

Abstract
With the rapid development of high-throughput quantitative proteomic and transcriptomic approaches, the molecular mechanisms of cancers have been comprehensively explored. However, cancer is a multi-dimensional disease with sophisticated regulations, and few studies focus on the crosstalk among multiomics. In order to explore the molecular mechanisms of gastric cancer (GC), particularly in the process of lymph node metastasis (LNM), we investigated dynamic profiling changes as well as crosstalk between long non-coding RNAs (lncRNAs), the proteome, and the lysine succinylome. Our study reports the first qualitative and quantitative profile of lysine succinylation in GC. We identified a novel mechanism through which the TCA cycle and pentose phosphate pathway might be regulated through lysine succinylation in their core enzymes. We then examined the potential of using lysine succinylation as a biomarker for GC and successfully developed a succinylation-dependent antibody for the K569 site in Caldesmon as putative biomarker. Finally, we investigated the relationship between the lysine succinylome and lncRNAs, identifying potential crosstalks between two lncRNAs and one succinylation site. These results expand our understanding of the mechanisms of tumorigenesis and provide new information for the diagnosis and prognosis of GC.
On a global scale, gastric cancer (GC) ranks as one of the most common malignant tumors and one of the leading causes of cancer death, especially in less developed countries1. In 2012, there were approximately 951,600 new GC cases and 723,100 associated deaths around the world1. GC is a complicated disease due to its histological and etiological heterogeneity2 and high tendency for lymph node metastasis (LNM), which remains the most important prognostic factor for GC patients. More than 50% of GC patients have LNM when initially diagnosed, which lead to a 5-year survival rate <30%3. Besides, the prognosis of LNM-negative patients was significantly better than that of positive patients4. Although the number of metastatic lymph nodes is considered the most significant evaluation of overall survival (OS), the negative lymph node count can play important roles of prognostic evaluation and clinical treatment of GC5. Not only in advanced gastric cancer (AGC), LNM in early gastric cancer (EGC) is also a critical factor for assessment of prognosis and therapeutic strategy6. Although decreasing trends in GC incidence and mortality rates have been observed in many countries, significant challenges continue to impede our understanding of GC, especially in the process of LNM. Proteomic and protein functional studies have found that numerous protein candidates may play important roles in the LNM of different cancers7.
Recently, based on the rapid development of quantitative proteomic approaches by tandem mass spectrometry (MS/MS), differential protein expression levels have been revealed in various tumors8,9,10,11,12. This may help identify feasible protein candidates as novel diagnostic biomarkers and prognostic targets. Protein post-translational modifications (PTMs) have also been shown to play important roles in regulating protein functions13, with specific PTMs on certain substrate residues diversifying and regulating the cellular proteome14. Multiple well-known modifications including acetylation15 and SUMOylation16, as well as several novel PTMs including succinylation17 and malonylation18, were recently investigated using MS/MS, utilizing high-quality antibodies against PTMs and biochemistry methods. These PTMs widely exist in eukaryotic and prokaryotic organisms19. Moreover, PTMs are deeply involved in human diseases20 including malignant tumors21,22, and proteins that mediate epigenetic regulations through histone acetylation and methylation are proven to be associated with the progression of cancers23. Furthermore, O-glycosylation changes in the serum of GC patients suggest that it, too, might correlate with GC tumorigenesis24.
Among these PTMs, lysine succinylation (Ksucc) is a novel PTM first identified in Escherichia coli17. As an evolutionarily conserved modification existing in multiple species25,26,27,28,29,30,31,32, it may affect various cellular functions including the tricarboxylic acid (TCA) cycle and fatty acid metabolism33. Therefore, Ksucc may play significant roles in the regulation of cellular metabolism, yet its role in cancer has not been explored. Tumor cells make complicated metabolic alterations characterized by changes in multiple metabolic pathways such as energy production and biosynthetic processes34, and protein acetylation, a well-known PTM, is known to regulate metabolism in pancreatic cancer22. Based on this, we hypothesized that Ksucc might play a role in cancer by regulating important cellular metabolic pathways. It has been accepted that the pathogenesis of GC involves multi-dimensional aspects due to the complexity and heterogeneity of the disease. Recently, long non-coding RNAs (lncRNAs) were proven to function as potential biomarkers in GC35. Interestingly, lncRNAs were reported to link with several protein modifications including acetylation36 and phosphorylation37, suggesting potential crosstalk between lncRNAs and PTMs.
Here, we have performed multiomics studies to explore the profiling of lncRNAs, the quantitative proteome, and the lysine succinylome in GC for the first time, using the combined advantages of transcriptomics and proteomics. This study, therefore, may increase our understanding of cancer biology and provide a way to screen novel biomarkers and targets for GC diagnostics and treatments.
Results
The workflow and strategy for quantitative multiomics in gastric cancer
To comprehensively map the multiomics of gastric cancer (GC), we performed quantitative transcriptomics by microarray and proteomics by LC-MS/MS analyses to explore the profiling of lncRNAs, the global proteome, and the lysine succinylome in four pairs of GC clinical samples. Bioinformatics analyses were employed to make deep multiomics studies in GC. GC clinical samples were obtained via biopsy from patients undergoing surgical resection at the First Hospital of China Medical University (Shenyang, China). Samples included one lymph node-negative case (N0 stage) and three lymph node-positive cases (N1 and N2 stage) according to the 7th edition of the UICC/AJCC TNM staging system. The detailed clinical and pathological data are shown in Supplementary Table 1. The study design and workflow are briefly illustrated in Fig. 1.
Figure 1. Workflow and strategy for quantitative multiomics in four pairs of gastric cancer tissues and matched normal tissues.
