

Abstract
Background and Objectives
The studies were designed to test the efficacy of two peroxisome proliferator-activated receptor γ (PPARγ) agonists in two rodent models of polycystic kidney disease (PKD).
Materials and Methods
The PCK rat is a slowly progressing cystic model while the Wpk-/- rat is a rapidly progressing model. PCK rats were fed with a pharmacological (0.4 mg/kg body weight [BW]) and a sub-pharmacological (0.04 mg/kg BW) dose of rosiglitazone (week 4–28). Wpk-/- rats were fed with pharmacological (2.0 mg/kg BW) and sub-pharmacologic (0.2 mg/kg BW) doses of pioglitazone from day 5 to 18. At termination, kidney weights of treated versus untreated cystic animals were used to determine efficacy. The current studies were also compared with previous studies containing higher doses of PPARγ agonists. The concentrations used in the animals were calculated with reference to equivalent human doses for both drugs.
Results
The current studies demonstrate: 1) that low, pharmacologically relevant, doses of the PPARγ agonists effectively inhibit cyst growth; 2) there is a class action of the drugs with both commercially available PPARγ agonists, rosiglitazone, and pioglitazone, inhibiting cyst growth; 3) the drugs showed efficacy in two different preclinical cystic models. In the PCK rat, animals fed with a sub-pharmacological dose of rosiglitazone for 24 weeks had significantly lower kidney weights than untreated animals (3.68 ± 0.13 g vs. 4.17 ± 0. 11 g, respectively, P < 0.01) while treatment with a pharmacologic dose had no significant effect on kidney weight. The rapidly progressing Wpk-/- rats were fed with pharmacological and sub-pharmacologic doses of pioglitazone from day 5 to 18 and the kidneys were compared with non-treated, cystic animals. Kidney weights on the pharmacologic dose were not statistically lower than the untreated animals while rats fed a sub-pharmacologic dose showed a significant decrease compared with untreated animals (3.35 ± 0.15 g vs. 4.55 ± 0.46 g, respectively, P = 0.045).
Conclusion
Concentrations of PPARγ agonists below the human equivalent diabetic doses are effective in slowing cyst growth in two rodent models of PKD.
Keywords: rosiglitazone, pioglitazone, cystic fibrosis transmembrane conductance regulator, polycystic kidney disease, cystic kidneys
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder characterized by the growth of fluid-filled cysts predominately in kidney tubules and liver bile ducts. ADPKD has an estimated incidence of 1:400 to 1:1000, making it one of the most common genetic diseases [1–3]. ADPKD progresses slowly and the fluid-filled cysts grow in size due to increased rates of epithelial cell proliferation together with increased fluid and electrolyte secretion into the cyst lumen. The expanding cysts compromise the normal kidney function and result in a decrease of renal function to the point of end-stage renal failure in midlife in approximately 50% of patients[1, 4]. In the US, therapy is limited to cyst aspiration, dialysis, and eventually, renal transplantation after organ failure. Clinical trials for a variety of agents have failed to produce an FDA-approved drug due to side effects that preclude life-long therapy.
In 85% of ADPKD patients, the genetic mutation occurs on chromosome 16, at the PKD1 gene locus that encodes the protein polycystin 1 (PC1). The other 15% of genetic mutations occur on chromosome 4, at the PKD2 gene locus, encoding the protein polycystin 2 (PC2)[5]. Polycystins 1 and 2 are found in the primary cilia, endoplasmic reticulum, junctional complexes, and the plasma membrane. In the primary cilia, the PCs form a complex that functions as a mechanosensitive Ca2+ channel with the characteristics of a transient receptor potential (TRP) channel[6–8]. While PC2 is regarded as the TRP channel, mutations in either PC1 or PC2 cause remarkably similar disease progression.
Loss of function of either PC1 or PC2 results in a decreased influx of Ca2+. In the renal epithelial cells of the distal nephron and cortical collecting duct, Ca2+ influx inhibits adenylyl cyclase VI and activates the Ca2+-stimulated phosphodiesterase 1. The decreased intracellular Ca2+ in ADPKD, therefore, leads to an increase in intracellular cAMP, a finding widely documented in cystic renal epithelia derived from preclinical cystic models and ADPKD patients. In the short term, the subsequent increase in protein kinase A (PKA) alters ion secretion, predominately via the cystic fibrosis transmembrane conductance regulator (CFTR). In the long term, an increase in intracellular cAMP stimulates increased cellular proliferation[2, 3].
