

Abstract
Selective modification of proteins at cysteine residues by reactive oxygen, nitrogen or sulfur species formed under physiological and pathological states is emerging as a critical regulator of protein activity impacting cellular function. This review focuses primarily on protein sulfenylation (-SOH), a metastable reversible modification connecting reduced cysteine thiols to many products of cysteine oxidation. An overview is first provided on the chemistry principles underlining synthesis, stability and reactivity of sulfenic acids in model compounds and proteins, followed by a brief description of analytical methods currently employed to characterize these oxidative species. The following chapters present a selection of redox-regulated proteins for which the -SOH formation was experimentally confirmed and linked to protein function. These chapters are organized based on the participation of these proteins in the regulation of signaling, metabolism and epigenetics. The last chapter discusses the therapeutic implications of altered redox microenvironment and protein oxidation in disease.
INTRODUCTION
Alteration of redox homeostasis underlies many pathological conditions impacting human health. The predominant small molecule effectors of redox regulation are the classically recognized reactive oxygen and nitrogen species (ROS/RNS), and the emerging reactive sulfur species (RSS). While controlled and localized production of ROS/RNS/RSS (e.g., H2O2, NO, H2S) by dedicated systems is critical to normal physiological processes, acute or chronic accumulation of these species contributes to disease development and response to therapies. Decades of studies have linked oxidative stress to metabolic and inflammatory diseases, cancer, aging, and aging-related pathologies, such as cardiovascular and neurodegenerative diseases. However, the detailed mechanisms linking redox imbalance to disease initiation, progression and treatment are still poorly understood. In this review, we discuss the complex mechanisms of protein redox regulation by focusing on key examples of enzymes involved in metabolism, signaling, and epigenetics. As demonstrated by the individual proteins selected for discussion here and summarized in Table 1, cysteine oxidation products are critically important for protein folding, function and cellular trafficking. Due to reversibility of many oxidative modifications, these qualify as protein molecular switches providing an additional layer of control to the regulation of cellular processes. In this review, we consider primarily the oxidative modification of protein cysteine thiols (-SH) to sulfenic acids (-SOH), critical metastable species connected to many other oxidative modifications (Figure 1). The first chapter provides the background information on factors controlling the stability of these species and methods of analysis. The second chapter presents critical enzymes involved in ROS metabolism and their own regulation by oxidation. Particular attention is paid to enzymes relying on thiol chemistry for their enzymatic activity and for controlling localized ROS levels within the cellular environment. The third, fourth and fifth chapters describe the mechanisms of thiol-based redox regulation of a selected number of signaling, metabolic and epigenetic proteins, respectively. Perspectives on current challenges and implications of protein redox regulation in drug discovery and development are provided in the concluding chapter.
Table 1.
Summary of proteins for which the -SOH formation was experimentally confirmed and linked to protein function.
| Protein | E.C. number or UniprotKB ID | Family or Class | Origin | Cys location | Posttranslational modification | Reference |
|---|---|---|---|---|---|---|
| Bcl-2 | P10415 | Adaptor protein | H. sapiens | 158 and 229 | -SOH | [131] |
| DJ-1 | Q99497 | Adaptor protein | H. sapiens | 106 | -SOH | [133], [134] |
| IQGAP | P46940 | Adaptor protein | H. sapiens; M. musculus | unknown | -SOH | [94] |
| Calbindin D28K | P05937 | Ca2+ binding protein | H. sapiens | 187, 219, 257 | -SOH and -SSG | [203] |
| Recoverin | P35243 | Ca2+ binding protein | H. sapiens | 39 | -SOH | [197] |
| DLDH | 1.8.1.4 | Dehydrogenase | R. norvegicus | unknown | -SOH | [185] |
| GAPDH | 1.2.1.12 | Dehydrogenase | H. sapiens | 152, 156* | -SOH and –SSG | [193], [187] |
| APE1 | 4.2.99.18 | DNA modifying enzyme | H. sapiens | 99 | -SOH and -SSG | [214] |
| SFGH | 3.1.2.12 | Hydrolase | S. cerevisiae | 60 | -SOH | [182] |
| MGL | 3.1.1.23 | Hydrolase | R. norvegicus | 201 and 208 | -SOH | [179] |
| TRPA1 | O75762 | Ion channel | M. musculus | unknown | -SOH | [199] |
| Kv1.5 | P22460 | Ion channel | H. sapiens | 581 | -SOH | [207] |
| Pin1≠ | 5.2.1.8 | Isomerase | H. sapiens | 113 | -SOH | [163] |
| Src≠ | 2.7.10.2 | Kinase | H. sapiens | Possibly 277 | -SOH | [95], [109] |
| IKKα | 2.7.11.10 | Kinase | M. musculus | unknown | -SOH and -SSG | [129] |
| Akt2 | 2.7.11.1 | Kinase | H. sapiens | 124, 297, 311 | -SOH | [120] |
| aaRS (ThrRS) | 6.1.1.3 | Ligase | E. coli | 182 | -SOH | [155] |
| MsrA | 1.8.4.11 | Oxidoreductase | B. taurus | 72 | -SOH | [65] |
| Aldose Reductase | 1.1.1.21 | Oxidoreductase |
R. norvegicus M. musculus |
298, 303 | -SOH and -SSG | [68], [70] |
| SOD1≠ | 1.15.1.1 | Oxidoreductase | H. sapiens | 111/112* | -SOH and -SSG | [58], [44] |
| Prx-1 | 1.11.1.15 | Oxidoreductase | H. sapiens | 52, 83, 173 | -SOH and -SSG | [50], [53] |
| PTP1B | 3.1.3.48 | Phosphatase | H. sapiens | 121, 215 | -SOH and -SSG | [82], [83] |
| SHP-1/2 | 3.1.3.48 | Phosphatase | H. sapiens; M. musculus | 455 459 |
-SOH | [103], [104] |
| PTEN | 3.1.3.48 | Phosphatase | H. sapiens | 124 | -SOH | [103] |
| PTPσ | 3.1.3.48 | Phosphatase | H. sapiens | 1589 | -SOH | [101] |
| A20 | 3.4.19.12 | Protease | H. sapiens | 103 | -SOH | [168] |
| USP1/7 | 3.4.19.12 | Protease | H. sapiens | unknown | -SOH | [169] |
| Cathepsin K | 3.4.22.38 | Protease | H. sapiens | 25 | -SOH | [170] |
| Ulp1 | 3.4.22.68 | Protease | S. cerevisiae | 580 | -SOH | [174] |
| EGFR1≠ | 2.7.10.1 | Receptor Kinase | H. sapiens | 797 | -SOH | [90] |
| VEGFR2 | 2.7.10.1 | Receptor Kinase | H. sapiens; M. musculus | unknown | -SOH | [94] |
| MTAP | 2.4.2.28 | Transferase | H. sapiens | 136 and 223 | -SOH | [137] |
| MST or MPST | 2.8.1.2 | Transferase | R. norvegicus | 247 | -SOH | [139] |
| hBCATm | 2.6.1.42 | Transferase | H. sapiens | 315 | -SOH | [141] |
| PRMT1 | 2.1.1.125 | Transferase | R. norvegicus | 101 and 208 | -SOH | [150] |
| NF-kB (p50) | P19838 | Transcription Factor | H. sapiens | 62 | -SOH and -SSG | [220] |
| RelA (p65) | 2.7.11.10 | Transcription Factor | M. musculus | unknown | -SOH and -SSG | [129] |
Unknown = cases where reversible oxidative effects on protein activity have been measured but the precise cysteine site targeted by oxidation has not been identified. In some cases, the nature of oxidative modification was derived from chemical labeling with selective probes (e.g., pulldown of -SOH proteins labeled with biotin-tagged 1,3-diketo reagents) or other methods, but follow-up studies with site-directed mutagenesis or MS to identify the -SOH site have not been performed.
Currently subject of clinical trials
Figure 1. Sulfenic acid as a central oxidative posttranslational modification at protein Cys sites.
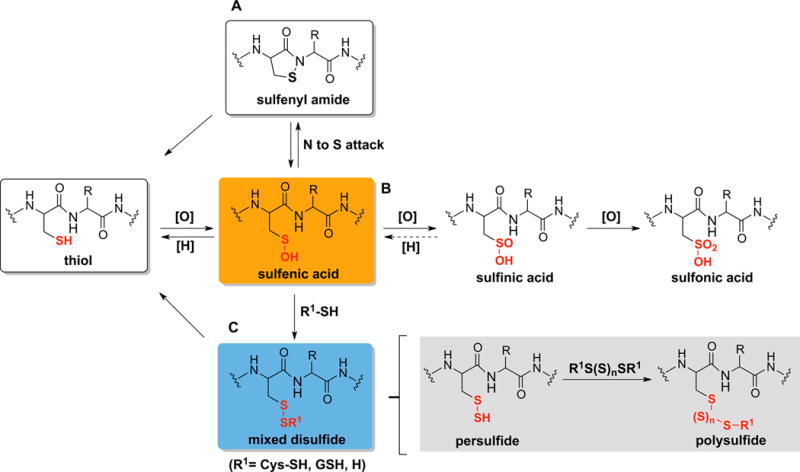
Reaction of -SOH with biological oxidants, reductants or nucleophiles yields various thiol oxoforms including: (A) intramolecular N-amide to -SOH attack to produce sulfenyl amide (-SN), (B) oxidation to sulfinic acid (-SO2H) or sulfonic acid (-SO3H), (C) mixed disulfide formation: S-thiolation (–SSCys or –SSG), S-sulfhydration (-SSH) and S-polysulfination (-S(S)nSR).
1. SYNTHESIS, STABILITY, REACTIVITY AND CHARACTERIZATION OF SULFENIC ACIDS
1.1 SYNTHESIS OF SULFENIC ACIDS
Sulfenic acid (-SOH) is an oxoacid rarely found in organic molecules. A general mechanism by which sulfenic acids are generated in vivo or synthesized chemically is through the one- or two-electron oxidation of a reduced thiol. Under certain conditions, a reduced thiol can interact with ROS/RNS as well as transition metals to produce -SOH. The factors driving the reactivity of thiols towards oxidants have been eloquently reviewed and are not discussed here [1]. In biological systems the pool of oxidants responsible for -SOH formation include hydroxyl radical (HO•) [2], hydrogen peroxide (H2O2) [3], peroxynitrite (ONOO−) [3], hypochlorous acid (HOCl) [4], and hypothiocyanous acid (HOSCN) [5]. Oxygen atom transfer from nitrite to –SH during the reduction of Fe(III)-NO2 complex to Fe(II)-NO can also generate sulfenic acid [6,7].
In addition to these direct mechanisms of oxidation, -SOH species are generated from other oxidized thiol species. For example, Cys thiosulfinate esters can react with low molecular weight thiols such as Cys or GSH to displace the -SCys counterpart and release -SOH [8] (Figure 2A). Another example of –SOH formation is through hydrolysis of a cyclic Cys sulfenate ester intermediate generated during the reaction of nitroxyl (HNO) with a C-terminal Cys [9] (Figure 2B). S100A1 and S100B are two examples of proteins with redox sensitive C-terminal Cys inhibited by oxidation [10].
Figure 2. Formation of -SOH from other cysteine oxoforms.
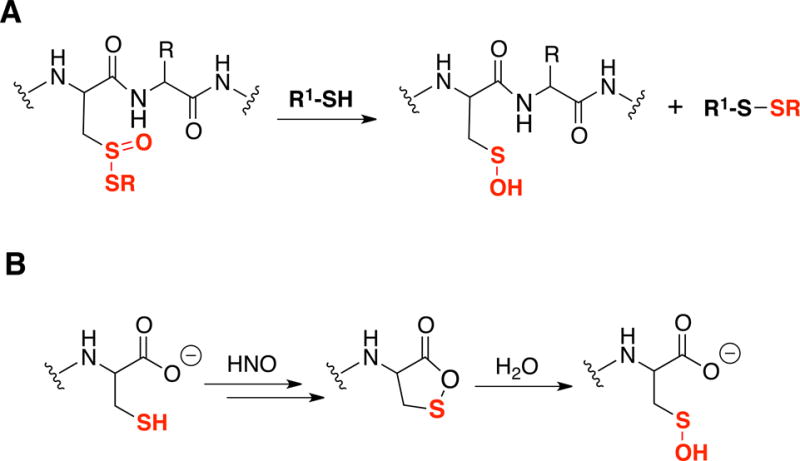
(A) Reaction of thiosulfinates and small molecular thiols; (B) Proposed mechanism for the formation of C-terminal cysteine -SOH.
1.2 STABILITY AND REACTIVITY OF SULFENIC ACIDS
The stability and reactivity of -SOH is controlled by several factors including electronic effects, electrostatic interactions, inductive effects and steric effects. In model compounds of -SOH these properties are derived primarily from the substitution pattern of the α-carbon bearing the sulfur atom (Figure 3), while in proteins these are controlled by the protein microenvironment and the amino acids surrounding the -SOH moiety.
Figure 3. Representative low molecular weight (LMW) sulfenic acid compounds.
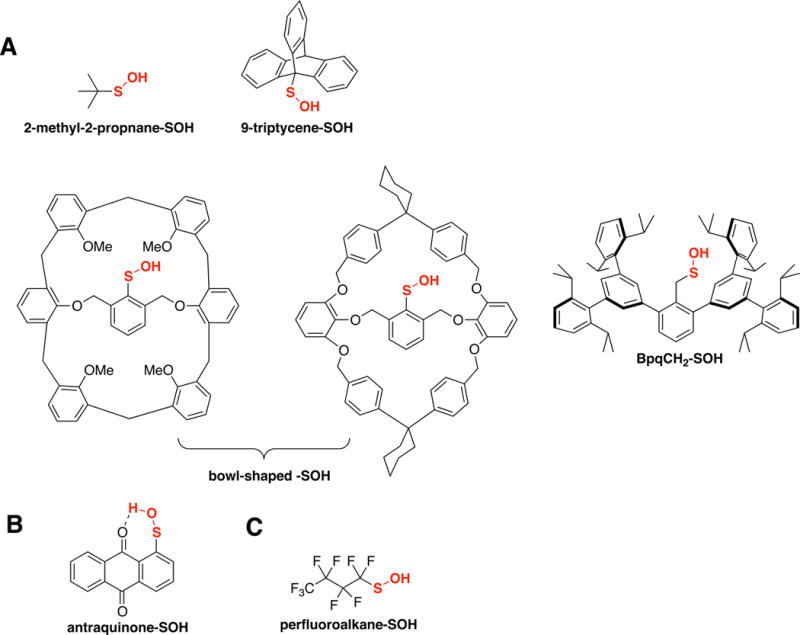
These model compounds are stabilized by various physicochemical factors: sterics (A), hydrogen bonding (B), and electronic (C).