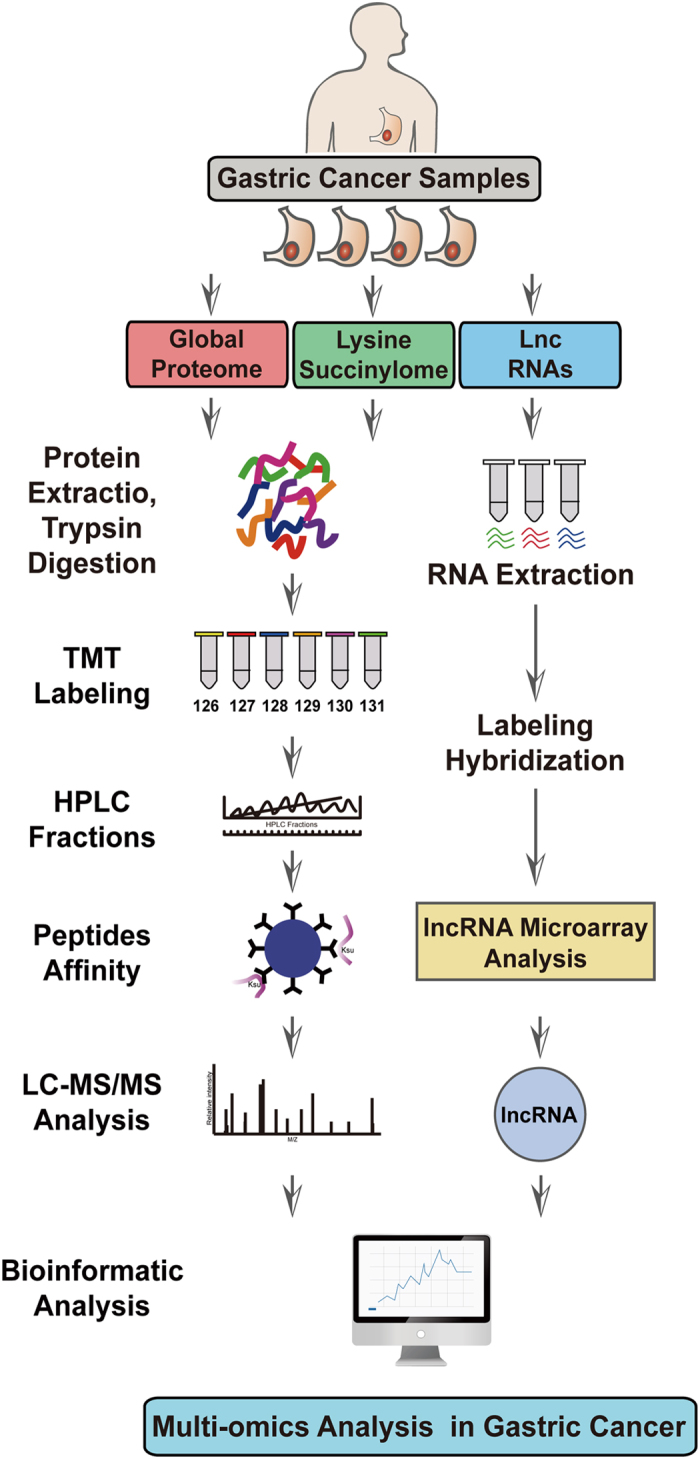
LC-MS/MS was used for quantitative proteomic and lysine succinylation analysis, while microarray was performed for global lncRNA profiles.
Identification and functional analysis of the global proteome in gastric cancer
To investigate global proteome profiling in GC, we first performed quantitative proteomic studies using TMT labeling, HPLC fractionation, and high-resolution LC-MS/MS analysis (See Supplementary Table 2 for proteomic study summary). Global proteome data generation quality control indicated that proteomics analysis system was robust (See Supplementary Fig. 1).
A total of 4,524 proteins were identified from these pairs of GC tissues, among which 3,147 proteins had quantitative changes. Differentially expressed proteins were selected by filtered for an average cut-off change of 1.3-fold38,39 and p value ≤ 0.05 when comparing tumor samples with their corresponding normal tissues. A Venn diagram illustrating the number of differentially expressed proteins in each pair is shown in Fig. 2a and Supplementary Table 2. In our further analyses, we found the global protein expression profile of four samples (named S2, S3, S4 and S5) were similar despite differences in their clinical and pathological information (Supplementary Fig. 2). We used these four pairs as biological replicates for total-proteomic analysis, aiming to find global differentially expressed proteins. A total of 236 proteins qualified as differentially expressed, including 110 down- and 126 up-regulated proteins. Intensive bioinformatics analyses were then carried out to annotate these differentially expressed proteins (Supplementary Table 2).
Figure 2. Identification and functional analysis of global proteomics in gastric cancer.

(a) Venn diagram illustrating differentially expressed proteins in each pair of samples. GO and KEGG pathway functional enrichment of (b) down- and (c) up-regulated proteins are examined. (d) Up- and down-regulated proteins in each pair (tumor vs. normal) are clustered by enriched functional terms discovered in (b) and (c). Functional enrichment clusters in different biological categories including nucleus, mitochondrion, catabolic process and ATPase are highlighted.
Proteins were annotated using Gene Ontology (GO) classification and the KEGG pathway database to further investigate their functions. We then performed enrichment tests to identify GO terms and pathways that were significantly enriched (Fig. 2b and Supplementary Table 2). Many down-regulated proteins were enriched in respiration-related metabolic process terms including oxidation-reduction activity, respiratory chain, and aerobic respiration (Fig. 2b). The KEGG pathway enrichment result was consistent with GO analysis, showing both the TCA cycle and oxidative phosphorylation as negatively enriched (Fig. 2b). The above enrichment results suggested that a hypoxic microenvironment is a central and typical characteristic during cancer development40. Other known cancer associated pathways including focal adhesion and the PI3K-Akt signaling pathway41,42 were enriched in down-regulated proteins. Most of the up-regulated terms were related to nuclear activity including nucleic acid binding, RNA splicing, and DNA replication pathway, suggesting more nuclear activities in GC than in normal cells (Fig. 2c).
As the lymph node statuses differed between the four GC samples (one N0 sample S2, two N1 samples S3 & S4, and one N2 sample S5), we were interested in exploring protein regulation patterns in the process of disease development, especially in lymph node metastasis (LNM) of GC. To do that, we applied GO enrichment based clustering analyses between all four pairs, then classified clusters into several sub-categories based on their functional relevance (Fig. 2d). Consistent with our previous finding that nuclear activities are positively correlated with the development of GC, we found that up-regulated proteins related to the nucleus showed a more obvious enrichment in the process of LNM, while there was no enrichment observed in down-regulated nuclear proteins. On the contrary, in the sub-category of mitochondrion, only down-regulated proteins were significantly enriched, confirming that decreased aerobic respiration is observed in cancer cells. Moreover, down-regulated proteins related to catabolic processes were also obviously enriched in the late stage of LNM. Interestingly, we found ATPase activities to be enriched in both up- and down-regulated proteins in the LNM process. This is not surprising since ATPase proteins take part in multiple essential cellular processes. Up-regulated ATPases included DNA- and RNA-dependent proteins as well as helicase-related APTases involved in nuclear activity such as DNA replication, mRNA processing, and transcription. On the other hand, the down-regulated ATPases were all related to the transmembrane movement of ions and protons that are likely to correlate with oxidative phosphorylation. Taken together, the above clustering results highlight physiological processes which may play important roles in the development of GC, especially in LNM process.
Dynamic clusters analysis of the protein expression profile in gastric cancer
Protein regulation is likely to be a dynamic process during tumorigenesis as the immune system continuously monitors and fights against cancer progression. As a result, essential proteins likely are initially up-regulated before being inhibited at later stages, as more regulation machineries are involved to control their expression, or vice versa. In order to explore these types of dynamic changes, we ran dynamic clusters analyses based on protein expression quantitation. Compared with traditional single protein analyses, changes in the protein clusters can be utilized to reveal further physiological functions in the dynamic cancer system.
