

Abstract
The anaerobic metabolism of phenol in the beta-proteobacterium Thauera aromatica proceeds via para-carboxylation of phenol (biological Kolbe-Schmitt carboxylation). In the first step, phenol is converted to phenylphosphate which is then carboxylated to 4-hydroxybenzoate in the second step. Phenylphosphate formation is catalyzed by the novel enzyme phenylphosphate synthase, which was studied. Phenylphosphate synthase consists of three proteins whose genes are located adjacent to each other on the phenol operon and were overproduced in Escherichia coli. The promoter region and operon structure of the phenol gene cluster were investigated. Protein 1 (70 kDa) resembles the central part of classical phosphoenolpyruvate synthase which contains a conserved histidine residue. It catalyzes the exchange of free [14C]phenol and the phenol moiety of phenylphosphate but not the phosphorylation of phenol. Phosphorylation of phenol requires protein 1, MgATP, and another protein, protein 2 (40 kDa), which resembles the N-terminal part of phosphoenol pyruvate synthase. Proteins 1 and 2 catalyze the following reaction: phenol + MgATP + H2O→phenylphosphate + MgAMP + orthophosphate. The phosphoryl group in phenylphosphate is derived from the β-phosphate group of ATP. The free energy of ATP hydrolysis obviously favors the trapping of phenol (Km, 0.04 mM), even at a low ambient substrate concentration. The reaction is stimulated severalfold by another protein, protein 3 (24 kDa), which contains two cystathionine-β-synthase domains of unknown function but does not show significant overall similarity to known proteins. The molecular and catalytic features of phenylphosphate synthase resemble those of phosphoenolpyruvate synthase, albeit with interesting modifications.
Phenol is a natural substrate which is formed from a variety of natural compounds. Phenol arises from tyrosine by tyrosine phenol lyase, but phenol also arises during the degradation of many secondary phenolic plant constituents, notably in the course of the degradation of lignin and phenylpropanoid compounds. Besides phenol, there are many other phenolic compounds, both natural and synthetic ones. Their mineralization proceeds via completely different pathways, depending on whether oxygen is available or not. For instance, groundwater and landfills are free of oxygen. Therefore, anaerobic metabolism of phenolic compounds is of general interest, from both scientific and applied aspects.
The initial steps in aerobic phenol metabolism are catalyzed by oxygenases. Phenol is oxidized to catechol (1,2-dihydroxybenzene) by phenol monooxygenases followed by oxygenolytic ring cleavage catalyzed by catechol dioxygenase. Hence, aerobic metabolism of phenol requires molecular oxygen for both ring hydroxylation and ring cleavage. In contrast to aerobic metabolism, anaerobic metabolism cannot rely on oxygen- and oxygenase-dependent steps. Therefore, anaerobic metabolism of phenol and related phenolic compounds promises unprecedented biochemistry. Anaerobic growth of pure cultures on phenol has been shown for sulfate-reducing (5), denitrifying (47, 50, 51), and iron-reducing (35) bacteria. The list of bacteria growing anaerobically with phenolic compounds is steadily growing (see references in reference 43). In all cases studied, anaerobic growth on phenol requires the presence of CO2 (50); CO2 is required as a cosubstrate for phenol carboxylation which results in the formation of 4-hydroxybenzoate. Phenol carboxylation has been known in chemistry for more than 100 years and is referred to as Kolbe-Schmitt carboxylation.
Anaerobic phenol metabolism by pure cultures has been studied in some detail only in the denitrifying beta-proteobacterium Thauera aromatica (2, 3, 16, 24, 29-31, 43, 50, 51). It involves two initial steps (Fig. 1). In the first step, phenol is converted to phenylphosphate. This reaction is catalyzed by an enzyme, E1, tentatively named phenylphosphate synthase (30). Phenylphosphate is the substrate of a second enzyme, E2, or phenylphosphate carboxylase, which catalyzes the carboxylation of phenylphosphate to 4-hydroxybenzoate (43). The corresponding enzymes are encoded by a large gene cluster which is phenol induced (Fig. 2A) (16). The phenol-induced gene products found in phenol-grown (induced) cells are missing in 4-hydroxybenzoate-grown (noninduced) cells which served as a control.
FIG. 1.

Proposed pathway of anaerobic metabolism of phenol in the denitrifying bacterium T. aromatica. E1, phenylphosphate synthase (reaction shown in the boxed area [studied in this work]) (EC 2.7.9.x); E2, phenylphosphate carboxylase (EC 4.1.1.x, studied in previous work) (43). The enzymes catalyzing the subsequent conversion of 4-hydroxybenzoate to three molecules of acetyl-CoA and one molecule of CO2 have been studied previously (8, 10, 11, 13, 15, 22, 23). The proposed enzyme-bound intermediates are shown in brackets.
FIG. 2.

A. Organization of the phenol gene cluster of T. aromatica (GenBank accession number AJ272115). The numbers refer to the ORF numbers of T. aromatica. Arrows indicate the direction of transcription. Black, genes encoding subunits of phenylphosphate synthase; grey, genes encoding subunits of phenylphosphate carboxylase. Genes coding for phenol-induced proteins identified in two-dimensional gel electrophoresis are indicated by stars (16, 43). ORF11 is a putative σ54-dependent regulator. ORFs 1 to 3 are subunits of phenylphosphate synthase. ORFs 4 to 6 and 12 are subunits of phenylphosphate carboxylase. ORFs 7 and 8 are putative ubiD/ubiX type (de)carboxylases (16, 43). ORFs 9, 10, 13, X, and 16 are hypothetical. ORF14 is similar to aromatic acid:CoA ligases. ORF15 is similar to proteins involved in the meta pathway of aerobic phenol metabolism in various species of bacteria. Black bars below the ORFs (b to o) represent the locations of fragments amplified in RT-PCR experiments. B. Results of RT-PCR experiments. a: negative control; fragment a could not be amplified on the RNA level; M: molecular mass standard; e: weakness of signal is due to suboptimal primer binding and is also observed in PCR experiments with chromosomal DNA. For details, see Materials and Methods.
Phenylphosphate synthase E1 (equation 1) was postulated to consist of two proteins, proteins 1 and 2, based on a sequence comparison of two phenol-induced genes (16). Putative protein 1 resembles the central part and putative protein 2 resembles the N-terminal part of classical phosphoenolpyruvate (PEP) synthase (EC 2.7.9.2) of, e.g., Escherichia coli. Phenol-induced cells, rather than noninduced cells, catalyzed an exchange of free [14C]phenol and the phenol moiety of phenylphosphate (30) (a kind of esterase reaction [equation 2]). This suggests that enzyme E1 is phosphorylated by phenylphosphate in the course of this exchange reaction (equations 3 and 4) and that, in the course of phenol phosphorylation, the enzyme is phosphorylated by another phosphoryl donor in an essentially irreversible step (equation 5). The phosphorylated enzyme E1 is supposed to subsequently transform phenol to phenylphosphate in a reversible reaction (equation 6). These features are consistent with a ping-pong mechanism. The whole reaction (equation 1) is understood as the sum of equations 5 and 6. The phosphoryl donor X∼P of enzyme E1 was unknown.
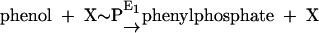 |
(1) |
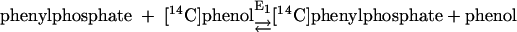 |
(2) |
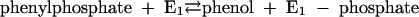 |
(3) |
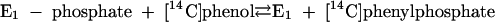 |
(4) |
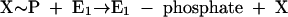 |
(5) |
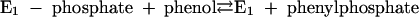 |
(6) |
It was the aim of this work to purify and study the first enzyme (E1) of the pathway that transforms phenol into phenylphosphate. Notably, despite many attempts, the phosphorylation of phenol could not be brought about in cell extracts, and the phosphoryl donor was therefore unknown. Furthermore, it was to be shown whether the enzyme catalyzing the exchange of free [14C]phenol and the phenol moiety of phenylphosphate (exchange reaction) and the enzyme catalyzing the X-P-dependent phosphorylation of phenol (net phosphorylation) are identical and whether the exchange of [14C]phenol is catalyzed by a partial fraction of the native enzyme.
MATERIALS AND METHODS
Gases, chemicals, biochemicals, materials for chromatography, and radioisotopes.
[U-14C]phenol (73 mCi mmol−1) was obtained from American Radiolabeled Chemicals/Biotrend (Cologne, Germany) and Hartmann Analytic (Braunschweig, Germany); [γ-32P]ATP (6,000 Ci mmol−1) was obtained from Amersham Pharmacia Biotech (Freiburg, Germany); Aminex-HPX-87H high-performance liquid chromatography (HPLC) columns were obtained from Bio-Rad (Munich, Germany); Nucleosil 100-10SB HPLC columns were obtained from Machery-Nagel (Düren, Germany); LiChroCART 125-4 HPLC columns filled with LiChrospher 100 PR-18 (C18) and silica gel 60 F254 thin-layer chromatography (TLC) plates were obtained from Merck (Darmstadt, Germany); amylose affinity columns (fast-performance liquid chromatography [FPLC]) were obtained from New England BioLabs (Frankfurt, Germany); and DEAE-Sepharose (FPLC) was obtained from Amersham Pharmacia Biotech. Chemicals were reagent grade and purchased from Fluka (Neu-Ulm, Germany), Merck, Serva (Heidelberg, Germany), and Sigma-Aldrich (Deisenhofen, Germany). Scintillation cocktail and acrylamide stock solution were purchased from Roth (Karlsruhe, Germany). Nitrogen, helium, and an N2-H2 gas mixture (95:5, vol/vol) were purchased from Sauerstoffwerke Friedrichshafen (Friedrichshafen, Germany). Restriction enzymes were obtained from MBI Fermentas (St. Leon-Rot, Germany).
Growth conditions of T. aromatica, cell harvesting, and storage.