A major issue in development of -SOH model compounds has been the need to achieve a balance between stability (e.g., preventing self-condensation) and reactivity. It was recognized early on that sterically hindered sulfenic acids such as those in bowl shaped structures are less prone to self-condensation, and also disulfide bond formation [11,12], enabling development of a series of -SOH model compounds (e.g., triptycenylsulfenic acid [13] and Bpq-SOH [14]). A classic and widely used -SOH compound is also the Fries acid, an antraquinone-functionalized arenesulfenic acid. In this case, the stability of -SOH is derived from its planar structure, which allows stabilization via six-membered hydrogen bonding interaction. The synthesis of stable primary -SOH compounds was more recently achieved by substituting the protons on the α-carbon with fluoro atoms [15]. In this case, the electron withdrawing property of the perfluoroalkyl chain stabilizes the sulfenyl moiety by reducing its nucleophilicity, and thus limiting self-condensation. Many of these -SOH compounds are sufficiently stable to allow characterization by analytical methods and display reactivity towards amines, thiols and Michael acceptors similar to protein -SOH. Thus, they provide opportunities to interrogate the chemistry of -SOH under conditions mimicking biological settings.
Based on the substitution pattern of known stable -SOH molecules, one could conclude that, similar to nitrosothiols (-SNO), the tertiary -SOH would be more stable than secondary and primary -SOH, if other stabilizing factors were excluded. However, within the protein context –SOH species are connected to a primary carbon in cysteine residues and thus other factors must contribute to the stability of -SOH. In addition to steric and electrostatic interactions mentioned above, these include intrinsic protein motions and dynamic structural changes induced by oxidation. Computational studies, for example, found that the lowest energy conformer of cysteine -SOH harbors four hydrogen-bonding interactions, which were proposed to impact both -SOH formation and stability [16]. Similar to small molecule -SOH, protein -SOH species can exist as stable products of thiol oxidation or can react with other moieties to produce disulfides (e.g., reaction with free or protein thiols, -SSR), thiosulfinate (reaction with other -SOH, -S(=O)SCys), sulfinic or sulfonic states (hyperoxidation, -SO2/3H), and sulfenyl amides (-SN). Sulfenyl amides are formed when the sulfenyl group is attacked by the peptidyl amide, N-terminal amine or side chain amines to produce cyclic sulfenyl amides [17–20]. Similar to small molecule -SOH compounds, the dynamic equilibrium between -SN and -SOH states contributes to the perceived stability of sulfenic acids in aqueous environments.
1.3 ANALYTICAL CHARACTERIZATION OF SULFENIC ACIDS
In general, small molecule –SOH compounds are relatively unstable and only a handful of them (mentioned above) have been fully characterized using analytical methods [13–15] (Figure 3). Although -SOH species display a signature absorption band at 367 nm, the extinction coefficient is relatively low and thus it can only be observed at higher concentrations [21]. As result other analytical methods are typically used to identify and characterize these species including infrared (IR) spectroscopy, 1H and 13C NMR, and mass spectrometry (MS). These methods were reviewed recently, and only a brief overview is provided here [22].
The chemical functional group of -SOH gives stretching bands at ~ 3,200–3,400 cm−1 (S-OH) and 900–1,100 cm−1 (S-O) in IR, which are absent in thiols. The modification of thiolates to sulfenic acids can be monitored by MS and 1H NMR, following the increase in Δmass = +16 m/z and the hydroxyl signal at δ= 2–3 ppm, respectively [13–15]. The use of these analytical techniques is contingent on the type and inherent stability of the -SOH during the course of analysis. In contrast, intact protein -SOH is mainly characterized by MS, as other techniques will require significant sample manipulation and complex data analysis. In particular, electrospray ionization (ESI) has been used as a softer ionization method for monitoring protein -SOH content of a given protein sample. Our group has employed ESI-MS extensively to examine the kinetics of peroxiredoxin hyperoxidation and repair and to test the reactivity and selectivity of chemical probes targeting the -SOH species [23–28]. 13C NMR can also be used to analyze protein -SOH and was applied to identify -SOH formation at Cys42 in the flavoprotein NADH peroxidase from Enterococcus [29]. In this case, the protein was expressed in E. coli using growth media supplemented with [3–13C] Cys. As expected, the signal of the β-carbon appeared to be deshielded for the –SOH and –SO2H oxoforms, when compared with the reduced protein. Sulfenic acids were also identified in several proteins whose structures were solved by X-ray crystallography. However, in many cases their origin is uncertain (e.g., true -SOH formed in solution and preserved during crystallization process or formed in the crystal upon exposure to X-rays) [22].
Because of the caveats associated with direct detection of protein -SOH in particular when applied to complex samples, several alternative methods for monitoring protein –SOH species were developed over the past years (Figure 4). These rely on either the derivatization of -SOH to a stable conjugate with the use of selective chemical probes or tag-switch methods of analysis consisting of a blocking step, selective -SOH reduction to -SH state, and reaction of newly formed thiols with a distinct chemical reagent (reviewed in [30]). Direct chemical derivatization of -SOH is possible by exploiting both the electrophilicity and nucleophilicity of the sulfenyl moiety. Early work using the relatively stable -SOH compounds 2-methyl-2-propanesulfenic acid or 9-triptycenesulfenic acid provided evidence for the nucleophilicity of the sulfenyl group towards carbon electrophiles as they reacted with methyl propiolate, a Michael acceptor [13,31] to generate corresponding sulfoxide-carbon adducts. This property of -SOH forms the basis of their reactivity with NBD-Cl a widely used reagent for the detection and quantification of protein -SOH [32]. In this case, while both the -SH and -SOH react with NBD-Cl, the reaction of -SOH with NBD-Cl produce alkylsulfoxide-7-nitrobenzofurazan, a product with a maximum absorbance (λmax= 347 nm), distinct from the product of NBD-Cl reaction with thiols (λmax= 420 nm). The reported side reaction of NBD-Cl with tyrosines and amines also generates species that do not overlap with the absorption profile of -SOH/NBD-Cl product (λmax= 382 nm and 480 nm, respectively) [32]. In addition, 2-nitro-5-thiobenzoic acid, the active chromogenic species in the TNB assay, is also utilized for the quantification of -SOH by monitoring the decrease in absorbance at 412 nm [21]. Other chemical derivatization strategies exploit the electrophilic character of the -SOH group reacting with nucleophilic entities such as carbon nucleophiles. Early work by Allison et al. demonstrated the use of 1,3-dicarbonyl reagents such as dimedone (5,5-dimethyl-1,3-cyclohexanedione) as nucleophilic traps for the sulfenyl moiety [33]. A series of 1,3-dicarbonyl based derivatives have since been synthesized to enable analysis of sulfenic acids in complex biological samples by fluorescence and immunofluorescence imaging, Western blot and MS. These reagents include the DCP-alkyne and DCP-biotin derivatives [34–37], biotin-cyclopentane-1,3-dione probe BP1 [24,38], linear β-ketoesters [28] and dimedone-based reagents [39–42] such as DYn [43]. These compounds have comparable reactivity towards sulfenic acids and offer specific advantages to fit a variety of experimental setups. Nevertheless, a limitation of these reagents is the slow reaction rates with protein -SOH, which impacts the detection and quantitative analysis of these species in biological settings. For example, a DYn probe (DYn-2) was applied recently for the global analysis of protein -SOH in RKO cells and resulted in the detection of over 1,000 protein sulfenylation sites [44]. Two critical requirements for the success of this analysis were the amount of starting biological material (minimum 30 mg, [45]), and the long incubation time of DYn-2 with cells (5 mM DYn-2, 2 h at 37°C). The addition of DYn-2 to cells likely increased -SOH levels due to covalent inhibition and possibly hyperoxidation of peroxiredoxins resulting in intracellular accumulation of effector ROS species such as H2O2. Thus, despite the numerous milestones achieved using 1,3-diketo nucleophilic probes, there is a clear need for new chemical functionalities to capture protein -SOH under conditions that are compatible with biological and clinical settings. New chemical probes (benzoxaborazoles, boronic acids and strained cycloalkynes) were introduced for labeling protein sulfenic acids [27,46] showing significantly higher (>100-fold) reactivity towards these oxidative species. For example, the strained cycloalkyne BCN-Bio1 had ~300-fold higher reactivity with protein -SOH compared with previous series of reagents. While potential reaction of strained cycloalkynes with -SSH has been revealed recently [47], these can be easily handled by effective blocking of these species as well as at the stage of MS analysis given the difference in the nature of products of BCN-Bio1 reaction with these species. This newly discovered reaction also offers the opportunity for simultaneous quantitative measurement of both -SSH and -SOH species within the same experiment, addressing a limitation with current approaches. The future development of these and other -SOH targeting probes will undoubtedly lead to improved methods for detection and quantitation of protein -SOH species in biological specimens of interest to basic and clinical research.
Figure 4. Chemical derivatization methods to detect protein sulfenylation.
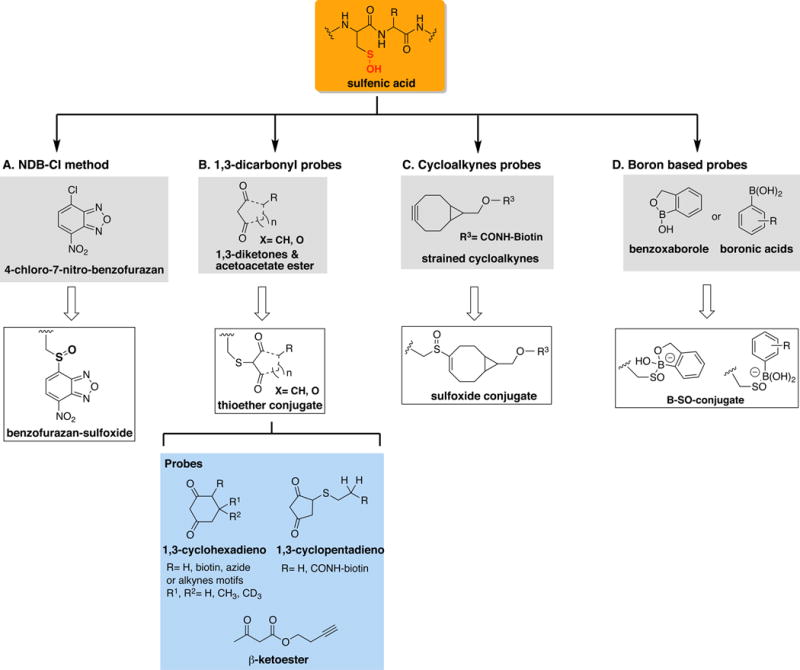
(A) NDB-Cl method gives a spectroscopic detectable signal at 347 nm; (B) 1,3-dicarbonyl probes produce irreversible thioether-conjugate amenable for various analytical detection including mass spectrometry (MS), Western blot and imaging; (C) Cycloalkynes probes produces a detectable sulfoxide conjugate applicable to Western blot and MS techniques; and, (D) Benzoxaboroles and boronic acids generate a covalent adduct detectable in 11B NMR and MS.
2. THIOL-BASED CATALYSIS AND REDOX REGULATION OF ENZYMES INVOLVED IN ROS METABOLISM
Both signaling and metabolism produce intracellular ROS, primarily through two mechanisms involving activation of the membrane-associated NADPH oxidases (NOX enzymes) and mitochondrial electron transport chain (ETC). A connection between these two sources of ROS was demonstrated for platelet-derived growth factor (PDGF) stimulation, which induced upregulation of NOX1 expression in a process dependent on mitochondrial ETC [48]. Stress conditions as result of serum withdrawal also increase mitochondrial ROS, leading to activation of PI3K/AKT, Rac, and NOX1, and ultimately cell death due to sustained production of ROS [49]. Thus, the same pathways connecting mitochondrial and cell membrane processes are responsible for both cell growth and cell death. What determines the outcome is the cellular capacity to reduce ROS and reverse the oxidative processes induced by ROS accumulation. The levels of ROS and their protein oxidized products are tightly controlled in cells through the actions of antioxidant systems, which are often localized to specific subcellular compartments. These include superoxide dismutases (SOD’s), catalase, peroxiredoxins (Prx’s), glutathione peroxidases (Gpx), thioredoxins/thioredoxin reductases (Trx/TrxR), and others (Figure 5). Many of these enzymes contain a reactive cysteine as catalytic center and are themselves susceptible to reversible or irreversible inactivation by oxidation (e.g., Prx, Trx, Grx, Ero1, Erv1). In the case of Prx proteins, the reactive cysteine cycles between reduced thiol and reversible oxidation states such as -SOH and intermolecular disulfide during catalysis. Hyperoxidation of Prx to the catalytically inactive -SO2H state controls localized ROS increase during signaling, as illustrated by the floodgate model [50,51]. The activity of some of the Prx isoforms is further regulated by phosphorylation, acetylation and glutathionylation. In the case of PrxI, EGF or PDGF-induced phosphorylation of Tyr194 as well as Cdc2-mediated phosphorylation at Thr90 decrease its activity, allowing for localized accumulation of ROS to promote growth factor signaling and progression through cell cycle [52–55]. Acetylation at the C-terminal by HDAC6 increases PrxI activity, thus connecting epigenetics to ROS metabolism. Glutathionylation at the non-catalytic Cys83 inhibits the chaperone function of PrxI by promoting the dimer state relative to other higher oligomeric structures that support the chaperone activity of this enzyme [53]. The complex functions of peroxiredoxins regulated by redox and other posttranslational modifications exemplify the critical role of ROS and ROS-induced protein oxidation as unifying regulatory element connecting signaling, metabolism and epigenetics. Additional examples of redox regulated enzymes involved in ROS metabolism and repair of oxidation products are described next. The focus is primarily on enzymes where there is evidence of -SOH formation, but other proteins for which the intermediacy of -SOH is expected are discussed as well.
Figure 5. Mammalian antioxidant defense system.