We obtained a series of increased or decreased protein clusters in the process of LNM, with a total of 15 clusters (Supplementary Fig. 3 and Supplementary Table 3) detected. We chose six types of protein clusters for interaction analysis (Figs 3 and 4), aiming to find functionally-related interaction networks from clusters with similar patterns. Clusters #1, #12, and #14 represented a series of proteins whose fold-changes tend to increase as the disease develops (Fig. 3a), while #2, #9 and #15 were protein clusters with decreasing fold-changes (Fig. 4a). We used the STRING database (http://string-db.org/) to analyze correlations between these proteins, and then interconnected them into sub-networks. Proteins in clusters #1, #12 and #14 were involved in biological processes including RNA processing (red), glycolysis/gluconeogenesis (green), focal adhesion (lavender), extracellular structure organization (modena), heat shock proteins system (yellow), ubiquitination (dark blue) and catabolic process (light blue) (Fig. 3b). Proteins in the “fold-change decreased” clusters were likely involved in oxidation-reduction process (green), RNA splicing (pink), protein processing system (blue) and response to stress (red) (Fig. 4b). We also noticed that several significant biological processes or pathways were involved in both increased and decreased protein clusters, such as response to stress, catabolic, and small molecule metabolic process.
Figure 3. Increased fold-change protein dynamic clusters in gastric cancer development.

(a) Expression pattern of three increased fold-change protein dynamic clusters. (b) The protein-protein interaction network of three increased fold-change protein dynamic clusters. Nodes stand for individual proteins, lines connecting to proteins stand for the correlation between them, and different colors represent different protein functional clusters. Network analyses were performed based on STRING database (http://string-db.org/).
Figure 4. Decreased fold-change protein dynamic clusters in gastric cancer development.

(a) Expression pattern of three decreased fold-change protein dynamic clusters. (b) The protein-protein interaction network of three decreased fold-change protein dynamic clusters. Nodes stand for individual proteins, lines connecting to proteins stand for the correlation between them, and different colors represent different protein functional clusters. Network analyses were performed based on STRING database (http://string-db.org/).
Identification and functional analysis of the lysine succinylome in gastric cancer
As one type of novel discovered PTMs, lysine succinylation (Ksucc) has been proven to be essential for regulating molecular functions such as cellular metabolism in physiological and pathophysiological states43. Its metabolic regulation in tumor cells has been intensively studied, as multiple metabolic pathways are altered during tumorigenesis including energy production and biosynthetic processes. However, its roles in cancers have not yet been explored. Protein acetylation, a well-known lysine acylation modification, has been demonstrated to regulate metabolism in pancreatic cancer22. We hypothesized that this newly identified lysine acylation type, lysine succinylation, may play a role in cancer by regulating important cellular metabolic pathways.
First, we tested if the Ksucc levels in GC samples were different from their pairing controls, using western blotting to detect global Ksucc levels. Abundant Ksucc expression levels were observed in GC, with different patterns of Ksucc observed between tumor tissues (designated as T) and normal tissues (designated as N) (Fig. 5a, full-length and multiple exposures gels of global lysine succinylation in Supplementary Fig. 4). To discover differentially regulated Ksucc sites, we applied an integrated quantitative Ksucc approach, using antibody-based affinity enrichment followed by LC-MS/MS analysis. Lysine succinylation data generation quality control indicated that the analysis system was robust (See Supplementary Fig. 5). A total of 1,241 Ksucc sites were identified, among which 986 sites were quantitative (Fig. 5b and Supplementary Table 4).
Figure 5. Identification and functional analysis of the lysine succinylome in gastric cancer.

(a) The expression profile of lysine succinylation in GC tissues by western blotting using a pan anti-succinyl lysine polyclonal antibody (full-length and multiple exposures gels are presented in Supplementary Fig. 4). (b) Summary of lysine succinylated sites in GC. (c) GO enrichment analysis of down-regulated succinylated proteins. (d) Three major pathways (focal adhesion, regulation of actin cytoskeleton and tight junction) of succinylated proteins. Red oval represents lysine succinylated proteins.
Among all identified succinylated proteins, approximately 75.8% (640/844) contained only one succinylated site, 15.2% (128/844) possessed two succinylated sites, and 2.5% (21/844) contained five or more Ksucc sites. Notably, six proteins were found to have ten or more Ksucc sites: spectrin alpha chain (17 sites), myosin-11 (12 sites), myosin-9 (13 sites), filamin-A (10 sites), calreticulin (10 sites) and spectrin beta chain (10 sites).
A total of 40 differentially expressed succinylated sites (with a cut-off change of 1.3-fold and p value ≤ 0.05) in 38 proteins were chosen for downstream analyses (see top ten differentially up- and down-regulated Ksucc sites in Table 1). Most of these succinylated proteins were localized in cytoplasm (47%), and 11% of them were distributed in the mitochondria (Supplementary Fig. 6a). As shown in Fig. 5c, down-regulated succinylated proteins were significantly enriched in cytoskeleton associated activities. Differential succinylated proteins in pathways related to cancer metastasis (focal adhesion, regulation of actin cytoskeleton and tight junction) are marked in Fig. 5d. These down-regulated succinylated proteins with functions closely linked with cellular cytoskeleton activities (ezrin (ZER), alpha-actinin-1 (ACTN1), alpha-actinin-4 (ACTN4) and caldesmon (CALD1)) probably contribute to metastasis in GC.
Table 1. Top ten differentially up- and down-regulated lysine succinylation sites.