Peroxisome proliferator activator receptor gamma (PPARγ) agonists are insulin-sensitizing agents used in the treatment of type 2 diabetes mellitus[9–11]. PPARγ is expressed in the renal collecting duct [12]. Studies in cell culture models of the principal cell type of the distal nephron have shown the PPARγ agonists inhibit cAMP-stimulated anion transport by inhibiting the synthesis of the CFTR protein[13]. The dose response relationships for PPARγ agonist inhibition of ADH-stimulated Cl- secretion via CFTR are an order of magnitude lower than the receptor transactivation profiles that characterize the drug’s effects on insulin sensitivity. Thus, it is reasonable to hypothesize that PPARγ agonists could be used as treatment for polycystic kidney disease (PKD). Previously, we [5] and others [14] have shown that a relatively high dose of the PPARγ agonist, pioglitazone, was effective in decreasing the renal cystic burden in the PCK rat model. However, high doses of PPARγ agonists have documented side effects, including edema and weight gain, leading to the suggestion that this class of drug may not be ideal for life-long treatment of a slowly progressing disease.
The current studies were designed to further test the class action effectiveness of PPARγ agonists by examining another clinically used agonist, rosiglitazone, in the PCK rat model. In addition, to further explore the potential clinical relevance of these compounds, preclinical testing was completed using doses equivalent to sub-pharmacological concentrations of PPARγ agonists in the PCK rat as well as in a genetically distinct, rapidly progressing cystic model, the Wpk-/- rat.
Materials and Methods
Animals
PCK rats were purchased from Charles River Laboratories, Inc. (Wilmington, MA, USA). Heterozygous Wistar Wpk+/- rats were provided by Dr. Nauta (Erasmus Medical Center Rotterdam, Rotterdam, Netherlands). The breeding colonies were established, pathogen-free, and maintained at Indiana University School of Medicine (IUSM) Laboratory Animal Resource Center. All animal studies were performed with approval of the IUSM IACUC and adhered to the NIH Guide for the Care and Use of Laboratory Animals. The animal protocols were designed to minimize pain or discomfort to the animals.
Methods
Both rosiglitazone (Avandia®) and pioglitazone (Actos®) were purchased in tablet form. PCK rats were fed rosiglitazone as part of their chow starting at weaning (4 weeks of age). Treatment groups were fed Purina no. 5002 LabDiet (control diet) supplemented with rosiglitazone calculated to provide the stated doses. Wpk-/- rats with cystic disease do not survive past weaning and, therefore, the pioglitazone was dissolved in grape juice to make it palatable for the suckling animals. Litters from heterozygous crosses were treated orally once daily from day 5 until day 18. The treated animals received either 2.0 mg/kg body weight (BW) or 0.2 mg/kg BW based on each animal’s daily weight.
On the day of sacrifice, the animals were anesthetized with 100 mg/kg intraperitoneally injected sodium pentobarbital. Blood was collected via intracardiac puncture and analyzed at IU Health Pathology Laboratory for serum electrolytes, glucose, and liver enzymes. Body weights were measured and the left kidney and right liver lobe were collected and frozen via liquid nitrogen. The body was flushed with saline and infused with 4% paraformaldehyde. The remaining kidney, liver, and heart were removed and stored in 4% paraformaldehyde. For histological analyses, the kidneys were embedded in paraffin. The paraffin samples were transversely sectioned (4 mmol/L sections) and stained with hematoxylin and eosin.
Statistical analysis
All statistical analyses were performed using SigmaPlot (version 12.5, Systat Software Inc.). In all comparisons, each group of treated animals was compared with the control (untreated) group. Normality of data distribution was tested using the F-test for unequal variance. Normally distributed data were analyzed using Student’s t-test for unpaired samples. For data that were heteroscedastic, a Mann—Whitney U statistical text was used. In all cases, data were expressed as average ± standard error of the mean (SEM) and were considered significant at the P ≤ 0.05 level.