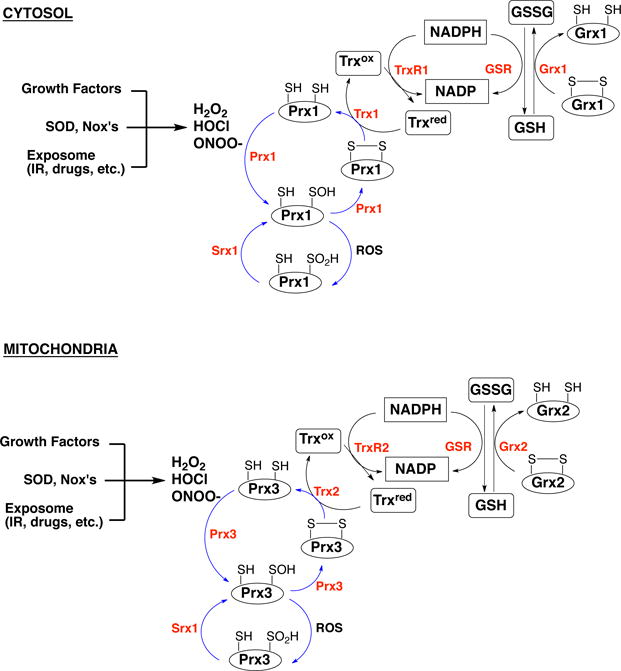
Cytosolic and mitochondrial antioxidant proteins involved in ROS/RNS metabolism. Prx - peroxiredoxins; Srx - sulfiredoxin; Trxox/Trxred - oxidized and reduced thioredoxin; GSSG/GSH - oxidized and reduced glutathione; Grx - glutaredoxin; GSR - glutathione reductase; TrxR - thioredoxin reductase.
Cu,Zn-Superoxide Dismutase (SOD1)
SOD1 is the cytoplasmic isoform of mitochondrial SOD2. SOD1 exists as a homodimer with each subunit containing one copper-binding site, one zinc-binding site, an intramolecular disulfide between Cys57 and Cys146, and two free cysteines Cys6 and Cys111 [56,57]. Mutations in this enzyme induce protein misfolding and aggregation and are associated with the neurodegenerative disease FALS (fetal amyotrophic lateral sclerosis) [58,59]. The oxidative modification of mutant or wild-type SOD1 by glutathionylation or hyperoxidation to -SO2H and -SO3H at Cys111 (Cys112 in human UniprotKB database) further induces destabilization, misfolding and/or aggregation of SOD1 [58–61]. Consistent with these findings, both mutation of Cys111 or overexpression of glutaredoxin-1 (Grx1) protect against SOD1 glutathionylation and improve solubility of both mutant and wild-type SOD1 [62].
Methionine Sulfoxide Reductase (Msr)
Msr catalyzes the reduction of protein methionine sulfoxides (MetSO). There are two classes of Msr enzymes, MsrA and MsrB, which act on the S and R stereoisoforms of MetSO, respectively [63]. Despite lacking sequence or structure homology, these enzymes undergo a similar three-step reaction mechanism: 1) formation of -SOH at the catalytic Cys, 2) formation of an intramolecular disulfide bond with a recycling Cys, and 3) disulfide reduction by the Trx/TrxR system or in vitro by reducing agents like DTT (dithiothreitol) [64,65].
Aldose Reductase (AR)
AR catalyzes the reduction of aldehydes and glutathione conjugates of unsaturated aldehydes derived from lipid peroxidation [66,67]. AR is involved in the antioxidant defense, and its activity is enhanced under oxidative stress conditions. Ischemia or treatment with H2O2 induce a 2–4 fold increase in AR activity through mechanisms involving -SOH formation at Cys298 and Cys303 [68], -SNO [69] or -SG modification [70].
Xanthine Dehydrogenase (XDH)/Xanthine Oxidase (XO)
Xanthine Dehydrogenase (XDH) / Xanthine Oxidase (XO) is an oxidoreductase regulated by changes in the redox environment. Multiple studies have implicated four Cys residues (Cys535, Cys992, Cys1316 and Cys1324) in the reversible XDH/XO conversion, with most data pointing to the disulfide bond formation between Cys535 and Cys992 as leading mechanism for locking the enzyme in the XO form [71,72]. The -SOH modification at these or other cysteines in XDH/XO has not been confirmed thus far.
3. REDOX REGULATION OF SIGNALING
The complex relationship between ROS and cellular signaling has been investigated for decades. While targeted studies have revealed oxidative mechanisms for regulation of protein activity in many kinases, phosphatases, and other signaling proteins, it is through the advancement of omics technologies that we came to appreciate the full breadth of these regulatory modifications in biological systems. The case studies presented in this chapter clearly show the significance of oxidation in regulation of enzymatic activity and highlight the need for considering these effects in the design of inhibitors targeting redox-regulated proteins.
3.1 RECEPTOR TYROSINE KINASES AS INDUCERS AND TARGETS OF ROS
Receptor tyrosine kinases (RTK’s) are transmembrane proteins composed of an N-terminal extracellular domain, a single transmembrane domain and an intracellular domain containing the kinase domain and other C-terminal regulatory elements. The completion of the Human Genome Project and subsequent analysis revealed 58 RTK’s grouped into 20 families [73,74].
Breakthrough articles in 1995 have established for the first time the important function of cell membrane receptors in production of endogenous ROS regulating signaling induced by ligands such as lysophosphatidic acid (LPA) [75] and platelet-derived growth factor (PDGF) [76]. It is widely acknowledged now that many other growth factors and cytokines employ ROS to control signaling events at cell membrane or within specialized cytoplasmic structures such as redoxosomes [77–79].
Localized ROS participate in regulation of RTK’s and their downstream signaling by: 1) direct oxidative modification of the receptor itself (e.g., EGFR, c-RET) [80,81], 2) inhibition of proteins acting as negative regulators of receptor activity (e.g., PTP1B) [82,83], 3) activation of intracellular proteins acting on RTK’s (e.g., PKC, Src) [84], and 4) activation or inhibition of proteins involved in downstream signaling (e.g., STAT3) [85]. These effects occur through the direct action of ROS on protein thiols or through relay mechanisms (e.g., Prx2/STAT3) [85]. Other cell membrane-localized receptors such as G-protein coupled proteins [86,87], integrins [88] and ion channels [89] are similarly regulated by ROS. Here we focus on several examples of signaling proteins to illustrate the broad variety of mechanisms involved in their regulation by ROS.
Epidermal Growth Factor Receptor (EGFR)
EGFR is one of the four members of the RTK family, which also contains ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4 proteins [80,90]. EGFR is composed of an N-terminal extracellular domain (~620 amino acids), a 23-amino acid transmembrane domain, a tyrosine kinase domain (amino acids 685–952), and a C-terminal tail (amino acids 953–1186) within which the tyrosine autophosphorylation sites are located [91]. Cysteines and their oxidation products play critical roles in the structural organization of EGFR and its kinase function. Cys rich modules are present both in the EGFR extracellular domain (CR1 and CR2) and in the ligands binding to the extracellular domain of EGFR. For example, EGF contains six cyteines forming three disulfide bonds (Cys60-Cys20, Cys14-Cys31, and Cys33-Cys42). Nevertheless, the crystal structure of the EGFR extracellular domain in complex with EGF shows that the pattern of disulfides in either EGFR or EGF is not changed upon binding of EGF to EGFR [92]. Early studies focusing on development of EGFR inhibitors have also identified a reactive cysteine site (Cys797) in the EGFR kinase domain. This site is located in the hinge region of the ATP binding pocket and was shown to undergo oxidation to -SOH [90], -SO2H [93], or generate a glutathionylated adduct (-SSG) in the presence of oxidized glutathione (GSSG) [93]. While oxidation of Cys797 to -SOH was described as activating [90], the other oxidative modifications were shown to have no effect on catalysis but impact the potency of inhibitors targeting the ATP binding pocket of EGFR [93].
Vascular Endothelial Growth Factor Receptor 2 (VEGFR2)
VEGFR2, and other endothelium-derived proteins are also oxidatively modified upon stimulation with VEGF. VEGF as well as H2O2 treatment of HUVEC cells induced -SOH formation in VEGFR2, ERK1/2, actin, PTP1B, and IQGAP1 [94]. These findings were validated in vivo using a mouse model of hindlimb ischemia [94]. In this study, imaging with –SOH chemical probes showed localized –SOH formation during endothelial cell migration.
Fibroblast Growth Factor Receptor 1 (FGFR1) and c-RET
FGFR1 contains a redox sensitive Cys residue, which is located in a Gly loop conserved in several families of protein tyrosine kinases. It has been shown that oxidation of FGFR1 at Cys488 inactivates its kinase activity and this is restored in the presence of DTT. The oxidative inactivation of FGFR1 was prevented by mutation of Cys488 to Ala [95]. Studies have also demonstrated the activation of c-RET activity by UV through mechanisms involving oxidation of Cys376 in the kinase domain [81].
3.2 REDOX REGULATED TYROSINE PHOSPHATASES
The regulation of protein tyrosine phosphatases by oxidation has been expertly reviewed in the literature [96]. The phosphatase activity of these proteins relies on a catalytic Cys residue, which is inhibited by oxidation under both physiological and pathological conditions [77]. Initial studies with various PTP’s, such as PTP1B, demonstrated Cys215 -SOH formation upon stimulation with growth factors or exogenous administration of H2O2 [82,97]. Subsequent reports showed PTP1B glutathionylation at Cys215 in the presence of diamide/GSH or GSSG [83]. Structural studies of oxidized PTP1B have identified an additional mechanism by which the enzyme protects itself from irreversible oxidation at Cys215 [17,19]. The nucleophilic addition of the protein backbone N-amide to the cysteine sulfenyl group produces a cyclic five-membered ring sulfenyl amide, a reaction mechanism also supported by chemical models [20]. The cyclic sulfenyl amide can regenerate active Cys215 (-SH) by hydrolysis to –SOH, reaction of -SOH with GSH or other thiols to form mixed disulfides, which are then reduced to generate Cys215 (-SH) and GSH. PTP1B regulation through oxidation at Cys215 has been linked to cancer, diabetes and cardiovascular conditions [98]. Because of the crosstalk between PTPs and kinases in regulation of diverse biological processes, studies have also emerged in understanding the nature of endogenous oxidants responsible for the inactivation of this class of enzymes. For instance, relatively stable H2O2 inactivates PTP1B at ~1,000-fold slower rates than peroxymonophosphate (PMP), when the reactions were performed under similar conditions [99,100]. There is the question of whether PMP is actually formed in vivo as potential metabolizing enzymes such as catalase were found ineffective towards this reactive species.
In addition to PTP1B, a number of other phosphatases undergo oxidation to -SOH or other oxidized states and with similar consequence on enzyme activity. These include the protein tyrosine phosphatase sigma [101], SHP1/2 (Src homology region 2 domain-containing phosphatase; also a tyrosine phosphatase) [102–104], and PTEN (phosphatase and tensin homolog; structurally resembles dual specificity protein tyrosine phosphatases) [103].
3.3 REDOX REGULATION OF OTHER SIGNALING KINASES AND ADAPTOR PROTEINS
Tyr KINASES
Src and c-Abl
Src is a non-receptor kinase with critical function in growth factor and integrin signaling. The redox regulation of Src activity is still under debate with some studies demonstrating inhibition of Src activity by H2O2 [95,105], while others showing the contrary, activation of Src by oxidation [106]. Individual mutations of all nine Cys residues in Src to Ala identified Cys277 as the redox sensitive site. Follow-up studies confirmed the participation of Cys27 in intermolecular disulfide formation with another Cys277 leading to homodimer formation and loss of kinase activity [95]. Other studies have proposed activation of Src through intramolecular disulfide bond formation between Cys245 and Cys487 induced by H2O2 [106], and possibly by nitric oxide [107] and peroxynitrite [108], though the NO and ONOO− mechanisms of Src activation have not been fully elucidated. In a recent study, increased Src sulfenylation and activity were detected and proposed to contribute to cartilage degradation in osteoarthritis [109]. Thus, it is likely that both mechanisms exist and come into play depending on stimuli, timing, and presence of other co-regulatory events. c-Abl is a member of Src-family of tyrosine kinases involved in cell division, adhesion, apoptosis and response to oxidative stress [110]. In addition to the kinase domain, c-Abl contains two small autoinhibitory domains, and additional DNA- and actin-binding domains located in the C-terminal region. Substrate binding to c-Abl releases the autoinhibition and activates the kinase function. Interestingly, one of these c-Abl substrates is catalase, an enzyme involved in H2O2 metabolism. c-Abl phosphorylates catalase at Tyr231 and Tyr386 enhancing its activity [111]. Both ionizing radiation and addition of extracellular H2O2 increase c-Abl activity which is critical to the initiation of pathways involved in the DNA damage response [112,113]. However, glutathionylation of recombinant c-Abl was shown to inhibit the kinase activity, a process which was reversed by addition of glutaredoxin [114]. Clearly, both Src and c-Abl are subject to diverse layers of redox regulation which would need to be clarified in future studies.
Janus Kinase 2 (JAK2)
JAK2 tyrosine kinase activity is regulated by four Cys residues, Cys866, Cys917, Cys1094, Cys1105, located in the catalytic domain [115]. Out of these Cys residues, Cys866 and Cys917 act as a redox sensitive switch and mutation of these residues was shown to decrease the autocatalytic kinase activity compared with the wild type JAK2 [116]. The current proposed mechanism involves a -SOH intermediate, which then triggers the intramolecular disulfide bond formation inducing inactivation of JAK2. However, the formation of -SOH at these residues has not been demonstrated.
