


Abstract
Prediction of gene expression levels driven by regulatory sequences is pivotal in genomic biology. A major focus in transcriptional regulation is sequence-to-expression modeling, which interprets the enhancer sequence based on transcription factor concentrations and DNA binding specificities and predicts precise gene expression levels in varying cellular contexts. Such models largely rely on the position weight matrix (PWM) model for DNA binding, and the effect of alternative models based on DNA shape remains unexplored. Here, we propose a statistical thermodynamics model of gene expression using DNA shape features of binding sites. We used rigorous methods to evaluate the fits of expression readouts of 37 enhancers regulating spatial gene expression patterns in Drosophila embryo, and show that DNA shape-based models perform arguably better than PWM-based models. We also observed DNA shape captures information complimentary to the PWM, in a way that is useful for expression modeling. Furthermore, we tested if combining shape and PWM-based features provides better predictions than using either binding model alone. Our work demonstrates that the increasingly popular DNA-binding models based on local DNA shape can be useful in sequence-to-expression modeling. It also provides a framework for future studies to predict gene expression better than with PWM models alone.
INTRODUCTION
Gene regulation is one of the major challenges of genomic biology, and among the different levels at which genes may be regulated the one that has received most attention to date is transcriptional regulation (1,2). A key aspect of transcriptional regulation is the sequence-specific DNA-binding of transcription factors, and in recent years there has been a strong push towards precise characterization of transcription factor (TF)-DNA binding and its underlying mechanisms (3). The extent to which a TF's binding specificity at a site is dictated by the TF directly interpreting the nucleotide sequence (‘base readout’) or DNA shape at the site (‘shape readout’) is a topic of considerable debate (4,5), as is the role played by secondary TFs (6–8) that cooperatively or competitively influence in vivo DNA-binding. Chromatin state is another major determinant of TF-DNA binding that has been discussed in numerous studies (6,9–11).
A number of high throughput assays have been developed to generate data sets on which our understanding of TF-DNA binding can be rigorously tested (12). To support the study of biochemical mechanisms underlying TF-DNA binding, various computational models have emerged to describe these mechanisms and use them to fit experimental data (13). The de facto leader of this pack is the ‘position weight matrix’ or PWM model, which prescribes a multinomial distribution over four nucleotides for each position of the binding site, the distributions at different positions being independent of each other (14,15). The PWM model has been extensively used in regulatory sequence analysis and numerous algorithms are available for inferring a PWM model from a TF's binding sites (16–20). At the same time, several reports have pointed out deficiencies in the model and presented alternative models that are claimed to be in greater agreement with binding data (21–23). In short, the high intensity of ongoing experimental and computational work in this field has taken us much closer to a quantitative and predictive model of a TF's DNA-binding specificity.
The ultimate goal in modeling TF-DNA binding is to use this ability to understand gene regulation. Achieving this goal will allow us to ‘read’ and interpret non-coding sequences and hence their relationship to organismal form and function (24), and their evolution (25). It will enable major advances in the genomics of human health, by providing accurate predictions of the effects of single nucleotide polymorphisms at the cellular level. Precise models of in vitro and in vivo binding take us only part of the way to this grand goal, and must be incorporated into sequence-specific models of gene expression (‘sequence-to-expression’ models heretofore) for their value to be truly realized. Sequence-to-expression models are steadily gaining popularity, and have been used, among other things, to predict precise levels of gene expression in different regions of the developing embryo (26–32) or to predict tissue-specific gene expression in humans (33–35). However, there is a disconnect today between these models of gene expression and the burgeoning body of work on TF-DNA binding specificity. Sequence-to-expression models exclusively rely on the PWM model of DNA-binding, and it is unknown if alternative, emerging models of DNA-binding can substantially improve prediction of gene expression. This is the gap that we attempt to fill in this work.
We considered a model of TF-DNA binding that incorporates local DNA shape at the binding site and asked if it performs as well as a PWM model in predicting gene expression. To answer this question we considered one of the best-studied regulatory systems today – the set of genes and respective enhancers (also called cis-regulatory modules or CRMs) responsible for anterior-posterior (A/P) patterning of the blastoderm-stage Drosophila embryo (27,28,31). We used the thermodynamics-based GEMSTAT model (28) to predict gene expression levels from enhancer sequence and TF concentrations, using the DNA-binding model to parse the enhancer sequence in terms of the types, strengths and arrangements of binding sites within. We used rigorous methods of comparing model fits (36), to find that a DNA-binding model based on ‘shape readout’ (37) performs at least as well as, and arguably better than, the PWM model. We performed additional tests to examine if integrating shape readout and PWM into a single model would achieve better predictions than using either binding model independently. To our knowledge, this is the first successful attempt at quantitatively modeling the function of an enhancer sequence using a description of TF-DNA binding specificity other than the PWM. The shape-based model used here was trained on (binding site) data from the Bacterial 1-hybrid system (38). With the growing availability of data sets describing TF-DNA binding affinities more comprehensively (3,12), we expect that it will be possible to train such models more accurately and to demonstrate their ability to predict gene expression better than with PWM models alone.