T. aromatica type strain K172 (DSMZ 6984) (2, 50) was cultured anaerobically in a fed-batch mode (21) at 30°C in a modified mineral salt medium (13, 50) which initially contained 0.5 mM phenol, 10 mM bicarbonate, and 2 mM nitrate. As a control, cells grown on 4-hydroxybenzoate (5 mM) and nitrate (15 mM) were used. The medium (pH 7.8) contained 32 mM K2HPO4, 4.3 mM NaH2PO4, 10 mM NH4Cl, 0.8 mM MgCl2, 0.1 mM CaCl2, and carbon and electron sources as indicated. Vitamins (B12, 0.1 μg ml−1; B6, 0.3 μg ml−1; pantothenate, 0.1 μg ml−1; B1, 0.2 μg ml−1; nicotinic acid, 0.2 μg ml−1; 4-aminobenzoate, 0.08 μg ml−1; biotin, 0.02 μg ml−1) (40) and trace elements (ZnCl2, 0.5 μM; MnCl2, 0.5 μM; CoCl2, 0.8 μM; CuCl2, 0.04 μM; NiCl2, 0.1 μM; Na2MoO4, 0.15 μM; H3BO3, 0.01 μM; FeCl2, 15 μM; nitrilotriacetate, 750 μM) were added from 1,000-fold stock solutions. Phenol and nitrate were fed from a 0.5 M phenol-1.8 M KNO3 stock solution, and 4-hydroxybenzoate was fed from a 0.5 M sodium 4-hydroybenzoate-1.8 M KNO3 stock solution. Growth was measured by determining the optical density at 578 nm (OD578). An OD578 of 1.0 (1-cm light path) corresponded to a cell concentration of approximately 0.3 g of dry cell mass per liter. The harvested cells were immediately frozen and stored in liquid nitrogen.
Plasmids, primers, and E. coli strains and their growth conditions.
The plasmid pETBlue-1 (Ampr lacZ PT7) was from Novagen (Bad Soden, Germany); pMal-c2X (Ampr lacIq lacZ Rtac malE) was from New England BioLabs. The oligonucleotide primers used were obtained from G. Igloi (University of Freiburg, Freiburg, Germany) or from MWG Biotech (Ebersberg, Germany) and are summarized in Table 1. E. coli SURE {e14− (McrA−) Δ(mcrCB-hsdSMR-mrr)171 endA1 supE44 thi-1 gyrA96 relA1 lac recB recJ sbcC umuC::Tn5 (Kanr) uvrC [F′ proAB lacIqZ ΔM15 Tn10 (Tetr)]} was obtained from Stratagene (Amsterdam, The Netherlands), and E. coli TUNER(DE3)pLacI cells [F− ompT hsdSB (rB− mB−) gal dcm lacY1 (DE3) pLacI(Cmr)] were obtained from Novagen. E. coli strains were grown aerobically at 37°C in Luria-Bertani medium (42) and, if necessary, with 0.1 mg of ampicillin ml−1.
TABLE 1.
Oligonucleotide primers used in this worka
| Primer no. | Primer | Sequence | Characteristics and use |
|---|---|---|---|
| 1 | ORF1pMalfor | 5′-GAAGATCTATGAA-GTTTCCTGTTCCG-3′ | 5′ primer for DNA fragment containing orf1 used for pMal-c2X vector |
| 2 | ORF1polyTfor | 5′-ATGAAGTTTCCTGTTCCGCAC-3′ | 5′ primer for DNA fragment containing orf1 used for pETBlue-1 vector |
| 3 | ORF1SalIrev | 5′-CCATGTCGACCACTCCCTCAATCC-3′ | 3′ primer for DNA fragment containing orf1 used for pETBlue-1 and pMal-c2X vectors |
| 4 | ORF2BglIIfor | 5′-GAGGGAAGATCTGACATGGGAAG-3′ | 5′ primer for DNA fragment containing orf2 used for pMal-c2X vector |
| 5 | ORF2polyTfor | 5′-ATGGGAAGTATCGTTTCCAC-3′ | 5′ primer for DNA fragment containing orf2 used for pETBlue-1 vector |
| 6 | PEPSSalIrev | 5′-GATCGGATTGGTCGACATCCA-3′ | 3′ primer for DNA fragment containing orf2 used for pETBlue-1 and pMal-c2X vectors |
| 7 | ORF3polyTfor | 5′-ATGATCGTACGCAACTGGATG-3′ | 5′ primer for DNA fragment containing orf3 used for pETBlue-1 vector |
| 8 | ORF3polyTrev | 5′-GACCTCGGTGGTGATTCTCTT-3′ | 3′ primer for DNA fragment containing orf3 used for pETBlue-1 vector |
| 9 | 5′orf1for | 5′-CACCACGGGTCGTATTGA-3′ | RT-PCR, negative control |
| 10 | orf1Cy5rev | 5′-CTCCGGGTAATGCAATCCGTCG-3′ | RT-PCR, negative control and primer extension experiment |
| 11 | orf1for | 5′-GCTCTGGGGGGTCACCA-3′ | RT-PCR, amplification of intergenic region between ORFs 1 and 2 |
| 12 | orf2rev | 5′-CGCACTTGCCGCCCACCA-3′ | RT-PCR, amplification of intergenic region between ORFs 1 and 2 |
| 13 | orf2for | 5′-GACAACTTCGTCGTCAACAAG-3′ | RT-PCR, amplification of intergenic region between ORFs 2 and 3 |
| 14 | orf3rev | 5′-GTGGATATTGGCTTCGGAAA-3′ | RT-PCR, amplification of intergenic region between ORFs 2 and 3 |
| 15 | orf3for | 5′-TGTTCTCGCTGGCGGCGCAT-3′ | RT-PCR, amplification of intergenic region between ORFs 3 and 4 |
| 16 | orf4rev | 5′-AAGAGAATCACCACCGAGGTC-3′ | RT-PCR, amplification of intergenic region between ORFs 3 and 4 |
| 17 | orf4for | 5′-CCTACGAGTGGAAGCAGAAGC-3′ | RT-PCR, amplification of intergenic region between ORFs 4 and 5 |
| 18 | orf5rev | 5′-TGTTCATCTCCTCGAGCA-3′ | RT-PCR, amplification of intergenic region between ORFs 4 and 5 |
| 19 | orf5for | 5′-CGGCATGGGCGTGATCGTGCTGG-3′ | RT-PCR, amplification of intergenic region between ORFs 5 and 6 |
| 20 | orf6rev | 5′-CCCAGTCCACTTCCTGTCTGAT-3′ | RT-PCR, amplification of intergenic region between ORFs 5 and 6 |
| 21 | orf6for | 5′-GGAGCGCGGCATCCACGTGTTC-3′ | RT-PCR, amplification of intergenic region between ORFs 6 and 12 |
| 22 | orf12rev | 5′-GCCGCGGGCGGACAAAAGGC-3′ | RT-PCR, amplification of intergenic region between ORFs 6 and 12 |
| 23 | orf12for | 5′-CCGCATCATCGAGACCGTCCA-3′ | RT-PCR, amplification of intergenic region between ORFs 7 and 12 |
| 24 | orf7rev | 5′-AAGGAAGTGATCCACCGCC-3′ | RT-PCR, amplification of intergenic region between ORFs 7 and 12 |
| 25 | orf7for | 5′-GGCACATCGAAAGTCGGGTT-3′ | RT-PCR; amplification of intergenic region between ORFs 7 and 8 |
| 26 | orf8rev | 5′-TGGTGATGTCGGATTCGG-3′ | RT-PCR, amplification of intergenic region between ORFs 7 and 8 |
| 27 | orf8for | 5′-GGCATGATCATCGCGCCCTGT T-3′ | RT-PCR, amplification of intergenic region between ORFs 8 and 9 |
| 28 | orf9rev | 5′-GATGCCACCGATCGCCCTTTCC-3′ | RT-PCR, amplification of intergenic region between ORFs 8 and 9 |
| 29 | orf9for | 5′-GGGATACAGCATCCGCAT-3′ | RT-PCR, amplification of intergenic region between ORFs 9 and 10 |
| 30 | orf10rev | 5′-GCCATGTGAATCTCCTTGCTGACG-3′ | RT-PCR, amplification of intergenic region between ORFs 9 and 10 |
| 31 | orf10for | 5′-GCATACCGATCGAACACTGG-3′ | RT-PCR, amplification of intergenic region between ORFs 10 and 13 |
| 32 | orf13rev | 5′-CGTTCCCTTGACGAGGAAGG-3′ | RT-PCR, amplification of intergenic region between ORFs 10 and 13 |
| 33 | orf13for | 5′-GGCGCTGACGAAGATGGAA-3′ | RT-PCR, amplification of intergenic region between ORFs 13 and 14 |
| 34 | orf14rev | 5′-TCCACTTGTTGGTGTAGAAGC-3′ | RT-PCR, amplification of intergenic region between ORFs 13 and 14 |
| 35 | orf14for | 5′-GAGTTCCGCATCAAGGTCAG-3′ | RT-PCR, amplification of intergenic region between ORFs 14 and 15 |
| 36 | orf15rev | 5′-CTCCGTTCGGATACTGGTCG-3′ | RT-PCR, amplification of intergenic region between ORFs 14 and 15 |
| 37 | orf15for | 5′-CATTGCGCAATGGCAGCACG-3′ | RT-PCR, amplification of intergenic region between ORFs 15 and X |
| 38 | orfXrev | 5′-CTTGAGCCGCTCCTTGCTCG-3′ | RT-PCR, amplification of intergenic region between orfs 15 and X |
The mutated nucleotides are marked in italic type, and the restriction sites are marked in boldface type. For gene order, see Fig. 2A.
Preparation of cell extract and protein determination.
Cells were broken by two different methods (1). One part of cells (fresh weight) was suspended in 1 volume of a solution containing 15% (vol/vol) glycerol, 40 mM mercaptoethanol, and 0.5 mg DNase I per ml, followed by French press treatment at 137 MPa at 4°C (2). One part of cells (fresh weight) was suspended in 1.5 volumes of a solution containing 15% (vol/vol) glycerol, 1 mM EDTA, 40 mM mercaptoethanol, and 0.5 mg of DNase I per ml, followed by incubation with 90 μg of lysozyme per g of cells for 10 min at 20°C. This method was used for T. aromatica cells exclusively.