Results
Human equivalent doses used in animal feeding studies
Rodent studies examining the mechanism of action of PPARγ agonists in normal animals have used supra-pharmacologic levels of the drugs [15–17]. The current studies have used more relevant concentrations that are analogous to those that would be used in humans. Table 1 provides the human equivalent doses (HED) of the PPARγ agonists based on FDA-recommended conversion metrics [18].
Table 1.
Summary of drug treatment comparing rat and human doses
| Animal model | Pioglitazone |
|||
|---|---|---|---|---|
| Drug treatment duration | Dose in rats mg/kg BW | Equivalent human dose mg/day | % decrease in KW/BW * | |
| PCK rat - females | 14 weeks (weeks 4-18) | 20 | 227 | 20.7 |
| PCK rat - males | 7 weeks (weeks 3-10) | 20 | 227 | 15.8 |
| PCK rat - females | 7 weeks (week 3-10) | 4 | 45 | 28.7 |
| PCK rat - males | 7 weeks (week 3-10) | 4 | 45 | 26.3 |
| Wpk-/- rats | Day 5-18 | 2.0 | 22.7 | -7.6 |
| Wpk-/- rats | Day 5-18 | 0.2 | 2.27 | 16.6 |
| Rosiglitazone |
||||
| PCK rat (females) | 24 weeks (week 4-28) | 0.4 | 4.5 | -4.1 |
| PCK rat (females) | 24 weeks (week 4-28) | 0.04 | 0.45 | 10.6 |
Notes: The equivalent human dose was calculated using the metric established by the FDA [18] and normalized to a 70 kg human. For comparison, standard dosing in humans for the treatment of diabetes is 15, 30, and 45 mg/day for pioglitazone and 4 or 8 mg/day for rosiglitazone. Where no gender is specified, both male and female animals were combined. Positive values denote a lower kidney mass as a percentage of body weight of treated animals when compared to untreated animals. The calculated values for the PCK rats treated with pioglitazone were taken from Blazer-Yost et al. [5] and the values for Wpk:-/- rats treated with pioglitazone and for PCK rats treated with rosiglitazone were calculated from the data obtained in the current study.
Effect of rosiglitazone in the PCK rat model of renal cystic disease
To test the effects of rosiglitazone, PCK rats were chosen to facilitate comparison with previous studies using pioglitazone. The PCK rat is orthologous to human autosomal recessive PKD, but is phenotypically more similar to human ADPKD. Like humans, the rats show a slowly progressing disease with gender dimorphism[19]. Females tend to develop less severe kidney disease and more severe liver disease.
PCK rats were treated with three doses of rosiglitazone, a supra-pharmacological dose of 4 mg/kg BW (HED = 45 mg/day), a pharmacological dose of 0.4 mg/kg BW (HED = 4.5 mg/day) or a sub-pharmacological dose of 0.04 mg/kg BW (HED = 0.45 mg/day). For comparison, human diabetic patients are typically treated with 4 or 8 mg/day.
Eight animals were randomly assigned to each treatment group with 11 rats serving as control. Only female animals were used because of gender dimorphism and to facilitate comparison with previously published data. The rats were fed either a control diet or a rosiglitazone-supplemented diet beginning at weaning and continuing for 24 weeks. In the cohort that received the high dose of rosiglitazone, five animals had to be sacrificed early due to the development of cholangitis, an infection of the common bile duct. Due to the small number of animals remaining, the fact that they were being administered an unrealistically high HED, and the finding that these remaining animals had an increase in BW (378.5 ± 2.47 g as compared to an average of 340.68 ± 5.5 g in animals on control diet), the decision was made not to include these animals in the summarized data.
Figure 1 illustrates representative transverse sections of the control and treated animals. Two sections from different animals were selected to show the high and low extremes of cystic burden. Interestingly, the pharmacologic dose did not substantially alter the number or size of renal cysts while the treatment with the sub-pharmacologic dose significantly decreased cyst size and apparent number.
Figure 1.