| Gene Name | UniProt Accession | Ksucc Position | MaxQuant Score | Modified sequence | Log2 C/N Ratio | Regulation |
|---|---|---|---|---|---|---|
| STAT1 | P42224 | 279 | 93.649 | _K(su)LEELEQK_ | 1.614 | up |
| EPCAM | P16422 | 129 | 109.07 | _RTDK(su)DTEITCSER_ | 1.491 | up |
| SFN | P31947 | 77 | 120.63 | _SNEEGSEEK(su)GPEVR_ | 1.198 | up |
| OAS2 | P29728 | 239 | 63.091 | _K(su)DNFDIAEGVR_ | 1.054 | up |
| GBP1 | P32455 | 487 | 63.624 | _EK(su)EIEVER_ | 1.051 | up |
| ALDH18A1 | P54886 | 409 | 72.34 | _K(su)DLEEAEGR_ | 0.899 | up |
| POR | P16435 | 291 | 56.916 | _K(su)LNQGTER_ | 0.816 | up |
| EEF1G | P26641 | 243 | 63.565 | _QK(su)PQAER_ | 0.787 | up |
| HSP90AB1 | P08238 | 550 | 112.44 | _EGLELPEDEEEK(su)K_ | 0.644 | up |
| CALD1 | Q05682 | 569 | 128.06 | _QQEAALELEELK(su)K_ | −1.112 | down |
| DYNC1H1 | Q14204 | 3284 | 124.29 | _EDLDK(su)VEPAVIEAQNAVK_ | −1.184 | down |
| ANXA6 | P08133 | 478 | 119.39 | _AINEAYK(su)EDYHK_ | −1.210 | down |
| TXNL1 | O43396 | 226 | 120.33 | _SEPTQALELTEDDIK(su)EDGIVPLR_ | −1.244 | down |
| FLNA | P21333 | 865 | 66.989 | _VK(su)VEPSHDASK_ | −1.312 | down |
| COL6A3 | P12111 | 869 | 119.96 | _IIDELNVK(su)PEGTR_ | −1.427 | down |
| TAGLN | Q01995 | 21 | 144.65 | _K(su)YDEELEER_ | −1.662 | down |
| VCL | P18206 | 387 | 90.475 | _K(su)IDAAQNWLADPNGGPEGEEQIR_ | −1.809 | down |
| HSP90B1 | P14625 | 800 | 98.582 | _ESTAEK(su)DEL_ | −2.170 | down |
| MYH11 | P35749 | 1499 | 117 | _ALEEALEAK(su)EELER_ | −2.437 | down |
The potential regulatory roles of lysine succinylation in the TCA cycle and pentose phosphate pathway in gastric cancer
In the differential expressed protein enrichment analysis, we found that proteins functioning in aerobic respiration processes including the TCA cycle and oxidative phosphorylation were down-regulated in disease development, indicating that aerobic respiration is suppressed in the mitochondria of cancer cells. Ksucc has been shown to be closely associated with the regulation of metabolism, leading us to test whether essential enzymes in cellular respiration have Ksucc modifications in cancer cells. We examined protein and Ksucc expression levels of enzymes in two respiration-related pathways: the TCA cycle and the pentose phosphate pathway (PPP) (Fig. 6 and Supplementary Table 5). The TCA cycle is an essential part of aerobic respiration that occurs in mitochondria. PPP uses the intermediate component from glycolysis to generate the reducing substrate NADPH that is essential for oxidative stress response. The activation of PPP has long been studied in cancer cells44,45.
Figure 6. Proteins and Ksucc expression profile in the TCA cycle and pentose phosphate pathway in gastric cancer.

(a) Protein expression profile in the TCA cycle. (b) Proteins expression profile in the pentose phosphate pathway. (c) Ksucc expression profile (normalized against protein level) in the TCA cycle. (d) Ksucc expression profile (normalized against protein level) in the pentose phosphate pathway. Color gray represents no quantitative data in this specific term.
As shown in Fig. 6a, TCA cycle-related enzymes decrease expression in the progression of LNM. On the contrary, several proteins in PPP, such as glucose-6-phosphate 1-dehydrogenasepresent (UniProt ID: P11413), 6-phosphofructokinase type C (UniProt ID: Q01813), and deoxyribose-phosphate aldolase (UniProt ID: Q9Y315), are gradually increased in the process of LNM (Fig. 6b).
Interestingly, we found obvious trends of Ksucc expression levels in these two important pathways. Although most of the enzymes in the TCA cycle pathway were gradually decreased, their Ksucc levels (normalized to their own protein level) were increased in the process of LNM (Fig. 6c). Interestingly, enzymes with increased Ksucc sites localized to the second step of PPP (Fig. 6d). Among them, succinylation of the K48 and K140 sites in fructose-bisphosphate aldolase B (UniProt ID: P05062), and the K6 site in transketolase (UniProt ID: P29401) were significantly increased in the N2 stage. Based on above results and previous reported works of Ksucc in metabolism, we believe that lysine succinylation may play significant negative roles in the TCA cycle pathway while playing positive roles in the pentose phosphate pathway.
Discovery of potential clinical biomarkers of PCNA and lysine succinylation in gastric cancer
Quantitative proteomic approaches enable us to find potential and feasible protein candidates that may function as novel biomarkers or targets. Proliferating cell nuclear antigen (PCNA) has been shown to be closely associated with several cancers, such as breast46, cervical47 and gastric cancer48. In order to confirm the accuracy of MS/MS data and experimental evidences, we performed validation study of PCNA in other 32 pairs of GC tissues using western blotting analysis (Fig. 7a, see remaining 16 pairs in Supplementary Fig. 7a and full-length gels in Supplementary Fig. 7b). We selected four pairs from N0, N1, N2 and N3 stage, respectively. The expression of PCNA was significantly up-regulated in cancer tissues when compared with their matched normal ones that may reveal its roles of potential biomarker. This result was absolutely coincident with our MS/MS data illustrating the accuracy of our results mentioned above.
Figure 7. The expression of PCNA and Ksucc-K569 of CALD1 in gastric cancer clinical tissues.

(a) 16 pairs of GC tissues were involved in the validation experiment by western blotting. We found the expression level of PCNA in cancer tissues was significantly up-regulated when compared with normal ones (see remaining 16 pairs in Supplementary Fig. 7a and full-length gels in Supplementary Fig. 7b). (b) Dot blotting analysis on indicated amount of peptides using purified succinyl-CALD1 (K569) antibody diluted 1:10,000. The corresponding peptides and sequences are CLELEEL-(succinyl) K-KKREER and CLELEELKKKREER, respectively. (c) The Ksucc expression level of K569 in CALD1 protein shows a significantly decreased level when compared to matched normal tissues by western blotting (full-length gels are presented in Supplementary Fig. 8). (d) Quantification of eight pairs show that the decrease of Ksucc-K569 in CALD1 is statistically significant (p = 0.010). (e) Quantification shows that Ksucc-K569 is statistically decreased when normalized against its protein level (p = 0.007).
In order to explore the possibility of Ksucc being a clinical biomarker in GC, we investigated one top-differentially succinylated protein, caldesmon (CALD1), from the above PTM analysis. CALD1 has been reported to be associated with tumor metastasis49 and drug resistance50, and its potential role in GC metastasis was illustrated previously51. This actin-binding protein functions as a significant regulator of podosome formation, thus regulating cancer cell invasion52. Its Ksucc levels are significantly differentially regulated in GC, while its protein expression level is not altered. Thus, we were interested in whether the lysine succinylation of CALD1 can be used as potential biomarker.