Cell extracts of T. aromatica and of E. coli containing an open reading frame 1 (ORF1) protein were centrifuged at 100,000 × g for 30 min at 4°C. Twenty percent (vol/vol) polyethyleneglycol (PEG 600) was added to the supernatant of T. aromatica cell extract, and ultracentrifugation was repeated. The resulting polyethyleneglycol-containing supernatant was used in the assays. E. coli cell extracts containing ORF2 and ORF3 proteins were used for the preparation of inclusion bodies. The approximate protein concentrations, determined by the method of Bradford (14), were 80 mg ml−1 (method 1) and 10 mg ml−1 (method 2).
Enzyme assays and assays of individual ORFs.
The radioactive assays followed the incorporation of radioactivity from [U-14C]phenol into phenylphosphate in a 0.1-ml assay mixture at 30°C; the reaction was started by adding cell extract or protein preparation. After 10 min of incubation, the reaction was stopped by adding 15 μl of 2 M H2SO4 followed by centrifugation. The supernatant was analyzed by thin-layer chromatography (10- to 20-μl samples) and radioautography or by HPLC (20 or 50 μl samples) and radiodetection by solid-state scintillation counting. Routinely, 0.2 mg of ORF1 and/or 0.2 mg of ORF2 was used. When ORF3 was tested, 0.025 to 0.2 mg of ORF3 was added.
(i) [14C]phenol exchange reaction.
The exchange of [14C]phenol and the phenol moiety of phenylphosphate was routinely tested by following the phenylphosphate-dependent formation of labeled products from [U-14C]phenol. The routinely used assay mixture contained 100 mM Tris-HCl (pH 8.4), 20 mM MgCl2, 1 mM MnCl2, 40 mM mercaptoethanol, 0.125 mM [U-14C]phenol (1.8 kBq), 1 mM phenylphosphate, and cell extract or protein sample (in 0.1 ml).
(ii) Net phenol phosphorylation reaction.
The formation of phenylphosphate was routinely tested by monitoring the MgATP-dependent formation of labeled products from [U-14C]phenol. The routinely used assay mixture contained 100 mM Tris-HCl (pH 8.6), 20 mM MgCl2, 1 mM MnCl2, 10 mM ATP, 40 mM mercaptoethanol, 0.125 mM [U-14C]phenol (1.8 kBq), and cell extract or protein sample (in 0.1 ml).
(iii) Test for other possible phosphoryl donors.
The following compounds (5 mM) were tested in the net phenol phosphorylation assay for their ability to transfer a phosphoryl group to [U-14C]phenol: ATP, GTP, ITP, UTP, CTP, phosphoenolpyruvate, acetylphosphate, carbamoylphosphate, creatine phosphate, ATP plus Pi, and ADP plus PPi.
(iv) Analysis of adenine nucleotides.
To identify the products formed from ATP during net phenol phosphorylation, a modified net phosphorylation assay mixture (100 μl) containing 2 mM unlabeled phenol, 2 mM MgATP, 20 μl (each) of ORF1 and ORF2 protein solution (0.2 mg of each protein), and 40 μl of ORF3 protein solution (0.2 mg) was used. The assays were incubated for 15 h at 30°C, and then the reaction was stopped by adding 15 μl of 2 M H2SO4. After centrifugation, a sample of the supernatant (100 μl) was analyzed for adenine nucleotides by HPLC (see below). To investigate whether the subunits of phenylphosphate synthase have ATPase activity in the absence of the substrate phenol, the same assay was used with the exception that phenol was omitted and the subunits were tested individually or in combination.
(v) Study of enzyme requirements and kinetics.
The oxygen sensitivity of phenylphosphate synthase was checked. Therefore, the preparation of cell extracts and assays was performed under aerobic and strict anaerobic conditions. The pH dependence of the reactions was tested by varying the pH values in the range of 7 to 10 for net phosphorylation by using 100 mM Tris-HCl and glycine-KOH. The exchange reaction was tested in the pH range of 4.5 to 10 by using 100 mM histidine-KOH, TES [N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid]-KOH, Tris-HCl, and glycine-KOH. The Mn2+ dependence was tested by altering the concentration of Mn2+ ions in the assay between 0 and 9 mM. For the determination of the apparent Km values for the substrates of net phenol phosphorylation, the (co)substrate concentrations were varied at saturating concentrations of the other (co)substrates (10 mM ATP, 0.4 mM phenol, 20 mM MgCl2). Assays were stopped after 5 min of incubation.
Experiments with [32P]ATP and determination of products of ATP hydrolysis. (i) Preparation of β-32P-labeled ATP.
β-32P-labeled ATP was enzymatically synthesized from γ-32P-labeled ATP. In the first step, a myokinase reaction (5 U of myokinase) (equation 7) was performed at 35°C for 15 min with a 270-μl reaction mixture containing 20 mM MgCl2, 10 mM AMP, 2 mM [γ-32P]ATP (100 μCi), and 150 mM K-morpholinepropanesulfonic acid (K-MOPS) buffer (pH 7.4).
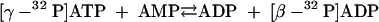 |
(7) |
The assay was stopped by adding 27 μl of 10% perchloric acid following centrifugation. Next, 5.5 μl of a 5 M potassium carbonate solution was added, followed by centrifugation to remove precipitated potassium perchlorate. The supernatant (300 μl) was loaded onto a Q-Sepharose fast-flow column (0.5 ml) previously equilibrated with buffer A (50 mM K-phosphate, pH 6.7). After washing with 1 bed volume of buffer A, AMP was eluted with 3 bed volumes of buffer A containing 30 mM KCl, ADP was eluted with 3 bed volumes of buffer A containing 80 mM KCl, and ATP together with some remaining ADP was eluted with 3 bed volumes of buffer A containing 500 mM KCl. The purity of the ADP-containing fraction (1.5 ml) was proved by HPLC analysis. In a second step, [β-32P]ADP was completely converted in 15 min at 35°C into [β-32P]ATP by pyruvate kinase (8 U) (equation 8) in an assay mixture containing 5 mM MgCl2 and 1 mM phosphoenolpyruvate.
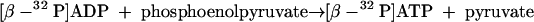 |
(8) |
The assay was stopped by adding 45 μl of 30% (wt/vol) perchloric acid following centrifugation, and neutralization was done with 23 μl of a 5 M potassium carbonate solution following centrifugation. The radioactivity content of the supernatant was determined by liquid scintillation counting (1.1 kBq μl−1). This solution was used directly in the assays after adding appropriate amounts of unlabeled ATP.
(ii) Detection of labeled phenylphosphate.
A modified net phenol phosphorylation assay (0.2-ml assay mixture) was used in which 2 mM labeled ATP ([γ-32P]ATP and [β-32P]ATP, respectively [5 kBq each]) and 1 mM unlabeled phenol were added. The reaction was started by the addition of 40 μl (each) of ORF1 and ORF2 protein solution (0.4 mg of each protein) or a combination of both. After 10 min of incubation at 30°C, the reaction was stopped by adding 30 μl of 2 M H2SO4 followed by centrifugation. Aliquots of the supernatant (20 μl, 500 Bq) were analyzed by TLC and radioautography.
HPLC, TLC, and detection methods.
Both HPLC and TLC were used to analyze the products of the reactions in which [U-14C]phenol or [32P]ATP was used.
(i) HPLC.
Detection was done by using a photodiode array detector (260 nm) and/or a solid scintillation radioactivity detector. For the separation of 14C-labeled products, an Aminex HPX-87H column (300 by 7.8 mm; Bio-Rad) was used with solvent 10 mM H2SO4 (isocratic) and a flow rate of 0.6 ml min−1 for a 50-μl sample; the retention time for phenol was 67 min, and the retention time for phenylphosphate was 8.5 min. For the separation of nucleotides, a Mono-Q column (1 ml; Pharmacia) with a flow rate of 1 ml min−1 and a sample size of 100 μl was used; solvents included buffer A (50 mM K-phosphate, pH 6.7) and buffer A plus 500 mM KCl. Separation was achieved by using a linear gradient over 25 min from 0 to 500 mM KCl. Retention times were as follows: AMP, 3 min; phenylphosphate, 5 min; ADP, 6 min; ATP, 8 min.
(ii) TLC.
For the separation of 14C- and 32P-labeled products, Silica gel 60 F254 (20 × 20 cm) plates with a solvent of ethanol-dichloromethane-water (80:10:10, vol/vol/vol) was used; the sample size was 10 to 20 μl (180 Bq of 14C or 500 Bq of 32P); development was done for 3 to 3.5 h at room temperature, followed by UV detection (254 nm; unlabeled standards), and radioactivity detection was done by phosphoimaging (Fujix BAS-IP MP 2040S, Fuji, Tokyo, Japan). Radioactivity was quantified after a 24-h incubation by scanning the radioautogram with a resolution of 100 μm (Molecular Imager FX; Bio-Rad), and radioactivity evaluation was done by using the program Quantity One (Bio-Rad). The method was calibrated by using samples whose radioactivity content was determined by liquid scintillation counting.
Molecular biological techniques.
DNA was isolated and purified according to the method described by Murray and Thomson (38). Plasmid DNA was isolated according to the method described by Birnboim and Doly (9). Standard procedures were used for concentration and buffer change of plasmid DNA and DNA fragments (42). Total RNA used in primer extension experiments was isolated via the hot-phenol method (1). Total RNA used for reverse transcription (RT)-PCR was isolated by using an RNeasy Total RNA kit (QIAGEN, Hilden, Germany). DNA and RNA concentrations were determined photometrically (260 nm).
(i) Cloning of gene fragments.