MATERIALS AND METHODS
Data collection
We trained our model on enhancers of genes that pattern the A/P axis in the blastoderm-stage of the Drosophila embryo. The core of the data set, collected in the original GEMSTAT publication (28), comprises the following: 37 experimentally characterized enhancers, 37 quantitative profiles of gene expression driven by each enhancer and quantitative concentration profiles of six TFs - bicoid (bcd), caudal (cad), giant (gt), hunchback (hb), knirps (kni) and Kruppel (Kr). To supplement these data, we added three additional TF concentration profiles: vielfaltig (vfl), Dstat and sloppy-paired (slp), which were obtained from FlyEx database (39). Similar to He et al. (28), we limited the gene expression modeling to the 20–80% region of the A/P axis, resulting in 60 ‘bins’ of gene expression and TF concentration values. PWMs of all TFs were constructed with MEME (18) applied to binding sites obtained via bacterial one hybrid (B1H) experiments, downloaded from the Fly Factor Survey database (38). To increase the quality of PWMs, we trimmed MEME-predicted PWMs to have nearly the same length as PWMs in Factor Survey database (38), by removing 0 to 3 degenerate (low information content) positions on either ends (Supplementary Table S1). The DNA shape readouts for all binding sites were obtained by DNAShape (37), which predicts values of minor groove width (MGW) and propeller twist (ProT) at base pair (bp) resolution and values of roll (Roll) and helix twist (HelT) at base pair step resolution; the values are calculated using a window approach around each base pair, which will score all base pairs except for the one or two base pairs at each end of the sequence for which we do not have sufficient flanking residues.
TFBS DNA shape score predicted by Random Forest classifier
Given a TF, we trained a Random Forest classifier (40), using the R package ‘randomForest’ (41), to predict the shape scores of its binding sites. As shown in Figure 1A, a TF binding site is characterized by a set of four ‘shape vectors’ (MGW, ProT, Roll and HelT); each vector has 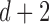 dimensions: d dimensions corresponding to a DNA shape readout at each position except for the terminal one or two base pairs, and two corresponding to the mean and standard deviation of DNA shape readouts over all positions in the binding site. The final feature vector fed into the Random Forest classifier was the concatenation of all four shape vectors, a representation we chose partly based on the work of Zhou et al. (37).
dimensions: d dimensions corresponding to a DNA shape readout at each position except for the terminal one or two base pairs, and two corresponding to the mean and standard deviation of DNA shape readouts over all positions in the binding site. The final feature vector fed into the Random Forest classifier was the concatenation of all four shape vectors, a representation we chose partly based on the work of Zhou et al. (37).
Figure 1.

DNA shape-based model of gene expression. A TF binding site is described by four shape feature vectors: MGW, ProT, Roll and HelT. Each vector includes the corresponding shape feature at every position of the site, along with the mean and standard deviation over all positions. For a given TF, a Random Forest classifier is trained on a sample of binding sites from Fly Factor Survey database to predict shape scores for putative binding sites.
To train each Random Forest, we sampled a set of binding sites for a given TF as the positive data and a set of random non-coding genomic regions, each with the same length as the TF's sites, as the negative data. To capture the numerous ways that random sequences can deviate from the TF's preferred binding sequences, we trained each classifier on 10 times as many negative examples as positive examples. We kept the multiplicative factor low as we wanted to prevent the Random Forest from being deluged by negative data to the extent that it suffers from the class imbalance problem (42). The output of the Random Forest is a probability of the input sequence being ‘positive’, meaning a TF binding site (TFBS). We denote this probability as the ‘DNA shape score’ in this study.
DNA shape-based quantitative sequence to expression model
The DNA shape-based sequence to expression model was adapted from the statistical thermodynamics model GEMSTAT (28). We briefly review the main ideas of GEMSTAT in Supplementary Note S1 and formulate below the key modification to its architecture that allows it to utilize DNA shape information. The contribution of each binding site to the enhancer's regulatory function is dictated by its ‘statistical weight’ 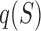 , given by the following equation:
, given by the following equation:
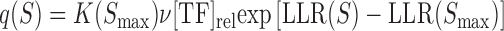 |
In this formulation, 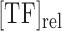 represents the relative TF concentration up to some constant
represents the relative TF concentration up to some constant 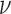 .
. 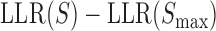 represents the difference in the log likelihood ratio between the site S and the consensus binding site
represents the difference in the log likelihood ratio between the site S and the consensus binding site 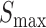 , and
, and 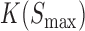 represents the association constant of TF-DNA binding. Since both
represents the association constant of TF-DNA binding. Since both 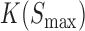 and
and 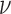 are unknown constants, GEMSTAT treats the product of the two as a free parameter.
are unknown constants, GEMSTAT treats the product of the two as a free parameter.
In constructing an analogous measure based on DNA shape data and not PWM data, only a single modification needs to be made to the binding site contribution formula, 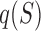 . In particular, the arguments of the exponent are changed to use DNA shape data. In the following formulation, Shape(S) represents the DNA shape score predicted by a Random Forest classifier and k represents a free scaling parameter.
. In particular, the arguments of the exponent are changed to use DNA shape data. In the following formulation, Shape(S) represents the DNA shape score predicted by a Random Forest classifier and k represents a free scaling parameter.