Ser/Thr KINASES
Protein Kinase B (Akt)
Akt is a family of three isoforms (Akt1, Akt2 and Akt3) with diverse biological functions despite the high level of conserved amino acid sequence [117–119]. These enzymes belong to the serine/threonine kinase family and are major signaling hubs in cytokine, growth factor, and integrin pathways. Redox proteomics studies have identified increased oxidation of Akt2 in NIH3T3 cells stimulated with PDGF or H2O2 [120]. These conditions caused activation of all isoforms through phosphorylation, but resulted in inhibition of only Akt2 kinase activity by ~66%, which was found to be reversible in the presence of reductant or upon removal of ROS-inducing stimuli (e.g., PDGF). MS analysis of oxidized Akt2 with and without chemical labeling of -SOH identified critical redox sensitive cysteines and a number of intramolecular disulfides formed during Akt2 oxidation. Oxidation to -SOH was identified at Cys124, a Cys residue unique to Akt2 and located in the linker region between the N-terminal pleckstrin homology domain and the catalytic kinase domain, and at Cys297 and Cys311, both located in the kinase domain and conserved in all three Akt isoforms. Disulfide mapping revealed that Cys124-Cys297 and Cys124-Cys311 are potential disulfides controlling H2O2-induced inhibition of Akt2. These results are consistent with decreased sensitivity to oxidation in the Cys124Ser, Cys297Ser, and Cys311Ser Akt2 mutants. Furthermore, the oxidative inactivation of Akt2 was dependent on cell type and stimulus employed. For example, while insulin treatment in myoblasts induced oxidation of Akt2, this was not observed in insulin-treated C2C12 myotubes, the differentiated state of myoblasts. These results are consistent with the Akt2 function in glucose uptake [121–124] and with the known insulin resistant phenotype of myoblasts [125,126]. Akt2 is an important target for drug design given its potential functions in pathologies associated with oxidative stress, impairment of glucose uptake (type 2 diabetes-like phenotype), and cancer [127].
Inhibitory κβ Kinase α (IKKα)
IL-17A stimulation induces NF-κB activation pathways and expression of proinflammatory mediators [128]. IL-17A treated C10 mouse alveolar epithelial cells or primary mouse tracheal epithelial cells display an increase in the formation of -SOH with concominant glutathionylation of proteins [129]. Further analysis of the proteins associated with the NF-κB pathway revealed Grx1 controlled glutathionylation of RelA (p65) and IKKα after treatment. Additional studies are needed to identify the source of ROS and the Cys sites of sulfenylation [129].
ADAPTOR PROTEINS
B-Cell Lymphoma-2 (Bcl-2)
Bcl-2 is an oxidative stress-response protein and a key regulator of apoptosis [130]. A study on the effect of H2O2 on Bcl-2 in human lung epithelial cells showed that exposure to H2O2 induces oxidation of Cys158 and Cys229 resulting in downregulation of Bcl-2 and apoptosis [131]. The effects of oxidation were reversible with antioxidant treatment and chemical labeling demonstrated the formation of -SOH at these sites.
Protein Deglycase (PARK7/DJ-1)
DJ-1 is an oncogenic protein that binds to the DNA-binding domain of p53, in a redox regulated process. The oxidative modification of Cys106 in DJ-1 to the -SOH, -SO2H, or -SO3H states increases the binding of DJ-1 to p53, while reducing conditions or mutation of Cys106 decrease this interaction [132–135]. The binding of DJ-1 to p53 was independent of the phosphorylation status of p53, however other modifications such as acetylation at lysine residues in p53 were found necessary to enhance DJ-1 binding to this protein.
4. REDOX REGULATION OF METABOLISM
In addition to the enzymes involved in ROS metabolism, a number of other metabolic enzymes are redox-regulated through -SOH - based mechanisms. These are discussed below and organized based on enzyme classification.
TRANFERASES
Methylthioadenosine Phosphorylase (MTAP)
Several transferase enzymes are targets of Cys oxidation, including sulfenylation. MTAP catalyzes the degradation of 5′-methylthioadenosine (MTA) to adenine and 5′-methylthioribose-1-phosphate (MTRP) [136] and undergoes oxidative inactivation by ROS [137]. Site-directed mutagenesis studies revealed two Cys residues, Cys136 and Cys223, to be essential for the decreased enzymatic conversion of MTA to MTAP with oxidation. Treatment of MTAP enzyme with H2O2 and subsequently with NBD-Cl confirmed the presence of -SOH at both residues. MS analysis also identified a disulfide bridge between Cys145 and Cys211; however, this oxidized form did not prove to be responsible for the oxidative inactivation of MTAP [137].
Methylpyruvate Sulfurtransferase (MST or MPST)
MST is a redox-regulated enzyme involved in the transulfuration pathway of cysteine metabolism [138]. Treatment with H2O2 inhibits the activity of this enzyme by inducing –SOH formation at Cys247 [139]. The –SOH formation at Cys247 was confirmed by MS, chemical labeling and site-directed mutagenesis. The sulfurtransferase activity of MST is effectively restored in the presence of reducing agents such as DTT or the Trx/TrxR system. However, under excess of oxidizing agent the inhibition of MST is irreversible due to the formation of higher oxidized species, in this case -SO3H.
Mitochondrial Branched Chain Aminotransferase (hBCATm)
Human BCATm (hBCATm) is a pyridoxal phosphate-dependent enzyme catalyzing the transfer of an amino group to non-polar amino acids such as leucine, isoleucine and valine [140]. Both thiol blocking agents and H2O2 were shown to inactivate hBCATm by acting on Cys315 and/or Cys318 [141,142]. These two functional Cys residues are proximal to the active site and form an intramolecular disulfide bridge under oxidizing conditions. Site-directed mutagenesis, chemical labeling and MS have established Cys315 as the reactive Cys site undergoing oxidation to –SOH and driving disulfide bond formation with the resolving Cys318 [141].
Methionine Adenosyltransferase (MAT)
MAT is a metabolic liver enzyme responsible for the catabolism of methionine in mammals and is regulated by H2O2 [143]. Both MAT I (homotetramer) and MAT III (homodimer) isoforms were found susceptible to H2O2-induced inactivation, presumably through reversible oxidative modification of Cys, as the activity of each isoform was recovered by GSH treatment. MAT III activity was fully reversed by physiological levels of GSH. In contrast, MAT I required GSH concentrations above physiological levels for full reactivation, suggesting that other reducing systems may be responsible for maintaining this protein in active state. Site-directed mutagenesis of ten Cys residues located in the α-subunit of the recombinant rat liver MAT identified Cys121 as the residue controlling H2O2-induced inactivation and potential site for -SOH formation [144].
Protein Arginine Methyltransferase 1 (PRMT1)
PRMTs are a family of nine enzymes responsible for methylation of proteins at arginine residues. Given their critical function in histone methylation, deregulation of these enzymes is associated with numerous diseases including cardiovascular and neurological conditions and cancer [145–147]. The human isoform PRMT1 is the major source of asymmetric dimethylarginine (ADMA) and its activity is inhibited by oxidation [148,149]. Site-directed mutagenesis, chemical labeling with -SOH directed probes, and MS analysis revealed -SOH formation at Cys101 and Cys208 correlating with decreased activity of PRMT1 [150]. The mechanisms by which these oxidation events impact the activity of PRMT1 and whether other PRMT isoforms are similarly redox regulated, given that these sites are conserved in all PRMTs, remain to be determined.
Other proteins with putative -SOH - based mechanisms of regulation
Pyruvate kinase M2 (PKM2) and transketolase (TK2) are two other transferases regulated by oxidation. Though the Cys sites responsible for redox sensitivity have been identified in both cases (Cys358 in PKM2 [151], Cys189 and Cys264 in TK2 [152,153]), it is currently unknown if these involve -SOH formation in vivo.
LIGASES
Aminoacyl-tRNA Synthetases (aaRS)
aaRS are enzymes pairing amino acids and tRNA during protein synthesis. These enzymes are prone to misactivation of amino acids that are structurally similar to the targeted amino acid. For example, threonyl-tRNA synthetase (ThrRS) misactivates Ser and then uses a Cys as editing site to hydrolyze Ser-tRNA to allow for the incorporation of the correct amino acid at Thr codons [154]. Inability to correct for misincorporation of Ser at Thr sites causes protein misfolding, leading to detrimental consequences in all species from bacteria to humans [86]. In E. coli, oxidative stress impairs the editing site of ThrRS by posttranslational oxidation of the conserved Cys182 [155–157] to -SOH. The ThrRS -SOH is formed under various oxidative conditions including air, H2O2 or HOCl and results in Ser misincorporation at Thr codons [158]. Sulfenic acid modifications in other aaRS such as glutamyl-tRNA synthetase, and methionyl-tRNA synthetase were also identified in HeLa cells using chemical labeling and MS analysis [39].
In this class of enzymes, carbamoyl phosphate synthetase (CPS) also showed inhibition of activity upon treatment with H2O2 [159,160]. CPS forms an intramolecular disulfide bond between Cys1327 and Cys1337, which has been proposed to occur through the intermediacy of -SOH.
ISOMERASES
Peptidyl-Prolyl Isomerase (Pin1)
Pin1 catalyzes the cis-trans interconversion of the phosphoSer/phosphoThr-Pro peptide bond, inducing conformational changes in cellular proteins [161]. This enzyme has neuroprotective properties, and patients with mild cognitive impairment show increased oxidation and decreased activity of Pin1 [162]. X-ray crystallography studies of recombinant Pin1 incubated with H2O2, revealed –SOH formation at Cys113 located in the active site of the enzyme [163]. Higher concentrations of H2O2 as well as prolonged exposures generated –SO2H and –SO3H oxidized proteins. Activity assays performed in the presence of H2O2, reaction with 4,4′‐dithiodipyrimidine (DTDP) and site-directed mutagenesis (by substituting Cys113 to Asp) showed reduced activity of Pin1, indicating that oxidation at Cys113 causes the inactivation of protein. Treatment of oxidized protein with DTT recovered approximately 40% of the activity, suggesting that 60% of the protein was in the hyperoxidized -SO2/3H state. Cumulatively these and other studies (e.g., [164]) underline the significance of Pin1 oxidation in neurological disorders and presumably other diseases as well.
CYSTEINE PROTEASES
Deubiquitinases (DUB)
Deubiquitinases hydrolyze ubiquitin modifications regulating proteostasis and activity of many signaling, metabolic and epigenetic proteins. There are five DUB families separated into cysteine protease and metalloprotease classes of enzymes [165]. To date, only cysteine-based DUBs are known to be regulated by oxidation [166]. One of these cysteine-based DUBs is A20, a tumor suppressor involved in regulation of NF-κB signaling, which belongs to the family of ovarian tumor deubiquitinases (OTUs) [167]. X-ray crystallography, chemical labeling, and functional studies identified protein sulfenylation at Cys103 in the catalytic domain of A20. Protein -SOH formation was also found in other OTUs such as OTUD7B (at Cys194), OTUB1, OTUD1, OTUD2, OTUD3, OTUD5 and OTUD6A [168].
The ubiquitin-specific protease USP1 (also a cysteine-based DUB) is reversibly inactivated upon oxidative stress in vivo and is associated with monoubiquitination-dependent DNA damage response during S-phase of the cell cycle. The oxidation status of USP1 was examined in U-2 OS cells by chemical labeling with -SOH probes, Western blot analysis and activity assays. These studies demonstrated -SOH formation and decreased activity in the presence of H2O2. Similarly, the USP7 isoform was reversibly inactivated by oxidation. Though the reversible –SOH modification site was not identified, MS analysis showed evidence of –SO3H formation at the catalytic cysteine site, pointing to increased sensitivity to hyperoxidation in USP7 and perhaps a more transient –SOH state [169].
Cathepsin K
Cathepsin K is a redox sensitive protease containing a catalytic Cys25 residue that is susceptible to oxidation by hydroperoxides and nitric oxide donors [170]. Treatment of recombinant protein with H2O2 induces time and concentration dependent inhibition of enzyme activity, which was partly (~30%) reversed by reduction with DTT. Further analysis using NDB-Cl showed approximately 35% -SOH content, consistent with the proportion of activity recovered by DTT treatment. The biological function of the –SOH and hyperoxidized forms of this enzyme in cells remains to be determined, but could serve as mechanism of protection against oxidative stress under inflammatory conditions [171].
Other proteases with putative -SOH - based mechanisms of regulation
Other redox-regulated proteases include caspase 3 and SENP1. Glutathionylation [172] and nitrosylation [173] studies confirmed Cys135 in caspase 3 as the target site of oxidation. Although sulfenylation is a possible intermediate in the path to glutathionylation, -SOH formation was not experimentally confirmed for this enzyme. In human SENP1, oxidation induced the formation of an intermolecular disulfide bond between Cys603 located in the active site, and Cys613 (a unique residue of SENP1) both in vivo and in vitro [174]. X-ray crystallography studies of Ulp1, the yeast homolog of SENP1, showed –SOH, -SO2H, and –SO3H modification at the catalytic Cys580 [174] suggesting potential –SOH formation in SENP1 as well.
HYDROLASES
Monoacylglycerol Lipase (MGL)
MGL is a serine hydrolase, and its function in brain is to inactivate the endocannabinoid neurotransmitter 2-arachidonoyl-sn-glycerol (2-AG) by hydrolyzing it into arachidonic acid and glycerol [175]. The catalytic site of MGL contains two regulatory Cys residues, Cys201 and Cys208, which are targeted by thiol-reacting MGL inhibitors. A third cysteine (Cys242) located within the active site in close proximity to the catalytic nucleophile Ser122 directly influences catalysis [176–178]. A recent study reported that inactivation of MGL by sulfenylation at Cys201 and Cys208 causes accumulation of 2-AG, and an increase in 2-AG-mediated endocannabinoid signaling in neurons [179]. In this study, the –SOH formation at Cys201 and 208 was demonstrated by multiple assays involving site-directed mutagenesis, chemical labeling and MS analysis.
S-Formylglutathione Hydrolase (SFGH)
In yeast, S-formylglutathione is converted enzymatically to GSH and formate by the action of SFGH, a homolog of human esterase D [180,181]. X-ray crystallography analysis [182], NBD-Cl treatment and site-directed mutagenesis [183] demonstrated -SOH formation at Cys60 (human Cys56 [180]), located in close proximity to the catalytic site. Activity assays linked the oxidative modification at Cys60 with inhibition of enzyme activity [183]. Interestingly, the oxidative inactivation of SFGH was found to be dependent on a closely located His160 residue which acts as hydrogen bond donor during the oxidative inactivation of the protein [182,183].
DEHYDROGENASES
Dihydrolipoamide Dehydrogenase (DLDH)
DLDH catalyzes the oxidation of dihydrolipoamide using NAD+ as electron acceptor [184]. Chemical labeling of DLDH with –SOH probes linked the increase in sulfenylation with loss in enzymatic activity. Attempts to determine the sites of sulfenylation were unsuccessful and only hyperoxidized Cys449 and Cys277 were identified, which ultimately were found not to be responsible for the inhibition of activity. It was suggested that one of the two cysteines (Cys45 or Cys50) in the active site could be the target of oxidation resulting in DLDH inactivation; however, this was not confirmed experimentally [185].
Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
The dehydrogenase activity of GAPDH was shown decades ago to be controlled by oxidation at reactive Cys sites [186]. Sulfenic acid formation in GAPDH turns off dehydrogenase activity and turns on the acylphosphatase activity [33]. Treatment of the -SOH form of GAPDH with carbon nucleophiles such as alkenes and dimedone inhibits both dehydrogenase and acylphosphatase activities [33]. Multiple studies including sequencing analysis of 14C-dimedone labeled protein digest [33], Western blot analysis with an anti-dimedone antibody [187], X-ray crystallography [188,189], and others [190–193] support the formation of -SOH at the catalytic Cys152 site.
PROTEINS INVOLVED IN METAL ION SENSING, BUFFERING AND TRANSPORT
Recoverin
Recoverin is a Ca2+ sensor protein posttranslationally modified by N-terminal myristoylation [194]. Mild oxidative environments regulate the activity of recoverin by acting on a conserved Cys residue (Cys39 in the human homolog) [195–197]. Experiments using site-directed mutagenesis of Cys39 to Asp (-SO2H mimetic) showed that hyperoxidation of myristoylated protein did not change the affinity for Ca2+ but drastically decreased the binding of recoverin to photoreceptor cell membranes [196]. On the other hand, the Cys39Ala mutant increased the cooperativity of Ca2+ binding to non-myristoylated protein [197]. Structural analysis further identified –SOH modification at Cys39 [197]. Thus, the oxidative modification of this protein controls its Ca2+ binding properties, intracellular location and potentially the interaction with other proteins involved in the visual cycle of photoreceptor cells.
Transient Receptor Potential Channel A1 (TRPA-1)
Redox regulation of TRPA-1 was found to produce vasodilation effects acting in the release of calcitocin-gene related peptide (CGRP) [198]. Recent studies demonstrated activation of TRPA-1 in a mouse model of streptozotocin (STZ)-induced diabetes, which is characterized by an increase in peroxynitrite [199]. Analysis of a custom made N-terminal peptide of human TRPA-1 containing six essential Cys residues (Cys621, Cys641, Cys665 and other three nearby Cys residues) using –SOH chemical probes confirmed the formation of this species in TRPA-1. Additional MS analysis of the modified peptide demonstrated formation of two possible disulfides bridges, involving Cys633 and Cys641, and Cys641 and Cys651. Other functional studies using triple (Cys621Ser, Cys642Ser and Cys665Ser) and quadruple (Cys621Ser, Cys642Ser, Cys665Ser and Lys710Arg) mutants showed that modification at both Cys and Lys residues (modified as Nε-(carboxymethyl)lysine) are responsible for the activation of TRPA1. Although the STZ-induced activation of TRPA-1 is increased by modifications at lysine residues, oxidation is indeed regulatory, as experiments performed in HEK-293T cells transfected with hTRPA-1 showed that the STZ-induced activation of TRPA1 can be significantly decreased by reducing agents.
Calbindin D28K
The human calbindin D28K protein serves as both Ca2+ buffer [200] and sensor [201] and contains five cysteine residues that are regulated by changes in cellular redox environment [202–204]. Protein incubation with GSH under aerobic conditions resulted in glutathionylation at Cys187, Cys219 and Cys257 [203]. MS studies also found Cys257 as -SO2H, which may imply a sulfenic acid intermediate leading to hyperoxidation or glutathionylation [203]. Cys100 was also found to form an intramolecular disulfide with Cys94 leading to decreased Ca2+ affinity [203,204]. The redox regulation of calbindin D28K could have implications in the modulation of cellular apoptosis by controlling the activity of IMPase [205] and caspase 3, among others [206].
Vascular Potassium-ATP Channels (KATP channels)
The voltage-gated potassium channel Kv1.5 was found to undergo oxidation to –SOH at Cys581, a modification linked to downregulation of the channel [207]. Other potassium channels are also known to be inhibited by glutathionylation at reactive Cys, including Kir6.1 at Cys 176 [208], the Kv4 channel at Cys13 [209], and Kir5.1 at Cys158 [210]. However, oxidation to -SOH has not been confirmed in these cases.
5. REDOX REGULATION OF EPIGENETICS
The connection between the central redox metabolism (e.g., mitochondrial ETC) and epigenetic modulation in in many diseases, but cancer in particular, is now well-established [211]. Many epigenetic enzymes are directly regulated by oxidative modifications or use substrates (e.g., S-adenosyl methionine - a methyl donor; acetyl coenzyme A - an acetyl donor) that are products of redox-regulated metabolic pathways. A few redox-regulated enzymes with epigenetic function are discussed next.
Apurinic/Apyrimidinic Endonuclease (APE1)
APE1 is a DNA repair enzyme that is also known as redox effector factor 1 [212]. APE1 displays its redox effector function by reducing disulfides or -SOH in target proteins to promote DNA binding and transcriptional activities and is inhibited by glutathionylation [213]. Site-directed mutagenesis of Cys residues within the redox regulatory portion of the protein (Cys65Ala, Cys93Ala and Cys99Ala) revealed that mutants containing Cys99Ala were not affected by oxidation, indicating that the oxidative inactivation of APE1 activity requires Cys99. The redox active Cys99 site was further confirmed by MS analysis, which identified glutathionylation with GSH/diamide treatment and –SOH, -SO2H and disulfides with H2O2 treatment [214,215].
Transcription Factors
A classic example of redox-regulated transcription factors is the NF-κB/Rel family [216,217]. Early studies have shown that the p50 subunit of NF-κB can undergo nitrosylation [218] and glutathionylation [219] at the Cys62 residue. Cys62 is part of the DNA binding domain, and its modification inhibits the DNA-binding activity of p50 [218,220]. Chemical labeling with dimedone followed by MS analysis also revealed the formation of -SOH at Cys62 [220].
DNA Methyltransferases (DNMT)
Changes in the cellular redox status are implicated in the regulation of epigenetic processes contributing to the development and progression of cancer [221–223]. One such epigenetic modification is DNA methylation, which is tightly regulated by the action of DNA methyltransferases enzymes such as DNMT1, DNMT3A and DNMT3B [224]. DNMT3A and DNMT3B but not DNMT1 are found to convert DNA modified 5-hydroxymethyl cytosine (5-hmC) to cytosine (C) in the presence of H2O2, and this dehydromethylase activity can be reversed in the presence of reducing agents, suggesting that DNA methylation is controlled by the oxidative state of the DNA methyltransferases [225]. Though the -SOH formation in these proteins has not been confirmed, DNMTs have a reactive catalytic Cys, which could be susceptible to oxidation.
6. CONCLUDING REMARKS AND PERSPECTIVES
The large body of literature that was partly reviewed here provides clear evidence for the significance of redox regulation in cellular signaling, metabolism, and epigenetics impacting the initiation, development and progression of many pathologies ranging from cardiovascular and neurological diseases, to diabetes, aging and cancer. With several of the redox regulated proteins being investigated in clinical trials for treatment of various diseases (Table 1), the consideration of specific oxidative modifications is of outmost importance to improve the efficacy of therapies targeting these proteins. This is a highly complex task that requires a holistic approach based on comprehensive understanding of the redox microenvironment and proteins’ oxidative state at the time of treatment along with considerations of how these might change during the course of treatment due to the effects of targeted drug itself and/or concurrent redox-altering treatments (e.g., ionizing radiation, chemotherapies).
It is evident from the examples highlighted in this review that, to a significant extent, most of the available knowledge in this field has been supported and disclosed by combined utilization of chemical technologies, classic molecular biology tools (e.g., site-directed mutagenesis), and advanced analytical instrumentation. These have contributed to the discovery and elucidation of mechanisms driving oxidative activation or inactivation of target proteins from variable sample sourcing including intact proteins, cells and tissues. While classical chemical probes like those containing the 1,3-diketo functionality (e.g., dimedone-like) and tag-switch assays have proven valuable for qualitative and semi-quantitative identification of protein sulfenic acids, significant work remains to achieve comprehensive quantitative measurement of oxidative protein modifications. The development of new -SOH trapping agents with enhanced cellular uptake, targeted subcellular localization, increased reaction kinetics and compatibility with a variety of downstream applications is ongoing and expected to further advance the field of thiol redox regulation. Also, it is clear from the examples included here that multiple oxidative modifications could exist at the same Cys site depending on the availability of ROS, RNS, or RSS effectors. However, with few exceptions [226–228], most of current workflows focus on the identification of single oxidative species (e.g., -SOH, -SNO, -SSG, or protein disulfides, etc). More studies are needed to address the site occupancy of various oxidative modifications in a given biological context and identify the key drivers of protein function. It is possible that within the cellular milieu, a particular microenvironment (e.g., that of protein within the cell or of reactive Cys within the protein) may selectively lead to formation of a single or few predominant oxidative species. However, it is also possible that multiple oxidative modifications are employed as a means to diversify the function and subcellular localization of redox-regulated proteins. Integration of experimental and computational approaches is further needed to identify structural motifs that allow prediction of –SOH (or other oxidative states of interest) formation and stability [229]. Ultimately, such computational strategies should enable classification of sulfenic acid species in categories depending on site occupancy and stability, which can have significant impact in predicting Cys residues that may serve as redox sensors or potential handle for attachment of covalent inhibitors.
In conclusion, redox regulation of protein activity is a critical component of the complex machinery driving cell function under both physiological and pathological conditions. Deciphering the timing of oxidative modification, location at protein and cellular level, abundance relative to unmodified protein or other oxidized species, nature, stability and functional consequence of oxidative modifications will continue to remain a significant focus for the foreseeable future.
Acknowledgments
Financial support was provided by the National Institutes of Health under award number R01 CA136810 and R33 CA177461 (IMAT) to CMF, Wake Forest Postdoctoral Program in Translational Radiation Oncology (T32 CA113267 to NODB), and PRIME-IRACDA postdoctoral fellowship (K12 GM102773 to EISL).
CITED REFERENCES
- 1.Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24(4):434–50. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 2.Meng FG, Zhang ZY. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta. 2013;1834(1):464–469. doi: 10.1016/j.bbapap.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42(33):9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 4.Nagy P, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the oxidation of cysteine by hypohalous acid to give cysteine sulfenic acid. J Am Chem Soc. 2007;129(45):14082–14091. doi: 10.1021/ja0737218. [DOI] [PubMed] [Google Scholar]
- 5.Barrett TJ, Pattison DI, Leonard SE, Carroll KS, Davies MJ, Hawkins CL. Inactivation of thiol-dependent enzymes by hypothiocyanous acid: role of sulfenyl thiocyanate and sulfenic acid intermediates. Free Radic Biol Med. 2012;52(6):1075–1085. doi: 10.1016/j.freeradbiomed.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinecke J, Ford PC. Formation of cysteine sulfenic acid by oxygen atom transfer from nitrite. J Am Chem Soc. 2010;132(27):9240–9243. doi: 10.1021/ja102221e. [DOI] [PubMed] [Google Scholar]
- 7.Heinecke JL, Khin C, Pereira JC, Suarez SA, Iretskii AV, Doctorovich F, Ford PC. Nitrite reduction mediated by heme models. Routes to NO and HNO? J Am Chem Soc. 2013;135(10):4007–4017. doi: 10.1021/ja312092x. [DOI] [PubMed] [Google Scholar]
- 8.Nagy P, Lemma K, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the reaction of cysteine thiosulfinate ester with cysteine to give cysteine sulfenic acid. J Org Chem. 2007;72(23):8838–8846. doi: 10.1021/jo701813f. [DOI] [PubMed] [Google Scholar]
- 9.Keceli G, Toscano JP. Reactivity of C-terminal cysteines with HNO. Biochemistry. 2014;53(22):3689–3698. doi: 10.1021/bi500360x. [DOI] [PubMed] [Google Scholar]
- 10.Zhukova L, Zhukov I, Bal W, Wyslouch-Cieszynska A. Redox modifications of the C-terminal cysteine residue cause structural changes in S100A1 and S100B proteins. Biochim Biophys Acta. 2004;1742(1–3):191–201. doi: 10.1016/j.bbamcr.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Goto K, Tokitoh N, Okazaki R. Synthesis of a stable arenesulfenic acid bearing a bowl-shaped macrobicyclic cyclophane skeleton. Angew Chem Int Ed Engl. 1995;34(10):1124–1126. [Google Scholar]
- 12.Saiki T, Goto K, Tokitoh N, Okazaki R. Synthesis and structure of a bridged calix[6]arene with a sulfenic acid functionality in the cavity. J Org Chem. 1996;61(9):2924–2925. doi: 10.1021/jo960068q. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura N. A stable sulfenic acid, 9-triptycenesulfenic acid: its isolation and characterization. J Am Chem Soc. 1983;105(24):7172–7173. [Google Scholar]
- 14.Ishihara M, Abe N, Sase S, Goto K. Synthesis, structure, and reactivities of a stable primary-alkyl-substituted sulfenic acid. Chem Lett. 2015;44(5):615–617. [Google Scholar]
- 15.Li X-B, Xu Z-F, Liu L-J, Liu J-T. Synthesis and identification of solution-stable sulfenic acids: perfluoroalkanesulfenic acids. Eur J Org Chem. 2014;2014(6):1182–1188. [Google Scholar]
- 16.Freeman F, Adesina IT, La JL, Lee JY, Poplawski AA. Conformers of cysteine and cysteine sulfenic acid and mechanisms of the reaction of cysteine sulfenic acid with 5,5-dimethyl-1,3-cyclohexanedione (dimedone) J Phys Chem B. 2013;117(50):16000–16012. doi: 10.1021/jp409022m. [DOI] [PubMed] [Google Scholar]