Primers were derived to amplify orf1, orf2, and orf3 via PCR (Table 1). PCR (41) was performed under the following conditions: 100 ng of target DNA; 200 nM each primer; 200 μM each dATP, dCTP, dTTP, and dGTP; 50 mM KCl; 1.5 mM MgCl2; 10 mM Tris-HCl (pH 9.0), and 1 U of Taq DNA polymerase (Amersham Biosciences). The assay mixture (100 μl) was overlaid with 75 μl of paraffin oil. PCR parameters were as follows: 45 s at 95°C and 45 s at 50°C and 30 cycles of 2 min (orf1 and orf2 fragment) and 1 min (orf3 fragment and colony PCR) at 72°C. Fragments containing orf1, orf2, or orf3 were cloned into the pETBlue-1 expression vector (Novagene, Darmstadt, Germany); fragments harboring orf1 or orf2 were also cloned into the pMal-c2X expression vector. Success of cloning was proved by colony PCR after transformation.
(ii) Overexpression of cloned genes.
Chemical transformation of bacterial cells was done according to standard protocols (18). The pETBlue-1 vector constructs were transformed and expressed in E. coli TUNER(DE3)pLacI cells, and the pMal-c2X vector constructs were transformed in E. coli SURE cells (in Luria-Bertani medium at 37°C under aerobic conditions). Growth was monitored by measuring the optical density at 578 nm. At an OD578 of 0.5, cultures were induced by adding 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Incubation continued for 3 h with shaking at 37°C. The formation of overproduced protein was monitored by analyzing samples of whole IPTG-induced cells at different time points by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
(iii) RT-PCR.
Contaminating DNA in total RNA preparations was removed with RNase-free DNase (5 U/μg, 30 min, 37°C; MBI Fermentas). Complete removal of DNA was tested by PCR (see Fig. 2B, lane a, and primer pair 5′orf1for-orf1Cy5rev in Table 1; expected fragment length, 555 bp). A cDNA library was obtained by an RT-PCR using random hexamer priming mix (MBI Fermentas). The cDNA was used in subsequent PCRs with specific primers. To confirm the expression of the gene cluster as an operon, primers were derived from the coding regions and used in PCR to amplify fragments including the intergenic regions between two ORFs (b, orf1for-orf2rev [530 bp]; c, orf2for-orf3rev [497 bp]; d, orf3for-orf4rev [450 bp]; e, orf4for-orf5rev [444 bp]; f, orf5for-orf6rev [526 bp]; g, orf6for-orf12rev [582 bp]; h, orf12for-orf7rev [491 bp]; i, orf7for-orf8rev; [242 bp]; j, orf8for-orf9rev [380 bp]; k, orf9for-orf10rev [277 bp], l, orf10for-orf13rev [388 bp]; m, orf13for-orf14rev [235 bp]; n, orf14for-orf15rev [427 bp]; o, orf15for-orfXrev [379 bp] [Table 1A and Fig. 2B]). For a molecular mass standard, a GeneRuler 1-kb DNA ladder (MBI Fermentas) was used.
(iv) Mapping of the 5′ end of mRNA.
The transcription start site of the mRNA encoded by the phenol gene cluster was mapped via primer extension (12, 34). A Cy5-labeled primer was derived from the 5′ end of orf1 (orf1rev). For a control, RNA of cells grown anaerobically on 4-hydroxybenzoate was used. Extension products were analyzed by use of an ALFexpress sequencer by J. Alt-Mörbe (Labor für DNA-Analytik, Freiburg, Germany).
Electrophoretic techniques.
DNA agarose gel electrophoresis (42), SDS-PAGE (10 and 12.5%) (32), and visualization of protein bands by staining with Coomassie blue R 250 was done according to standard methods.
Purification of overproduced ORF1 protein.
[U-14C]phenol exchange into phenylphosphate, followed by TLC separation of products and quantification of [14C]phenylphosphate spots, was used as an assay to monitor ORF1 activity throughout purification. All steps were performed at 4°C.
(i) DEAE-Sepharose chromatography.
A DEAE-Sepharose column (volume, 16 ml; diameter, 16 mm; Amersham Pharmacia Biotech) equilibrated with buffer B (20 mM Tris-Cl [pH 7.5], 20% [vol/vol] glycerol, 20 mM mercaptoethanol) was used at a flow rate of 2 ml min−1. The sample (20-ml French press cell extract, 400 mg of protein diluted with 20 ml of buffer B) was applied, and the column was washed with 3 bed volumes of buffer B, followed by a two-step elution with buffer B containing 75 and 150 mM ammonium sulfate, respectively. Fractions of 10 ml were collected and analyzed for enzyme activity. ORF1 eluted at a 150 mM salt concentration. All fractions containing predominantly ORF1 (∼40 ml, 50 mg of protein) were combined and applied to the Butyl-Sepharose column.
(ii) Butyl-Sepharose chromatography.
Ammonium sulfate was added to the combined fractions to a final concentration of 1 M. After a short centrifugation (5 min, 5,000 × g), the supernatant was loaded onto a Butyl-Sepharose column (volume, 28 ml; diameter, 26 mm; Amersham Pharmacia Biotech) previously equilibrated with buffer B plus 1 M ammonium sulfate and run at a flow rate of 2 ml min−1. After washing with 3 bed volumes of buffer B plus 1 M ammonium sulfate, the column was eluted in two steps with buffer B containing 150 and 0 mM ammonium sulfate, respectively. ORF1 protein eluted in the 0 mM salt fraction. The application of a linear salt gradient did not improve purification. Fractions of 10 ml were collected and analyzed. The fractions containing mainly ORF1 (30 ml, 20 mg of protein) were combined and applied to the Mono-Q column.
(iii) Mono-Q chromatography.
Pooled fractions of the previous chromatography techniques were loaded onto a Mono-Q FPLC column (1 ml; Amersham Pharmacia Biotech) equilibrated with buffer B plus 50 mM NaCl. After washing with 3 bed volumes, the column was developed with a linear gradient (100 ml) of 0 to 500 mM NaCl in buffer B. Fractions of 5 ml were collected and analyzed. ORF1 eluted at a 230 to 280 mM salt concentration. Fractions containing ORF1 (15 ml) were combined and concentrated via an Amicon concentrator cell (10-kDa exclusion size membrane) to a protein concentration of 10 mg ml−1.
Purification of MalE-ORF1 protein.
An Amylose column (volume, 12 ml; diameter, 16 mm; Amersham Pharmacia Biotech) equilibrated with buffer C (20 mM Tris-Cl [pH 7.4], 200 mM NaCl, 1 mM EDTA) was used at a flow rate of 1 ml min−1. The sample (20-ml French press cell extract, 120 to 160 mg of protein diluted with 20 ml of buffer C) was applied, and the column was washed with 12 bed volumes of buffer C followed by elution with buffer C containing 10 mM maltose. Active fractions were combined and concentrated via an Amicon concentrator cell to a protein concentration of 10 mg ml−1.
Refolding of the proteins ORF2 and ORF3. (i) Preparation of inclusion bodies.
Overproduced ORF2, ORF3, and MalE-ORF2 were almost exclusively present in inclusion bodies. Therefore, inclusion bodies formed in the pET system were prepared from extracts after French press treatment of the cells. Extraction and refolding of the proteins ORF2 and ORF3 followed in part the protocols described by Coligan et al. (19). The cell extract was centrifuged at 5,000 × g for 10 min (4°C), the supernatant was discarded, and the pellet (inclusion bodies) was washed (resuspended or centrifuged) in two successive steps. The first washing step (50 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1% [vol/vol] Triton X-100) is thought to remove lipids and membrane fragments, while the second washing step (50 mM Tris-Cl [pH 8.0], 1 mM EDTA, 0.5 M urea) should remove proteins adsorbed on the inclusion body surface.
(ii) Refolding of ORF2 and ORF3.
The resulting pellet was suspended in a 1.5- to 2-fold volume of buffered urea solution (100 mM Tris-Cl [pH 8.5], 8 M urea, 50 mM mercaptoethanol). After 1 h of stirring on ice and 30 min of centrifugation (40,000 × g), the supernatant was separated from the resulting pellet. The pellet was discarded, and the supernatant was added dropwise into cold refolding solution (15% [vol/vol] glycerol, 50 mM Tris-Cl [pH 8.5], 10 mM mercaptoethanol) while stirring on ice. The final protein concentration in the refolding buffer was 0.1 mg ml−1. This protein solution was subsequently concentrated via an Amicon concentrator cell (10-kDa exclusion size) to 10 mg ml−1 for ORF2 and 5 mg ml−1 for ORF3, as determined by the method of Lowry et al. (36). Successful refolding was monitored by reconstitution of net phosphorylation activity after complementation with purified ORF1 or ORF1 and ORF2. A standard assay, as described above, was used. Refolding of the insoluble proteins ORF2 and ORF3 rendered them soluble but possibly mostly still inactive. Significant amounts of both proteins were precipitating during the first minutes of the enzyme assay. It is likely that these proteins did not posses their native conformations.
UV-VIS spectroscopy.
UV-visible (VIS) spectra of solutions of proteins ORF1, ORF2, and ORF3 were recorded between 250 and 600 nm against buffer. The protein was dissolved in 25 mM Tris-HCl (pH 8.5) buffer containing 15% (vol/vol) glycerol at a concentration of approximately 0.5 mg ml−1.
RESULTS
[14C]phenol exchange reaction and phosphorylation of phenol by extracts of T. aromatica cells.
Extracts of cells grown under denitrifying conditions with phenol (induced cells) and 4-hydroxybenzoate (noninduced cells) were studied for the [14C]phenol exchange reaction and the nucleoside triphosphate-dependent phosphorylation of phenol, two reactions which are considered pertinent to phenol metabolism. Induced cells catalyzed the exchange of free [14C]phenol and the phenol moiety of phenylphosphate at a specific rate of 0.11 nmol min−1 mg−1 of protein. This phenylphosphate-dependent formation of [14C]phenylphosphate in the presence of [U-14C]phenol was not observed in noninduced cells. Performing the assays under anaerobic conditions did not enhance the overall enzyme activity. The reaction was stimulated by Mn2+ (1 mM), and the activity in the absence of added Mn2+ was 14%. In contrast to previous results (30), the optimal pH for the [14C]phenol exchange reaction in cell extract was 8.5; the rate at pH 7.5 was 10% (data not shown). The different pH response may be due to buffer effects on both the reaction rate and TLC separation of phenylphosphate. The reaction was linearly time dependent in the range of 0 to 8 min and linearly protein dependent in the range of 0 to 8 mg of protein ml−1. Induced cells did not catalyze the exchange of free [32P]phosphate and the phosphoryl group of phenylphosphate (16).