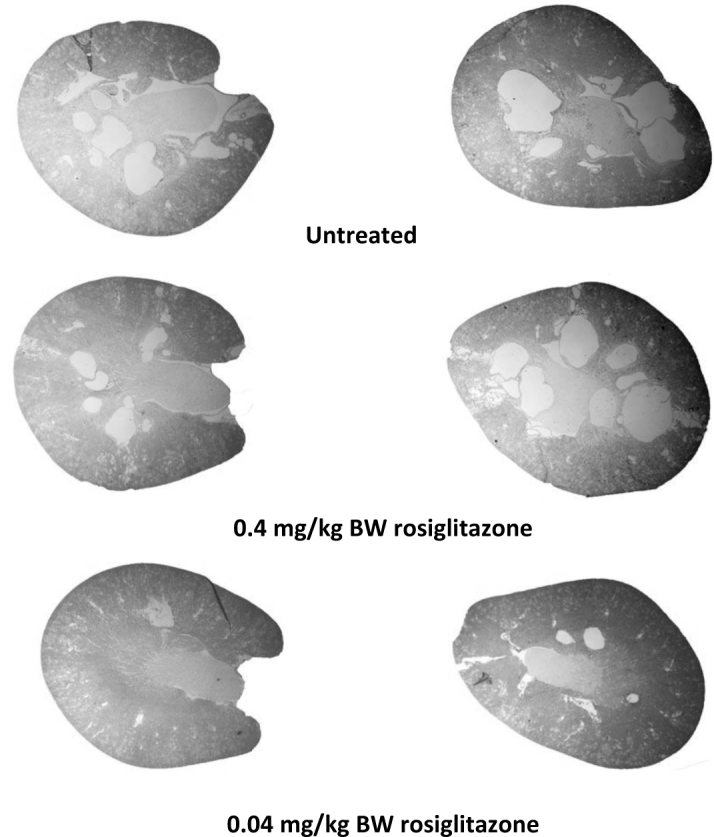
Histological kidney sections from naïve and rosiglitazone-treated PCK rats. The images show transverse histological sections of kidneys from the PCK rats after 24 weeks of feeding beginning in the post-weaning period. Sections taken from animals treated with a diet containing 0.4 mg/kg BW (HED = 4.5 mg/day) or 0.04 mg/ kg BW (HED = 0.45 mg/day) are compared to littermates fed a control (normal chow) diet. The two sections are taken from kidneys of different animals and illustrate the extremes of cystic burden in each of the groups.
The administration of either 0.4 or 0.04mg/kg BW rosiglitazone had no effect on BW, heart weight, or liver weight (Figure 2). No changes were observed in the hematocrits or serum and urine electrolytes (data not shown). The cystic burden, when measured as either total kidney weight or as kidney weight as a percentage of BW, was statistically decreased only by the lowest concentration of rosiglitazone (HED = 0.45 mg/day) (Figure 3). These data form a quantitative measure for the renal sections shown in Figure 1. While only the lowest concentration of rosiglitazone decreases the kidney weight, both the pharmacological and sub-pharmacological doses were effective in reducing serum creatinine. The PCK rats have a relatively normal rodent glucose concentration and this was moderately decreased by 0.4 mg/kg BW treatment, but not by the sub-pharmacologic dose.
Figure 2.

Effect of long-term rosiglitazone treatment on body weight, heart weight, and liver cyst burden in the PCK rat. Animals were fed rosiglitazone at the concentrations listed starting at the time of weaning (4 weeks) for 24 weeks. Data are plotted as means ± SEM with the number of animals in each cohort listed at the base of the column. None of the treatments resulted in a statistically significant change in the treated animals as compared to the rats on a control diet.
Figure 3.

Effect of long-term rosiglitazone treatment on kidney cyst burden, serum creatinine, and serum glucose in the PCK rat. Animals were fed rosiglitazone at the concentrations listed starting at the time of weaning (4 weeks) for 24 weeks. Data are plotted as means ± SEM with the number of animals in each cohort listed at the base of the column. * denotes mean values that were statistically lower than the control fed animals (P < 0.05) by the Student’s t-test.