We have successfully developed a site-specific primary antibody against succinylation at site K569 of CALD1. The modification specificity of the purified antibody was characterized by ELISA and dot blotting analyses (Fig. 7b). Taking advantage of this site-specific primary antibody, we collected a total of ten pairs of primary human gastric cancer and adjacent normal tissues and detected the level of K569 succinylated CALD1 via western blotting. The results revealed that the succinylation level of CALD1 K569 significantly decreases in GC compared to their matched normal tissues in nine pairs (Fig. 7c). Quantification of eight pairs (excluding two pairs with Ksucc expression levels in tumor tissues too low to be quantified) showed that the decreased level of Ksucc-K569 in CALD1 is statistically significant (p = 0.010) (Fig. 7d). When normalized against its protein level, most pairs of samples also showed a decreased ratio of Ksucc-K569 versus total protein expression of CALD1 in GC (p = 0.007) (Fig. 7e, full-length gels of Ksucc-K569 of CALD1 from ten pairs of GC tissues in Supplementary Fig. 8).
Combined with our MS/MS results, this indicates that Ksucc-K569 of CALD1 may function as a potential biomarker in GC. However, we still need experimental confirmation in more samples to validate this finding.
Potential crosstalk between lncRNAs and lysine succinylation by co-expression detection and protein-protein interaction analysis in gastric cancer
Previous reports identified that lncRNAs can regulate several protein modifications, including acetylation and phosphorylation. For example, lncRNA-DC, exclusively expressed in human conventional dendritic cells (DCs), was reported to directly bind to STAT3 in the cytoplasm and promoted STAT3 phosphorylation on tyrosine-70537. This inspired us to examine relationships between Ksucc and lncRNAs in GC.
In lncRNA microarray analyses, we quantified a total of 11,497 lncRNAs, (Supplementary Table 6). A total of 1,001 lncRNAs were identified to be significantly differentially expressed with fold-change cutoff at 2.053. We established a co-expression patterning-based method to explore the profiling of lncRNAs and Ksucc in GC, and 257 potential co-expressing lncRNA-Ksucc pairs were identified with a co-expression score ≥0.50 (Supplementary Table 6). To further filter the correlation list, we used the STRING interaction database to depict the crosstalks. We defined an association relationship as follows: one succinylated site co-expressing with one lncRNA, and the target protein of the lncRNA interacts with the succinylated protein. We considered that the lncRNA is associated with this succinylated site (Supplementary Fig. 9). With such criteria, we obtained two potential associations of lncRNA-Ksucc with co-expression score ≥0.50 and interaction confidence score ≥0.50. Interestingly, both lncRNA-AX748340 and lncRNA-LOC100507250 negatively associated with the K409 succinylation of delta-1-pyrroline-5-carboxylate synthase (ALDH18A1). Further experiments are required to confirm these two potential correlations between lncRNAs and Ksucc.
Discussion
In this study, we utilized a combination of quantitative transcriptomic and proteomic approaches to investigate the lncRNAs, global proteins, and post-translational modification profiles in GC samples. Quantitative proteomic analysis showed that proteins with up-regulated expression in tumors are enriched in nuclear activity processes such as cell cycle regulation, DNA replication machinery, and mRNA processing. This result is consistent with our knowledge on physiological changes in tumorigenesis, as deregulated DNA replication in cancers can contribute to genomic instability54 and abnormal DNA replication stress is considered a hallmark of cancer that drives cancer development55. Functional clustering between samples with different lymph node statuses also revealed that proteins related to the nucleus tend to be up-regulated in disease development and process of LNM. On the other hand, down-regulated proteins were enriched in cellular respiration and mitochondria activity, suggesting that aerobic respiration is inhibited in GC cells.
We did dynamic clustering to identify proteins which change expression patterns during disease progression. In clusters with protein expression tending to increase with the process of LNM, components in ubiquitination system, RNA processing, heat shock protein system, extracellular structure organization, and focal adhesion were enriched. Clusters with proteins whose expression gradually decrease tend to function in aerobic respiration and stress response. This does not completely overlap with functional enrichment results, as dynamic changes of a protein are often more subtle than overall fold changes. However, common terms between these two analyses reveal that genes in TCA cycle and oxidative phosphorylation are not simply inhibited in tumor cells, instead their expression levels tend to decrease as disease becomes more severe. Thus these common genes have potential not only as cancer biomarkers but also as distinguishing factors for lymph node staging.
Succinylation was recently discovered in many eukaryotic organisms as a conserved type of lysine post-translational modification. Several studies suggest that lysine succinylation regulates metabolism, yet the details of these regulations remain elusive. We selected two important glucose metabolic processes (TCA cycle and PPP) that were known to be significantly regulated in cancer cells and detected if their Ksucc level changes follow specific patterns. The TCA cycle is an essential part of cellular respiration downstream of glycolysis that is known to undergo inhibition in cancer, while the PPP is activated to produce ribonucleotides and NADPH to meet the demand of fast growth in cancer cells45. Essential enzymes in the TCA cycle or PPP indeed had similar regulation trends in our GC samples compared with reports in the literature. For example, increased expression level and activity of glucose-6-phosphate 1-dehydrogenase (G6PD) are observed in cancer cells56, which is consistent with our results. We examined the Ksucc changes in these genes to check for changing patterns. During cancer development, the Ksucc expression levels of enzymes involved in the TCA cycle showed an obvious increasing trend when normalized against dramatically decreasing protein levels, suggesting that Ksucc may play negative roles in TCA cycle enzymes in GC. On the contrary, proteins in PPP are more up-succinylated at later stages of cancers, indicating that activation of lysine succinylation may result in opposite protein expression changes. The TCA cycle generates and consumes succinyl-CoA, an intermediate substrate for lysine succinylation, so we speculate that the balance between succinyl-CoA generation and consumption in metabolic pathways might contribute to this sophisticated regulation of Ksucc and protein expression in the tumor-activated glucose metabolic tuning. Future studies using animal models containing both desuccinylation deficiency (SIRT5KO) and malignant tumors might be able to further study the mechanism of succinylation regulation in tumorigenesis33.
By using RNA microarray analyses, differentially expressed lncRNAs were determined between GC tissues and their matched normal tissues. Several known lncRNAs identified in our study, including H19, CCAT1, GAS5 and FER1L4, have previously been recognized to play significant roles in GC development and progression57. LncRNAs regulate several protein modifications including acetylation and phosphorylation. In addition, ncRNAs of different origins can regulate histone methylation, resulting in genome regulatory functions58. We established an integrated analysis to explore correlations between lncRNAs and lysine succinylation in GC. Surprisingly, only two pairs of lncRNA-Ksucc were identified, and both lncRNAs targeted to a single succinylation site (K409 site of ALDH18A1).
Using the advantages of proteomics approaches, we provide a complete and detailed picture of lncRNA, protein, and lysine succinylation profiles in GC samples. Our study may increase our understanding of tumorigenesis and LNM, especially within energy-related regulation in GC. Moreover, this study provides a novel type of potential biomarker for GC diagnostics.
