

Summary
Lnk is an SH2 domain–containing adaptor protein expressed preferentially in lymphocytes. To illuminate the importance of Lnk, we generated lnk−/− mice. Whereas T cell development was unaffected, pre-B and immature B cells accumulated in the spleens. In the bone marrow, B-lineage cells were proportionately increased, reflecting enhanced production of pro-B cells that resulted in part from hypersensitivity of precursors to SCF, the ligand for c-kit. Hence, Lnk ordinarily acts to regulate B cell production. Further characterization of lnk−/− mice also revealed that full-length Lnk is a 68 kDa protein containing a conserved proline-rich region and a PH domain. Lnk is a representative of a multigene adaptor protein family whose members act, by analogy with Lnk, to modulate intra-cellular signaling.
Introduction
Lymphocytes differentiate from hematopioetic precursor cells through a process whereby the coordinate regulation of cell proliferation, differentiation, and death directs development of functional cells. Strict controls ensure that defective or autoreactive lymphocytes are purged from the developing repertoire. Additional regulatory mechanisms promote the expansion and maturation of sufficient numbers of functional lymphocytes to guarantee the prompt elaboration of adaptive immune responses. Significant advances have been made in understanding the signal transduction pathways regulating both lymphocyte development and activation (reviewed by Alberola-Ila et al., 1997; DeFranco, 1997; Farrar et al., 1998; Killeen et al., 1998); however, the mechanisms underlying homeostasis in the lymphocyte compartment remain poorly defined.
Both cell-to-cell contact and soluble growth factors play important roles in regulating the development of lymphocytes. Self-antigens presented on stromal cells trigger antigen receptors, signals from which help to determine the fate of lymphoid precursors. Stem cell factor (SCF) and IL-7 assist in regulating the expansion of lymphoid precursors by binding to c-kit and the IL-7 receptor, respectively (reviewed by Baird et al., 1999). The binding of extracellular ligands to these polypeptide receptors initiates a cascade of events through the activation of intracellular protein kinases. The phosphorylation events that these kinases catalyze both modulate the catalytic activity of effector enzymes and mediate protein–protein interactions that juxtapose critical signal transduction elements (see the above reviews). In this context, it is increasingly appreciated that a group of cellular proteins called adaptor proteins regulate the interaction of effector enzymes with surface receptors and their target substrates.
Adaptor proteins lack catalytic function but still possess interaction domains; e.g., SH2 and PTB domains that bind tightly to phosphotyrosine residues, SH3 domains that bind proline-rich residues, or PH domains that appear to mediate interactions with lipid membranes (reviewed by Birge et al., 1996; Pawson and Scott, 1997). Growing evidence supporting the importance of adaptor proteins has been presented in numerous signal transduction cascades. For example, mutant mice lacking the adaptor proteins SLP-76 (SH2 domain-containing leukocyte protein of 76 kDa) or LAT (linker for activation of T cells) manifest severe defects in T lymphopoiesis due to impairment of pre-T receptor signaling (Clements et al., 1998; Pivniouk et al., 1998; Zhang et al., 1999). Similarly, mice lacking BLNK/SLP-65 exhibit defective maturation of pro-B to pre-B cells (Jumaa et al., 1999; Pappu et al., 1999). Disruption of the c-cbl gene results in augmented and inappropriate lymphocyte proliferative responses (Murphy et al., 1998; Naramura et al., 1999). Earlier studies demonstrated that lymphocytes from mice lacking the kinase substrate HS1 exhibit impaired proliferative responses and a disturbance in normal repertoire selection (Taniuchi et al., 1995). Thus, adaptor proteins play crucial roles in normal lymphocyte development.
Lnk has been reported as a 38 kDa adaptor protein containing an SH2 domain and several candidate tyrosine phosphorylation sites that might mediate interactions with Grb2, PLCγ, and PI3K in activated T cells (Huang et al., 1995). Experiments with transgenic mice, however, did not identify a limiting role for the 38 kDa Lnk protein in T cell development or function (Takaki et al., 1997). Here, we report the selective expansion of B-lineage cells in lnk-deficient mice. This increase in B lymphopoiesis resulted from enhanced proliferation of B cell progenitors, a phenomenon that could be ascribed, at least in part, to increased sensitivity to SCF. Further characterization of lnk−/− mice also revealed that the full-length Lnk protein is nearly twice the size of that which was reported and contains a previously unappreciated proline-rich region and a PH domain at its amino terminus. Together, these sequences define a conserved family of Lnk-related adaptor proteins.
Our results demonstrate a specific, cell-autonomous function for Lnk in limiting B lymphopoiesis and suggest that this family of adaptor proteins functions more generally to regulate responsiveness to growth factor stimulation.
Results
Generation of lnk-Deficient Mice
The exons encoding the previously defined Lnk protein were replaced by a neomycin phosphotransferase cassette via homologous recombination in mouse ES cells (Figure 1A). Successful homologous recombination and germline transmission were confirmed via genomic blot analysis of mouse tail DNA from offspring of heterozygous lnk+/− mice (Figure 1B). Transcripts corresponding to the lnk cDNA were no longer expressed in lnk−/− mice (Figure 1C). lnk−/− mice did not show any overt developmental abnormalities, were produced in Mendelian ratios, and were indistinguishable from lnk+/+ or lnk+/− littermates on the basis of size, activity, or fertility.
Figure 1. Targeted Disruption of the Murine lnk Gene.

(A) Targeting vector and homologous recombination at the lnk locus. Top: a partial restriction map of the murine lnk locus. Known exons are depicted as black boxes. Restriction sites for BamHI (B), BglII (Bg), and EcoRI (E) are indicated, as are the external and cDNA probe. Middle: the lnk gene targeting construct. A BglII fragment containing most of the reported coding sequence of lnk with a neomycin phosphotransferase cassette (Neo). Bottom: predicted structure of the disrupted lnk gene. The exon containing the true initiation codon identified in this study (see Results) is indicated by asterisks.
(B) Representative Southern blot analysis of genomic DNA of progeny mice born from a heterozygote cross. DNA was digested with BglII and probed with the external probe.
(C) Expression of the lnk transcript in spleen. Poly(A)+ RNA (3 μg) from splenocytes was separated, transferred, and hybridized with mouse lnk cDNA probe (upper) or β-actin probe (lower).
Normal Thymocyte Development in lnk−/− Mice
The association of Lnk with Grb2, PI3K, and PLCγ observed after T cell receptor (TCR) cross-linking (Huang et al., 1995) prompted us to analyze T cell development. Cellularity in the thymus and T cell development assessed by expression of CD4 and CD8 appeared normal in lnk−/− mice (data not shown). Expression levels of CD3 on thymocytes were also indistinguishable between lnk−/− and lnk+/+ mice. The tyrosine phosphorylation patterns of cellular proteins and calcium influx induced by anti-CD3 treatment were also identical in lnk−/− and lnk+/+ thymocytes (data not shown). We conclude that Lnk seems to be dispensable for thymocyte development.