Effect of pioglitazone in the Wpk rat model of renal cystic disease
To examine the effectiveness of PPARγ agonists in a genetically distinct model, the Wpk rats were used. Wpk rats have a mutation in TMEM67 and are an orthologous model for Meckel Gruber Syndrome type 3. The homozygous affected (Wpk-/-) animals exhibit rapidly progressing cystic disease and severe hydrocephalus while the heterozygous (Wpk+/- rats have no renal phenotype and live to adulthood[19–21]. The homozygous affected animals do not survive past three weeks of age. Wpk-/- rats were treated with pharmacologic (2.0 mg/kg BW; HED 22.7 mg/day) and sub-pharmacological (0.2 mg/kg BW) doses of pioglitazone from postnatal day 5 to 18.
As in the PCK rats, pioglitazone did not cause increases in BW (Figure 4), liver weight, liver weight as a percentage BW or hematocrits (data not shown). The heart weight was statistically lower in the animals treated with the low dose. In the Wpk-/- rat model, the glucose levels in the untreated animals were above the normal values for rodents. The HED of 22.7 mg/day pioglitazone returned these levels to more normal values while the sub-pharmacological dose of 0.2 mg/kg BW (HED = 2.27mg), trended lower, but did not reach statistical significance (P = 0.067; 2-tailed Student’s t-test).
Figure 4.

Effect of short-term pioglitazone treatment in the Wpk-/- rat. Animals were fed pioglitazone at the concentrations listed starting at postnatal day 5 and continuing until postnatal day 18. Data are plotted as means ± SEM with the number of animals in each cohort listed at the base of the column. * denotes mean values that were statistically lower than the control fed animals (P < 0.05) by the Student’s t-test.
With regard to the effectiveness of the PPARγ agonist on cystic burden in Wpk rat model, only the administration of the pioglitazone at the sub-pharmacologic dose was shown to be effective in statistically decreasing renal kidney weight (Figure 4). These data are consistent with the effect of low-dose rosiglitazone in the PCK rat model.
Discussion
ADPKD is often first diagnosed in young adults who experience flank pain due to the growth of multiple renal cysts. The cyst expansion is unrelenting and, in many patients, progresses to renal failure in midlife. In the U.S., the only approved therapies for this disease are cyst aspiration for pain management and dialysis or renal transplantation when the kidneys fail. Thus, there is a critical need for safe, inexpensive treatments that can delay the progression of cyst enlargement and the accompanying destruction of the renal parenchyma that can lead to renal failure. Several drugs, such as the V2 receptor agonist tolvaptan as well as immunosuppressive and chemotherapeutic agents, have entered or, in the case of tolvaptan, completed clinical trials[22–24]. However, these drugs have side effects that may limit patient compliance as well as safety of long-term use. Our overall goal is to develop safe, efficacious drugs that can be used as life-long medications starting at the time of diagnosis in ADPKD patients.
Renal cell culture studies showed that the PPARγ agonists’ inhibitory effect on CFTR, the ion channel responsible for cyst growth, occurs at a concentration that is approximately 10-fold lower than the EC50s for receptor transactivation and that PPARγ agonists decrease CFTR synthesis [13]. In vivo, in the bile duct, PPARγ agonists decrease the amount of CFTR on the apical membrane[5]. The decrease in CFTR channels or channel activity can be measured using electrophysiological techniques, but the low abundance in the kidney precludes the quantification of the apical channels localized in the principal cells. However, we postulate that the mechanism is likely the same in both the renal tubule and the bile duct, namely, a decrease in apical expression of CFTR.
Previous studies have examined the effectiveness of PPARγ agonists on PKD in rodent models. In the embryonic lethal PKD1 knockout mouse, homozygous embryos have hydrops, cardiac conotruncal defects, and renal cystogenesis[25]. Maternally administrated pioglitazone (80 mg/kg/day) improved the survival of the embryos by several days, with a decrease in renal cystogenesis, via an unknown mechanism[25]. A separate PC-PKD1-KO model presents as a rapidly progressing, but less severe phenotype in which the mice develop cystic disease at approximately 1 month of age and survive for 2 months. The dams were treated with 30 mg/kg food weight (estimated to be 4.8 mg/ kg BW/day) pioglitazone and the same concentration was continued after weaning. This dose did not decrease cyst burden or change renal function, but did decrease blood pressure and increase survival[26]. In a slow-progressing model, the Han:SPRD rat, which demonstrates proximal cystic disease, rosiglitazone at 10 mg/kg BW/day decreased the renal cystic burden, protected renal function, and prolonged survival in short-term (8 weeks post weaning) and long-term (6 months post weaning) studies [27].