Extracts of induced cells also catalyzed the MgATP-dependent formation of [14C]phenylphosphate from [U-14C]phenol at a specific rate of 1.5 nmol min−1 mg−1 of protein. Noninduced cells were inactive. Activity was reliably detected in cell extracts only if cells were lysed by lysozyme treatment followed by 20% (vol/vol) PEG 600 precipitation; the activity was recovered in the supernatant. This reaction was slightly stimulated (20%) by 1 mM Mn2+. No [14C]phenol phosphorylation was observed with GTP, CTP, ITP, phosphoenolpyruvate, acetylphosphate, carbamoylphosphate, or creatine phosphate. Inorganic phosphate or pyrophosphate did not stimulate the ATP-dependent reaction. The reaction had a narrow pH optimum at pH 9.0; below pH 7, no activity was detectable. The reaction slowly leveled off and was linearly dependent on time only in the first few minutes. Protein dependence was nonlinear and almost quadratic, suggesting that the net phosphorylation reaction was catalyzed by a multicomponent system, in contrast to the [14C]phenol exchange reaction. Half-maximal rates were obtained with 40 ± 10 μM phenol. The apparent Km value for ATP could not be determined because of interfering activities in the cell extract.
Overexpression of two putative phenylphosphate synthase genes in E. coli.
Two genes (orf1 and orf2) out of a cluster of up to 16 genes (Fig. 2A) are considered to play a role in phenol phosphorylation (16, 43). However, overexpression experiments with a recombinant pMal-c2 vector encoding both ORFs resulted only in the overproduction of ORF1; therefore, the two ORFs were cloned and expressed separately. Attempts to purify protein ORF2 from T. aromatica cell extract were unsuccessful. The ORF2 assay was based on the complementation of phenylphosphate synthase activity when ORF1 was present (see below). Therefore, both ORFs were cloned and overexpressed in two different vector systems in E. coli. While a considerable amount of protein ORF1 was soluble (30 to 50% of overproduced protein), protein ORF2 consistently formed inclusion bodies when the pMal or the pETBlue vector system was used. ORF1 could be purified by several chromatographic steps, while ORF2 had to be refolded from the inclusion bodies formed when the pETBlue-1 vector was used.
Purification of phenylphosphate synthase proteins ORF1 and ORF2.
ORF1 without a tag was purified from 400 mg of cell extract protein, yielding 10 mg of ORF1 which was more than 80% pure (SDS-PAGE) (Table 2 and Fig. 3). In the case of the N-terminal fusion protein MalE-ORF1, an amylose affinity column was used, and the purified fusion protein was not proteolytically processed. From 120 mg of cell extract protein, 8 mg of MalE-ORF1 which was 70% pure was obtained (SDS-PAGE) (data not shown). Inclusion bodies of ORF2 formed in the pET system were prepared from cell extract. From the urea extract of 350 mg of protein of inclusion bodies, 180 mg of soluble ORF2 protein which was more than 90% pure was obtained (SDS-PAGE) (Fig. 4).
TABLE 2.
Purification of overproduced ORF1 from 10 g of cells (fresh cell mass) of E. coli TUNER (pET vector system)a
| Purification step | Total activity (nmol min−1) | Total protein (mg) | Sp ac (nmol min−1 mg−1) | Yield (%) | Enrich- ment (fold) |
|---|---|---|---|---|---|
| Soluble extract | 24 | 400 | 0.06 | 100 | 1 |
| DEAE-Sepharose | 27.5 | 50 | 0.55 | 114 | 9.2 |
| Butyl-Sepharose | 16.1 | 18.7 | 0.86 | 67 | 14.3 |
| Mono-Q | 10.5 | 9.5 | 1.1 | 43 | 18.3 |
The exchange of radioactivity from [14C]phenol into phenylphosphate was measured.
FIG. 3.

Purification of ORF1 protein overproduced in E. coli TUNER. SDS-PAGE (10%) of various steps of the purification is shown. Lanes: 1, molecular mass standard; 2, total cell protein of induced E. coli cells (30 μg of protein); 3, soluble fraction of cell extract (induced cells) (20 μg of protein); 4, DEAE-Sepharose fraction (6 μg of protein); 5, Butyl-Sepharose fraction (5 μg of protein); 6, Mono-Q fraction (2 μg of protein).
FIG. 4.

Purification of overproduced proteins ORF2 and ORF3. SDS-PAGE (12.5%) of ORF2 and ORF3 after refolding is shown. Lanes: 1, ORF2 (5 μg of protein); 2, ORF3 (5 μg of protein); 3, molecular mass standard.
[U-14C]phenol exchange into phenylphosphate catalyzed by protein ORF1.
Overproduced and purified wild-type ORF1 protein and the fusion protein MalE-ORF1 were tested for enzyme activities related to phenol phosphorylation. Two reactions were studied, the exchange of free [U-14C]phenol and the phenol moiety of phenylphosphate (exchange reaction) and the ATP-dependent formation of [14C]phenylphosphate from [U-14C]phenol (net phenol phosphorylation). Both ORF1 preparations (with and without a MalE tag) catalyzed the exchange reaction with a specific activity of 1.1 nmol min−1 mg−1 of protein; however, they were unable to catalyze the ATP-dependent net phenol phosphorylation. The reaction was dependent on Mn2+ (1 mM). No exchange activity was detectable with 1 mM Ca2+, Mg2+, Zn2+, or Co2+. The optimum pH of the exchange reaction was around 8.7, with half-maximal rates at pHs 8.2 and 9.4.
Reconstitution of phenylphosphate synthase activity by combining proteins ORF1 and ORF2.
ORF2 alone showed neither exchange nor net phenol phosphorylation activity (Fig. 5A). A combination of both ORF1 (without tag) and ORF2 exhibited both exchange activity and net phosphorylation activity (1.1 nmol min−1 mg−1 of protein) (Fig. 5A). In contrast, the combination of the MalE-ORF1 fusion protein and ORF2 did not reconstitute net phosphorylation activity (data not shown). Mn2+ (1 mM) stimulated the rate of the net phosphorylation reaction fourfold.
FIG. 5.

Characterization of phenylphosphate synthase. A. Study of ATP-dependent net phosphorylation of [14C]phenol to phenylphosphate (P) and of [14C]phenol exchange into phenylphosphate (E). From left to right: either ORF1 (0.2 mg) alone, ORF2 alone (0.2 mg), or ORF1 and ORF2 combined (0.2 mg each) were tested in the two assays (P and E). B. Study of the incorporation of 32P into phenylphosphate from either γ-32P-labeled ATP ([γ-32P]) or β-32P-labeled ATP ([β-32P]). Autoradiograms of TLC plates are shown. For experimental details, see Materials and Methods.
Determination of products of ATP hydrolysis.
The products of ATP hydrolysis formed by the reconstituted phenylphosphate synthase (ORF1 and ORF2) in the presence of 2 mM phenol were determined. AMP was the only product formed, besides phenylphosphate and trace amounts of ADP (data not shown).
ORF2 shows sequence similarity to the N-terminal part of phosphoenolpyruvate synthase which is supposed to contain the ATP binding site (25). Moreover, ORF2 was needed to reconstitute net phenol phosphorylation activity. This finding implies that ORF2 is involved in the binding and cleavage of ATP. When ORF2 alone was incubated in the absence of phenol, ATP was very slowly hydrolyzed, resulting mostly in the formation of ADP, but small amounts of AMP were also formed (Fig. 6B). This result suggests that ORF2 has low ATPase activity. Note that a small amount of ADP was also formed in the absence of enzyme (Fig. 6A). The combined subunits ORF1 and ORF2 catalyzed the formation of AMP and trace amounts of ADP from ATP under the same conditions (Fig. 6C). The addition of ORF3 to ORF1 and ORF2 gave the same result (Fig. 6D), indicating that the reaction catalyzed by phenylphosphate synthase was as follows: phenol + MgATP + H2O→phenylphosphate + MgAMP + inorganic phosphate.
FIG. 6.

HPLC analysis of products derived from 2 mM MgATP which were formed by subunits of phenylphosphate synthase in the absence of phenol. A. Control in which ATP was incubated for 15 h in the assay mixture in the absence of protein. B. Assay containing purified ORF2 (0.2 mg). C. Assay containing purified ORF1 (0.2 mg) plus ORF2 (0.2 mg). D. Assay containing purified ORF1 (0.2 mg) plus ORF2 (0.2 mg) plus ORF3 (0.2 mg). Elution times: AMP, 3 min; ADP, 6 min; ATP, 8 min.
Further catalytic properties of phenylphosphate synthase.
Purified phenylphosphate synthase consisting of ORF1 and ORF2 was characterized with respect to cosubstrate specificity and kinetic properties. Besides ATP, UTP (50% activity) was the only nucleoside triphosphate which could be used as a cosubstrate for phenol phosphorylation. The divalent metal ions Mg2+ and Mn2+ were required as cofactors. Mg2+ was strictly required, as expected for an ATP-consuming enzyme. Without Mn2+, the rate of net phosphorylation dropped to 25% of the rate with Mn2+. The addition of monovalent cations such as K+ (50 mM) or NH4+ (30 mM) did not stimulate enzyme activity. Mercaptoethanol (40 mM) stimulated phenylphosphate formation (70% increase) and resulted in better reproducibility of the results. Apparent Km values were 0.04 mM for phenol and 2 mM for ATP. The reaction was linearly time dependent in the range of 0 to 5 min maximally. The dependence of the rate of net phosphorylation on the amount of ORF2 is shown in Fig. 7A; a constant amount of ORF1 (4.2 mg/ml) was added. The net phenol phosphorylation reaction had a pH optimum of around 8.5, with half-maximal rates at pHs 7.5 and 9.2. UV-VIS spectra of the proteins ORF1, ORF2, and ORF3 showed only protein absorption maxima at around 280 nm. ORF1 showed a shoulder at 290 nm, which is most likely due to its relatively high (3.8%) tryptophan content.