Accumulation of B Cells in Spleens of lnk−/− Mice
While thymocyte development was not demonstrably affected, the spleens of lnk−/− mice were enlarged and contained about twice as many cells as those of lnk+/+ mice (Figure 2A). The B cell compartment was the main population contributing to the increased cellularity of lnk−/− spleens. In particular, B220loIgMhi cells, which represent relatively immature and newly generated B cells, accumulated in the spleens of lnk−/− mice. Most of the IgM+ cells also expressed reduced amounts of CD23 and IgD, which is consistent with their immaturity (Figure 2A). Remarkably, lnk−/− spleens also contained significant numbers of B220loIgM− cells. These B220loIgM− cells were CD19+ CD43− IgG− (data not shown), indicating that they most likely represent pre-B cells. The absolute number of B220hi mature B cells was maintained or slightly increased in lnk−/− mice. Consistent with their normal thymocyte development, the numbers of splenic CD4+ or CD8+ T cells in lnk−/− mice were comparable to those found in lnk+/+ mice. Therefore, the increase in cell number observed in the spleens of lnk−/− mice results largely from an increase in relatively immature B cells. Notably, B and pre-B cell numbers were moderately increased in the bone marrow and spleens of heterozygous mice (Figures 2A and 2B). This finding of haploinsufficiency in lnk+/− animals indicates that B cell production is normally constrained by the intracellular accumulation of Lnk protein.
Figure 2. Accumulation of B-Lineage Cells in lnk−/− Mice.

(A) Splenic B cells are increased and show immature phenotypes in lnk−/− mice. Representative two-color fluorescence plots showing expression of B220 and IgM (upper panels) on splenocytes and expression of IgD (middle left) or CD23 (middle right) on IgM+ B cells. Percentages represent the fractions of the total gated live cells that fall into the indicated boxes. Cellularity of pre-B (B220+IgM−), B (B220+IgM+), CD4+ T, and CD8+ T in spleen (lower panels). lnk+/+, lnk+/−, and lnk−/− mice are represented by open, shaded, and closed symbols, respectively. *p < 0.01, †p < 0.05 by the Student’s t test.
(B) B cell precursors are accumulated in the bone marrow of lnk−/− mice. Representative two-color fluorescence plots showing expression of B220 and IgM (upper panels), as well as expression of B220 and CD43 (middle panels) on bone marrow cells. Percentages represent the fractions of the total gated live cells within the indicated boxes. Cellularity of pro-B: B220+CD43+, pre-B: B220+CD43−IgM−, immature B: B220loIgM+, mature B: B220hiIgM+, myeloid: B220−CD43+ in bone marrow (lower panels). lnk+/+, lnk+/−, and lnk−/− mice are represented by open, shaded, or closed symbols, respectively. *p < 0.01, †p < 0.05.
Proliferation and Apoptosis of lnk−/− B Cells
To investigate whether the accumulation of B cells in the spleens of lnk−/− mice was linked to augmented proliferation or decreased apoptosis, we compared the responses of splenocytes to various stimuli. Splenic B cells from lnk−/− mice showed a markedly reduced proliferative response following B cell receptor (BCR) cross-linking by anti-IgM, while they proliferated almost as well as lnk+/+ B cells in response to anti-CD40 or LPS treatment (Table 1A). The time course of proliferation induced by BCR cross-linking was not significantly altered by the absence of Lnk (data not shown). IgM cross-linking induces both proliferation and apoptosis of B cells (Mayumi et al., 1995; DeFranco, 1997). Even in the absence of stimulation, lnk−/− B cells tended to die more readily in culture than did control B cells (Table 1B). IgM cross-linking induced apoptosis of splenic B cells in a dose-dependent manner, and generally, the amount of cell death was higher in lnk−/− cells than in lnk+/+ cells. Taken together, lnk−/− B cells exhibit characteristics of immature B cells (Norvell et al., 1995; Norvell and Monroe, 1996), and the augmented accumulation of B cells in the spleens of lnk−/− mice probably does not result from enhanced B cell proliferation or survival, in situ, but rather from increased production of B-lineage cells.
Table 1.
Responses of Splenic B Cells and Humoral Immunity in lnk−/− Mice
| (A) Mitogenic Responses of lnk−/− Splenic B Cells | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mouse | RPMI | Anti-IgM | Anti-CD40 | LPS | ||
| Experiment 1 | +/+ | 6.6 ± 0.9 | 34.3 ± 0.8 | 110.7 ± 5.0 | 172.4 ± 8.0 | |
| −/− | 9.1 ± 1.1 | 17.5 ± 2.9 | 105.1 ± 1.6 | 208.3 ± 1.1 | ||
| Experiment 2 | +/+ | 5.2 ± 0.1 | 49.2 ± 1.8 | 122.6 ± 7.8 | 229.4 ± 7.2 | |
| −/− | 4.2 ± 0.4 | 24.4 ± 6.9 | 107.4 ± 5.4 | 200.7 ± 6.6 | ||
| −/− | 4.9 ± 0.3 | 17.7 ± 1.5 | 131.6 ± 8.7 | 267.9 ± 21.4 | ||
|
| ||||||
| (B) Anti-IgM-Induced Apoptosis of lnk−/− Splenic B Cells | ||||||
|
| ||||||
| Anti-IgM (μg/ml) | ||||||
| Mouse | 0 | 3.3 | 10 | 30 | ||
|
| ||||||
| +/+ (n = 4) | 6.4 ± 0.4 | 14.0 ± 2.0 | 16.9 ± 1.4 | 16.9 ± 1.7 | ||
| −/− (n = 4) | 10.7 ± 0.4* | 22.9 ± 1.6* | 31.4 ± 1.3* | 31.8 ± 1.1* | ||
|
| ||||||
| (C) Serum Immunoglobulin Level (μg/ml) | ||||||
|
| ||||||
| Mouse | IgM | IgG3 | IgG1 | IgG2a | IgG2b | IgA |
|
| ||||||
| +/+ (n = 10) | 290 ± 34 | 154 ± 61 | 1008 ± 429 | 329 ± 91 | 764 ± 166 | 226 ± 41 |
| −/− (n = 12) | 949 ± 191* | 130 ± 28 | 469 ± 117 | 429 ± 146 | 1032 ± 246 | 190 ± 60 |
|
| ||||||
| (D) Antibody Production against Thymus-Independent Antigen, TNP-Ficoll (Relative Titer × 10−2) | ||||||
|
| ||||||
| Mouse | IgM | IgG3 | ||||
|
| ||||||
| +/+ (n = 6) | 41 ± 10 | 52 ± 30 | ||||
| −/− (n = 6) | 26 ± 7 | 30 ± 7 | ||||
|
| ||||||
| (E) Antibody Production against Thymus-Dependent Antigen, DNP-KLH (Relative Titer × 10−2) | ||||||
|
| ||||||
| Mouse | IgM | IgG3 | IgG1 | IgG2a | IgG2b | |
|
| ||||||
| +/+ (n = 4) | 8.4 ± 2.6 | 21 ± 19 | 1600 ± 830 | 110 ± 81 | 1600 ± 820 | |
| −/− (n = 7) | 7.2 ± 4.0 | 43 ± 35 | 810 ± 380 | 230 ± 130 | 1500 ± 800 | |
(A) Splenocytes from lnk−/− mice or wild-type littermates were stimulated with anti-IgM (10 μg/ml), anti-CD40 (3 μg/ml), or LPS (10 μg/ml), and proliferation was measured at day 3 by [3H]thymidine incorporation. The values are the mean cpm × 103 (± SD) of triplicate determinations normalized with respect to the representation of IgM+ B cells in the spleen.
(B) Splenocytes were stimulated with the indicated concentration of anti-IgM antibodies for 16 hr. Dead B cells were identified by staining with FITC-anti-B220 and 7-amino-actinomycin-D, and the percentages of dead B cells among the total B220+ cell population were calculated. Results are compiled from two experiments, and the mean values (± SEM) of indicated groups of mice are shown. *p < 0.01 compared with +/+ mice.