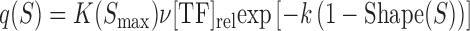 |
Model training and evaluation
The DNA shape-based model and the PWM-based model, i.e. GEMSTAT, were trained on 37 experimentally characterized enhancers regulating A/P patterning genes in Drosophila embryos, using the same training strategy as described in our previous work (29). In order to fairly compare DNA shape-based models with PWM-based models, we used the same GEMSTAT interaction mode (direct) and only considered self-cooperativity of bcd and cad. Following He et al. (28), in the PWM-based model, we annotated a site S with an 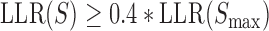 as a binding site. To yield a similar number of binding sites for the DNA shape-based model, a site with shape score greater than 0.6 was annotated as a binding site.
as a binding site. To yield a similar number of binding sites for the DNA shape-based model, a site with shape score greater than 0.6 was annotated as a binding site.
To measure the goodness of fit between the real and predicted gene expression, we used the scoring function called ‘weighted pattern generating potential’ (wPGP) (43), which essentially rewards the agreement between endogenous and predicted readouts and penalizes the disagreement. The wPGP score ranges between 0 and 1, with higher values indicating better fits. By choosing wPGP as the measurement, we were able to avoid the following issues common to widely used methods such as correlation or root mean square error: biases from overly narrow or overly board predicted expression and insensitivity to shift and scaling of the expression profiles as previously reported (44).
RESULTS
DNA shape-based model predicts gene expression at least as well as PWM-based model
The main purpose of this work was to test if a quantitative sequence-to-expression model based on DNA shape at putative binding sites provides better fits to expression data than the PWM-based model that has been tested successfully in multiple prior studies (26–29). For this, we used the GEMSTAT model that relates enhancer sequence to its expression (28), with one major modification. The binding energy of any site S is normally computed by GEMSTAT as 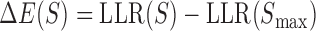 , where
, where 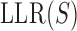 is the log-likelihood ratio of site S under the PWM model (compared to a background model) and
is the log-likelihood ratio of site S under the PWM model (compared to a background model) and 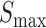 is the consensus binding site. We replaced this PWM-based score of a binding site to instead be
is the consensus binding site. We replaced this PWM-based score of a binding site to instead be 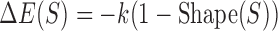 , where k is a free parameter and
, where k is a free parameter and 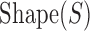 is a score in the range 0–1, computed based on the DNA shape features at site S. This computation is done by a Random Forest classifier that is separately trained on shape readouts (37) of a sample of binding sites for a given TF. (The TF's PWM used in the comparator model is trained on the same set of binding sites.) See Materials and Methods for details (Figure 1), and Supplementary Note S2 for details of and an alternative method for incorporating the shape scores into GEMSTAT.
is a score in the range 0–1, computed based on the DNA shape features at site S. This computation is done by a Random Forest classifier that is separately trained on shape readouts (37) of a sample of binding sites for a given TF. (The TF's PWM used in the comparator model is trained on the same set of binding sites.) See Materials and Methods for details (Figure 1), and Supplementary Note S2 for details of and an alternative method for incorporating the shape scores into GEMSTAT.
For a fair comparison, we focused on the same data set used in one of the original PWM-based modeling studies (28), which includes 37 experimentally characterized enhancers regulating A/P patterning genes in Drosophila embryos. Each enhancer is characterized by the relative expression level (on a scale of 0 to 1) driven by that enhancer at distinct positions (‘bins’) along the A/P axis of the embryo. We used wPGP scores (36) to evaluate the goodness of fit between experimentally observed and predicted expression profiles.
On the whole, the DNA shape-based model performed as well as and arguably better than the PWM-based model, as shown in Table 1 and Figure 2. The DNA shape-based model achieved a wPGP score of 0.784, averaged over the 37 enhancers in the training data set while PWM-based model averaged at 0.755. This difference, being taken over averages of scores, is significant based on our prior experience (29) and direct statistical testing (Wilcoxon signed-rank test P-value of 0.003). For 14 out of 37 enhancers we noted better fits using the shape-based model (wPGP score improved by >0.05), whereas for 8 out of 37 enhancers the shape-based model produced worse fits (Supplementary Table S2). These results provided clear evidence that DNA shape readout at putative binding sites can lead to accurate quantitative modeling of gene expression, and suggest that it yields arguably better fits than nucleotide readout.
Table 1. 10-fold cross-validation assessment of various models.
| Model | #Pars | Avg. wPGP (Training) | Avg. wPGP± std (CV) |
|---|---|---|---|
| Shape-based model | 22 | 0.784 | 0.727 ± 0.020 |
| PWM-based model | 21 | 0.755 | 0.677 ± 0.004 |
| PWM-based model, Perturbed LLR scores | 21 | 0.643 | 0.603 ± 0.021 |
For each model, shown are the number of free parameters used (‘#Pars’), the average wPGP scores from parameter optimization over all 37 enhancers (‘Avg. wPGP (Training)’), and the wPGP scores from cross-validation (‘Avg. wPGP (CV)’), averaged over ten repeats of cross validation with different (random) definitions of the ten folds. Standard deviations over the ten repeats are also shown.
Figure 2.