- 17.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423(6941):769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 18.Sarma BK, Mugesh G. Redox regulation of protein tyrosine phosphatase 1B (PTP1B): a biomimetic study on the unexpected formation of a sulfenyl amide intermediate. J Am Chem Soc. 2007;129(28):8872–8881. doi: 10.1021/ja070410o. [DOI] [PubMed] [Google Scholar]
- 19.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423(6941):773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 20.Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for redox regulation of protein tyrosine phosphatase 1B (PTP1B) activity. J Am Chem Soc. 2005;127(31):10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 21.Poole LB, Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Methods Enzymol. 2002;348:122–36. doi: 10.1016/s0076-6879(02)48632-6. [DOI] [PubMed] [Google Scholar]
- 22.Furdui CM, Poole LB. Chemical approaches to detect and analyze protein sulfenic acids. Mass Spectrom Rev. 2014;33(2):126–146. doi: 10.1002/mas.21384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reisz JA, Bechtold E, King SB, Poole LB, Furdui CM. Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 2013;280(23):6150–6161. doi: 10.1111/febs.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Simple synthesis of 1,3-cyclopentanedione derived probes for labeling sulfenic acid proteins. Chem Commun (Camb) 2011;47(32):9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonsson TJ, Tsang AW, Lowther WT, Furdui CM. Identification of intact protein thiosulfinate intermediate in the reduction of cysteine sulfinic acid in peroxiredoxin by human sulfiredoxin. J Biol Chem. 2008;283(34):22890–22894. doi: 10.1074/jbc.C800124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haynes AC, Qian J, Reisz JA, Furdui CM, Lowther WT. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J Biol Chem. 2013;288(41):29714–29723. doi: 10.1074/jbc.M113.473470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole TH, Reisz JA, Zhao W, Poole LB, Furdui CM, King SB. Strained cycloalkynes as new protein sulfenic acid traps. J Am Chem Soc. 2014;136(17):6167–6170. doi: 10.1021/ja500364r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian J, Wani R, Klomsiri C, Poole LB, Tsang AW, Furdui CM. A simple and effective strategy for labeling cysteine sulfenic acid in proteins by utilization of beta-ketoesters as cleavable probes. Chem Commun (Camb) 2012;48(34):4091–4093. doi: 10.1039/c2cc17868k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crane EJ, 3rd, Vervoort J, Claiborne A. 13C NMR analysis of the cysteine-sulfenic acid redox center of enterococcal NADH peroxidase. Biochemistry. 1997;36(28):8611–8618. doi: 10.1021/bi9707990. [DOI] [PubMed] [Google Scholar]
- 30.Baez NO, Reisz JA, Furdui CM. Mass spectrometry in studies of protein thiol chemistry and signaling: opportunities and caveats. Free Radic Biol Med. 2015;80:191–211. doi: 10.1016/j.freeradbiomed.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis FA, Billmers RL. Chemistry of sulfenic acids. 4. The first direct evidence for the involvement of sulfenic acids in the oxidation of thiols. J Am Chem Soc. 1981;103(23):7016–7018. [Google Scholar]
- 32.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36(48):15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 33.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249(19):6234–6243. [PubMed] [Google Scholar]
- 34.Poole LB, Zeng BB, Knaggs SA, Yakubu M, King SB. Synthesis of chemical probes to map sulfenic acid modifications on proteins. Bioconjug Chem. 2005;16(6):1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- 35.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18(6):2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu SE, King SB, Furdui CM, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol. 2010;473:77–94. doi: 10.1016/S0076-6879(10)73003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bansal N, Mims J, Kuremsky JG, Olex AL, Zhao W, Yin L, Wani R, Qian J, Center B, Marrs GS, et al. Broad phenotypic changes associated with gain of radiation resistance in head and neck squamous cell cancer. Antioxid Redox Signal. 2014;21(2):221–236. doi: 10.1089/ars.2013.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4(9):783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 40.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A. 2009;106(38):16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charles RL, Schroder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6(9):1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Truong TH, Garcia FJ, Seo YH, Carroll KS. Isotope-coded chemical reporter and acid-cleavable affinity reagents for monitoring protein sulfenic acids. Bioorg Med Chem Lett. 2011;21(17):5015–5020. doi: 10.1016/j.bmcl.2011.04.115. [DOI] [PubMed] [Google Scholar]
- 43.Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew Chem Int Ed Engl. 2011;50(6):1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- 44.Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Gupta V, Tallman KA, Porter NA, Carroll KS, Liebler DC. Global, in situ, site-specific analysis of protein S-sulfenylation. Nat Protoc. 2015;10(7):1022–37. doi: 10.1038/nprot.2015.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu CT, Benkovic SJ. Capturing a sulfenic acid with arylboronic acids and benzoxaborole. J Am Chem Soc. 2013;135(39):14544–14547. doi: 10.1021/ja407628a. [DOI] [PubMed] [Google Scholar]
- 47.Galardon E, Padovani D. Reactivity of persulfides toward strained bicyclo[6.1.0]nonyne derivatives: relevance to chemical tagging of proteins. Bioconjug Chem. 2015;26(6):1013–1016. doi: 10.1021/acs.bioconjchem.5b00243. [DOI] [PubMed] [Google Scholar]
- 48.Katsuyama M, Fan C, Arakawa N, Nishinaka T, Miyagishi M, Taira K, Yabe-Nishimura C. Essential role of ATF-1 in induction of NOX1, a catalytic subunit of NADPH oxidase: involvement of mitochondrial respiratory chain. Biochem J. 2005;386(Pt 2):255–261. doi: 10.1042/BJ20041180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006;281(47):36228–36235. doi: 10.1074/jbc.M606702200. [DOI] [PubMed] [Google Scholar]
- 50.Hall A, Karplus PA, Poole LB. Typical 2-Cys peroxiredoxins–structures, mechanisms and functions. FEBS J. 2009;276(9):2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 52.Jang HH, Kim SY, Park SK, Jeon HS, Lee YM, Jung JH, Lee SY, Chae HB, Jung YJ, Lee KO, et al. Phosphorylation and concomitant structural changes in human 2-Cys peroxiredoxin isotype I differentially regulate its peroxidase and molecular chaperone functions. FEBS Lett. 2006;580(1):351–355. doi: 10.1016/j.febslet.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 53.Chae HZ, Oubrahim H, Park JW, Rhee SG, Chock PB. Protein glutathionylation in the regulation of peroxiredoxins: a family of thiol-specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antioxid Redox Signal. 2012;16(6):506–523. doi: 10.1089/ars.2011.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140(4):517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 55.Chang TS, Jeong W, Choi SY, Yu S, Kang SW, Rhee SG. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J Biol Chem. 2002;277(28):25370–25376. doi: 10.1074/jbc.M110432200. [DOI] [PubMed] [Google Scholar]
- 56.Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller AF, Teixeira M, Valentine JS. Superoxide dismutases and superoxide reductases. Chem Rev. 2014;114(7):3854–3918. doi: 10.1021/cr4005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15(6):1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilcox KC, Zhou L, Jordon JK, Huang Y, Yu Y, Redler RL, Chen X, Caplow M, Dokholyan NV. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem. 2009;284(20):13940–13947. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Redler RL, Wilcox KC, Proctor EA, Fee L, Caplow M, Dokholyan NV. Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry. 2011;50(32):7057–7066. doi: 10.1021/bi200614y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leinartaite L, Johansson AS. Disulfide scrambling in superoxide dismutase 1 reduces its cytotoxic effect in cultured cells and promotes protein aggregation. PLoS One. 2013;8(10):e78060. doi: 10.1371/journal.pone.0078060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujiwara N, Nakano M, Kato S, Yoshihara D, Ookawara T, Eguchi H, Taniguchi N, Suzuki K. Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J Biol Chem. 2007;282(49):35933–35944. doi: 10.1074/jbc.M702941200. [DOI] [PubMed] [Google Scholar]
- 62.Cozzolino M, Amori I, Pesaresi MG, Ferri A, Nencini M, Carri MT. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283(2):866–874. doi: 10.1074/jbc.M705657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boschi-Muller S, Olry A, Antoine M, Branlant G. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim Biophys Acta. 2005;1703(2):231–238. doi: 10.1016/j.bbapap.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 64.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J Biol Chem. 2000;275(46):35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 65.Lowther WT, Brot N, Weissbach H, Honek JF, Matthews BW. Thiol-disulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proc Natl Acad Sci U S A. 2000;97(12):6463–6468. doi: 10.1073/pnas.97.12.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Srivastava S, Watowich SJ, Petrash JM, Srivastava SK, Bhatnagar A. Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry. 1999;38(1):42–54. doi: 10.1021/bi981794l. [DOI] [PubMed] [Google Scholar]
- 67.Ramana KV, Dixit BL, Srivastava S, Balendiran GK, Srivastava SK, Bhatnagar A. Selective recognition of glutathiolated aldehydes by aldose reductase. Biochemistry. 2000;39(40):12172–12180. doi: 10.1021/bi000796e. [DOI] [PubMed] [Google Scholar]
- 68.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. J Biol Chem. 2006;281(22):15110–15120. doi: 10.1074/jbc.M600837200. [DOI] [PubMed] [Google Scholar]
- 69.Kaiserova K, Tang XL, Srivastava S, Bhatnagar A. Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. J Biol Chem. 2008;283(14):9101–9112. doi: 10.1074/jbc.M709671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wetzelberger K, Baba SP, Thirunavukkarasu M, Ho YS, Maulik N, Barski OA, Conklin DJ, Bhatnagar A. Postischemic deactivation of cardiac aldose reductase: role of glutathione S-transferase P and glutaredoxin in regeneration of reduced thiols from sulfenic acids. J Biol Chem. 2010;285(34):26135–26148. doi: 10.1074/jbc.M110.146423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuwabara Y, Nishino T, Okamoto K, Matsumura T, Eger BT, Pai EF. Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proc Natl Acad Sci U S A. 2003;100(14):8170–8175. doi: 10.1073/pnas.1431485100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishino T, Okamoto K, Eger BT, Pai EF. Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008;275(13):3278–3289. doi: 10.1111/j.1742-4658.2008.06489.x. [DOI] [PubMed] [Google Scholar]
- 73.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19(49):5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 74.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Q, Olashaw N, Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J Biol Chem. 1995;270(48):28499–28502. doi: 10.1074/jbc.270.48.28499. [DOI] [PubMed] [Google Scholar]
- 76.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 77.Klomsiri C, Rogers LC, Soito L, McCauley AK, King SB, Nelson KJ, Poole LB, Daniel LW. Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radic Biol Med. 2014;71:49–60. doi: 10.1016/j.freeradbiomed.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11(6):1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spencer NY, Engelhardt JF. The basic biology of redoxosomes in cytokine-mediated signal transduction and implications for disease-specific therapies. Biochemistry. 2014;53(10):1551–1564. doi: 10.1021/bi401719r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Truong TH, Carroll KS. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry. 2012;51(50):9954–9965. doi: 10.1021/bi301441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kato M, Iwashita T, Takeda K, Akhand AA, Liu W, Yoshihara M, Asai N, Suzuki H, Takahashi M, Nakashima I. Ultraviolet light induces redox reaction-mediated dimerization and superactivation of oncogenic Ret tyrosine kinases. Mol Biol Cell. 2000;11(1):93–101. doi: 10.1091/mbc.11.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273(25):15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 83.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38(20):6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 84.Saito S, Frank GD, Mifune M, Ohba M, Utsunomiya H, Motley ED, Inagami T, Eguchi S. Ligand-independent trans-activation of the platelet-derived growth factor receptor by reactive oxygen species requires protein kinase C-d and c-Src. J Biol Chem. 2002;277(47):44695–44700. doi: 10.1074/jbc.M208332200. [DOI] [PubMed] [Google Scholar]
- 85.Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol. 2015;11(1):64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 86.Ushio-Fukai M. Vascular signaling through G protein-coupled receptors: new concepts. Curr Opin Nephrol Hypertens. 2009;18(2):153–159. doi: 10.1097/MNH.0b013e3283252efe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacol Sin. 2012;33(3):342–350. doi: 10.1038/aps.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Svineng G, Ravuri C, Rikardsen O, Huseby NE, Winberg JO. The role of reactive oxygen species in integrin and matrix metalloproteinase expression and function. Connect Tissue Res. 2008;49(3):197–202. doi: 10.1080/03008200802143166. [DOI] [PubMed] [Google Scholar]
- 89.Kozai D, Ogawa N, Mori Y. Redox regulation of transient receptor potential channels. Antioxid Redox Signal. 2014;21(6):971–986. doi: 10.1089/ars.2013.5616. [DOI] [PubMed] [Google Scholar]