The PCK rat model is a slowly progressing model that has been used extensively for preclinical testing [28, 29]. In a previous study, the PCK rat was treated with pioglitazone for either 7 or 14 weeks directly after weaning [5] (Table 1). The results showed an improvement in renal and liver cyst burden accompanied by a decrease in apical expression of CFTR in bile duct epithelia[5]. The current studies were designed to extend these findings into pharmacologically relevant doses in an alternative model and to determine the class action of the PPARγ agonists on the cyst growth in the PCK rat model.
In PCK rats, an extended treatment of 24 weeks with rosiglitazone reduced renal cystic burden. However, the highest dose, 4.0 mg/kg BW, resulted in the development of cholangitis in the majority of the animals. This dose of rosiglitazone was chosen to match the previous pioglitazone study, which was conducted for 7 or 14 weeks[5]. In the pioglitazone study, no animals developed cholangitis and it is unknown whether the pioglitazone would have the same effect if the studies were of a similar duration. The etiology of the cholangitis is unknown, but is likely related to the higher dose treatment since none of the control or lower dose-treated animals developed this problem. The animals on 4.0 mg/kg BW also showed statistically significant weight gain, likely from the propensity of rosiglitazone to cause fluid retention. Regardless, this concentration of the PPARγ agonist is unrealistically high and the data regarding the remaining three rats on the highest dose are not included.
At the pharmacological (0.4 mg/kg BW) or subpharmacological (0.04 mg/kg BW) rosiglitazone doses, the known side effect of fluid retention does not appear to be relevant. There were no statistical changes in hematocrits (data not shown), total BW, or heart weight. As an insulin-sensitizing agent, the rosiglitazone lowered the serum glucose only when administered at a pharmacological level of 0.4 mg/kg BW. Neither dose was effective in mitigating the liver cyst burden. Renal cyst burden as assessed by total kidney weight or by kidney weight as a percentage of BW was diminished by only the lowest dose of rosiglitazone (HED = 0.45 mg/day). In contrast, serum creatinine was statistically lower after treatment with either 0.4 or 0.04 mg/kg BW indicating a beneficial effect on renal function that measurement of kidney weight is likely not sensitive enough to detect. These data together with previously published studies using pioglitazone[5], indicate that the effects of PPARγ agonists on the reduction of cyst burden in the PCK rat is a class action effect. In agreement with the tissue culture studies, the current studies show that the effect is manifested at sub-pharmacological doses.
The Wpk rat model was used to extend the studies to an additional, unrelated, preclinical model. This is a rapidly progressing model in which the kidneys represent approximately 15% of the BW by day 18. The Wpk-/- rats were treated with pioglitazone from postnatal day 5 to 18. The two treatment doses represent a pharmacologic dose compared to a sub-pharmacologic dose. The pharmacologic dose was not effective in reducing the cystic burden in the short-term treatment necessitated by the rapidly progressing Wpk model.
For unknown reasons, the blood glucose in the Wpk-/- rats was unusually high. The increased blood glucose was significantly decreased by pharmacological levels of pioglitazone, but was not significantly decreased by the lower dose. The average heart weight was significantly decreased by the low-dose treatment, but this significance was lost when expressed as a percentage of BW (data not shown). Likewise, the hematocrits were unaffected by the drug treatment, indicating that the PPARγ agonist-induced fluid retention is not seen at either concentration.
Total kidney weight was significantly decreased only in the low-dose treated animals. This is remarkably similar to results obtained in response to sub-pharmacological treatment with rosiglitazone in the PCK rats.
In conclusion, the efficacy of PPARγ agonist concentrations acting as insulin sensitizing agents by reducing elevated serum glucose, is in agreement with previous rodent and human studies. Interestingly, there is consistency in the current studies that sub-pharmacologic levels of both pioglitazone and rosiglitazone are more effective in reducing cyst burden in the rodent models than higher concentrations. The reported side effects of the PPARγ agonists are dose-related and, therefore, it is expected that minimal adverse effects will be associated with low-dose treatment.