Performance of DNA shape-based model compared to PWM-based model on 37 Drosophila enhancers. The goodness of fit between predicted and real expression for each enhancer was assessed by wPGP scores. Dotted lines delineate regions where the difference in wPGP score between the two models is <0.05.
To better appreciate the differences between fits of enhancer readouts from the two models, we plotted the predicted expression profiles of the two models along with real expression patterns for a selection of six enhancers (Figure 3). It was evident that the DNA shape-based model improved the expression prediction by predicting more accurately defined boundaries of spatial expression domains. For example, for the enhancers ‘eve_stripe5’ as well as ‘run_stripe1’, the shape-based model accurately predicts the posterior and anterior boundary, respectively. Qualitative refinements were observed on other enhancers. For instance, the shape-based model reduced a spurious anterior domain predicted by the PWM-based model for the enhancer ‘eve_37ext_ru’, correctly modeled the anterior peak in ‘ftz_+3’ which the PWM-based model failed to predict, and correctly suppressed an ectopic posterior domain of ‘slp_(-3)’ expression predicted by the PWM-based model. More complete comparisons of gene expression profiles where the DNA shape-based model produced better or worse fits than the PWM-based model are shown in Supplementary Figure S1.
Figure 3.

Fits between model and data. Predicted expression profiles of DNA shape-based model (orange lines) and PWM-based model (purple lines) are compared to experimentally determined expression profiles (black lines), for six selected Drosophila enhancers. Each expression profile is on a relative scale of 0 to 1 (y-axis), and shown for the regions between 20% and 80% of the A/P axis of the embryo. Title in each panel is in the format of “enhancer name, wPGP by DNA shape-based model (‘S’), wPGP by PWM-based model (‘P’).” See more enhancers fits in Supplementary Figure S1.
The results above have indicated, both quantitatively and qualitatively, that a DNA shape-based characterization of binding sites performed at least as well as the more conventional PWM-based model in sequence-to-expression modeling. It should be noted that while both models used the same set of parameters, the DNA shape-based model had one additional parameter (‘k’, see Materials and Methods) to map the site score computed by the Random Forest-based classifier to a pseudo-energy term in GEMSTAT. (The PWM-based model has 21 parameters while the shape-based model has 22 parameters.) A widely accepted method to compare models with different complexities is to assess goodness of fit under cross validation. We therefore performed 10-fold cross-validation on all 37 enhancers for each model. Since the partition of the data set into training and test sets was decided randomly, we repeated the exercise 10 times with either model. The DNA shape-based model reported a wPGP score (averaged over all 37 enhancers, and over the ten repeats) of 0.727 with standard deviation 0.020 and PWM-based model led to an average wPGP of 0.677 with standard deviation 0.004 (Table 1). Thus, we confirmed that the improved fits from the DNA shape-based model are not due to its additional free parameter.
We considered the possibility that the improved fits with the shape-based model are primarily due to a single TF (or a minority of TFs) for which the PWM is not an appropriate model of binding specificity, while for other TFs the PWM model is more suited for use in expression modeling. We tested this possibility and found it to be false. In particular, we repeated the model fitting exercise with the shape-based scoring of binding sites for every TF except one, for which PWM-based scoring was used. We compared the goodness of fit (average wPGP across enhancers) of such hybrid models with that of the purely shape-based model, and noted that for all TFs except cad, the fits deteriorated upon substituting shape-based scores with PWM-based LLR scores for that TF's sites. (Figure 4A) (The goodness of fit was almost unchanged upon switching from the shape model to the PWM model for cad.) This suggests that for every TF in this analysis the shape-based score is as good or better than the PWM-based score for the purpose of expression modeling.
Figure 4.

DNA shape is characterized differently from PWM (A) Change of goodness of fit (avg. wPGP) of DNA shape-based model predictions when binding sites of a specific TF were forced to use LLR rather than shape scores. (B) Visualization of kni binding sites correlation between shape scores and LLR. (C) Pearson correlations of binding sites for each of nine TF in this study and all TFs.
We wondered if the difference between shape-based and sequence-based models arises from the difference in how the binding site scoring method was trained – as a PWM trained on sample sites versus a Random Forest classifier trained on samples of sites and non-sites. To make the models more similar in this aspect, we trained a Random Forest classifier on 1-mer sequence features (the so-called ‘1-hot encoding’ (45)), using the same training data sets as for shape model. We then incorporated scores predicted by Random Forest into GEMSTAT in the same way as for the DNA shape model. The average wPGP score of this alternative sequence-based model was 0.756 (Supplementary Tables S3 and S4), nearly the same as the PWM-based model. We repeated 10-fold cross-validation ten times, and obtained an average wPGP score of 0.673 with standard deviation 0.014, again very similar to that of the PWM-based model, suggesting that the gap between shape-based and sequence-based models is not merely due to a difference in how underlying binding site scoring methods were trained.
In our direct comparisons between the shape-based and PWM-based models, all other aspects of modeling were identical, including the set of putative sites considered by either model. However, one point of difference was that the site length used to compute shape readouts was in some cases different from the site length used to score for PWM matches. This was motivated by our observation that the PWM-based model yielded better fits when using shorter (‘trimmed’) PWMs than those constructed directly from the available sample of binding sites. We systematically examined the effect of motif length on our claims above (Supplementary Note S3, Figure S2, Table S5) and confirmed that the comparisons and claims reported above involve a fair treatment of the PWM model. That is, the gap between shape-based and PWM-based models is even greater when the same site lengths are used for both models and PWMs are not ‘trimmed’.