(C–E) Antibody production in lnk−/− mice. Serum concentrations of immunoglobulin subclasses determined by isotype-specific ELISA (C). Mice were injected with TNP-ficoll (D) or DNP-KLH (E), and the amounts of hapten-specific antibodies were measured by ELISA. Data shown are the mean (± SEM) of indicated groups of mice. *p<0.01 compared with +/+ mice.
Mature lnk−/− B Cells Are Functionally Intact
BCR cross-linking induced identical patterns of tyrosine phosphorylation of intracellular proteins in wild-type and lnk−/− splenic B cells and augmented intracellular free calcium levels identically in the more mature, B220hi B cells of both types of animals (data not shown). The accumulation of immature B cells in lnk−/− spleens was associated with a mild disturbance of splenic architecture, typified by enlargement and fibrosis of marginal zone areas and irregularities in the shape of primary follicles (data not shown). With these observations in mind, we examined whether lnk−/− mice are capable of producing normal humoral immune responses. Serum levels of IgM were elevated about 5-fold in lnk−/− mice, whereas the levels of all other immunoglobulin isotypes were comparable to those found in littermate controls (Table 1C). Immunization with the thymus-independent type 2 (TI-2) or thymus-dependent (TD) antigens produced comparable levels of hapten-specific antibodies in both types of animals (Tables 1D and 1E). We conclude that peripheral lnk−/− B cells display relatively normal functional characteristics.
B Cell Precursors Are Increased in the Bone Marrow of lnk−/− Mice
In lnk−/− mice, cellularity in the bone marrow was increased about 1.5-fold (Figure 2B). Subpopulations representing different stages of B cell development in the bone marrow can be distinguished as fractions (Fr.) A to E on the basis of expression of various surface markers (Hardy et al., 1991). The bone marrow of lnk−/− mice contains an increased proportion of the B220+IgM− population (from Fr. A to D), representing pro-B and pre-B cells (Figure 2B). B220loIgM+ immature B cells (Fr. E) were also overrepresented in lnk−/− mice. In contrast, B220hiIgM+ mature B cells (Fr. F) recirculating from the periphery were less abundant in lnk−/− mice. The proportion of B220+CD43+ pro-B cells (from Fr. A to C) was increased 1.5-fold in lnk−/− mice. Viewed in the context of the 1.5-fold increase in the total number of bone marrow cells, this corresponds to a more than 2-fold increase in the number of pro-B cells in lnk−/− mice. Four-color staining of pro-B cells revealed that the HSA+BP-1− subset (Fr. B) is the least mature population of abnormally abundant cells in lnk−/− bone marrow (data not shown). All cell subsets representing subsequent differentiation steps in B lymphopoiesis were proportionally increased. Importantly, B cell maturation was not blocked at any stage. In contrast to the marked expansion of B cell precursors, the total number of myeloid cells was normal in lnk−/− mice (Figure 2B), and these animals did not show any signs of anemia, or hemorrhage (data not shown). These results demonstrate that the absence of Lnk either specifically promotes pro-B cell production or prevents apoptosis of B-lineage cells more generally.
Proliferation and Apoptosis of lnk−/− B Cell Precursors
To assess the proliferative state of B cell precursors in vivo, mice were fed BrdU and proliferating cells in B220+ compartments were enumerated. During a 40 hr exposure period, the fraction of BrdU-incorporating B220lo B cell precursors was significantly increased in bone marrow cells from lnk−/− mice (Figures 3A and 3B). In the spleen, however, only a small percentage of B220+ B cells incorporated BrdU in both groups of animals. A kinetic analysis of BrdU+ B-lineage cells after 10 days exposure to BrdU demonstrated enhanced turnover of B-lineage cells in the bone marrow and spleen and augmented entry of newly generated B cells into the spleen B cell compartment in lnk−/− mice (Figure 3B). We found no differences in in vitro survival for any of the B cell precursor populations of lnk−/− as compared with wild-type mice (data not shown). Taken together, the accumulation of lnk−/− B-lineage cells in the bone marrow mainly results from enhanced proliferation of B cell precursors and not prolonged survival of bone marrow lymphocytes or abnormal proliferation of peripheral B cells. As reported for wild-type mice (Hardy et al., 1991), cycling cells were detected only among B220+CD43+ pro-B cells, not in B220+CD43− pre-B or immature B cells in lnk−/− mice (data not shown). Hence, the enhanced proliferation of lnk−/− B cell progenitors is confined to populations that normally proliferate in wild-type mice.
Figure 3. In Vivo Incorporation of BrdU by lnk−/− B Cell Precursors and Splenic B Cells.
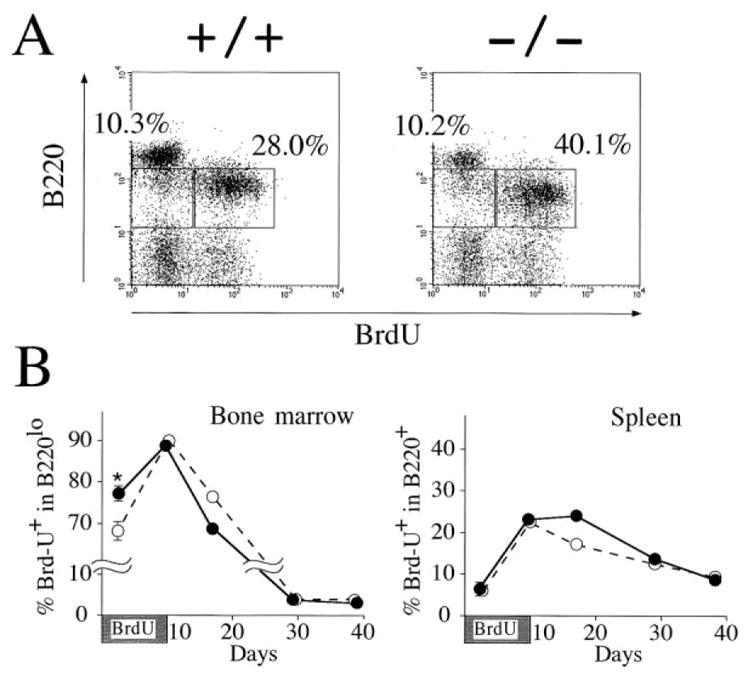
(A) Mice were fed BrdU in drinking water for 40 hr, and bone marrow cells were stained for B220 expression and BrdU content. Percentages represent the fractions of the gated lymphocytes that fall into the indicated boxes. The data shown are representative results from three independent experiments.
(B) Mice were fed BrdU for 10 days. Two lnk−/− (closed circles) and lnk−/− (open circles) were killed and analyzed at each time point (except day 2). The mean percentage of BrdU+ cells in B220lo bone marrow cells (left) or the mean percentage of BrdU+ cells in B220+ splenocytes (right) is shown. At day2, 4 lnk−/− and 4 lnk+/+ mice were analyzed and the mean ± SEM is shown. *p < 0.01 compared with +/+ mice.
Defects in B Cell Precursors Are Responsible for B Cell Overproduction
The B cell overproduction observed in lnk−/− mice could in principle result from an intrinsic defect within precursor B cells or from defects within bone marrow stromal cells that are known to support B cell development via direct and indirect interactions. To distinguish between these possibilities, we transferred bone marrow cells from lnk−/− or lnk+/+ mice into lethally irradiated wild-type mice or from wild-type mice into irradiated lnk−/− or lnk+/+ mice. Total bone marrow and spleen cell populations were significantly augmented in wild-type mice reconstituted with lnk−/− rather than lnk+/+ bone marrow cells. This increase in cell number resulted from a 2-fold enhancement in the size of the B220+ compartment (Figure 4A). Importantly, enhanced production of B cells was not observed when wild-type bone marrow cells were transferred into lethally irradiated lnk−/− mice. In fact, reconstituted lnk−/− recipient mice had slightly fewer B220+ bone marrow cells than did lnk+/+ recipient mice (Figure 4B). Hence, an intrinsic defect within B cell precursors, and not in resident bone marrow stromal cells, is responsible for the B cell overproduction observed in lnk−/− mice.