Methods
Gastric Cancer Sample Collection and Preparation
The four pairs of human tissue samples used in our study were obtained from patients diagnosed with GC who underwent surgical resection at the First Hospital of China Medical University (Shenyang, China). The matched adjacent non-tumor tissues were obtained from the furthest part of the resected specimens away from the tumor (>5 cm). The samples were washed with ice-cold phosphate buffered saline (PBS) (GE Healthcare, Beijing, China) to remove residual blood after surgical resection, and then immediately snap-frozen in liquid nitrogen and stored at −80 °C for further analyses. There were no preoperative radiotherapy, chemotherapy, or other therapies done on these four GC patients. This study was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the Research Ethics Committee of China Medical University (Shenyang, China). Informed consents were obtained from all patients. A summary of the clinical and pathological data for these GC patients is shown in Supplementary Table 1. Patients were staged according to the 7th edition of the UICC/AJCC TNM staging system.
Protein Extraction, Trypsin Digestion, TMT Labeling, HPLC Fractionation and Lysine Succinylated Peptides Affinity Enrichment
GC tissues were firstly grounded into power with liquid nitrogen and then suspended in ice-cold lysis reagent (8 M urea, 5 mM dithiothreitol (DTT), 1% (v/v) protease inhibitor cocktail, 3 μM trichostatin A (TSA), 50 mM nicotinamide (NAM)) with occasional sonication. Cell lysates were centrifuged at 12,000 g at 4 °C for 10 min, and the resulting supernatants were collected. The protein concentration was determined by the 2-D Quant kit (GE Healthcare, USA) according to the manufacturer’s instructions. Afterwards, proteins were precipitated with 15% trichloroacetic acid (TCA) for 4 h at 4 °C and the resulting precipitate was washed for three times with −20 °C acetone. Dried protein pellets were re-suspended in 100 mM tetraethylammonium bromide (TEAB) and digested with trypsin at an enzyme-to-substrate ratio of 1:50 for 12 h at 37 °C. Then the peptides were reduced with DTT and alkylated with iodoacetamide (IAA) in darkness. To ensure complete digestion, the second digestion was conducted by adding trypsin at enzyme-to-substrate ration of 1:100 for 4 h at 37 °C.
Then, tandem-mass-tag (TMT-6 plex) labeling was performed for the quantification of global proteome and lysine succinylome. Four pairs of gastric cancer clinical tissues were involved in our analysis and we divided them into two independent experiments. In the first experiment, the tumor and normal tissues of S2 were labeled with TMT-130 and 131, respectively. In the second experiment, the tumor and normal tissues of S3, S4 and S5 were labeled with TMT-126, 127, 128, 129, 130 and 131, respectively. To lower the complexity of the peptides mixture and enhance the accuracy and throughout of protein identification, the tryptic peptides were fractionated by high pH reverse-phase HPLC using Agilent 300Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length) with solvent A of 98% H2O and 2% acetonitrile containing 10 mM ammonium formate (pH 10) and solvent B of 2% H2O and 98% acetonitrile containing 10 mM ammonium formate. The LC gradient was from 2% to 60% solvent B for 80 min to generate 80 fractions with 1 min per fraction and then combined into 18 fractions for global proteome and 8 fractions for lysine succylome. The fractionated samples were dried by vacuum centrifugation and stored at −20 °C for further usage.
For lysine succinylome analyses, antibody-based affinity enrichment was performed. Briefly, the tryptic peptides were re-dissolved in NETN buffer (100 mM NaCl, 1 mM ethylene diamine tetraacetic acid (EDTA), 50 mM Tris-HCl, 0.5% NP-40, pH 8.0) and incubated with anti-succinyllysine antibody agarose conjugated beads (PTM Biolabs, Chciago, United States) in a ratio of 15 μL beads/mg protein at 4 °C overnight with gentle shaking. After incubation, the beads were washed four times with NETN buffer followed by twice washes with ddH2O. The bound peptides were eluted with 1% trifluoroacetic acid (TFA) and dried under a vacuum.
LC-MS/MS Analysis
Peptides were dissolved in solvent A (0.1% formic acid in 2% acetonitrile (ACN), 98% H2O), directly loaded onto a reversed-phase pre-column (Acclaim PepMap100 C18 column, 3 μm, 75 μm × 2 mm, 100 Å, Thermo Scientific). Peptide separation was performed using a reversed-phase analytical column (Acclaim PepMap RSLC C18 column, 50 μm × 15 mm, 2 μm, 100 Å, Thermo Scientific). The gradient was comprised of an increase from 6% to 22% solvent B (0.1% FA in 98% ACN) for 24 min, 22% to 36% for 10 min and climbing to 80% in 3 min then holding at 80% for the last 3 min, at a constant flow rate of 300 nl/min on an EASY-nLC 1000 UPLC system, the resulting peptides were analyzed by Q Exactive Plus hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific).
The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q Exactive Plus coupled online to the UPLC. Intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using NCE setting as 28; ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 1.5E4 in the MS survey scan with 15.0 s dynamic exclusion. The electrospray voltage applied was 2.0 kV. Automatic gain control (AGC) was used to prevent overfilling of the ion trap; 5E4 ions were accumulated for generation of MS/MS spectra. For MS scans, the m/z scan range was 350 to 1600. For TMT quantification, the first mass was set as 100 m/z.
Quantification Method
Peptide quantitation ratio based on the relative intensities of reporter ions within an MS/MS spectrum. A peptide match will only be used for quantitation if it meets this criterion: Minimum score >20, Maximum expect <0.05, at least homology, unique to one protein hit. For each peptide ratio, a correction factor is applied such that the median of the ratios for all peptide matches that pass the quality tests is unity. The median unique peptide ratio is selected to represent the protein ratio with at least two unique peptide ratios. And the statistic algorithm will give each protein a p value calculated from the peptide ratios which could indicate the accuracy of protein quantitation. And only proteins with p value that < 0.05 were considered accurate quantitation.
Bioinformatics Analyses
Protein functional annotation, enrichment, motif, enrichment-based clustering, dynamic clustering, protein-protein interaction analysis, and other analyses were used in our study. Detailed descriptions of these analyses are described in Supplementary Information.
Antibody Generation and Western Blotting
The polyclonal antibody specific for lysine succinylation at K569 of caldesmon (CALD1) was produced in collaboration with PTM BioLab, Inc. Eight week old rabbits were immunized with the succinylated antigen peptide (CLELEEL-(succinyl) K-KKREER) of CALD1. General procedures of antisera screening, purification and antibody characterization were followed. The specificity of the purified antibody against this specific epitope was characterized by ELISA and dot blot analyses.