DNA shape models capture information different from PWM
In light of our aforementioned conclusion that shape-based models perform at least as well as PWM-based models in predicting enhancer readouts, we next asked if the PWM-based score and DNA shape-based score are simply two ways to quantify exactly the same information, differing only procedurally. They are closely related scores, since both are computed from the primary sequence of a binding site. At the same time, each has its own intuitive biophysical explanation: the PWM-based score is related directly to the binding energy of a site (14,16) assuming positional independence, while the shape-based score reflects how similar a putative site's local DNA shape is to that of the training set of binding sites.
To objectively characterize the relationship between the two scores, we examined their mutual correlation over all putative binding sites for each TF. Figure 4B shows the scatter plot of the two scores across all binding sites for the TF kni, where we noted Pearson correlation of 0.623. Figure 4C shows Pearson correlation for each of the nine TFs; these correlations are typically around 0.5, ranging between 0.211 (gt) to 0.677 (vfl), with the correlation over putative binding sites of all TFs being 0.525 (Figure 4C, ‘All’). We interpreted these observations to mean that the shape-based score, while being closely related to the PWM-based score of a site, is not redundant with the latter and contains additional information not captured by the direct sequence readout. Our tests above (Figure 2) further indicated that the additional information captured by the shape score is useful for predicting gene expression profiles as well as and arguably better than with PWM scores. However, we considered the possibility that this improvement (average wPGP of 0.784 for the shape-based model compared to 0.755 for the PWM-based model) is an artifact of our procedure. Specifically, it was possible that our modeling is fundamentally incapable of discerning an accurate TF-DNA binding model from a noisy version thereof, either due to noise in the data or over-parameterization, or for an unknown reason. To test this possibility, we repeated the PWM-based model-fitting exercise after artificially perturbing the LLR scores of binding sites, and found the PWM model to perform worse with these slightly perturbed LLR scores of sites, ruling out the concern raised above (Table 1 and Supplementary Note S4).
Previous work has found sequence models that consider nucleotide inter-dependencies to better fit binding affinity data than the PWM model (22,23). We therefore tested if a Random Forest trained to classify sites based on their k-mer profile can lead to improved expression predictions. A ‘1-mer+2-mer’ sequence-based model achieved a wPGP score of 0.770 on average, and a 1-mer+2-mer+3-mer model yielded an average wPGP of 0.765. (Supplementary Tables S3 and S4; the wPGP score of each enhancer, under either model, can be found in Supplementary Table S6.) Thus, the fits achieved with higher order k-mer models were better than those from a 1-hot encoding or the PWM model, but not better than fits of the shape-based model. This is consistent with the view that DNA shape features provide an alternative and more compact representation of positional interdependencies in binding sites (46).
Combining shape and sequence readout into a single model does not improve fits
The literature suggests that models integrating DNA shape with PWM-based sequence readout can improve prediction of TF-DNA binding over models that use either representation independently (42,46,47). However, sequence-to-expression modeling requires not just the prediction of TF binding strengths, but also quantifying how different configurations of DNA-bound TFs relate to gene expression levels. Given this, it is not entirely clear whether integrating DNA shape and sequence would significantly improve expression modeling. We tested this hypothesis by first comparing a model that integrates DNA shape scores into PWM-based models (referred to henceforth as ‘integrative PWM-based’ models) with PWM-based models. To incorporate DNA shape information into the PWM-based model, we replaced the term for binding energy of a site in GEMSTAT to be 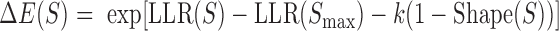 where
where 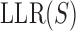 is the log likelihood ratio score of site S under the PWM model,
is the log likelihood ratio score of site S under the PWM model, 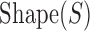 is the score of site S computed by a Random Forest classifier using the site's shape readout, and k is a free parameter.
is the score of site S computed by a Random Forest classifier using the site's shape readout, and k is a free parameter.
As shown in Figure 5A, for most enhancers the integrative PWM-based model fits expression data nearly as well as the PWM-based model. The wPGP scores are nearly identical with the average over all 37 enhancers being 0.752 and 0.755, respectively. The integrative PWM-based model outperforms the PWM-based model (a wPGP score difference of 0.05 or more) for six of the enhancers, while the PWM-based model outperforms the integrative PWM-based model for five of the enhancers. (The wPGP scores of each enhancer, under either model, can be found in Supplementary Table S7.) Since the integrative model did not perform consistently better than the sequence based model, we did not explore other formulas for incorporating DNA shape scores into PWM-based models.
Figure 5.

Performance of integrative models compared to (A) PWM-based model and (B) DNA shape-based model on 37 Drosophila enhancers assessed by wPGP scores. Dotted lines delineate regions where the difference in wPGP between the two models is greater than 0.05.