Figure 4. Defects in B Cell Precursors Are Responsible for B Cell Overproduction.

(A) Irradiated C57BL/6 mice were transferred with bone marrow cells from lnk+/+ (open symbols) or lnk−/− (closed symbols). The resulting chimeric mice were analyzed at 7–8 weeks after bone marrow transfer. Data shown are cell numbers of indicated cell populations in the bone marrow (per femur) or the spleen. *p < 0.01.
(B) lnk+/+ (open symbols) or lnk−/− mice (closed symbols) were irradiated and transferred with C57BL/6 bone marrow cells. †p < 0.05.
(C) Successful repopulation of lnk−/− precursor cells into B cell compartment in a competitive environment. Bone marrow cells negative for the lineage makers (Lin; B220, CD3ε, Gr-1, Mac-1, and TER-119) were isolated from lnk−/− or lnk+/+ mice and were injected (2 × 105 per head) into RAG2−/− mice without irradiation. Splenocytes from the injected mice were analyzed at 7–8 weeks after injection. Displayed are representative two-color fluorescence plots showing expression of IgM and B220 (upper panels) or expression of B220 and CD3 (lower panels). Percentages represent the fractions of the total gated live cells that fall into the indicated boxes. The data shown are representative results from three independent experiments.
lnk−/− Precursor Cells Can Competitively Repopulate B Cell Compartments
We tested whether the loss of lnk confers a growth advantage upon B cell precursors even in a highly competitive environment. Although RAG2−/− mice lack mature lymphocytes, lymphoid precursors and myeloid cells arise normally in the bone marrow (Shinkai et al., 1992). Therefore, upon transfer into nonirradiated RAG2−/− mice, donor hematopoietic precursor cells must compete with endogenous RAG2−/− stem cells to populate mature lymphocyte compartments. Bone marrow precursor cells negative for expression of lineage-specific markers were isolated from lnk−/− or lnk+/+ mice, and identical numbers of these precursor cells were subsequently injected into RAG2−/− recipients without prior irradiation. We found that lnk−/− donor precursors efficiently generated B cells in the spleens of RAG2−/− recipients (Figure 4C). In contrast, lnk+/+ donor hematopoietic precursors, while satisfactorily reconstituting the T cell compartment, failed to generate B cells (Figure 4C), even if five times more lnk+/+ donor cells were injected (data not shown). We conclude that the absence of Lnk confers upon immature bone marrow cells an enhanced ability to support B lymphopoiesis, even in a competitive environment.
Augmented Proliferation of lnk−/− Precursors Induced by SCF, the Ligand for C-kit
The accumulation of B cell precursors in lnk−/− mice resembles the phenotypes observed in mice injected with IL-7 (Morrissey et al., 1991). Bone marrow cells from lnk−/− mice produced more colonies in response to IL-7 than did those from lnk+/+ mice, reflecting the increased B cell precursor frequency in lnk−/− bone marrow (Figure 5A). The minimum concentration of IL-7 that produces apparent colonies was, however, comparable in animals of both genotypes (1 ng/ml). We conclude that the response of pro-B cells to IL-7 is not materially affected by the absence of Lnk.
Figure 5. Colony Formation by Bone Marrow Cells in Response to IL-7 and SCF.

(A) The numbers of colonies appearing in clonal cultures of bone marrow cells in semi-solid medium containing various concentrations of IL-7 (left) or SCF (middle) or containing both SCF and IL-7 (right) were compared between lnk+/+ (open bars) and lnk−/− (solid bars) mice. The values are the mean colony number (± SD) of triplicate cultures. *p < 0.005, †p < 0.05 compared with +/+ mice. The data shown are representative results from two independent experiments.
(B) Lymphoid colonies formed in the presence of SCF (4 ng/ml) and IL-7 (100 ng/ml).
In bone marrow cells, SCF is known to support the proliferation of B cell precursors when tested in combination with IL-7. Colony numbers were increased in lnk−/− as compared with wild-type bone marrow cells when exposed to any concentration of SCF (Figure 5A). Importantly, at low concentrations of SCF (e.g., 0.8 ng/ml) where lnk+/+ bone marrow cells were unable to form colonies, lnk−/− precursors were clearly responsive. Moreover, lnk−/− colonies were both larger in size and contained more cells than did lnk+/+ colonies (data not shown). In the presence of both SCF and IL-7, lnk−/− precursors also produced more colonies than did lnk+/+ precursors (Figure 5A). The lymphoid colonies (ascertained by morphology as well as cell surface phenotype) generated from lnk−/− precursors were larger in size and contained more cells than lnk+/+ colonies (Figure 5B). These results indicate that lnk−/− precursors are hypersensitive to SCF. Hence, in B cell precursors, c-kit-mediated proliferation is augmented as a consequence of Lnk deficiency.
Lnk Is a 68 kDa Protein that Is Absent in lnk−/− Splenocytes
Lnk has been reported to encode a 38 kDa protein (Huang et al., 1995; Takaki et al., 1997). Unexpectedly, a 68 kDa protein recognized by antibodies against the C-terminal region of Lnk was decreased in lnk+/− mice and disappeared in lnk−/− animals (Figure 6A), suggesting that previously reported lnk sequence was incomplete. We isolated lnk cDNAs containing additional 5′ sequences and assembled what we believe to be a full-length lnk cDNA. The deduced amino acid sequence depicts full-length Lnk as a 548 amino acid protein with a calculated molecular mass of 60.5 kDa (Figure 6B).
Figure 6. Revised Structure of Lnk and Its Family Members.

(A) Western blot analysis of splenocytes by anti-Lnk antibodies. Splenocyte lysates prepared from lnk+/+, lnk+/−, or lnk−/− mice were analyzed by immunoblotting probed with anti-Lnk-C-terminal antibodies (left panel). Immunoprecipitates from splenocyte lysates by anti-Lnk-N-terminal antibodies were immunoblotted using biotinylated anti-Lnk-N-terminal antibodies directed against a newly identified N-terminal portion of full-length Lnk protein (right panel). Arrows indicate a 68 kDa band representing Lnk.
(B) Amino acid sequence of mouse Lnk predicted from a combined sequence of lnk cDNAs. N-terminal region rich with proline residues and homologous to APS and SH2-B (dotted line), PH domain (solid line), SH2 domain (shaded), a conserved tyrosine phosphorylation site at the C-terminal end (boxed) are indicated.
(C) Amino acid sequence alignments of proline-rich N-terminal region, PH domain, SH2 domain, and conserved tyrosine phosphorylation site. Amino acid residues identical in at least two of three mammalian Lnk family members (Lnk, APS, and SH2-B) are indicated by shaded boxes. Proline residues in the N-terminal region are highlighted by boldface.
(D) Schematic representation of Lnk-family members.
Antibodies against the N terminus of the full-length Lnk protein were raised and used to examine Lnk expression from the mutated allele of our lnk−/− mice. The 68 kDa Lnk was detected by the anti-N-terminal Lnk antibodies in lnk+/+ or lnk+/− splenocyte extracts immunoprecipitated with anti-C-terminal (data not shown) or anti-N-terminal antibodies (Figure 6A). No accumulation of smaller proteins was observed in lnk−/− or lnk+/− mice (Figure 6A). By these criteria, the lnk gene disruption that we have engineered is a true null mutation, at least with respect to Lnk protein production.