FIG. 7.

Dependence of phenylphosphate synthase activity on (A) increasing amounts of ORF2 and (B) increasing amounts of ORF3. A. A constant amount of 840 μg of ORF1 was present in a 200-μl assay mixture (5-min incubation). B. Constant amounts of 100 μg of ORF1 and 200 μg of ORF2 were present in a 100-μl assay mixture (10-min incubation).
Demonstration that the β-phosphoryl group of ATP is incorporated into phenylphosphate.
Whether the β- or the γ-phosphoryl group of ATP was transferred to phenol was studied. When [γ-32P]ATP was used in the assay, no radioactive phenylphosphate was formed. In contrast, labeled phenylphosphate was formed when β-32P-labeled ATP was used (Fig. 5B). This result showed that the β-phosphate of ATP is transferred to phenol in the course of the reaction and that the γ-phosphate must have been released as phosphate.
Stimulation of net phenol phosphorylation by ORF3 protein.
The previous experiments indicated that proteins encoded by orf1 and orf2 are basically sufficient to constitute net phosphorylation activity of phenylphosphate synthase. Nevertheless, the observed rates of this activity were very low. This result might indicate that one or both of the overexpressed proteins were largely inactive and/or that a component was missing. A plausible candidate for a missing component was the product of gene orf3 which is adjacent to orf2. Therefore, orf3 was cloned and overexpressed in E. coli, and whether it had an effect on phenol phosphorylation rate was tested. The procedure for cloning, overexpression, and refolding from inclusion bodies followed the protocol given for ORF2, and purification resulted in 70% pure ORF3 (Fig. 4). When increasing amounts of purified ORF3 were added to assays containing ORF1 and ORF2, an increase of up to fourfold in ATP-dependent phenylphosphate synthase activity was observed (Fig. 7B). This effect was not due to an increase of the protein concentration since bovine serum albumin did not show such a stimulatory effect. ORF3 had no effect on the rate of the [14C]phenol exchange reaction.
Possible promoter region and primer extension analysis.
orf1 is the first gene of the phenol gene cluster (Fig. 2A). A transcription start site upstream of orf1 was identified via a primer extension experiment. A labeled primer was derived from the 5′ region of orf1 and used in a reverse transcription assay with total RNA isolated from cells grown anaerobically on phenol and 4-hydroxybenzoate. The cDNA products were analyzed on a sequencing gel. With RNA isolated from phenol-grown cells, a specific cDNA product corresponding to a transcription start at a guanosine 65 bp upstream of the ATG translation start codon of orf1 was observed (Fig. 8). In the area 12 to 24 bp 5′ of the transcription start, a σ54-RNA polymerase-specific promoter sequence was found (GTGGCACACCCTTTGCA) (6) (Fig. 8). No cDNA product was observed with total RNA isolated from 4-hydroxybenzoate-grown cells, indicating that the transcription of the phenol gene cluster is phenol induced.
FIG. 8.

Promoter structure of the phenol gene clusters of (A) T. aromatica and (B) G. metallireducens. The intergenic sequence between orf1 and orf11 (regulator gene) in T. aromatica and in G. metallireducens is shown. The ATG start codons are marked in boldface type. Numbers in front of the lines indicate the nucleotide positions within the phenol gene clusters (T. aromatica [GenBank accession number AJ272115] and G. metallireducens, intergenic region between putative ORFs [Joint Genome Institute accession numbers gi|23053807 and gi|23053806]). Shine-Dalgarno sequences are underlined. −24/−12 σ54 promoters are shown in grey. Arrows: inverted repeats; box: T. aromatica transcription start; dashed box: G. metallireducens proposed transcription start (bp + 1). The proposed ATG start codon for orf1 of Geobacter was corrected (see the text).
Expression of the gene cluster as an operon.
RT-PCR was carried out to identify which of the genes encoded by the phenol gene cluster are coordinately expressed. A cDNA library of total RNA isolated from cells grown anoxically on phenol was prepared and used in PCR. Primers were derived from the coding regions of various genes and used to amplify PCR fragments covering the intergenic regions between two respective ORFs. These experiments demonstrated that ORFs 1 to 12, whose products are directly involved in phenol carboxylation, are transcribed as one mRNA product (Fig. 2B). Interestingly, ORFs 7 to 10 and 13 to 15, some of which encode phenol-induced proteins, are also located on the transcript. Furthermore, the operon seems not to be complete with orf15. The 5′ part of an additional ORF (orfX) was found downstream of orf15. RT-PCR experiments indicate that this ORF is also cotranscribed. Hence, at least 15 genes of the cluster probably form an operon and are encoded on one transcript.
DISCUSSION
General properties of phenylphosphate synthase.
We have described an enzyme that catalyzes the following reaction: phenol + MgATP + H2O→phenylphosphate + MgAMP + orthophosphate. Two phosphate groups are transferred from the donor ATP to two different acceptors, phenol and water. The new enzyme, therefore, has the EC number 2.7.9.x and belongs to phosphotransferases with paired acceptors. The recommended name is phenylphosphate synthase or phenol:water dikinase. The systematic name is ATP:phenol:water phosphotransferase. The reaction catalyzed by phenylphosphate synthase is similar to that of phosphoenolpyruvate synthase (pyruvate:water dikinase) (EC 2.7.9.2), and so are the primary structures of the two 70- and 40-kDa subunits of the enzyme (see below). In contrast to phosphoenolpyruvate synthase, phenylphosphate synthase is stimulated by another small 23-kDa protein which does not have any counterpart in the literature. Enzyme activity is stimulated by Mn2+ and is not strongly affected by molecular oxygen, if at all.
Molecular composition and catalytic properties of phenylphosphate synthase.
Phenylphosphate synthase activity clearly depends on ORF1 and ORF2 and is stimulated severalfold by ORF3. A strict dependence on ORF3 was not observed. Unfortunately, the overproduction of ORF2 and ORF3 resulted in the formation of inclusion bodies, and refolding of the insoluble proteins rendered them soluble but possibly mostly still inactive. Large portions of both proteins were precipitating during the first minutes of the enzyme assay. This result indicates that these proteins did not posses their native conformations. Therefore, the turnover rate of the reconstituted enzyme was much lower than expected. Also, no reliable information about the native subunit composition of the enzyme could be obtained. Interestingly, even in concentrated cell extracts, the observed phenylphosphate synthase activity was extremely low and could be reliably measured only when cell extract was prepared by lysozyme treatment of cells, the extract was precipitated by polyethyleneglycol, and the protein content in the assay mixture was high. This finding suggests that the native enzyme easily decomposes into its subunits and becomes inactive. A similar behavior is known for some other enzymes; for instance, acetyl-coenzyme A (CoA) carboxylase is notorious for being completely inactive in cell extracts of most bacteria, and low enzyme activity can be achieved only by recombining the three separately purified subunits (20). The natural substrates of the enzyme are phenol (apparent Km, 0.04 mM) and MgATP (apparent Km, 2 mM). The low apparent Km value for phenol obviously allows the bacterium to grow at rather low ambient concentrations of phenol.
Function of ORF1, ORF2, and ORF3 of phenylphosphate synthase.
The conclusion that ORF1 alone is sufficient to catalyze the [14C]phenol exchange reaction was derived from experiments with ORF1 (without and with fusion protein) overproduced in E. coli. ATP-dependent net phosphorylation of phenol required the addition of ORF2, indicating that the active holoenzyme can be reconstituted from its subunits and therefore may not form a stable enzyme complex. The N-terminal 42-kDa MalE protein fusion to ORF1 seemed to disturb ORF1 and ORF2 interaction since this combination was inactive in ATP-dependent phenol phosphorylation, whereas the fusion protein was active in the [14C]phenol exchange reaction.
The dependence of phenol phosphorylation on the amount of ORF2 added to a constant amount of ORF1 resulted in a saturation curve (Fig. 7A) rather than in a sharp titration curve, again indicating that the two subunits may not form a stable complex. For unknown reasons, the rate of phenylphosphate formation rapidly decreased with time. This rate decrease cannot be explained by rapidly reaching an equilibrium between [14C]phenol and [14C]phenylphosphate.
The role of ORF3 is at issue. Adding protein ORF3 to the standard net phosphorylation assay improves the overall rate of phenylphosphate formation. ORF3 contains a pair of cystathionine-β-synthase domains (7). These domains of so-far-unknown function are proposed to participate in the binding of the adenosyl moiety or may play a role as a structural or regulatory component (27, 44). A similar function could be proposed for ORF3.
Proposed catalytic mechanism and similarity of phenylphosphate synthase to PEP synthase.
Phenylphosphate synthase transfers the β-phosphoryl rather than the γ group from ATP (donor) to the acceptor phenol. This behavior has been reported for PEP synthase, which catalyzes a similar reaction (equation 9).