Presumably, most of the agents in clinical and preclinical trials target ADPKD treatment beginning at the time of diagnosis with the rationale that the earlier the intervention to slow cyst proliferation and expansion, the better the prognosis. The development of pharmaceutical interventions to be initiated at the time of diagnosis, typically in young adulthood or earlier, imposes several considerations. Most importantly, the side-effect profile must be highly favorable. There is no benefit to treating a slowly progressing disease with drugs whose side effects are likely to cause cumulative damage to patients that is equal in severity to the disease progression. In addition, since patient compliance is of utmost importance, it is crucial that a potential drug does not have an impact on the patient’s life-style, particularly in young, asymptomatic patients. The polyuria and potential for dehydration while using V2 receptor antagonists such as tolvaptan are of concern for patient compliance. Finally, the financial burden of life-long drug treatment has to be considered for both the patient and the healthcare systems world-wide. PPARγ agonists represent a relatively low-cost alternative to more expensive drugs that are being developed for ADPKD treatment[30, 31].
Conclusion
PPARγ agonists may be useful as life-long treatment for ADPKD.
Acknowledgements
The authors thank Dr. Brian Decker, Indiana University School of Medicine for advice in converting the animal doses to equivalent human doses and Alexander Carr for expert technical assistance.
Website: www.intern-med.com
Footnotes
Conflict of interest The authors declare that they have no conflicts of interest.
Source of Support
These studies were supported by a Project Development Team grant within the Indiana Clinical and Translational Sciences Institute funded by a National Institutes of Health, National Center for Research Resources grant, number RR025761.
Declaration
All animal studies were performed with approval of the Indiana University School of Medicine Institutional Animal Care and Use Committee and adhered to the NIH Guide for the Care and Use of Laboratory Animals.
References
- 1.Chapman AB.. Approaches to testing new treatments in autosomal dominant polycystic kidney disease: Insights from the CRISP and HALT-PKD studies. Clin J Am Soc Nephrol. 2008;3:1197–1204. doi: 10.2215/CJN.00060108. [DOI] [PubMed] [Google Scholar]
- 2.Harris PC, Torres VE.. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124:2315–22. doi: 10.1172/JCI72272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres VE, Harris PC.. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–68. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel V, Chowdhury R, Igarashi P.. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009;18:99–106. doi: 10.1097/MNH.0b013e3283262ab0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blazer-Yost BL, Haydon J, Eggleston-Gulyas T, Chen J-H, Wang X, Gattone V. et al. Pioglitazone attenuates cystic burden in the PCK rodent model of polycystic kidney disease. PPARγ Res. 2010 doi: 10.1155/2010/274376. Article ID 274376, 8 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delmas P.. Polycystins: polymodal receptor/ion-channel cellular sensors. Pflugers Arch - Eur J Physiol. 2005;451:264–76. doi: 10.1007/s00424-005-1431-5. [DOI] [PubMed] [Google Scholar]
- 7.Giamarchi A, Padilla B, Coste B, Radoux M, Crest M, Honore E. et al. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006;7:787–93. doi: 10.1038/sj.embor.7400745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilius B, Owsianik G.. The transient receptor potential family of ion channels. Genome Biol. 2011;12:218–29. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA.. Peroxisome proliferator-activated receptor (PPARγ) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinol. 1994;135:798–800. doi: 10.1210/endo.135.2.8033830. [DOI] [PubMed] [Google Scholar]
- 10.Chetty VT, Sharma AM.. Can PPARγ agonists have a role in the management of obesity-related hypertension? Vascul Pharm. 2006;45:46–53. doi: 10.1016/j.vph.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Willson TM, Brown PJ, Sternbach DD, Henke BR.. The PPARγs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–50. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 12.Guan Y.. Targeting peroxisome proliferator-activated receptors (PPARγ’s) in kidney and urologic disease. J Nephrol Urol. 2002;54:65–79. [PubMed] [Google Scholar]