Lnk Is a Member of a Conserved Multigene Adaptor Protein Family
With the additional N-terminal Lnk sequence available for comparison, we found that the overall structure of Lnk closely resembles that of two other adaptor proteins, APS (Yokouchi et al., 1997) and SH2-B (Osborne et al., 1995; Riedel et al., 1997). All three of these proteins contain a conserved N-terminal domain that includes a proline-rich stretch, followed by PH and SH2 domains (Figures 6C and 6D). A potential tyrosine phosphorylation site in the C-terminal region is also conserved among these proteins except in SH2-Bβ, a variant form of SH2-B resulting from alternative splicing of the SH2-B mRNA (Rui et al., 1997). Bioinformatic analysis identified a sequence containing the entire human LNK gene. The putative exon/intron boundaries align perfectly with the mouse lnk gene. By fusion of hLNK exons, we assembled a tentative hLNK cDNA sequence. While this manuscript was in preparation, a bona fide hLNK cDNA was reported (Li et al., 2000); the sequence differs only subtly from our assembled hLNK cDNA.
Discussion
Lnk Is Dispensable for T Cell Development and TCR Signaling
Lnk was originally described as a 38 kDa adaptor protein expressed in T cells that displays characteristics that very closely resemble those of pp36/LAT. The apparent phosphorylation of Lnk upon TCR cross-linking, and its association with Grb2, PI3K, and PLCγ (Huang et al., 1995), suggested a role for Lnk in TCR signal transduction. However, we found that Lnk expression was more abundant in B cells than in T cells and that overexpression of a 38 kDa form of Lnk (which we now know to be truncated with respect to the wild-type protein) in transgenic mice under control of the proximal lck promoter did not compromise T cell development or function (Takaki et al., 1997).
Although the effect of Lnk overexpression in T-lineage cells must be reevaluated using the full-length lnk cDNA identified in the present study, analysis of lnk−/− mice again suggests that Lnk has no nonredundant function in T cell development or TCR-mediated signaling. Instead, the lnk gene disruption affects the cellular compartment in which Lnk is ordinarily expressed at highest levels.
Lnk Constrains Production of B Cells
The absence of Lnk resulted in a substantial accumulation of B-lineage cells in the bone marrow and spleens of lnk−/− mice, primarily as a result of overproduction of pro-B cells in the bone marrow. The results from our bone marrow transfer experiments support a firm conclusion: lnk gene disruption yields a cell autonomous defect in the B cell compartment. lnk−/− hematopoietic progenitors produce more B cells than do wild-type progenitors when adoptively transferred into an irradiated wild-type, or a nonirradiated RAG2-deficient, microenvironment. Reciprocally, B cell production by wild-type progenitors developing in the lnk−/− microenvironment was not enhanced but in fact slightly suppressed. This observation might also reflect the action of a feedback mechanism working within stromal cells in response to the enhanced B lymphopoiesis by lnk−/− hematopoietic progenitors.
Serum levels of IgM, but not other immunoglobulin isotypes, were substantially elevated in lnk−/− mice. However, the peritoneal B-1 cell compartment, one major source of serum IgM in normal mice, was not expanded in lnk−/− animals (data not shown). It is conceivable that continuous oversupply of immature B cells from the bone marrow may gradually result in the accumulation of mature B cells in peripheral lymphoid tissues and may permit more frequent maturation of B cells into IgM-producing cells. Aged lnk−/− mice tend to have more mature B cells in lymph nodes (data not shown).
Lnk Does Not Play a Role in BCR or Pre-BCR Signaling
Signals from the pre-B cell receptor (pre-BCR) are critical for B cell development, promoting the transition of pro-B cells to pre-B cells (Karasuyama et al., 1996). B cell development in mice lacking pre-BCR components is blocked at the pro-B cell stage (reviewed by Benschop and Cambier, 1999). This pro-B to pre-B transition is accompanied by rounds of cell expansion, in which Lnk could conceivably play a role. However, our observations in splenic B cells argue against the involvement of Lnk in signal transduction from the BCR and, by inference, from the pre-BCR. Although the phenotype of splenic B cells from lnk−/− mice closely resembles that of newly generated immature B cells, the biochemical consequences of BCR signaling appeared normal in stimulated lnk−/− B cells and lnk−/− mice produced normal antibody responses following immunization. Hence, it seems unlikely that Lnk plays a substantive role in either pre-BCR or BCR signal transduction.
Lnk Negatively Regulates Signaling from the Receptor Tyrosine Kinase, C-kit
The most salient property of lnk−/− B cell precursors is their enhanced sensitivity to SCF. It is of interest that the Lnk family member APS was identified through a yeast two-hybrid screen using the c-kit kinase domain as bait (Yokouchi et al., 1997). The tyrosine phosphorylated C-terminal region of APS, which is conserved among the Lnk family, binds to c-Cbl, a negative regulator of tyrosine kinases (Wakioka et al., 1999; Yokouchi et al., 1999). In transfected COS cells, Lnk becomes phosphorylated by, and is coimmunoprecipitated with, c-kit (S.T., unpublished data). Considering those observations, we speculate that Lnk associates with c-kit and simultaneously recruits other proteins that act either as negative regulators of the kinase itself (e.g., phosphotyrosine phosphatases) or can accelerate degradation of the activated receptor complex, as has been demonstrated for c-Cbl (Miyake et al., 1998; Joazeiro et al., 1999). Identification of these Lnk-associated molecules will provide insights into the functional behavior of the entire Lnk family.
The c-kit kinase is expressed in hematopoietic stem cells but gradually disappears during B-lineage differentiation (Ghia et al., 1998; Payne et al., 1999). Pro-B cells (Fr. B) are the earliest population whose expansion is apparent in the bone marrow of lnk−/− mice. However, we cannot exclude the possibility that augmented B-lineage cell production initiates in the pre-pro-B cells, which represent only a subset of cells in Fr. A (Li et al., 1996). Lnk is highly expressed in B-lineage cells (Takaki et al., 1997). This presumably accounts for the selective accumulation of B cells in lnk−/− mice despite the importance of SCF and c-kit in directing the development of a wide-range of cell types.
Lnk Family Adaptor Proteins
Analysis of Lnk immunoreactive species in lnk−/− mice stimulated reassessment of published Lnk sequences, which in turn revealed that Lnk forms an adaptor protein family together with APS and SH2-B. APS, identified as a substrate of c-kit, becomes tyrosine phosphorylated after BCR ligation (Yokouchi et al., 1997). SH2-B was identified by its ability to associate with the immunoreceptor tyrosine-based activation motif (ITAM) of the FcεRγ chain (Osborne et al., 1995). APS and SH2-B are also potential substrates for other protein tyrosine kinases including the insulin receptor (Riedel et al., 1997; Kotani et al., 1998; Ahmed et al., 1999; Moodie et al., 1999), Trk family receptors (Qian et al., 1998; Rui et al., 1999), the PDGF receptor (Rui and Carter-Su, 1998; Yokouchi et al., 1999), and JAK2 (Rui et al. 1997; Wakioka et al., 1999). Nevertheless, the functions of APS and SH2-B remain unknown. Considering the role of Lnk in c-kit-mediated B-lymphopoiesis, we propose that APS and SH2-B also serve as governors for receptor tyrosine kinase signaling, both in hematopoietic cells and in other cell types. Consistent with this speculation, high-level expression of APS inhibits PDGF-induced mitogenesis of fibroblasts, and signaling via the JAK-STAT pathway, in concert with c-Cbl (Wakioka et al., 1999; Yokouchi et al., 1999). In Trk-mediated signaling in developing neurons, however, APS and SH2-B seem to work as positive regulators, activating the MAPK pathway through association with Grb2 (Qian et al., 1998). SH2-Bβ, a variant form of SH2-B that lacks a conserved tyrosine residue at the C-terminal end, enhanced neurite outgrowth in PC12 cells when overexpressed (Rui et al., 1999). Hence, the putative positive regulatory function of Lnk family members may not require phosphorylation of the C-terminal tyrosine residue conserved among this group of proteins.