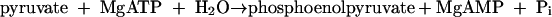 |
(9) |
The catalytic features of phenylphosphate synthase are consistent with a ping-pong mechanism, as in PEP synthase (39). ORF2 contains an ATP binding domain, suggesting that this protein catalyzes the transfer of the pyrophosphoryl group of ATP to the conserved target histidine residue (His569) in ORF1; this residue corresponds to the conserved histidine 421 in E. coli PEP synthase which is thought to be reversibly transformed into a pyrophosphorylated histidine-β-phosphate-γ-phosphate. The terminal γ-phosphate group is proposed to be irreversibly hydrolysed off (transferred to water) from ORF1, giving rise to free γ-phosphate, whereas the histidine-β-phosphate is reversibly transferred to phenol (note that phenol formally corresponds to the enol form of pyruvate). The phosphoryl group transfer from phosphorylated ORF1 to phenol giving rise to phenylphosphate and nonphosphorylated ORF1 must be reversible, resulting in the observed exchange reaction of [14C]phenol and the phenol moiety of phenylphosphate. This means that the group transfer potential of phosphohistidine of ORF1 and that of phenylphosphate should not be too dissimilar. The role of Mn2+ in this catalytic cycle is at issue. Maybe Mn2+ is required to stabilize the enzyme-phosphate intermediate or the interaction between ORF1 and phenylphosphate, since the exchange reaction was mainly affected by Mn2+. Some kinases require another divalent cation for full activity in addition to Mg2+, which coordinates with the nucleotide. Examples are pyruvate kinase and phosphoenolpyruvate carboxykinase (4, 33, 49). The second divalent metal cation has a separate distinct binding site (37). Hence, the proposed mechanism for phenylphosphate synthase shown in Fig. 9 is completely analogous to that of PEP synthase. This scheme does not account for the role of ORF3, however, and therefore must be incomplete.
FIG. 9.

Hypothetical catalytic mechanism of the complete phenylphosphate synthase reaction catalyzed by the combined action of ORF1 and ORF2 and of the partial reaction, the exchange of free [14C]phenol, and the phenol moiety of phenylphosphate (exchange reaction) catalyzed by ORF1 alone. Note that the role of ORF3 is not explained by this scheme.
Similar entries in databases.
Geobacter metallireducens is the only one of the bacteria which reportedly grows with phenol under anaerobic conditions whose genome has been partially sequenced (NCBI accession no. gi|48851373|ref|NZ_AAAS00000000.12). This genome contains orthologues of ORFs 1, 2, and 3 located near each other (discussed in reference 43). This suggests that phenol metabolism in this bacterium is similar to that in T. aromatica. The following similar proteins were reported in the database (Fig. 10).
FIG. 10.

Comparison of the proteins ORF1 (A), ORF2 (B), and ORF3 (C) with proteins in databases. The numbers refer to the positions of the amino acids. A. Highly similar sequences are marked in black, and sequences with weak or no similarities are in white. Dark grey indicates an extended conserved region in the proteins of T. aromatica, B. subtilis, and G. metallireducens, and light grey indicates an additional sequence similarity between the proteins of T. aromatica and G. metallireducens. H marks a conserved histidine residue in all four proteins. Striping (B) and cross-hatching (C) indicate sequence similarities. For details, see Discussion. CBS, cystathionine-β-synthase; PEPS, PEP synthase.
(i) ORF1.
ORF1 (612 amino acids [aa], 70.26 kDa) has the highest similarity (percentage of identical or similar amino acids) with a hypothetical, ORF1-like protein of similar size from G. metallireducens (NCBI accession no. gi|48845085|ref|ZP_00299374.1) (78% identity and 88% similarity). The N-terminal part of approximately 500 amino acids does not show strong similarity with other known proteins. However, the approximately 100-amino-acid C-terminal part of the protein has high similarity with the central part of PEP synthase from E. coli (accession number gi|16129658|ref|NP_416217) (37% identity and 55% similarity). Interestingly, an extended C-terminal region of ORF1 (approximately 240 aa) has a counterpart in the C-terminal region of the PEP synthase of Bacillus subtilis (accession number gi|16078943|ref|NP_389764) (28% identity and 47% similarity). The highly conserved 100-amino-acid C-terminal part of ORF1, which corresponds to the central or C-terminal part of most PEP synthases, contains a conserved histidine which was shown for PEP synthase of E. coli and is considered to represent the phosphorylation site for phenylphosphate synthase.
(ii) ORF2.
ORF2 (374 aa, 40.42 kDa) has the highest similarity with a hypothetical protein of similar size from G. metallireducens (NCBI accession no. gi|488450861|ref|ZP_00299375.1) (70% identity and 83% similarity) and with the N-terminal part (approximately 350 aa) of PEP synthase of E. coli (44% identity and 61% similarity) and other (putative) PEP synthases from different bacteria (e.g., B. subtilis and Staphylothermus marinus).
(iii) ORF3.
ORF3 (223 aa, 24.24 kDa) has the highest similarity with a hypothetical (NCBI Microbial Genomes Annotation Project) protein of similar size from G. metallireducens (NCBI accession no. gi|48845089|ref|ZP_00299378.1) (60% identity and 77% similarity). Both proteins contain a pair of cystathionine-β-synthase domains (pfam00571) (7) with so-far-unknown function.
Possible regulator of the gene cluster.
A gene for a putative regulator protein, ORF11, is located directly upstream of orf1 but is transcribed in the opposite direction (16, 43). ORF11 shows high similarity to σ54-dependent transcription activators of gene clusters involved in the aerobic degradation of phenolic [e.g., (dimethyl)phenol] compounds (16). Examples are PoxR of Ralstonia sp. (Joint Genome Institute accession number gi|3445531) (54% identity and 70% similarity) and DmpR of Pseudomonas sp. (accession number gi|483552) (47% identity and 68% similarity) (26, 45, 46). Orf11 also has high similarity with three putative σ54-dependent activators of G. metallireducens (accession numbers gi|23053791, gi|23053793, and gi|23053807) (66% identity and 79% similarity). One of these regulator genes is located as orf11 in T. aromatica (Fig. 2A) (43). They all contain an XylR domain (pfam06505) in the N-terminal part; the binding of the respective effector molecule is expected to lead to their activation (reviewed in references 17 and 48).
Promoter structure.
σ54-RNA polymerase specifically binds to promoters with the consensus sequence YTGGCACGN4TTGCW in the area 12 to 24 bp 5′ of the transcription start (6). A putative −12/−24 promoter consensus sequence was found 5′ of the transcription start of T. aromatica orf1 (16) (Fig. 8A). A similar putative σ54-dependent promoter sequence was also found in the Geobacter gene cluster but was overlapping with the proposed ATG start codon for the orf1 orthologue (Fig. 8B), which is rather unlikely. Also, no Shine-Dalgarno consensus sequence is found in the proposed −8-bp region, and the ORF1 orthologue would be N-terminally prolonged by 38 amino acids compared with the T. aromatica ORF1 sequence. We therefore propose a corrected ATG start codon for Geobacter ORF gi|23053806 (Fig. 8B), which shows a Shine-Dalgarno consensus sequence, and the derived protein has full sequence similarity to the Thauera ORF1 protein. The Geobacter −12/−24 σ54 promoter sequence is located about 120 bp upstream of the corrected start of orf gi|23053806.
The σ54-dependent activator proteins usually bind to enhancer sequences that are mostly inverted repeats located 100 to 200 bp upstream of the promoter (28). Inverted repeats representing possible binding sites for the regulator protein are found about 260 bp upstream of the Thauera −12/−24 promoter consensus sequence and in the region between −220 and −370 bp upstream of the Geobacter orthologue (Fig. 8), respectively. Contact between regulator protein and RNA polymerase is mediated by the formation of a DNA loop. In some cases, DNA is bent via the binding of the integration host factor to AT-rich DNA sequences located between the promoter and enhancer regions. This feature also applies to the 5′ region of the phenol gene cluster of Thauera and to the orthologue of Geobacter (accession number gi|23053806) (Fig. 8). We therefore conclude that the phenol gene cluster of T. aromatica and the putative phenol gene cluster of G. metallireducens are under the transcriptional control of a σ54-dependent regulator, most probably encoded by orf11 and an orf11 orthologue of G. metallireducens, respectively.
Postulation of operon structure.
ORF11 was proposed to control the expression of the phenol gene cluster, acting as a transcriptional activator when binding to phenol or a similar compound (16). It was previously shown (43) that only the 5′ part of the phenol gene cluster of T. aromatica (ORFs 1 to 6 and 12) is needed for anaerobic degradation of phenol to 4-hydroxybenzoate, and we proposed that the 3′ part of the cluster (ORFs 7 to 10 and 13 to 15) may play a role in the degradation of a phenol derivative. Nevertheless, both parts of the gene cluster seemed to be coordinately expressed, as indicated by identification of phenol-induced proteins and RT-PCR experiments. It was shown for DmpR (45, 46) that this regulator protein was activated not only by phenol but also by some (methyl) derivates. Therefore, it is possible that not only phenol but also similar phenolic compounds may activate ORF11 and lead to the transcription of the gene cluster; the degradation of those compounds may need the proteins encoded by the 3′ part of the cluster.
Acknowledgments
This work was supported by a grant of the Deutsche Forschungsgemeinschaft, by a fellowship of Deutsche Forschungsgemeinschaft/Mongolian Academy of Sciences to A.N. in the frame of Graduiertenkolleg “Biochemie der Enzyme,” and by the Fonds der Chemischen Industrie.