- 90.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2011;8(1):57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2014;6(4):a020768. doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110(6):775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 93.Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Natl Acad Sci U S A. 2014;111(1):173–178. doi: 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaplan N, Urao N, Furuta E, Kim SJ, Razvi M, Nakamura Y, McKinney RD, Poole LB, Fukai T, Ushio-Fukai M. Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free Radic Res. 2011;45(10):1124–1135. doi: 10.3109/10715762.2011.602073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci U S A. 2009;106(13):5070–5075. doi: 10.1073/pnas.0806117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meng T-C, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular Cell. 2002;9(2):387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 97.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37(16):5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 98.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150(4):345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 99.LaButti JN, Chowdhury G, Reilly TJ, Gates KS. Redox regulation of protein tyrosine phosphatase 1B by peroxymonophosphate (=O3POOH) J Am Chem Soc. 2007;129(17):5320–5321. doi: 10.1021/ja070194j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.LaButti JN, Gates KS. Biologically relevant chemical properties of peroxymonophosphate (=O3POOH) Bioorg Med Chem Lett. 2009;19(1):218–221. doi: 10.1016/j.bmcl.2008.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeon TJ, Chien PN, Chun HJ, Ryu SE. Structure of the catalytic domain of protein tyrosine phosphatase sigma in the sulfenic acid form. Mol Cells. 2013;36(1):55–61. doi: 10.1007/s10059-013-0033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179(10):6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 103.Crump KE, Juneau DG, Poole LB, Haas KM, Grayson JM. The reversible formation of cysteine sulfenic acid promotes B-cell activation and proliferation. Eur J Immunol. 2012;42(8):2152–2164. doi: 10.1002/eji.201142289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen CY, Willard D, Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48(6):1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 105.Tang H, Hao Q, Rutherford SA, Low B, Zhao ZJ. Inactivation of SRC family tyrosine kinases by reactive oxygen species in vivo. J Biol Chem. 2005;280(25):23918–23925. doi: 10.1074/jbc.M503498200. [DOI] [PubMed] [Google Scholar]
- 106.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25(15):6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Akhand AA, Pu M, Senga T, Kato M, Suzuki H, Miyata T, Hamaguchi M, Nakashima I. Nitric oxide controls Src kinase activity through a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J Biol Chem. 1999;274(36):25821–25826. doi: 10.1074/jbc.274.36.25821. [DOI] [PubMed] [Google Scholar]
- 108.Minetti M, Mallozzi C, Di Stasi AM. Peroxynitrite activates kinases of the Src family and upregulates tyrosine phosphorylation signaling. Free Radic Biol Med. 2002;33(6):744–754. doi: 10.1016/s0891-5849(02)00891-2. [DOI] [PubMed] [Google Scholar]
- 109.Wood ST, Long DL, Reisz JA, Yammani RR, Burke EA, Klomsiri C, Poole LB, Furdui CM, Loeser RF. Cysteine-mediated redox regulation of cell signaling in chondrocytes stimulated with fibronectin fragments. Arthritis Rheumatol. 2015 doi: 10.1002/art.39326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shaul Y. c-Abl: activation and nuclear targets. Cell Death Differ. 2000;7(1):10–16. doi: 10.1038/sj.cdd.4400626. [DOI] [PubMed] [Google Scholar]
- 111.Cao C, Leng Y, Kufe D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J Biol Chem. 2003;278(32):29667–29675. doi: 10.1074/jbc.M301292200. [DOI] [PubMed] [Google Scholar]
- 112.Wang X, Zeng L, Wang J, Chau JF, Lai KP, Jia D, Poonepalli A, Hande MP, Liu H, He G, et al. A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Differ. 2011;18(1):5–15. doi: 10.1038/cdd.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun X, Majumder P, Shioya H, Wu F, Kumar S, Weichselbaum R, Kharbanda S, Kufe D. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J Biol Chem. 2000;275(23):17237–17240. doi: 10.1074/jbc.C000099200. [DOI] [PubMed] [Google Scholar]
- 114.Leonberg AK, Chai YC. The functional role of cysteine residues for c-Abl kinase activity. Mol Cell Biochem. 2007;304(1–2):207–212. doi: 10.1007/s11010-007-9501-y. [DOI] [PubMed] [Google Scholar]
- 115.Mamoon NM, Smith JK, Chatti K, Lee S, Kundrapu K, Duhe RJ. Multiple cysteine residues are implicated in Janus kinase 2-mediated catalysis. Biochemistry. 2007;46(51):14810–14818. doi: 10.1021/bi701118u. [DOI] [PubMed] [Google Scholar]
- 116.Smith JK, Patil CN, Patlolla S, Gunter BW, Booz GW, Duhe RJ. Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic Biol Med. 2012;52(6):1101–1110. doi: 10.1016/j.freeradbiomed.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8(16):2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci U S A. 2009;106(17):7004–7009. doi: 10.1073/pnas.0901933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007;35(Pt 2):231–235. doi: 10.1042/BST0350231. [DOI] [PubMed] [Google Scholar]
- 120.Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, Furdui CM. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci U S A. 2011;108(26):10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brozinick JT, Jr, Roberts BR, Dohm GL. Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes. 2003;52(4):935–941. doi: 10.2337/diabetes.52.4.935. [DOI] [PubMed] [Google Scholar]
- 122.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBb) Science. 2001;292(5522):1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 123.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBb. J Clin Invest. 2003;112(2):197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ardite E, Barbera JA, Roca J, Fernandez-Checa JC. Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-kB activation. Am J Pathol. 2004;165(3):719–728. doi: 10.1016/s0002-9440(10)63335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hansen JM, Klass M, Harris C, Csete M. A reducing redox environment promotes C2C12 myogenesis: implications for regeneration in aged muscle. Cell Biol Int. 2007;31(6):546–553. doi: 10.1016/j.cellbi.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Clark AR, Toker A. Signalling specificity in the Akt pathway in breast cancer. Biochem Soc Trans. 2014;42(5):1349–1355. doi: 10.1042/BST20140160. [DOI] [PubMed] [Google Scholar]
- 128.Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 129.Nolin JD, Tully JE, Hoffman SM, Guala AS, van der Velden JL, Poynter ME, van der Vliet A, Anathy V, Janssen-Heininger YM. The glutaredoxin/S-glutathionylation axis regulates interleukin-17A-induced proinflammatory responses in lung epithelial cells in association with S-glutathionylation of nuclear factor kB family proteins. Free Radic Biol Med. 2014;73:143–153. doi: 10.1016/j.freeradbiomed.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gupta S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review) Int J Oncol. 2003;22(1):15–20. [PubMed] [Google Scholar]
- 131.Luanpitpong S, Chanvorachote P, Stehlik C, Tse W, Callery PS, Wang L, Rojanasakul Y. Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells. Mol Biol Cell. 2013;24(6):858–869. doi: 10.1091/mbc.E12-10-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101(24):9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kato I, Maita H, Takahashi-Niki K, Saito Y, Noguchi N, Iguchi-Ariga SM, Ariga H. Oxidized DJ-1 inhibits p53 by sequestering p53 from promoters in a DNA-binding affinity-dependent manner. Mol Cell Biol. 2013;33(2):340–359. doi: 10.1128/MCB.01350-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;317(3):722–728. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 135.Takahashi-Niki K, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson’s disease patients. Biochem Biophys Res Commun. 2004;320(2):389–397. doi: 10.1016/j.bbrc.2004.05.187. [DOI] [PubMed] [Google Scholar]
- 136.Williams-Ashman HG, Seidenfeld J, Galletti P. Trends in the biochemical pharmacology of 5′-deoxy-5′-methylthioadenosine. Biochem Pharmacol. 1982;31(3):277–288. doi: 10.1016/0006-2952(82)90171-x. [DOI] [PubMed] [Google Scholar]
- 137.Fernandez-Irigoyen J, Santamaria M, Sanchez-Quiles V, Latasa MU, Santamaria E, Munoz J, Sanchez Del Pino MM, Valero ML, Prieto J, Avila MA, et al. Redox regulation of methylthioadenosine phosphorylase in liver cells: molecular mechanism and functional implications. Biochem J. 2008;411(2):457–465. doi: 10.1042/BJ20071569. [DOI] [PubMed] [Google Scholar]
- 138.Nagahara N, Sawada N. The mercaptopyruvate pathway in cysteine catabolism: a physiologic role and related disease of the multifunctional 3-mercaptopyruvate sulfurtransferase. Curr Med Chem. 2006;13(10):1219–1230. doi: 10.2174/092986706776360914. [DOI] [PubMed] [Google Scholar]
- 139.Nagahara N, Katayama A. Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J Biol Chem. 2005;280(41):34569–34576. doi: 10.1074/jbc.M505643200. [DOI] [PubMed] [Google Scholar]
- 140.Conway ME, Yennawar N, Wallin R, Poole LB, Hutson SM. Human mitochondrial branched chain aminotransferase: structural basis for substrate specificity and role of redox active cysteines. Biochim Biophys Acta. 2003;1647(1–2):61–65. doi: 10.1016/s1570-9639(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 141.Conway ME, Poole LB, Hutson SM. Roles for cysteine residues in the regulatory CXXC motif of human mitochondrial branched chain aminotransferase enzyme. Biochemistry. 2004;43(23):7356–7364. doi: 10.1021/bi0498050. [DOI] [PubMed] [Google Scholar]
- 142.Conway ME, Yennawar N, Wallin R, Poole LB, Hutson SM. Identification of a peroxide-sensitive redox switch at the CXXC motif in the human mitochondrial branched chain aminotransferase. Biochemistry. 2002;41(29):9070–9078. doi: 10.1021/bi020200i. [DOI] [PubMed] [Google Scholar]
- 143.Markham GD, Pajares MA. Structure-function relationships in methionine adenosyltransferases. Cell Mol Life Sci. 2009;66(4):636–648. doi: 10.1007/s00018-008-8516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sanchez-Gongora E, Ruiz F, Mingorance J, An W, Corrales FJ, Mato JM. Interaction of liver methionine adenosyltransferase with hydroxyl radical. FASEB J. 1997;11(12):1013–1019. doi: 10.1096/fasebj.11.12.9337154. [DOI] [PubMed] [Google Scholar]
- 145.Morettin A, Baldwin RM, Cote J. Arginine methyltransferases as novel therapeutic targets for breast cancer. Mutagenesis. 2015;30(2):177–189. doi: 10.1093/mutage/geu039. [DOI] [PubMed] [Google Scholar]
- 146.Cha B, Jho EH. Protein arginine methyltransferases (PRMTs) as therapeutic targets. Expert Opin Ther Targets. 2012;16(7):651–664. doi: 10.1517/14728222.2012.688030. [DOI] [PubMed] [Google Scholar]
- 147.Kuhn P, Xu W. Protein arginine methyltransferases: nuclear receptor coregulators and beyond. Prog Mol Biol Transl Sci. 2009;87:299–342. doi: 10.1016/S1877-1173(09)87009-9. [DOI] [PubMed] [Google Scholar]
- 148.Sydow K, Munzel T. ADMA and oxidative stress. Atheroscler Suppl. 2003;4(4):41–51. doi: 10.1016/s1567-5688(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 149.Cooke JP. ADMA: its role in vascular disease. Vasc Med. 2005;10(Suppl 1):S11–17. doi: 10.1177/1358836X0501000103. [DOI] [PubMed] [Google Scholar]
- 150.Morales Y, Nitzel DV, Price OM, Gui S, Li J, Qu J, Hevel JM. Redox control of protein arginine methyltransferase 1 (PRMT1) activity. J Biol Chem. 2015;290(24):14915–14926. doi: 10.1074/jbc.M115.651380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Junod AF, Clement A, Jornot L, Petersen H. Differential effects of hyperoxia and hydrogen peroxide on thymidine kinase and adenosine kinase activities of cultured endothelial cells. Biochim Biophys Acta. 1985;847(1):20–24. doi: 10.1016/0167-4889(85)90147-8. [DOI] [PubMed] [Google Scholar]
- 153.Sun R, Eriksson S, Wang L. Oxidative stress induced S-glutathionylation and proteolytic degradation of mitochondrial thymidine kinase 2. J Biol Chem. 2012;287(29):24304–24312. doi: 10.1074/jbc.M112.381996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sankaranarayanan R, Moras D. The fidelity of the translation of the genetic code. Acta Biochim Pol. 2001;48(2):323–335. [PubMed] [Google Scholar]
- 155.Ling J, Soll D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc Natl Acad Sci U S A. 2010;107(9):4028–4033. doi: 10.1073/pnas.1000315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cura V, Moras D, Kern D. Sequence analysis and modular organization of threonyl-tRNA synthetase from Thermus thermophilus and its interrelation with threonyl-tRNA synthetases of other origins. Eur J Biochem. 2000;267(2):379–393. doi: 10.1046/j.1432-1327.2000.01011.x. [DOI] [PubMed] [Google Scholar]
- 157.Cruzen ME, Arfin SM. Nucleotide and deduced amino acid sequence of human threonyl-tRNA synthetase reveals extensive homology to the Escherichia coli and yeast enzymes. J Biol Chem. 1991;266(15):9919–9923. [PubMed] [Google Scholar]
- 158.Wu J, Fan Y, Ling J. Mechanism of oxidant-induced mistranslation by threonyl-tRNA synthetase. Nucleic Acids Res. 2014;42(10):6523–6531. doi: 10.1093/nar/gku271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Alonso E, Cervera J, Garcia-Espana A, Bendala E, Rubio V. Oxidative inactivation of carbamoyl phosphate synthetase (ammonia). Mechanism and sites of oxidation, degradation of the oxidized enzyme, and inactivation by glycerol, EDTA, and thiol protecting agents. J Biol Chem. 1992;267(7):4524–4532. [PubMed] [Google Scholar]
- 160.Hart EJ, Powers-Lee SG. Role of Cys-1327 and Cys-1337 in redox sensitivity and allosteric monitoring in human carbamoyl phosphate synthetase. J Biol Chem. 2009;284(9):5977–5985. doi: 10.1074/jbc.M808702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8(11):904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 162.Keeney JT, Swomley AM, Harris JL, Fiorini A, Mitov MI, Perluigi M, Sultana R, Butterfield DA. Cell cycle proteins in brain in mild cognitive impairment: insights into progression to Alzheimer disease. Neurotox Res. 2012;22(3):220–230. doi: 10.1007/s12640-011-9287-2. [DOI] [PubMed] [Google Scholar]
- 163.Innes BT, Sowole MA, Gyenis L, Dubinsky M, Konermann L, Litchfield DW, Brandl CJ, Shilton BH. Peroxide-mediated oxidation and inhibition of the peptidyl-prolyl isomerase Pin1. Biochim Biophys Acta. 2015;1852(5):905–912. doi: 10.1016/j.bbadis.2014.12.025. [DOI] [PubMed] [Google Scholar]
- 164.Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BM, Luo ML, Nechama M, Lin YM, et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol Dis. 2015;76:13–23. doi: 10.1016/j.nbd.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Heideker J, Wertz IE. DUBs, the regulation of cell identity and disease. Biochem J. 2015;465(1):1–26. doi: 10.1042/BJ20140496. [DOI] [PubMed] [Google Scholar]
- 166.Enesa K, Evans P. The biology of A20-like molecules. Adv Exp Med Biol. 2014;809:33–48. doi: 10.1007/978-1-4939-0398-6_3. [DOI] [PubMed] [Google Scholar]
- 167.Verstrepen L, Carpentier I, Beyaert R. The biology of A20-binding inhibitors of NF-kB activation (ABINs) Adv Exp Med Biol. 2014;809:13–31. doi: 10.1007/978-1-4939-0398-6_2. [DOI] [PubMed] [Google Scholar]