Although Lnk deficiency affects primarily c-kit-mediated proliferation of B cell progenitors, Lnk is expressed throughout B cell development. APS is expressed mainly in mature B cell lines but not in pro-B or pre-B cell lines (Iseki et al., 2000). It is possible that Lnk might regulate signaling through receptors other than c-kit in mature B-lineage cells; however, its regulatory function could be compensated by APS in the absence of Lnk. In this context, it will be interesting to examine the phenotypes imposed by disruption of the genes encoding other Lnk family members, separately or in combination.
The elucidation of Lnk function in developing B lymphocytes will provide a conceptual framework for understanding the importance of Lnk family members in other systems, where these adaptor proteins almost certainly play crucial regulatory roles. As a first step, our studies make plain that in the mouse Lnk ordinarily acts to constrain the growth of the B cell compartment and hence to maintain satisfactory lymphoid homeostasis. Viewed in this way, it will be interesting to learn whether Lnk expression is itself regulated, providing a mechanism for enhanced B lymphopoiesis that might prove important in combating infectious agents with high antigenic variation.
Experimental Procedures
Generation of lnk-Deficient Mice
The isolation of a clone harboring the lnk gene has been described previously (Takaki et al., 1997). Three BglII fragments containing exons of the published lnk coding region were replaced with a neomycin phosphotransferase (Neo) cassette, and a herpes simplex virus thymidine kinase gene was inserted in the 3′-end of the lnk-Neo construct. The targeting construct was linearized and introduced into AK7 ES cells (gift of Drs. K. Akiyama and P. Soriano) by electroporation. Conditions and G418/gancyclovir selection were performed as described previously (Appleby et al., 1992). Chimeric mice were generated from lnk+/− ES cell clones, and germline transmission of the mutant allele was confirmed by Southern blot analysis of DNA obtained from tail biopsies. All mice were housed in a specific pathogen-free facility.
Flow Cytometric Analysis
Single cell suspensions of lymphocytes were prepared from lymphoid tissues of 6- to 7-week-old mice. The cells were stained using predetermined optimal concentrations of the respective antibodies and were analyzed on a Becton-Dickinson FACScan instrument. The following monoclonal antibodies were used: fluorescein isothiocyanate (FITC)-conjugated anti-CD8, phycoerythrin (PE)-conjugated anti-CD4, PE- or biotin-conjugated anti-CD3ε, biotin-anti-CD69, PE-anti-CD43, FITC-anti-BP-1, PE-anti-Gr-1, biotin-anti-heat-stable antigen (HSA), FITC-anti-Mac-1, PE-anti-CD23, PE-anti-CD5 (all purchased from Pharmingen), FITC-, PE-, or TRICOLOR-anti-B220 (Caltag Laboratories), biotin-anti-IgD (Southern Biotechnology). FITC- or PE-conjugated F(ab′)2 fragments of polyclonal anti-IgM were purchased from Caltag Laboratories and PE-or TRI-COLOR-conjugated streptoavidin (Caltag Laboratories) was used to reveal biotin-coupled antibody staining.
Mitogenic and Apoptotic Responses of Splenic B Cells
Splenocytes (2 × 105) were cultivated in 200 μl of RPMI1640 medium supplemented with 10% fetal calf serum and 100 μM 2-mercapto-ethanol in a 96-well plate. Cells were stimulated with 10 μg/ml goat anti-mouse IgM F(ab′)2 (Organon Teknika), 3 μg/ml anti-CD40 (Pharmingen), or 10 μg/ml LPS (Difco Laboratories). Cells were pulse-labeled with [3H]thymidine (0.2 μCi per well) during the last 16 hr of culture period, and incorporated [3H]thymidine was measured using a Scintillation counter.
To measure IgM-induced apoptosis of splenic B cells, splenocytes (1 × 106) were cultured in 1 ml of medium containing various concentrations of anti-IgM F(ab′)2 fragment for 16 hr. Cells were collected and stained with FITC-conjugated anti-B220 antibody and 2 μg/ml of 7-amino-actinomycin D (7-AAD, Sigma) and were analyzed by flow cytometry. Live, apoptotic, and dead cells from a gated population of B220+ cells were identified by low-, intermediate-, and high-level 7-AAD staining (Barton et al., 1996).
Serology
Serum concentrations of each immunoglobulin isotype were determined in 6-week-old mice by isotype-specific enzyme-linked immunosorbent assay (ELISA) as described previously (Uehara et al., 1994). To examine the antibody production against TI-2 antigens, mice were intraperitoneally injected with 100 μg of TNP-ficoll in saline and were bled on day 10 after injection. To examine the response against TD antigens, mice were immunized intraperitoneally with 100 μg of DNP-KLH in a 1:1 homogenate with incomplete Freund’s adjuvant. A booster dose of 100 μg of DNP-KLH in saline was given at day 20, and mice were bled on day 30. Serial dilutions of serum were analyzed for TNP- or DNP-specific immunoglobulin isotypes by ELISA using DNP-coupled bovine serum albumin (cross-reacts with anti-TNP antibodies) as capture reagent.
Measurement of BrdU Incorporation
BrdU labeling of cells in vivo and BrdU staining were performed as described before (Tough and Sprent, 1994). In brief, mice were given drinking water containing BrdU (Sigma) at 0.8 mg/ml. Cells from bone marrow or spleen were stained with PE-coupled anti-B220 and then fixed, permeabilized, and treated with DNase I (Sigma). Cells were subsequently stained with FITC-conjugated anti-BrdU (Becton-Dickinson) and analyzed on a FACScan.
Bone Marrow Transfer
Bone marrow cells were isolated and CD3+ cells depleted using a MACS magnetic cell sorter system (Miltenyi Biotec, Germany). 1–2 × 106 cells were intravenously injected into lethally irradiated (9.5 Gy) recipient mice. For repopulation assays in RAG2−/− recipients, bone marrow cells were depleted of lineage-committed cells using a MACS system after incubation with a cocktail of biotin-conjugated antibodies against various lineage markers (Lin: B220, CD3, Gr-1, Mac-1, and TER-119) and streptavidin-coupled microbeads. 2–10 × 105 of the resulting Lin− cells were intravenously injected into RAG2−/− mice without irradiation. Bone marrow cells and splenocytes of chimeric animals were isolated and analyzed at 7–8 weeks after transfer.
Colony Formation Assay
Clonal cultures of bone marrow cells were performed as described previously (Akasaka et al., 1997). Cells (1 × 104) were placed in 35 mm standard culture dish (Becton-Dickinson), and colonies were scored on day 12 of culture. The size of lymphoid colonies identified in situ was assessed on day 9 of culture. To confirm the accuracy of the in situ identification, individual lymphoid colonies were lifted under microscopic visualization and examined for morphological appearance as well as B220 expression (Akasaka et al., 1997).
Isolation of cDNA and Sequence Analysis
Mouse lnk cDNA fragment was used as a probe to screen a mouse spleen cDNA library (Stratagene) by plaque hybridization. Three clones contained additional upstream sequence of the published lnk cDNA. The full-length lnk cDNA was assembled by fusing appropriate fragments of newly isolated cDNA clones with the published lnk cDNA, and accordingly GenBank accession number U89992 was updated.
