of an integrated prostate cancer research information system. Clinical genitourinary cancer. 2006;5(1):61–6. doi: 10.3816/CGC.2006.n.019. [DOI] [PubMed] [Google Scholar]
- 7.Smith MR, Sweeney CJ, Corn PG, Rathkopf DE, Smith DC, Hussain M, et al. Cabozantinib in chemotherapy-pretreated metastatic castration-resistant prostate cancer: results of a phase II nonrandomized expansion study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32(30):3391–9. doi: 10.1200/JCO.2013.54.5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith MR, De Bono JS, Sternberg CN, Le Moulec S, Oudard S, De Giorgi U, et al. Final analysis of COMET-1: Cabozantinib (Cabo) versus prednisone (Pred) in metastatic castration-resistant prostate cancer (mCRPC) patients (pts) previously treated with docetaxel (D) and abiraterone (A) and/or enzalutamide (E) AMER SOC CLINICAL ONCOLOGY; 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA: 2015. [Google Scholar]
- 9.Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(49):E4762–9. doi: 10.1073/pnas.1319948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faltermeier CM, Drake JM, Clark PM, Smith BA, Zong Y, Volpe C, et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(2):E172–81. doi: 10.1073/pnas.1521674112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer discovery. 2014;4(1):94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger R, Febbo PG, Majumder PK, Zhao JJ, Mukherjee S, Signoretti S, et al. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer research. 2004;64(24):8867–75. doi: 10.1158/0008-5472.CAN-04-2938. [DOI] [PubMed] [Google Scholar]
- 14.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nature protocols. 2008;3(10):1630–8. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirschner AN, Wang J, van der Meer R, Anderson PD, Franco-Coronel OE, Kushner MH, et al. PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer. Journal of the National Cancer Institute. 2015;107(2) doi: 10.1093/jnci/dju407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren K, Gou X, Xiao M, Wang M, Liu C, Tang Z, et al. The over-expression of Pim-2 promote the tumorigenesis of prostatic carcinoma through phosphorylating eIF4B. The Prostate. 2013;73(13):1462–9. doi: 10.1002/pros.22693. [DOI] [PubMed] [Google Scholar]
- 19.van Weerden WM, Bierings HG, van Steenbrugge GJ, de Jong FH, Schroder FH. Adrenal glands of mouse and rat do not synthesize androgens. Life sciences. 1992;50(12):857–61. doi: 10.1016/0024-3205(92)90204-3. [DOI] [PubMed] [Google Scholar]
- 20.O’Regan L, Fry AM. The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Molecular and cellular biology. 2009;29(14):3975–90. doi: 10.1128/MCB.01867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeon YJ, Lee KY, Cho YY, Pugliese A, Kim HG, Jeong CH, et al. Role of NEK6 in tumor promoter-induced transformation in JB6 C141 mouse skin epidermal cells. The Journal of biological chemistry. 2010;285(36):28126–33. doi: 10.1074/jbc.M110.137190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nassirpour R, Shao L, Flanagan P, Abrams T, Jallal B, Smeal T, et al. Nek6 mediates human cancer cell transformation and is a potential cancer therapeutic target. Molecular cancer research: MCR. 2010;8(5):717–28. doi: 10.1158/1541-7786.MCR-09-0291. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Lee YM, Baitsch L, Huang A, Xiang Y, Tong H, et al. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. eLife. 2014;3:e01763. doi: 10.7554/eLife.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Center BITGDA. SNP6 Copy number analysis (GISTIC2) Broad Institute of MIT and Harvard 2016 [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer cell. 2006;10(4):321–30. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Mendiratta P, Mostaghel E, Guinney J, Tewari AK, Porrello A, Barry WT, et al. Genomic strategy for targeting therapy in castration-resistant prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27(12):2022–9. doi: 10.1200/JCO.2008.17.2882. [DOI] [PubMed] [Google Scholar]