As integrating DNA shape information into PWM-based models did not significantly improve average wPGP scores over PWM-based models, we examined the utility of the converse methodology that adds sequence readout to a DNA shape-based model. In order to accomplish this, we added an additional feature to the Random Forest underlying the shape-based model: the LLR score of the binding site according to the TF's PWM. That is, the binding energy term of a site S in GEMSTAT was computed as 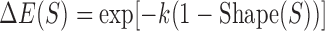 , where
, where 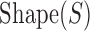 is now computed by a Random Forest classifier trained on pre-determined binding sites, using their shape readouts as well as LLR scores. This alternative integrative model (henceforth referred to as a ‘integrative shape-based’ model) performed as well as the shape-based models (Figure 5B), with average wPGP scores over all 37 enhancers being 0.776 and 0.784, respectively. Either model outperformed the other on six of the enhancers (Supplementary Table S7).
is now computed by a Random Forest classifier trained on pre-determined binding sites, using their shape readouts as well as LLR scores. This alternative integrative model (henceforth referred to as a ‘integrative shape-based’ model) performed as well as the shape-based models (Figure 5B), with average wPGP scores over all 37 enhancers being 0.776 and 0.784, respectively. Either model outperformed the other on six of the enhancers (Supplementary Table S7).
In recognition of the fact that there are alternative ways to encode the sequence, we repeated the above test by directly using k-mers of putative sites as features (in addition to shape features) of the Random Forest classifier, and using the resulting score in computing 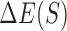 as in the previous paragraph. We evaluated three variants of integrative shape+k-mer models, increasing the complexity of models one by one. As listed in Supplementary Table S7, the integrative ‘shape+1-mer’, ‘shape+1-mer+2-mer’ and ‘shape+1-mer+2-mer+3-mer’ models achieved average wPGP scores of 0.777, 0.767 and 0.762, respectively (Supplementary Tables S3 and S4). In short, this subsection shows that the shape-based model is not improved upon by incorporating sequence-readout into it, nor is the PWM-based model improved upon by including shape readout.
as in the previous paragraph. We evaluated three variants of integrative shape+k-mer models, increasing the complexity of models one by one. As listed in Supplementary Table S7, the integrative ‘shape+1-mer’, ‘shape+1-mer+2-mer’ and ‘shape+1-mer+2-mer+3-mer’ models achieved average wPGP scores of 0.777, 0.767 and 0.762, respectively (Supplementary Tables S3 and S4). In short, this subsection shows that the shape-based model is not improved upon by incorporating sequence-readout into it, nor is the PWM-based model improved upon by including shape readout.
DISCUSSION
Sequence-to-expression models have been effectively used to understand the precise relationship between regulatory sequence and gene expression patterns (27,28,30–32). TF-DNA binding predictions in these quantitative models typically rely on the PWM representation that assumes every nucleotide in TF binding sites contributes additively and independently to the binding energy at thermodynamic equilibrium, an assumption that does not always hold. A mounting body of work on TF-DNA binding specificity has gone beyond the PWM model by considering the nucleotides dependencies (22,48), flanking sequences of binding sites (49,50) and DNA structural features (42,46,51) and shown highly promising results in TF-DNA recognition. At the same time, it is not well understood if alternative models of DNA binding can improve the prediction of gene expression. Our work aims at filling this gap by incorporating local DNA shape at the binding site to sequence-to-expression models and asking if it performs as well as a PWM-based model. We found the answer to be affirmative: the DNA shape-based model is arguably better than the PWM-based model in predicting expression. To our knowledge, this sequence-to-expression model based on DNA shape features is the first of its kind.
Previous work has demonstrated that DNA shape-based models compare favorably to sequence-based models for the simpler yet challenging task of modeling TF-DNA binding strength, and that integrative ‘shape+sequence’ models perform considerably better than sequence-only or shape-only models (42,46,47). However, in this study, we did not see further improvement in our integrative models utilizing both shape-readout and sequence-readout, over models using DNA shape only. This may be in part due to limitations of how our integrative models were constructed, or due to lack of comprehensive data for training our shape models, but it is also a possible indication that better prediction of TF-DNA binding may not always lead to better expression prediction.
Our model succeeds in quantifying the impact of DNA shape on prediction of precise spatiotemporal expression patterns, and also indicates an intuitive and simple approach to deal with DNA shape data. Prior work has suggested several approaches to aggregating shape features as well as learning models, including Random Forest (42) and support vector machine (SVM) (46,47,51). Our approach is in good agreement with the prior use of Random Forest as the learning model, and demonstrates the feasibility of simply using first-order local shape features. We also adopted SVM as an alternative learning model but this appeared to have no further improvement, and we did not pursue deeper investigations thereof.
It is also worth noting that we explored two choices of incorporating shape scores into the original GEMSTAT model. We initially treated the shape score (normalized to a scale of 0 to 1) as being directly related to the probability of a site being bound at a specific TF concentration condition (Supplementary Note S2, approach 1). This preliminary attempt at incorporating the shape score did not show promising results. The approach used in this study considered the binding affinity of a site, relative to that of the optimal site, as an exponentially decaying function of the shape score (Supplementary Note S2, approach 2). We expect that future work will continue to improve design of the shape score from the underlying features and integration of shape scores into sequence-to-expression models.