REFERENCES
- 1.Aiba, H., S. Adhya, and B. de Combrugge. 1981. Evidence for two functional gal promoters in intact Escherichia coli. J. Biol. Chem. 256:11905-11910. [PubMed] [Google Scholar]
- 2.Anders, H. J., A. Kaetzke, P. Kämpfer, W. Ludwig, and G. Fuchs. 1995. Taxonomic position of aromatic-degrading denitrifying pseudomonad strains K 172 and KB 740 and their description as new members of the genera Thauera, as Thauera aromatica sp. nov., and Azoarcus, as Azoarcus evansii sp. nov., respectively, members of the beta subclass of the Proteobacteria. Int. J. Syst. Bacteriol. 45:327-333. [DOI] [PubMed] [Google Scholar]
- 3.Aresta, M., and A. Dibenedetto. 2002. Development of environmentally friendly syntheses: use of enzymes and biomimetic systems for the direct carboxylation of organic substrates. J. Biotechnol. 90:113-128. [DOI] [PubMed] [Google Scholar]
- 4.Baek, Y. H., and T. Nowak. 1982. Kinetic evidence for a dual role for muscle pyruvate kinase. Arch. Biochem. Biophys. 217:491-497. [DOI] [PubMed] [Google Scholar]
- 5.Bak, F., and F. Widdel. 1986. Anaerobic degradation of phenol and phenol derivatives by Desulfobacterium phenolicum sp. nov. Arch. Microbiol. 146:177-180. [Google Scholar]
- 6.Barrios, H., B. Valderrama, and E. Morett. 1999. Compilation and analysis of σ54-dependent promoter sequences. Nucleic Acids Res. 27:4305-4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bateman, A. 1997. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22:12-13. [DOI] [PubMed] [Google Scholar]
- 8.Biegert, T., U. Altenschmidt, C. Eckerskorn, and G. Fuchs. 1993. Enzymes of anaerobic metabolism of phenolic compounds. 4-Hydroxybenzoate-CoA ligase from a denitrifying Pseudomonas species. Eur. J. Biochem. 213:555-561. [DOI] [PubMed] [Google Scholar]
- 9.Birnboim, H., and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7:1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boll, M., and G. Fuchs. 1995. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur. J. Biochem. 234:921-933. [DOI] [PubMed] [Google Scholar]
- 11.Boll, M., S. S. Albracht, and G. Fuchs. 1997. Benzoyl-CoA reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. A study of adenosinetriphosphatase activity, ATP stoichiometry of the reaction and EPR properties of the enzyme. Eur. J. Biochem. 244:840-851. [DOI] [PubMed] [Google Scholar]
- 12.Boorstein, W. R., and E. A. Craig. 1989. Primer extension analysis from RNA. Methods Enzymol. 180:347-369. [DOI] [PubMed] [Google Scholar]
- 13.Brackmann, R., and G. Fuchs. 1993. Enzymes of anaerobic metabolism of phenolic compounds. 4-Hydroxybenzoyl-CoA reductase (dehydroxylating) from a denitrifying Pseudomonas species. Eur. J. Biochem. 213:563-571. [DOI] [PubMed] [Google Scholar]
- 14.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 15.Breese, K., and G. Fuchs. 1998. 4-Hydroxybenzoyl-CoA reductase (dehydroxylating) from the denitrifying bacterium Thauera aromatica—prosthetic groups, electron donor, and genes of a member of the molybdenum-flavin-iron-sulfur proteins. Eur. J. Biochem. 251:916-923. [DOI] [PubMed] [Google Scholar]
- 16.Breinig, S., and G. Fuchs. 2000. Genes involved in the anaerobic metabolism of phenol in the bacterium Thauera aromatica. J. Bacteriol. 182:5849-5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck, M., M. T. Gallegos, D. Studholme, Y. Guo, and J. D. Gralla. 2000. The bacterial enhancer-dependent σ54 (σN) transcription factor. J. Bacteriol. 182:4129-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen, S. N., A. C. Y. Chang, and L. Hsu. 1972. Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. Proc. Natl. Acad. Sci. USA 69:2110-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coligan, J. E., B. M. Dunn, D. W. Speicher, and P. T. Wingfield. 2003. Current protocols in protein science, vol. 1. John Wiley & Sons Inc., Hoboken, N.J.
- 20.Cronan, J. E., Jr., and C. O. Rock. 1996. Biosynthesis of membrane lipids, p. 612-636. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed., vol. 1. ASM Press, Washington, D.C. [Google Scholar]
- 21.Ebenau-Jehle, C., M. Boll, and G. Fuchs. 2003. 2-Oxoglutarate:NADP+ oxidoreductase in Azoarcus evansii: properties and function in electron transfer reactions in aromatic ring reduction. J. Bacteriol. 185:6119-6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson, J., M. Dispensa, and C. S. Harwood. 1997. 4-Hydroxybenzoyl-coenzyme A reductase (dehydroxylating) is required for anaerobic degradation of 4-hydroxybenzoate by Rhodopseudomonas palustris and shares features with molybdenum-containing hydroxylases. J. Bacteriol. 179:634-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson, J., M. Dispensa, G. C. Fogg, D. T. Evans, and C. S. Harwood. 1994. 4-Hydroxybenzoate-coenzyme A ligase from Rhodopseudomonas palustris: purification, gene sequence, and role in anaerobic degradation. J. Bacteriol. 176:634-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heider, J., M. Boll, K. Breese, S. Breinig, C. Ebenau-Jehle, U. Feil, N. Gad'on, B. Laempe, B. Leuthner, M. E. Mohamed, S. Schneider, G. Burchhardt, and G. Fuchs. 1998. Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch. Microbiol. 170:120-131. [DOI] [PubMed] [Google Scholar]
- 25.Herzberg, O., C. C. H. Chen, G. Kapadia, M. McGuire, L. J. Caroll, S. J. Noh, and D. Dunaway-Mariano. 1996. Swiveling-domain mechanism for enzymatic phosphotransfer between remote reaction sites. Proc. Natl. Acad. Sci. USA 93:2652-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hino, S., K. Watanabe, and N. Takahashi. 1998. Phenol hydroxylase cloned from Ralstonia eutropha strain E2 exhibits novel kinetic properties. Microbiology 144:1765-1772. [DOI] [PubMed] [Google Scholar]
- 27.Kemp, B. E. 2004. Bateman domains and adenosine derivatives form a binding contract. J. Clin. Investig. 113:182-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kustu, S., A. K. North, and D. S. Weiss. 1991. Prokaryotic transcriptional enhancers and enhancer-binding proteins. Trends Biochem. Sci. 16:397-402. [DOI] [PubMed] [Google Scholar]
- 29.Lack, A., and G. Fuchs. 1992. Carboxylation of phenylphosphate by phenol carboxylase, an enzyme system of anaerobic phenol metabolism. J. Bacteriol. 174:3629-3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lack, A., and G. Fuchs. 1994. Evidence that phenol phosphorylation to phenylphosphate is the first step in anaerobic phenol metabolism in a denitrifying Pseudomonas sp. Arch. Microbiol. 161:132-139. [DOI] [PubMed] [Google Scholar]
- 31.Lack, A., I. Tommasi, M. Aresta, and G. Fuchs. 1991. Catalytic properties of phenol carboxylase. In vitro study of CO2:4-hydroxybenzoate isotope exchange reaction. Eur. J. Biochem. 197:473-479. [DOI] [PubMed] [Google Scholar]
- 32.Laemmli, U. K. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 33.Lee, M. H., C. A. Hebda, and T. Novak. 1981. The role of cations in avian liver phosphoenolpyruvate carboxykinase catalysis. Activation and regulation. J. Biol. Chem. 256:12793-12801. [PubMed] [Google Scholar]
- 34.Leuthner, B., and J. Heider. 2000. Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of β oxidation of the intermediate benzylsuccinate. J. Bacteriol. 182:272-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lovley, D. R., and D. J. Lonergan. 1990. Anaerobic oxidation of toluene, phenol, and p-cresol by the dissimilatory iron-reducing organism, GS-15. Appl. Environ. Microbiol. 56:1858-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowry, O. H., N. J. Rosenrough, A. L. Farr, and P. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 37.Matte, A., L. W. Tari, and L. T. Delbaere. 1998. How do kinases transfer phosphoryl groups? Structure 6:413-419. [DOI] [PubMed] [Google Scholar]
- 38.Murray, M. G., and W. F. Thompson. 1980. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8:4321-4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narindrasorasak, S., and W. A. Bridger. 1977. Phosphoenolypyruvate synthetase of Escherichia coli: molecular weight, subunit composition, and identification of phosphohistidine in phosphoenzyme intermediate. J. Biol. Chem. 252:3121-3127. [PubMed] [Google Scholar]
- 40.Pfennig, N. 1978. Rhodocyclus purpureus gen. nov. and sp. nov., a ring-shaped, vitamin B12-requiring member of the family Rhodospirillaceae. Int. J. Syst. Bacteriol. 28:283-288. [Google Scholar]
- 41.Saiki, R. K., S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich, and A. Arnheim. 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350-1354. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 43.Schühle, K., and G. Fuchs. 2004. Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic phenol metabolism in Thauera aromatica. J. Bacteriol. 186:4556-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott, J. W., S. A. Hawley, K. A. Green, M. Anis, G. Stewart, G. A. Scullion, D. G. Norman, and D. G. Hardie. 2004. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 113:274-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shingler, V., M. Bartilson, and T. Moore. 1993. Cloning and nucleotide sequence of the gene encoding the positive regulator (DmpR) of the phenol catabolic pathway encoded by pVI150 and identification of DmpR as a member of the NtrC family of transcriptional activators. J. Bacteriol. 175:1596-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shingler, V., and T. Moore. 1994. Sensing of aromatic compounds by the DmpR transcriptional activator of phenol-catabolizing Pseudomonas sp. strain CF600. J. Bacteriol. 176:1555-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shinoda, Y., Y. Sakai, M. Ue, A. Hiraishi, and N. Kato. 2000. Isolation and characterization of a new denitrifying spirillum capable of anaerobic degradation of phenol. Appl. Environ. Microbiol. 66:1286-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Studholme, D. J., and R. Dixon. 2003. Domain architectures of σ54-dependent transcriptional activators. J. Bacteriol. 185:1757-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tari, L. W., A. Matte, H. Goldie, and L. T. Delbaere. 1997. Mg2+-Mn2+ clusters in enzyme-catalyzed phosphoryl-transfer reactions. Nat. Struct. Biol. 4:990-994. [DOI] [PubMed] [Google Scholar]
- 50.Tschech, A., and G. Fuchs. 1987. Anaerobic degradation of phenol by pure cultures of newly isolated denitrifying pseudomonads. Arch. Microbiol. 148:213-217. [DOI] [PubMed] [Google Scholar]
- 51.Tschech, A., and G. Fuchs. 1989. Anaerobic degradation of phenol via carboxylation to 4-hydroxybenzoate: in vitro study of isotope exchange between 14CO2 and 4-hydroxybenzoate. Arch. Microbiol. 152:594-599. [Google Scholar]