- 13.Nofziger C, Brown KK, Smith CD, Harrington W, Murray D, Bisi J, Ashton TT, Maurio FF, Kalsi KK, West A, Baines D, Blazer-Yost BL.. PPARγ agonists inhibit vasopressin-mediated anion transport in the MDCK-C7 cell line. Am J Physiol Renal Physiol. 2009;297:F55–62. doi: 10.1152/ajprenal.00090.2009. [DOI] [PubMed] [Google Scholar]
- 14.Yoshihara D, Kurahashi H, Morita M, Kugita M, Hiki Y, Aukema HM. et al. PPAR-g agonist ameliorates kidney and liver disease in an ortholo-gousrat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. 2011;300:F465–74. doi: 10.1152/ajprenal.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Yang B, McNulty JA, Clifton LG, Binz JG, Grimes AM. et al. GI262570, a peroxisome proliferator-activated receptor γ agonist, changes electrolytes and water reabsorption from the distal nephron in rats. J Pharm Exper Therap. 2005;312:718–25. doi: 10.1124/jpet.104.074088. [DOI] [PubMed] [Google Scholar]
- 16.Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE. et al. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11:861–6. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- 17.Song J, Knepper MA, Hu X, Verbalis JG, Ecelbarger CA.. Rosiglitazone activates renal sodium and water reabsorptive pathways and lowers blood pressure in normal rats. J Pharm Exper Therap. 2004;308:426–33. doi: 10.1124/jpet.103.058008. [DOI] [PubMed] [Google Scholar]
- 18.Reagen-Shaw S, Nihal M, Ahmad N.. Dose translation from animal to human studies. FASEB J. 2007;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 19.Mason SB, Liang Y, Sinders RM, Miller CA, Eggleston-Gulyas T, Crisler-Roberts R. et al. Disease stage characterization of hepatorenal fibrocystic pathology in the PCK Rat model of ARPKD. Anat Rec. 2010;293:1279–88. doi: 10.1002/ar.21166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gattone VH 2nd, Tourkow BA, Trambaugh CM, Yu AC, Whelan S, Phillips CL. et al. Development of multiorgan pathology in the wpk rat model of polycystic kidney disease. Anat Record A. 2004;277:384–95. doi: 10.1002/ar.a.20022. [DOI] [PubMed] [Google Scholar]
- 21.Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S. et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Gen. 2006;38:191–6. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 22.Mahnensmith RL.. Novel treatments of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2014;9:831–6. doi: 10.2215/CJN.02480314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E. et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–18. doi: 10.1056/NEJMoa1205511. for the TEMPO 3:4 Trial Investigators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torres VE, Harris PC.. Strategies targeting cAMP signaling in the treatment of PKD. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muto S, Aiba A, Saito Y, Nakao K, Nakamura K, Tomita K. et al. Pioglita-zone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002;11:1731–42. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- 26.Raphael KL, Strait KA, Stricklett PK, Baird BC, Piotek K, Germino GG. et al. Effect of pioglitazone on survival and renal function in a mouse model of polycystic kidney disease. Am J Nephrol. 2009;30:468–73. doi: 10.1159/000242432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai B, Liu Y, Mei C, Fu L, Xiong X, Zhang Y. et al. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin Sci. 2010;119:323–33. doi: 10.1042/CS20100113. [DOI] [PubMed] [Google Scholar]
- 28.Nagao S, Kugita M, Yoshihara D, Yamaguchi T.. Animal models for human polycystic kidney disease. Exp Anim. 2012;61:477–88. doi: 10.1538/expanim.61.477. [DOI] [PubMed] [Google Scholar]
- 29.Torres VE, Harris PC.. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Internal Med. 2007;261:17–31. doi: 10.1111/j.1365-2796.2006.01743.x. [DOI] [PubMed] [Google Scholar]
- 30.Erickson KF, Glenn M, Chertow GM, Goldhaber-Fiebert JD.. Costeffectiveness of tolvaptan in autosomal dominant polycystic kidney disease. Ann Intern Med. 2013;159:382–9. doi: 10.7326/0003-4819-159-6-201309170-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lentine KL, Xiao H, Machnicki G, Gheorghian A, Schnitzer MA.. Renal function and healthcare costs in patients with polycystic kidney disease. Clin J Am Soc Nephrol. 2010;5:1471–9. doi: 10.2215/CJN.00780110. [DOI] [PMC free article] [PubMed] [Google Scholar]