The DNA shape data used in this study was obtained from computational processing of binding site sequences. This raises the concern that DNA shape scores differ only procedurally from LLR scores (derived from the PWM), but are intrinsically the same information. Our tests suggest that this is not the case and show that DNA shape score captures information different from LLR. It is worth noting that shape features at a single nucleotide position are determined by a pentamer sequence centered at the targeted nucleotide. We have limited information about the flanking sequences of the binding site, so that the shape feature values were unavailable at the terminal positions of some of our TF binding sites. Since it has been reported that DNA shape in the flanking regions of binding sites influences binding specificity (50), we believe that the advantage observed here is an underestimate of how well DNA shape-based models can be used in gene expression predictions. We expect that our modeling approach will be more accurate if and when we have more comprehensive TF binding affinity data sets available.
The reader may ask if the thermodynamics-based sequence-to-expression model was necessary for our study. In order to study the effects of particular aspects of data on a higher-level prediction task, one has to make several choices: a modeling or prediction framework, semantic features of the model and the precise way to quantify those features. In an investigation with so many moving parts, it is natural to first attempt to make reasonable choices about some of those parts, and having fixed them, examine the effect of the one remaining moving part. This is the rationale of our approach. We have extensive experience with the thermodynamic modeling framework and the biological system we utilized here, so we chose to ask questions about shape versus sequence readout in this context.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (NSF) [1136913 to S.S.]; National Institutes of Health (NIH) [R01GM114341 to S.S.]. Funding for open access charge: National Science Foundation (NSF) [1136913 to S.S.]; National Institutes of Health (NIH) [R01GM114341 to S.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Carroll S.B., Grenier J.K., Weatherbee S.D. From DNA to diversity: molecular genetics and the evolution of animal design. Hoboken: John Wiley & Sons; 2013. [Google Scholar]
- 2.Davidson E.H. The regulatory genome: gene regulatory networks in development and evolution. Cambridge: Academic Press; 2010. [Google Scholar]
- 3.Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordân R., Rohs R. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 2014;39:381–399. doi: 10.1016/j.tibs.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siggers T., Gordân R. Protein–DNA binding: complexities and multi-protein codes. Nucleic Acids Res. 2013;42:2099–2111. doi: 10.1093/nar/gkt1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng Q., Kazemian M., Pham H., Blatti C., Celniker S.E., Wolfe S.A., Brodsky M.H., Sinha S. Computational identification of diverse mechanisms underlying transcription factor-DNA occupancy. PLoS Genet. 2013;9:e1003571. doi: 10.1371/journal.pgen.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wasson T., Hartemink A.J. An ensemble model of competitive multi-factor binding of the genome. Genome Res. 2009;19:2101–2112. doi: 10.1101/gr.093450.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biggin M.D. Animal transcription networks as highly connected, quantitative continua. Dev. Cell. 2011;21:611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Pique-Regi R., Degner J.F., Pai A.A., Gaffney D.J., Gilad Y., Pritchard J.K. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011;21:447–455. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan T., Li X.-Y., Sabo P.J., Thomas S., Stamatoyannopoulos J.A., Biggin M.D., Eisen M.B. Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development. PLoS Genet. 2011;7:e1001290. doi: 10.1371/journal.pgen.1001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J., Zhuang J., Iyer S., Lin X., Whitfield T., Greven M., Pierce B., Dong X., Kundaje A., Cheng Y., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stormo G.D., Zhao Y. Determining the specificity of protein–DNA interactions. Nat. Rev. Genet. 2010;11:751–760. doi: 10.1038/nrg2845. [DOI] [PubMed] [Google Scholar]
- 13.Weirauch M., Cote A., Norel R., Annala M., Zhao Y., Riley T., Saez-Rodriguez J., Cokelaer T., Vedenko A., Talukder S., et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013;31:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berg O.G., von Hippel P.H. Selection of DNA binding sites by regulatory proteins. Trends Biochem. Sci. 1988;13:207–211. doi: 10.1016/0968-0004(88)90085-0. [DOI] [PubMed] [Google Scholar]
- 15.Stormo G.D., Schneider T.D., Gold L., Ehrenfeucht A. Use of the ‘Perceptron'algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 17.Stormo G.D. Modeling the specificity of protein-DNA interactions. Quant. Biol. 2013;1:115–130. doi: 10.1007/s40484-013-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey T.L., Elkan C. Proc. Second Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 19.Djordjevic M., Sengupta A.M., Shraiman B.I. A biophysical approach to transcription factor binding site discovery. Genome Res. 2003;13:2381–2390. doi: 10.1101/gr.1271603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orenstein Y., Shamir R. A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data. Nucleic Acids Res. 2014;42:e63. doi: 10.1093/nar/gku117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stringham J.L., Brown A.S., Drewell R.A., Dresch J.M. Flanking sequence context-dependent transcription factor binding in early Drosophila development. BMC Bioinformatics. 2013;14:298–310. doi: 10.1186/1471-2105-14-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y., Ruan S., Pandey M., Stormo G.D. Improved models for transcription factor binding site identification using nonindependent interactions. Genetics. 