For western blotting, a total of 20 ug of protein were separated by 10% SDS-PAGE then transferred to a polyvinylidene difluoride (PVDF) or nitrocellulose (NC) membrane. The membrane was blocked with 5% (w/v) skim milk in TBST and incubated overnight at 4 °C with the pan anti-succinyl lysine antibody (1:1000 dilution; PTM Biolabs), anti-GAPDH antibody (1:1000; Cell Signaling Technology), anti-PCNA antibody (1:500; HuaAn Biotechnology, Hangzhou) or anti-Caldesmon antibody (1:1000; Wanleibio, Shenyang). The membrane was then incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:5000; Thermo) for 1 h at room temperature. The membrane was washed with TBST buffer and visualized with enhanced chemiluminescence (ECL) immunoblotting detection reagents (Millipore). Quantitation of western blotting was performed using the measuring function in ImageJ software (http://rsbweb.nih.gov/ij/).
Additional Information
How to cite this article: Song, Y. et al. Quantitative global proteome and lysine succinylome analyses provide insights into metabolic regulation and lymph node metastasis in gastric cancer. Sci. Rep. 7, 42053; doi: 10.1038/srep42053 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank the department of Surgical Oncology of The First Hospital of China Medical University for providing human GC samples. We also thank the College of China Medical University and Jingjie PTM BioLab (Hangzhou) for technical assistance in experiments. This work was supported by National Science Foundation of China (81372549), the Special Prophase Program for National Key Basic Research Program of China (No. 2014CB560712) and Clinical Capability Construction Project for Liaoning Provincial Hospitals (LNCCC-A01-2014).
Footnotes
The authors declare no competing financial interests.
Author Contributions Y.S. and J.W. contributed equally to this work. Y.S. and J.W. designed experiments. Z.C. participated in the conception of the idea. J.S., X.C. and Y.W. contributed to the literature search. Y.S., J.W. and C.C. performed the experiments. Z.C., J.S., X.C. and Y.W. helped to perform the experiments. J.W. and P.G. analyzed data. Y.S. and J.W. contributed to drafting and editing of the manuscript. Z.W. participated in the conception and coordination. All authors have read and approved the final manuscript for publication.
References
- Torre L. A. et al. Global cancer statistics, 2012. CA: a cancer journal for clinicians 65, 87–108, doi: 10.3322/caac.21262 (2015). [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209, doi: 10.1038/nature13480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Y. et al. Peritoneal carcinomatosis and lymph node metastasis are prognostic indicators in patients with Borrmann type IV gastric carcinoma. Hepato-gastroenterology 49, 874–877 (2002). [PubMed] [Google Scholar]
- Hyung W. J., Lee J. H., Choi S. H., Min J. S. & Noh S. H. Prognostic impact of lymphatic and/or blood vessel invasion in patients with node-negative advanced gastric cancer. Annals of surgical oncology 9, 562–567 (2002). [DOI] [PubMed] [Google Scholar]
- Deng J. Y. & Liang H. Clinical significance of lymph node metastasis in gastric cancer. World journal of gastroenterology 20, 3967–3975, doi: 10.3748/wjg.v20.i14.3967 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., He Q., Cui J., Bian S. & Chen L. Lymph node metastasis in early gastric cancer. Chinese medical journal 127, 560–567 (2014). [PubMed] [Google Scholar]
- Zong J., Guo C., Liu S., Sun M. Z. & Tang J. Proteomic research progress in lymphatic metastases of cancers. Clinical & translational oncology: official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 14, 21–30, doi: 10.1007/s12094-012-0757-7 (2012). [DOI] [PubMed] [Google Scholar]
- Ran X. et al. A quantitative proteomics study on olfactomedin 4 in the development of gastric cancer. International journal of oncology 47, 1932–1944, doi: 10.3892/ijo.2015.3168 (2015). [DOI] [PubMed] [Google Scholar]
- Liu P. J. et al. In-depth proteomic analysis of six types of exudative pleural effusions for nonsmall cell lung cancer biomarker discovery. Molecular & cellular proteomics: MCP 14, 917–932, doi: 10.1074/mcp.M114.045914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S. et al. Proteomic maps of breast cancer subtypes. Nature communications 7, 10259, doi: 10.1038/ncomms10259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell, doi: 10.1016/j.cell.2016.05.069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62, doi: 10.1038/nature18003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran S., Lippens G., Steen H. & Gunawardena J. Post-translational modification: nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley interdisciplinary reviews. Systems biology and medicine 4, 565–583, doi: 10.1002/wsbm.1185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. & Jensen O. N. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 9, 4632–4641, doi: 10.1002/pmic.200900398 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. et al. Global analysis of lysine acetylation in strawberry leaves. Frontiers in plant science 6, 739, doi: 10.3389/fpls.2015.00739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoliatte F. et al. Large-scale analysis of lysine SUMOylation by SUMO remnant immunoaffinity profiling. Nature communications 5, 5409, doi: 10.1038/ncomms6409 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang Z. et al. Identification of lysine succinylation as a new post-translational modification. Nature chemical biology 7, 58–63, doi: 10.1038/nchembio.495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C. et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Molecular & cellular proteomics: MCP 10, M111 012658, doi: 10.1074/mcp.M111.012658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain J. A., Solis N. & Cordwell S. J. Beyond gene expression: the impact of protein post-translational modifications in bacteria. Journal of proteomics 97, 265–286, doi: 10.1016/j.jprot.2013.08.012 (2014). [DOI] [PubMed] [Google Scholar]
- Bouchut A., Chawla A. R., Jeffers V., Hudmon A. & Sullivan W. J. Jr. Proteome-wide lysine acetylation in cortical astrocytes and alterations that occur during infection with brain parasite Toxoplasma gondii. PloS one 10, e0117966, doi: 10.1371/journal.pone.0117966 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogachek M. V. et al. Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer cell 25, 748–761, doi: 10.1016/j.ccr.2014.04.008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D. et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer cell 23, 464–476, doi: 10.1016/j.ccr.2013.02.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning B., Li W., Zhao W. & Wang R. Targeting epigenetic regulations in cancer. Acta biochimica et biophysica Sinica 48, 97–109, doi: 10.1093/abbs/gmv116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. et al. Liquid chromatography mass spectrometry-based O-glycomics to evaluate glycosylation alterations in gastric cancer. Proteomics. Clinical applications 10, 206–215, doi: 10.1002/prca.201500041 (2016). [DOI] [PubMed] [Google Scholar]