Sequence of hLNK cDNA was deduced from the minus strand of HTGS in GenBank, accession number AC002395. The following fragments homologous to mlnk cDNA were used (numbers indicate positions of the corresponding bases in AC002395). Exon 1, 155,017 and probably upstream thereof; exon 2, 126145–126044; exon 3, 125956–125865; exon 4, 125773–125679; exon 5, 125568–125354; exon 6, 125242–125071; and exon 7, 124915–124569. All exons are flanked by consensus sequence of splice acceptor sites (AG) on their 5′-ends and splice donor sites (A/GGGT) on their 3′-ends.
Antibodies and Western Blotting
Polyclonal rabbit antisera were raised against the synthetic peptide NEPTVQPSRTSSAC that represents aa 2–14 of mLnk and cysteine residue added for coupling to carrier protein. The antibodies were affinity purified and biotinylated with N-hydroxysuccinimidobiotin (Pierce).
Splenocytes were lysed, and the lysates were clarified by centrifugation (Takaki et al., 1997). Total splenocyte lysate (derived from 5 × 106 cells) or immunoprecipitates by anti-Lnk-N-terminal antibodies (derived from 1 × 108 cells) were separated on SDS-PAGE gel and transferred onto nitrocellulose membranes. Membranes were probed with anti-Lnk-C-terminal antibodies (Takaki et al., 1997) or with biotinylated anti-Lnk-N-terminal antibodies. Bound antibodies were detected using HRP-conjugated second reagents via chemiluminescence.
Acknowledgments
We would like to thank K. Akiyama and P. Soriano for generously providing AK-7 ES cells, James D. Kerner for help with ES cell culture, Xiao-Cun Pan for assistance in maintaining our mouse colony, Katherine A. Forbush, Richard Peet, Byoung-Gon Moon, and Hai-Zhuan Zhang for technical assistance, Kathy Prewitt for expert secretarial assistance, Kathryn J. Allen and David Coder for assistance in flow cytometry analysis, and members of the MRL Bioinformatics department for their support. We thank our colleagues for helpful discussions and for critical reading of the manuscript. S. T. was supported in part by a Long-Term Research Fellowship from the Human Frontiers Science Program. B. M. I is supported by National Institutes of Health grants #T32 RR07019 and #1 K08 AJ01445-01. R. M. P. was an Investigator of the Howard Hughes Medical Institute. This work was supported in part by Grant-in-Aids from the Ministry of Education, Science, Sports, and Culture, and the Science and Technology Agency of Japan.
References
- Ahmed Z, Smith BJ, Kotani K, Wilden P, Pillay TS. APS, an adapter protein with a PH and SH2 domain, is a substrate for the insulin receptor kinase. Biochem J. 1999;341:665–668. [PMC free article] [PubMed] [Google Scholar]
- Akasaka T, Tsuji K, Kawahira H, Kanno M, Harigaya K, Hu L, Ebihara Y, Nakahata T, Tetsu O, Taniguchi M, Koseki H. The role of mel-18, a mammalian Polycomb group gene, during IL-7-dependent proliferation of lymphocyte precursors. Immunity. 1997;7:135–146. doi: 10.1016/s1074-7613(00)80516-6. [DOI] [PubMed] [Google Scholar]
- Alberola-Ila J, Takaki S, Kerner JD, Perlmutter RM. Differential signaling by lymphocyte antigen receptors. Annu Rev Immunol. 1997;15:125–154. doi: 10.1146/annurev.immunol.15.1.125. [DOI] [PubMed] [Google Scholar]
- Appleby MW, Gross JA, Cooke MP, Levin SD, Qian X, Perlmutter RM. Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell. 1992;70:751–763. doi: 10.1016/0092-8674(92)90309-z. [DOI] [PubMed] [Google Scholar]
- Baird AM, Gerstein RM, Berg LJ. The role of cytokine receptor signaling in lymphocyte development. Curr Opin Immunol. 1999;11:157–166. doi: 10.1016/s0952-7915(99)80027-2. [DOI] [PubMed] [Google Scholar]
- Barton K, Muthusamy N, Chanyangam M, Fischer C, Clendenin C, Leiden JM. Defective thymocyte proliferation and IL-2 production in transgenic mice expressing a dominant-negative form of CREB. Nature. 1996;379:81–85. doi: 10.1038/379081a0. [DOI] [PubMed] [Google Scholar]
- Benschop RJ, Cambier JC. B cell development: signal transduction by antigen receptors and their surrogates. Curr Opin Immunol. 1999;11:143–451. doi: 10.1016/s0952-7915(99)80025-9. [DOI] [PubMed] [Google Scholar]
- Birge RB, Knudsen BS, Besser D, Hanafusa H. SH2 and SH3-containing adaptor proteins: redundant or independent mediators of intracellular signal transduction. Genes Cells. 1996;1:595–613. doi: 10.1046/j.1365-2443.1996.00258.x. [DOI] [PubMed] [Google Scholar]
- Clements JL, Yang B, Ross-Barta SE, Eliason SL, Hrstka RF, Williamson RA, Koretzky GA. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science. 1998;281:416–419. doi: 10.1126/science.281.5375.416. [DOI] [PubMed] [Google Scholar]
- DeFranco AL. The complexity of signaling pathways activated by the BCR. Curr Opin Immunol. 1997;9:296–308. doi: 10.1016/s0952-7915(97)80074-x. [DOI] [PubMed] [Google Scholar]
- Farrar MF, Doerfler P, Sauer K. Signal transduction pathways regulating the development of αβ T cells. Biochem Biophys Acta. 1998;1377:F35–F78. doi: 10.1016/s0304-419x(97)00038-3. [DOI] [PubMed] [Google Scholar]
- Ghia P, ten Boekel E, Rolink AG, Melchers F. B-cell development: a comparison between mouse and man. Immunol Today. 1998;19:480–485. doi: 10.1016/s0167-5699(98)01330-9. [DOI] [PubMed] [Google Scholar]
- Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Li Y, Tanaka K, Moore KG, Hayashi JI. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase Cγ1, Grb2, and phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1995;92:11618–11622. doi: 10.1073/pnas.92.25.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseki M, Takaki S, Takatsu K. Molecular cloning of the mouse APS as a member of the Lnk family adaptor proteins. Biochem Biophys Res Commun. 2000;272:45–54. doi: 10.1006/bbrc.2000.2736. [DOI] [PubMed] [Google Scholar]
- Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286:309–312. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- Jumaa H, Wollscheid B, Mitterer M, Wienands J, Reth M, Nielsen PJ. Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity. 1999;11:547–554. doi: 10.1016/s1074-7613(00)80130-2. [DOI] [PubMed] [Google Scholar]
- Karasuyama H, Rolink A, Melchers F. Surrogate light chain in B cell development. Adv Immunol. 1996;63:1–41. doi: 10.1016/s0065-2776(08)60853-6. [DOI] [PubMed] [Google Scholar]
- Killeen N, Irving BA, Pippig S, Zingler K. Signaling checkpoints during the development of T lymphocytes. Curr Opin Immunol. 1998;10:360–367. doi: 10.1016/s0952-7915(98)80176-3. [DOI] [PubMed] [Google Scholar]