- 28.Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer cell. 2013;23(1):35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Stoyanova T, Cooper AR, Drake JM, Liu X, Armstrong AJ, Pienta KJ, et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):20111–6. doi: 10.1073/pnas.1320565110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belham C, Comb MJ, Avruch J. Identification of the NIMA family kinases NEK6/7 as regulators of the p70 ribosomal S6 kinase. Current biology: CB. 2001;11(15):1155–67. doi: 10.1016/s0960-9822(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 31.Zhang B, Zhang H, Wang D, Han S, Wang K, Yao A, et al. Never in mitosis gene A-related kinase 6 promotes cell proliferation of hepatocellular carcinoma via cyclin B modulation. Oncology letters. 2014;8(3):1163–68. doi: 10.3892/ol.2014.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jee HJ, Kim AJ, Song N, Kim HJ, Kim M, Koh H, et al. Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell cycle. 2010;9(23):4703–10. doi: 10.4161/cc.9.23.14059. [DOI] [PubMed] [Google Scholar]
- 33.Dirmeier U, Hoffmann R, Kilger E, Schultheiss U, Briseno C, Gires O, et al. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene. 2005;24(10):1711–7. doi: 10.1038/sj.onc.1208367. [DOI] [PubMed] [Google Scholar]
- 34.Bosco A, Ehteshami S, Stern DA, Martinez FD. Decreased activation of inflammatory networks during acute asthma exacerbations is associated with chronic airflow obstruction. Mucosal immunology. 2010;3(4):399–409. doi: 10.1038/mi.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun H, Fang H, Chen T, Perkins R, Tong W. GOFFA: gene ontology for functional analysis–a FDA gene ontology tool for analysis of genomic and proteomic data. BMC bioinformatics. 2006;7(Suppl 2):S23. doi: 10.1186/1471-2105-7-S2-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Dorst EB, van Muijen GN, Litvinov SV, Fleuren GJ. The limited difference between keratin patterns of squamous cell carcinomas and adenocarcinomas is explicable by both cell lineage and state of differentiation of tumour cells. Journal of clinical pathology. 1998;51(9):679–84. doi: 10.1136/jcp.51.9.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Mo JQ, Hu Q, Boivin G, Levin L, Lu S, et al. Targeted overexpression of vav3 oncogene in prostatic epithelium induces nonbacterial prostatitis and prostate cancer. Cancer research. 2008;68(15):6396–406. doi: 10.1158/0008-5472.CAN-08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer research. 2008;68(3):927–36. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 39.Vaz Meirelles G, Ferreira Lanza DC, da Silva JC, Santana Bernachi J, Paes Leme AF, Kobarg J. Characterization of hNek6 interactome reveals an important role for its short N-terminal domain and colocalization with proteins at the centrosome. Journal of proteome research. 2010;9(12):6298–316. doi: 10.1021/pr100562w. [DOI] [PubMed] [Google Scholar]
- 40.Tao S, He H, Chen Q. ChIP-seq analysis of androgen receptor in LNCaP cell line. Molecular biology reports. 2014;41(9):6291–6. doi: 10.1007/s11033-014-3511-0. [DOI] [PubMed] [Google Scholar]
- 41.Qin J, Lee HJ, Wu SP, Lin SC, Lanz RB, Creighton CJ, et al. Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. The Journal of clinical investigation. 2014;124(11):5013–26. doi: 10.1172/JCI76412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou W, Li J, Wang X, Hu R. Stable knockdown of TPPP3 by RNA interference in Lewis lung carcinoma cell inhibits tumor growth and metastasis. Molecular and cellular biochemistry. 2010;343(1–2):231–8. doi: 10.1007/s11010-010-0518-2. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Z, He J, Kang R, Zhao S, Liu L, Li F. RNA interference targeting PSCA suppresses primary tumor growth and metastasis formation of human prostate cancer xenografts in SCID mice. The Prostate. 2015 doi: 10.1002/pros.23110. [DOI] [PubMed] [Google Scholar]
- 44.Li C, Ma H, Wang Y, Cao Z, Graves-Deal R, Powell AE, et al. Excess PLAC8 promotes an unconventional ERK2-dependent EMT in colon cancer. The Journal of clinical investigation. 2014;124(5):2172–87. doi: 10.1172/JCI71103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuo J, Ma H, Cai H, Wu Y, Jiang W, Yu L. An inhibitory role of NEK6 in TGFbeta/Smad signaling pathway. BMB reports. 2015;48(8):473–8. doi: 10.5483/BMBRep.2015.48.8.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. The EMBO journal. 2013;32(20):2751–63. doi: 10.1038/emboj.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bishop JL, Sio A, Angeles A, Roberts ME, Azad AA, Chi KN, et al. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget. 2015;6(1):234–42. doi: 10.18632/oncotarget.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351–6. doi: 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012;60(1):59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.