- Li X. et al. Systematic identification of the lysine succinylation in the protozoan parasite Toxoplasma gondii. Journal of proteome research 13, 6087–6095, doi: 10.1021/pr500992r (2014). [DOI] [PubMed] [Google Scholar]
- Pan J., Chen R., Li C., Li W. & Ye Z. Global Analysis of Protein Lysine Succinylation Profiles and Their Overlap with Lysine Acetylation in the Marine Bacterium Vibrio parahemolyticus. Journal of proteome research 14, 4309–4318, doi: 10.1021/acs.jproteome.5b00485 (2015). [DOI] [PubMed] [Google Scholar]
- Kosono S. et al. Changes in the Acetylome and Succinylome of Bacillus subtilis in Response to Carbon Source. PloS one 10, e0131169, doi: 10.1371/journal.pone.0131169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L. et al. First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. Journal of proteome research 14, 107–119, doi: 10.1021/pr500859a (2015). [DOI] [PubMed] [Google Scholar]
- Yang M. et al. Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Molecular & cellular proteomics: MCP 14, 796–811, doi: 10.1074/mcp.M114.045922 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y. et al. Altered acetylation and succinylation profiles in Corynebacterium glutamicum in response to conditions inducing glutamate overproduction. MicrobiologyOpen 5, 152–173, doi: 10.1002/mbo3.320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D. et al. Global Proteome Analyses of Lysine Acetylation and Succinylation Reveal the Widespread Involvement of both Modification in Metabolism in the Embryo of Germinating Rice Seed. Journal of proteome research 15, 879–890, doi: 10.1021/acs.jproteome.5b00805 (2016). [DOI] [PubMed] [Google Scholar]
- Weinert B. T. et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports 4, 842–851, doi: 10.1016/j.celrep.2013.07.024 (2013). [DOI] [PubMed] [Google Scholar]
- Park J. et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Molecular cell 50, 919–930, doi: 10.1016/j.molcel.2013.06.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey M. D. et al. Dysregulated metabolism contributes to oncogenesis. Seminars in cancer biology 35 Suppl, S129–150, doi: 10.1016/j.semcancer.2015.10.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. Y., Pan H. F., Leng R. X. & Ye D. Q. Long noncoding RNAs: novel insights into gastric cancer. Cancer letters 356, 357–366, doi: 10.1016/j.canlet.2014.11.005 (2015). [DOI] [PubMed] [Google Scholar]
- Wan G. et al. A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. The EMBO journal 32, 2833–2847, doi: 10.1038/emboj.2013.221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 344, 310–313, doi: 10.1126/science.1251456 (2014). [DOI] [PubMed] [Google Scholar]
- Papaioannou M. D. et al. Loss of Dicer in Sertoli cells has a major impact on the testicular proteome of mice. Molecular & cellular proteomics: MCP 10, M900587MCP900200, doi: 10.1074/mcp.M900587-MCP200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. R. et al. Quantitative proteomic and genomic profiling reveals metastasis-related protein expression patterns in gastric cancer cells. Journal of proteome research 5, 2727–2742, doi: 10.1021/pr060212g (2006). [DOI] [PubMed] [Google Scholar]
- Huang D., Li C. & Zhang H. Hypoxia and cancer cell metabolism. Acta biochimica et biophysica Sinica 46, 214–219, doi: 10.1093/abbs/gmt148 (2014). [DOI] [PubMed] [Google Scholar]
- Cho D. C. Targeting the PI3K/Akt/mTOR pathway in malignancy: rationale and clinical outlook. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy 28, 373–381, doi: 10.1007/s40259-014-0090-5 (2014). [DOI] [PubMed] [Google Scholar]
- Jiang W. G. et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Seminars in cancer biology 35 Suppl, S244–275, doi: 10.1016/j.semcancer.2015.03.008 (2015). [DOI] [PubMed] [Google Scholar]
- Hirschey M. D. & Zhao Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Molecular & cellular proteomics: MCP 14, 2308–2315, doi: 10.1074/mcp.R114.046664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra K. C. & Hay N. The pentose phosphate pathway and cancer. Trends in biochemical sciences 39, 347–354, doi: 10.1016/j.tibs.2014.06.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P., Du W. & Wu M. Regulation of the pentose phosphate pathway in cancer. Protein & cell 5, 592–602, doi: 10.1007/s13238-014-0082-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurikova M., Danihel L., Polak S. & Varga I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta histochemica 118, 544–552, doi: 10.1016/j.acthis.2016.05.002 (2016). [DOI] [PubMed] [Google Scholar]
- Madhumati G., Kavita S., Anju M., Uma S. & Raj M. Immunohistochemical Expression of Cell Proliferating Nuclear Antigen (PCNA) and p53 Protein in Cervical Cancer. Journal of obstetrics and gynaecology of India 62, 557–561, doi: 10.1007/s13224-012-0180-6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N. et al. Prognostic evaluation of Nanog, Oct4, Sox2, PCNA, Ki67 and E-cadherin expression in gastric cancer. Medical oncology 32, 433, doi: 10.1007/s12032-014-0433-6 (2015). [DOI] [PubMed] [Google Scholar]
- Chang K. P. et al. Overexpression of caldesmon is associated with lymph node metastasis and poorer prognosis in patients with oral cavity squamous cell carcinoma. Cancer 119, 4003–4011, doi: 10.1002/cncr.28300 (2013). [DOI] [PubMed] [Google Scholar]
- De Marchi T. et al. Annexin-A1 and caldesmon are associated with resistance to tamoxifen in estrogen receptor positive recurrent breast cancer. Oncotarget 7, 3098–3110, doi: 10.18632/oncotarget.6521 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q. et al. Identification and functional validation of caldesmon as a potential gastric cancer metastasis-associated protein. Journal of proteome research 12, 980–990, doi: 10.1021/pr3010259 (2013). [DOI] [PubMed] [Google Scholar]
- Yoshio T. et al. Caldesmon suppresses cancer cell invasion by regulating podosome/invadopodium formation. FEBS letters 581, 3777–3782, doi: 10.1016/j.febslet.2007.06.073 (2007). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Microarray expression profile analysis of long non-coding RNAs in human gastric cardiac adenocarcinoma. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 33, 1225–1238, doi: 10.1159/000358692 (2014). [DOI] [PubMed] [Google Scholar]
- Kotsantis P., Jones R. M., Higgs M. R. & Petermann E. Cancer therapy and replication stress: forks on the road to perdition. Advances in clinical chemistry 69, 91–138, doi: 10.1016/bs.acc.2014.12.003 (2015). [DOI] [PubMed] [Google Scholar]
- Macheret M. & Halazonetis T. D. DNA replication stress as a hallmark of cancer. Annual review of pathology 10, 425–448, doi: 10.1146/annurev-pathol-012414-040424 (2015). [DOI] [PubMed] [Google Scholar]
- Furuta E., Okuda H., Kobayashi A. & Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochimica et biophysica acta 1805, 141–152, doi: 10.1016/j.bbcan.2010.01.005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y. et al. LncRNAs: emerging biomarkers in gastric cancer. Future oncology 11, 2427–2441, doi: 10.2217/fon.15.175 (2015). [DOI] [PubMed] [Google Scholar]
- Joh R. I., Palmieri C. M., Hill I. T. & Motamedi M. Regulation of histone methylation by noncoding RNAs. Biochimica et biophysica acta 1839, 1385–1394, doi: 10.1016/j.bbagrm.2014.06.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.