- 168.Kulathu Y, Garcia FJ, Mevissen TE, Busch M, Arnaudo N, Carroll KS, Barford D, Komander D. Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat Commun. 2013;4:1569. doi: 10.1038/ncomms2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cotto-Rios XM, Bekes M, Chapman J, Ueberheide B, Huang TT. Deubiquitinases as a signaling target of oxidative stress. Cell Rep. 2012;2(6):1475–1484. doi: 10.1016/j.celrep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Godat E, Herve-Grvepinet V, Veillard F, Lecaille F, Belghazi M, Bromme D, Lalmanach G. Regulation of cathepsin K activity by hydrogen peroxide. Biol Chem. 2008;389(8):1123–1126. doi: 10.1515/BC.2008.109. [DOI] [PubMed] [Google Scholar]
- 171.Lalmanach G, Diot E, Godat E, Lecaille F, Herve-Grepinet V. Cysteine cathepsins and caspases in silicosis. Biol Chem. 2006;387(7):863–870. doi: 10.1515/BC.2006.109. [DOI] [PubMed] [Google Scholar]
- 172.Huang Z, Pinto JT, Deng H, Richie JP., Jr Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem Pharmacol. 2008;75(11):2234–2244. doi: 10.1016/j.bcp.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zech B, Wilm M, van Eldik R, Brune B. Mass spectrometric analysis of nitric oxide-modified caspase-3. J Biol Chem. 1999;274(30):20931–20936. doi: 10.1074/jbc.274.30.20931. [DOI] [PubMed] [Google Scholar]
- 174.Xu Z, Lam LS, Lam LH, Chau SF, Ng TB, Au SW. Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008;22(1):127–137. doi: 10.1096/fj.06-7871com. [DOI] [PubMed] [Google Scholar]
- 175.Dinh TP, Freund TF, Piomelli D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem Phys Lipids. 2002;121(1–2):149–158. doi: 10.1016/s0009-3084(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 176.King AR, Dotsey EY, Lodola A, Jung KM, Ghomian A, Qiu Y, Fu J, Mor M, Piomelli D. Discovery of potent and reversible monoacylglycerol lipase inhibitors. Chem Biol. 2009;16(10):1045–1052. doi: 10.1016/j.chembiol.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.King AR, Lodola A, Carmi C, Fu J, Mor M, Piomelli D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. Br J Pharmacol. 2009;157(6):974–983. doi: 10.1111/j.1476-5381.2009.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM. Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. Chembiochem. 2010;11(2):218–227. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 179.Dotsey EY, Jung KM, Basit A, Wei D, Daglian J, Vacondio F, Armirotti A, Mor M, Piomelli D. Peroxide-dependent MGL sulfenylation regulates 2-AG-mediated endocannabinoid signaling in brain neurons. Chem Biol. 2015;22(5):619–628. doi: 10.1016/j.chembiol.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Harms N, Ras J, Reijnders WN, van Spanning RJ, Stouthamer AH. S-Formylglutathione hydrolase of Paracoccus denitrificans is homologous to human esterase D: a universal pathway for formaldehyde detoxification? J Bacteriol. 1996;178(21):6296–6299. doi: 10.1128/jb.178.21.6296-6299.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee WH, Wheatley W, Benedict WF, Huang CM, Lee EY. Purification, biochemical characterization, and biological function of human esterase D. Proc Natl Acad Sci U S A. 1986;83(18):6790–6794. doi: 10.1073/pnas.83.18.6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Legler PM, Kumaran D, Swaminathan S, Studier FW, Millard CB. Structural characterization and reversal of the natural organophosphate resistance of a D-type esterase, Saccharomyces cerevisiae S-formylglutathione hydrolase. Biochemistry. 2008;47(36):9592–9601. doi: 10.1021/bi8010016. [DOI] [PubMed] [Google Scholar]
- 183.Legler PM, Leary DH, Hervey WJt, Millard CB. A role for His-160 in peroxide inhibition of S. cerevisiae S-formylglutathione hydrolase: evidence for an oxidation sensitive motif. Arch Biochem Biophys. 2012;528(1):7–20. doi: 10.1016/j.abb.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 184.Patel MS, Vettakkorumakankav NN, Liu TC. Dihydrolipoamide dehydrogenase: activity assays. Methods Enzymol. 1995;252:186–195. doi: 10.1016/0076-6879(95)52022-8. [DOI] [PubMed] [Google Scholar]
- 185.Yan LJ, Sumien N, Thangthaeng N, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic Res. 2013;47(2):123–133. doi: 10.3109/10715762.2012.752078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hildebrandt T, Knuesting J, Berndt C, Morgan B, Scheibe R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol Chem. 2015;396(5):523–537. doi: 10.1515/hsz-2014-0295. [DOI] [PubMed] [Google Scholar]
- 187.Maller C, Schroder E, Eaton P. Glyceraldehyde 3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxid Redox Signal. 2011;14(1):49–60. doi: 10.1089/ars.2010.3149. [DOI] [PubMed] [Google Scholar]
- 188.Jenkins JL, Tanner JJ. High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 3):290–301. doi: 10.1107/S0907444905042289. [DOI] [PubMed] [Google Scholar]
- 189.Cowan-Jacob SW, Kaufmann M, Anselmo AN, Stark W, Grutter MG. Structure of rabbit-muscle glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 12):2218–2227. doi: 10.1107/s0907444903020493. [DOI] [PubMed] [Google Scholar]
- 190.Schuppe-Koistinen I, Moldeus P, Bergman T, Cotgreave IA. S-Thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment. Eur J Biochem. 1994;221(3):1033–1037. doi: 10.1111/j.1432-1033.1994.tb18821.x. [DOI] [PubMed] [Google Scholar]
- 191.Grant CM, Quinn KA, Dawes IW. Differential protein S-thiolation of glyceraldehyde-3-phosphate dehydrogenase isoenzymes influences sensitivity to oxidative stress. Mol Cell Biol. 1999;19(4):2650–2656. doi: 10.1128/mcb.19.4.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cotgreave IA, Gerdes R, Schuppe-Koistinen I, Lind C. S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase: role of thiol oxidation and catalysis by glutaredoxin. Methods Enzymol. 2002;348:175–182. doi: 10.1016/s0076-6879(02)48636-3. [DOI] [PubMed] [Google Scholar]
- 193.Peralta D, Bronowska AK, Morgan B, Doka E, Van Laer K, Nagy P, Grater F, Dick TP. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat Chem Biol. 2015;11(2):156–163. doi: 10.1038/nchembio.1720. [DOI] [PubMed] [Google Scholar]
- 194.Lim S, Dizhoor AM, Ames JB. Structural diversity of neuronal calcium sensor proteins and insights for activation of retinal guanylyl cyclase by GCAP1. Front Mol Neurosci. 2014;7:19. doi: 10.3389/fnmol.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Permyakov SE, Nazipova AA, Denesyuk AI, Bakunts AG, Zinchenko DV, Lipkin VM, Uversky VN, Permyakov EA. Recoverin as a redox-sensitive protein. J Proteome Res. 2007;6(5):1855–1863. doi: 10.1021/pr070015x. [DOI] [PubMed] [Google Scholar]
- 196.Permyakov SE, Zernii EY, Knyazeva EL, Denesyuk AI, Nazipova AA, Kolpakova TV, Zinchenko DV, Philippov PP, Permyakov EA, Senin II. Oxidation mimicking substitution of conservative cysteine in recoverin suppresses its membrane association. Amino Acids. 2012;42(4):1435–1442. doi: 10.1007/s00726-011-0843-0. [DOI] [PubMed] [Google Scholar]
- 197.Ranaghan MJ, Kumar RP, Chakrabarti KS, Buosi V, Kern D, Oprian DD. A highly conserved cysteine of neuronal calcium-sensing proteins controls cooperative binding of Ca2+ to recoverin. J Biol Chem. 2013;288(50):36160–36167. doi: 10.1074/jbc.M113.524355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Eberhardt M, Dux M, Namer B, Miljkovic J, Cordasic N, Will C, Kichko TI, de la Roche J, Fischer M, Suarez SA, et al. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat Commun. 2014;5:4381. doi: 10.1038/ncomms5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Andersson DA, Filipovic MR, Gentry C, Eberhardt M, Vastani N, Leffler A, Reeh P, Bevan S. Streptozotocin stimulates TRPA1 directly: involvement of peroxynitrite. J Biol Chem. 2015;290(24):15185–15196. doi: 10.1074/jbc.M115.644476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Christakos S, Barletta F, Huening M, Dhawan P, Liu Y, Porta A, Peng X. Vitamin D target proteins: function and regulation. J Cell Biochem. 2003;88(2):238–244. doi: 10.1002/jcb.10349. [DOI] [PubMed] [Google Scholar]
- 201.Berggard T, Miron S, Onnerfjord P, Thulin E, Akerfeldt KS, Enghild JJ, Akke M, Linse S. Calbindin D28k exhibits properties characteristic of a Ca2+ sensor. J Biol Chem. 2002;277(19):16662–16672. doi: 10.1074/jbc.M200415200. [DOI] [PubMed] [Google Scholar]
- 202.Tao L, Murphy ME, English AM. S-nitrosation of Ca2+-loaded and Ca2+-free recombinant calbindin D28K from human brain. Biochemistry. 2002;41(19):6185–6192. doi: 10.1021/bi015846+. [DOI] [PubMed] [Google Scholar]
- 203.Cedervall T, Berggård T, Borek V, Thulin E, Linse S, Akerfeldt KS. Redox sensitive cysteine residues in calbindin D28k are structurally and functionally important. Biochemistry. 2005;44(2):684–693. doi: 10.1021/bi049232r. [DOI] [PubMed] [Google Scholar]
- 204.Vanbelle C, Halgand F, Cedervall T, Thulin E, Akerfeldt KS, Laprevote O, Linse S. Deamidation and disulfide bridge formation in human calbindin D28k with effects on calcium binding. Protein Sci. 2005;14(4):968–979. doi: 10.1110/ps.041157705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Phillips RG, Meier TJ, Giuli LC, McLaughlin JR, Ho DY, Spolsky RM. Calbindin D28K gene transfer via Herpes Simplex Virus amplicon vector decreases hippocampal damage in vivo following neurotoxic insults. J Neurochem. 1999;73(3):1200–1205. doi: 10.1046/j.1471-4159.1999.0731200.x. [DOI] [PubMed] [Google Scholar]
- 206.Bellido T, Huening M, Raval-Pandya M, Manolagas SC, Christakos S. Calbindin-D28k is expressed in osteoblastic cells and suppresses their apoptosis by inhibiting caspase-3 activity. J Biol Chem. 2000;275(34):26328–26332. doi: 10.1074/jbc.M003600200. [DOI] [PubMed] [Google Scholar]
- 207.Svoboda LK, Reddie KG, Zhang L, Vesely ED, Williams ES, Schumacher SM, O’Connell RP, Shaw R, Day SM, Anumonwo JM, et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ Res. 2012;111(7):842–853. doi: 10.1161/CIRCRESAHA.111.263525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang Y, Shi W, Chen X, Cui N, Konduru AS, Shi Y, Trower TC, Zhang S, Jiang C. Molecular basis and structural insight of vascular KATP channel gating by S-glutathionylation. J Biol Chem. 2011;286(11):9298–9307. doi: 10.1074/jbc.M110.195123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jerng HH, Pfaffinger PJ. S-Glutathionylation of an auxiliary subunit confers redox sensitivity to Kv4 channel inactivation. PLoS One. 2014;9(3):e93315. doi: 10.1371/journal.pone.0093315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jin X, Yu L, Wu Y, Zhang S, Shi Z, Chen X, Yang Y, Zhang X, Jiang C. S-Glutathionylation underscores the modulation of the heteromeric Kir4.1-Kir5.1 channel in oxidative stress. J Physiol. 2012;590(Pt 21):5335–5348. doi: 10.1113/jphysiol.2012.236885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hitchler MJ, Domann FE. Redox regulation of the epigenetic landscape in cancer: a role for metabolic reprogramming in remodeling the epigenome. Free Radic Biol Med. 2012;53(11):2178–87. doi: 10.1016/j.freeradbiomed.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Li M, Wilson DM., 3rd Human apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal. 2014;20(4):678–707. doi: 10.1089/ars.2013.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5(1):36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kim YJ, Kim D, Illuzzi JL, Delaplane S, Su D, Bernier M, Gross ML, Georgiadis MM, Wilson DM., 3rd S-Glutathionylation of cysteine 99 in the APE1 protein impairs abasic endonuclease activity. J Mol Biol. 2011;414(3):313–326. doi: 10.1016/j.jmb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Luo M, Zhang J, He H, Su D, Chen Q, Gross ML, Kelley MR, Georgiadis MM. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2012;51(2):695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kB/IkB family: intimate tales of association and dissociation. Genes Dev. 1995;9(22):2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 217.Perkins ND, Schmid RM, Duckett CS, Leung K, Rice NR, Nabel GJ. Distinct combinations of NF-kB subunits determine the specificity of transcriptional activation. Proc Natl Acad Sci U S A. 1992;89(5):1529–1533. doi: 10.1073/pnas.89.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-kB DNA binding by nitric oxide. Nucleic Acids Res. 1996;24(12):2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Klatt P, Pineda Molina E, Perez-Sala D, Lamas S. Novel application of S-nitrosoglutathione-Sepharose to identify proteins that are potential targets for S-nitrosoglutathione-induced mixed-disulphide formation. Biochem J. 2000;349(Pt 2):567–578. doi: 10.1042/0264-6021:3490567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40(47):14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 221.Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16(1):13–19. doi: 10.2174/1389450116666150113121054. [DOI] [PubMed] [Google Scholar]
- 222.Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK, Cho JW, Park YM, Jung G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135(6):2128–2140. 2140 e1–8. doi: 10.1053/j.gastro.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 223.Szumiel I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol. 2015;91(1):1–12. doi: 10.3109/09553002.2014.934929. [DOI] [PubMed] [Google Scholar]
- 224.Wang KY, Chen CC, Shen CK. Active DNA demethylation of the vertebrate genomes by DNA methyltransferases: deaminase, dehydroxymethylase or demethylase? Epigenomics. 2014;6(3):353–363. doi: 10.2217/epi.14.21. [DOI] [PubMed] [Google Scholar]
- 225.Chen CC, Wang KY, Shen CK. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem. 2012;287(40):33116–33121. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wojdyla K, Williamson J, Roepstorff P, Rogowska-Wrzesinska A. The SNO/SOH TMT strategy for combinatorial analysis of reversible cysteine oxidations. J Proteomics. 2015;113:415–434. doi: 10.1016/j.jprot.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 227.Wang YT, Piyankarage SC, Williams DL, Thatcher GR. Proteomic profiling of nitrosative stress: protein S-oxidation accompanies S-nitrosylation. ACS Chem Biol. 2014;9(3):821–830. doi: 10.1021/cb400547u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Guo J, Gaffrey MJ, Su D, Liu T, Camp DG, 2nd, Smith RD, Qian WJ. Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat Protoc. 2014;9(1):64–75. doi: 10.1038/nprot.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Defelipe LA, Lanzarotti E, Gauto D, Marti MA, Turjanski AG. Protein topology determines cysteine oxidation fate: the case of sulfenyl amide formation among protein families. PLoS Comput Biol. 2015;11(3):e1004051. doi: 10.1371/journal.pcbi.1004051. [DOI] [PMC free article] [PubMed] [Google Scholar]