- Kotani K, Wilden P, Pillay TS. SH2-Bα is an insulin-receptor adapter protein and substrate that interacts with the activation loop of the insulin-receptor kinase. Biochem J. 1998;335:103–109. doi: 10.1042/bj3350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YS, Wasserman R, Hayakawa K, Hardy RR. Identification of the earliest B lineage stage in mouse bone marrow. Immunity. 1996;5:527–535. doi: 10.1016/s1074-7613(00)80268-x. [DOI] [PubMed] [Google Scholar]
- Li Y, He X, Schembri-King J, Jakes S, Hayashi J. Cloning and characterization of human Lnk, and adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J Immunol. 2000;164:5199–5206. doi: 10.4049/jimmunol.164.10.5199. [DOI] [PubMed] [Google Scholar]
- Mayumi M, Ohshima Y, Hata D, Kim KM, Heike T, Katamura K, Furusho K. IgM-mediated B cell apoptosis. Crit Rev Immunol. 1995;15:255–269. doi: 10.1615/critrevimmunol.v15.i3-4.40. [DOI] [PubMed] [Google Scholar]
- Miyake S, Lupher ML, Jr, Druker B, Band H. The tyrosine kinase regulator Cbl enhances the ubiquitination and degradation of the platelet-derived growth factor receptor alpha. Proc Natl Acad Sci USA. 1998;95:7927–7932. doi: 10.1073/pnas.95.14.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodie SA, Alleman-Sposeto J, Gustafson TA. Identification of the APS protein as a novel insulin receptor substrate. J Biol Chem. 1999;274:11186–11193. doi: 10.1074/jbc.274.16.11186. [DOI] [PubMed] [Google Scholar]
- Morrissey PJ, Conlon P, Charrier K, Braddy S, Alpert A, Williams D, Namen AE, Mochizuki D. Administration of IL-7 to normal mice stimulates B-lymphopoiesis and peripheral lymphadenopathy. J Immunol. 1991;147:561–568. [PubMed] [Google Scholar]
- Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, Langdon WY, Bowtell DD. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naramura M, Kole HK, Hu RJ, Gu H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci USA. 1999;22:15547–15552. doi: 10.1073/pnas.95.26.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norvell A, Monroe JG. Acquisition of surface IgD fails to protect from tolerance-induction. Both surface IgM- and surface IgD-mediated signals induce apoptosis of immature murine B lymphocytes. J Immunol. 1996;156:1328–1332. [PubMed] [Google Scholar]
- Norvell A, Mandik L, Monroe JG. Engagement of the antigen-receptor on immature murine B lymphocytes results in death by apoptosis. J Immunol. 1995;154:4404–4413. [PubMed] [Google Scholar]
- Osborne MA, Dalton S, Kochan JP. The yeast tribrid system: genetic detection of trans-phosphorylated ITAM-SH2-interactions. Biotechnology. 1995;13:1474–1478. doi: 10.1038/nbt1295-1474. [DOI] [PubMed] [Google Scholar]
- Pappu R, Cheng AM, Li B, Gong Q, Chiu C, Griffin N, White M, Sleckman BP, Chan AC. Requirement for B cell linker protein (BLNK) in B cell development. Science. 1999;286:1949–1954. doi: 10.1126/science.286.5446.1949. [DOI] [PubMed] [Google Scholar]
- Payne KJ, Medina KL, Kincade PW. Loss of c-kit accompanies B-lineage commitment and acquisition of CD45R by most murine B-lymphocyte precursors. Blood. 1999;94:713–723. [PubMed] [Google Scholar]
- Pivniouk V, Tsitsikov E, Swinton P, Rathbun G, Alt FW, Geha RS. Impaired viability and profound block in thymocyte development in mice lacking the adaptor protein SLP-76. Cell. 1998;94:229–238. doi: 10.1016/s0092-8674(00)81422-1. [DOI] [PubMed] [Google Scholar]
- Qian X, Riccio A, Zhang Y, Ginty DD. Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron. 1998;21:1017–1029. doi: 10.1016/s0896-6273(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Riedel H, Wang J, Hansen H, Yousaf N. PSM, an insulin-dependent, pro-rich, PH, SH2 domain containing partner of the insulin receptor. J Biochem. 1997;122:1102–1113. doi: 10.1093/oxfordjournals.jbchem.a021868. [DOI] [PubMed] [Google Scholar]
- Rui L, Carter-Su C. Platelet-derived growth factor (PDGF) stimulates the association of SH2-Bβ with PDGF receptor and phosphorylation of SH2-Bβ. J Biol Chem. 1998;273:21239–21245. doi: 10.1074/jbc.273.33.21239. [DOI] [PubMed] [Google Scholar]
- Rui L, Mathews LS, Hotta K, Gustafson TA, Carter-Su C. Identification of SH2-Bβ as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling. Mol Cell Biol. 1997;17:6633–6644. doi: 10.1128/mcb.17.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Herrington J, Carter-Su C. SH2-B is required for nerve growth factor-induced neuronal differentiation. J Biol Chem. 1999;274:10590–10594. doi: 10.1074/jbc.274.15.10590. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Takaki S, Watts JD, Forbush KA, Nguyen NT, Hayashi J, Alberola-Ila J, Aebersold R, Perlmutter RM. Characterization of Lnk. An adaptor protein expressed in lymphocytes. J Biol Chem. 1997;272:14562–14570. doi: 10.1074/jbc.272.23.14562. [DOI] [PubMed] [Google Scholar]
- Taniuchi I, Kitamura D, Maekawa Y, Fukuda T, Kishi H, Watanabe T. Antigen-receptor induced clonal expansion and deletion of lymphocytes are impaired in mice lacking HS1 protein, a substrate of the antigen-receptor-coupled tyrosine kinases. EMBO J. 1995;14:3664–3678. doi: 10.1002/j.1460-2075.1995.tb00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara S, Hitoshi Y, Numata F, Makino M, Howard M, Mizuochi T, Takatsu K. An IFNγ-dependent pathway plays a critical role in the pathogenesis of murine immunodeficiency syndrome induced by LP-BM5 murine leukemia virus. Int Immunol. 1994;6:1937–1947. doi: 10.1093/intimm/6.12.1937. [DOI] [PubMed] [Google Scholar]
- Wakioka T, Sasaki A, Mitsui K, Yokouchi M, Inoue A, Komiya S, Yoshimura A. APS, an adaptor protein containing Pleckstrin homology (PH) and Src homology-2 (SH2) domains inhibits the JAK-STAT pathway in collaboration with c-Cbl. Leukemia. 1999;13:760–767. doi: 10.1038/sj.leu.2401397. [DOI] [PubMed] [Google Scholar]
- Yokouchi M, Suzuki R, Masuhara M, Komiya S, Inoue A, Yoshimura A. Cloning and characterization of APS, an adaptor molecule containing PH and SH2 domains that is tyrosine phosphorylated upon B-cell receptor stimulation. Oncogene. 1997;15:7–15. doi: 10.1038/sj.onc.1201163. [DOI] [PubMed] [Google Scholar]
- Yokouchi M, Wakioka T, Sakamoto H, Yasukawa H, Ohtsuka S, Sasaki A, Ohtsubo M, Valius M, Inoue A, Komiya S, et al. APS, an adaptor protein containing PH and SH2 domains, is associated with the PDGF receptor and c-Cbl and inhibits PDGF-induced mitogenesis. Oncogene. 1999;18:759–767. doi: 10.1038/sj.onc.1202326. [DOI] [PubMed] [Google Scholar]
- Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, Grinberg A, Tsay HC, Jacobs HM, Kessler CM, et al. Essential role of LAT in T cell development. Immunity. 1999;10:323–332. doi: 10.1016/s1074-7613(00)80032-1. [DOI] [PubMed] [Google Scholar]