2012;191:781–790. doi: 10.1534/genetics.112.138685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santolini M., Mora T., Hakim V. A general pairwise interaction model provides an accurate description of in vivo transcription factor binding sites. PLoS One. 2014;9:e99015. doi: 10.1371/journal.pone.0099015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farnham P.J. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duque T., Samee M.A.H., Kazemian M., Pham H.N., Brodsky M.H., Sinha S. Simulations of enhancer evolution provide mechanistic insights into gene regulation. Mol. Biol. Evol. 2014;31:184–200. doi: 10.1093/molbev/mst170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fakhouri W.D., Ay A., Sayal R., Dresch J., Dayringer E., Arnosti D.N. Deciphering a transcriptional regulatory code: modeling short‐range repression in the Drosophila embryo. Mol. Syst. Biol. 2010;6:341–354. doi: 10.1038/msb.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Segal E., Raveh-Sadka T., Schroeder M., Unnerstall U., Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 28.He X., Samee M.A.H., Blatti C., Sinha S. Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression. PLoS Comput. Biol. 2010;6:e1000935. doi: 10.1371/journal.pcbi.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng P.-C., Samee M.A.H., Sinha S. Incorporating chromatin accessibility data into sequence-to-expression modeling. Biophys. J. 2015;108:1257–1267. doi: 10.1016/j.bpj.2014.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janssens H., Hou S., Jaeger J., Kim A.-R., Myasnikova E., Sharp D., Reinitz J. Quantitative and predictive model of transcriptional control of the Drosophila melanogaster even skipped gene. Nat. Genet. 2006;38:1159–1165. doi: 10.1038/ng1886. [DOI] [PubMed] [Google Scholar]
- 31.Zinzen R.P., Papatsenko D. Enhancer responses to similarly distributed antagonistic gradients in development. PLoS Comput. Biol. 2007;3:e84. doi: 10.1371/journal.pcbi.0030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gertz J., Siggia E.D., Cohen B.A. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature. 2009;457:215–218. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natarajan A., Yardımcı G.G., Sheffield N.C., Crawford G.E., Ohler U. Predicting cell-type–specific gene expression from regions of open chromatin. Genome Res. 2012;22:1711–1722. doi: 10.1101/gr.135129.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pennacchio L.A., Loots G.G., Nobrega M.A., Ovcharenko I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007;17:201–211. doi: 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blatti C., Kazemian M., Wolfe S., Brodsky M., Sinha S. Integrating motif, DNA accessibility and gene expression data to build regulatory maps in an organism. Nucleic Acids Res. 2015;43:3998–4012. doi: 10.1093/nar/gkv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samee M.A.H., Sinha S. Evaluating thermodynamic models of enhancer activity on cellular resolution gene expression data. Methods. 2013;62:79–90. doi: 10.1016/j.ymeth.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Zhou T., Yang L., Lu Y., Dror I., Machado A.C.D., Ghane T., Di Felice R., Rohs R. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013;41:W56–W62. doi: 10.1093/nar/gkt437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noyes M.B., Meng X., Wakabayashi A., Sinha S., Brodsky M.H., Wolfe S.A. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res. 2008;36:2547–2560. doi: 10.1093/nar/gkn048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pisarev A., Poustelnikova E., Samsonova M., Reinitz J. FlyEx, the quantitative atlas on segmentation gene expression at cellular resolution. Nucleic Acids Res. 2009;37:D560–D566. doi: 10.1093/nar/gkn717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 41.Liaw A., Wiener M. Classification and regression by random Forest. R. News. 2002;2:18–22. [Google Scholar]
- 42.Hooghe B., Broos S., Van Roy F., De Bleser P. A flexible integrative approach based on random forest improves prediction of transcription factor binding sites. Nucleic Acids Res. 2012;40:e106. doi: 10.1093/nar/gks283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong J.-W., Hendrix D.A., Levine M.S. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kazemian M., Blatti C., Richards A., McCutchan M., Wakabayashi-Ito N., Hammonds A., Celniker S., Kumar S., Wolfe S., Brodsky M., et al. Quantitative analysis of the Drosophila segmentation regulatory network using pattern generating potentials. PLoS Biol. 2010;8:e1000456. doi: 10.1371/journal.pbio.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt B. Bioinformatics: high performance parallel computer architectures. Boca Raton: CRC Press; 2010. [Google Scholar]
- 46.Zhou T., Shen N., Yang L., Abe N., Horton J., Mann R.S., Bussemaker H.J., Gordan R., Rohs R. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc. Natl. Acad. Sci. U.S.A. 2015;112:4654–4659. doi: 10.1073/pnas.1422023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J., Ramsey S.A. A DNA shape-based regulatory score improves position-weight matrix-based recognition of transcription factor binding sites. Bioinformatics. 2015;31:3445–3450. doi: 10.1093/bioinformatics/btv391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharon E., Lubliner S., Segal E. A feature-based approach to modeling protein–DNA interactions. PLoS Comput. Biol. 2008;4:e1000154. doi: 10.1371/journal.pcbi.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordân R., Shen N., Dror I., Zhou T., Horton J., Rohs R., Bulyk M.L. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3:1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levo M., Zalckvar E., Sharon E., Machado A.C.D., Kalma Y., Lotam-Pompan M., Weinberger A., Yakhini Z., Rohs R., Segal E. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res. 2015;25:1–12. doi: 10.1101/gr.185033.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maienschein-Cline M., Dinner A.R., Hlavacek W.S., Mu F. Improved predictions of transcription factor binding sites using physicochemical features of DNA. Nucleic Acids Res. 2012;40:e175. doi: 10.1093/nar/gks771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.