

ABSTRACT
Establishing and maintaining conformational integrity of monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs) during development and manufacturing is critical for ensuring their clinical efficacy. As presented here, we applied site-specific carboxyl group footprinting (CGF) for localized conformational interrogation of mAbs. The approach relies on covalent labeling that introduces glycine ethyl ester tags onto solvent-accessible side chains of protein carboxylates. Peptide mapping is used to monitor the labeling kinetics of carboxyl residues and the labeling kinetics reflects the conformation or solvent-accessibility of side chains. Our results for two case studies are shown here. The first study was aimed at defining the conformational changes of mAbs induced by deglycosylation. We found that two residues in CH2 domain (D268 and E297) show significantly enhanced side chain accessibility upon deglycosylation. This site-specific result highlighted the advantage of monitoring the labeling kinetics at the amino acid level as opposed to the peptide level, which would result in averaging out of highly localized conformational differences. The second study was designed to assess conformational effects brought on by conjugation of mAbs with drug-linkers. All 59 monitored carboxyl residues displayed similar solvent-accessibility between the ADC and mAb under native conditions, which suggests the ADC and mAb share similar side chain conformation. The findings are well correlated and complementary with results from other assays. This work illustrated that site-specific CGF is capable of pinpointing local conformational changes in mAbs or ADCs that might arise during development and manufacturing. The methodology can be readily implemented within the industry to provide comprehensive conformational assessment of these molecules.
KEYWORDS: Antibody-drug conjugate, carboxyl group footprinting, covalent labeling, hydrogen-deuterium exchange, mass spectrometry, monoclonal antibody, Protein conformation, rate constant, side chains, solvent accessibility
Abbreviations
- mAb
monoclonal antibody
- ADC
antibody-drug conjugate
- HDX
hydrogen/deuterium exchange
- MS
mass spectrometry
- vcMMAE
valine-citrulline-monomethyl auristatin E
- CD
circular dichroism
- DSC
differential scanning calorimetry
- IgG
Immunoglobulin G
- Fc
fragment crystallizable
- Fab
fragment antigen-binding
- LC/MS
liquid chromatography mass spectrometry
- RC
rate constant
- GEE
glycine ethyl ester
- EDC
1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride
Introduction
Monoclonal antibodies (mAbs) have played a substantial role in advancing options for treating many types of diseases, including cancer, infectious diseases and immune-mediated disorders, during the past two decades.1 More recently, antibody-drug conjugates (ADCs) consisting of a mAb scaffold that is chemically linked to a cytotoxic small-molecule drug have emerged as effective cancer therapies and therapeutic candidates.,2,3 Several properties of mAbs make them highly useful in a therapeutic setting: they bind specifically to a cognate epitope, have relatively long half lives in vivo and may elicit a clinically relevant immune response. In ADCs, the targeting specificity of the mAb component ensures that the cytotoxic payload is delivered only to cancer cells expressing the antigen, thereby avoiding some of the more debilitating side effects of traditional chemotherapy.4,5
Therapeutic mAbs and ADCs are complicated multimeric molecules, and chemical modifications to their primary amino acid sequence may be incurred during the manufacturing and storage. There is potential for the clinical efficacy of mAbs and ADCs to be compromised by post-translational modifications (PTMs) and chemical modifications due to their effects on antigen binding or biological clearance. Disruption of mAb/ADC conformation or higher-order structure also has the potential to impair the potency and stability of the therapeutic. While indirect evidence of conformational changes might be apparent in some analytical assays used for product release and characterization such as binding, potency assays and some chromatographic assays,6 more traditional biophysical assays such as circular dichroism (CD), fluorescence spectroscopy, and differential scanning calorimetry (DSC) have typically been used for conformational characterization.7-11 These assays can monitor the global state conformation of protein samples in a simple and rapid manner, but are not suitable for detecting small localized, but potentially significant, conformational differences that may exist between protein samples.
As use of high-resolution mass spectrometry (MS) has become more commonplace, chemical labeling coupled with MS has become a viable approach for investigating protein conformation. This general approach has contributed to the understanding of conformational features of globular proteins, membrane proteins and large protein complexes at the peptide or residue level.12-19 While MS-based technology has not been widely used for conformational elucidation of protein therapeutics yet, some applications demonstrating the utility of the approach can be found in the literature. Hydrogen/deuterium exchange (HDX) coupled with MS is the most common approach used in the studies of biotherapeutic proteins.20-30 Classic or “bottom-up” HDX MS monitors isotope exchange kinetics of amide hydrogens on a protein to gain conformational insights into the protein backbone. Solvent-exposed and non-structured amides will undergo rapid HDX, while those buried in protein tertiary structures or engaged in stable secondary structures will show much slower HDX. HDX MS has been successfully applied to reveal the impact of various chemical modifications and post-translational modifications on the conformation and dynamics of IgG molecules.20,21,26,27 The role of N-glycans on the function and stability of mAbs has also been characterized by HDX MS21,24 while others have used HDX MS to assess the structural changes associated with charge and size variants of mAbs.22,25 To achieve a peptide-level resolution, HDX MS typically utilizes proteolytic enzymes to cleave isotope-labeled protein samples into peptides that are subsequently separated by liquid chromatography and detected by MS. However, due to the labile nature of HDX, rapid back-exchange of deuterium to hydrogen during the sample handling is always a concern because the back-exchange can result in a false readout due to the method induced loss of deuterium labels. To minimize the back-exchange, HDX MS requires rapid enzymatic treatment and chromatographic separation (usually within 10 min) at quenched conditions (0°C and pH 2.5). This requirement limits its applications for studying protein samples that need extensive post-labeling purification or separation. Often protein therapeutics are formulated in a complex matrix that is essential to maintain the active conformation and stability, and the matrix may not be compatible with downstream MS analysis. To measure the HDX kinetics of individual peptides by MS, additional post-labeling purification may be needed. The inherent challenge of analyzing protein samples that require non-MS-compatible matrices tends to limit the application of HDX MS.
Covalent labeling is an alternative approach for studying therapeutic protein molecules. In principle, covalent labeling coupled with MS (termed “footprinting” technology)15-18,31-38 is orthogonal method to HDX MS because covalent labeling utilizes reagents that target and react with amino acid side chains instead of protein backbone. Solvent-exposed side chains are labeled readily in a protein solution and those buried in the protein higher-order structure or interacted with other partners/molecules are more protected and less-labeled. MS-based peptide mapping is then used to measure the labeling kinetics of individual side chains. The labeling kinetics reflect the side chain conformation or accessibility present in the solution. From the standpoint of technical execution, covalent labeling has some advantages over “labile” labeling techniques such as HDX because the chemical label is stable and not subject to back-exchange. Eliminating back-exchange allows for greater flexibility in sample handling and makes this approach attractive for studying protein samples in complex-challenging matrices. Extensive post-labeling protein purification and concentration steps can be included to minimize the interference of excipient molecules (such as polymers, detergents, membranes, etc.) with downstream MS analysis. Various post-labeling chromatographic methods and enzymatic treatments can be used to achieve good separation and detection of peptides and high overall sequence coverage.
Different covalent labeling strategies have been developed over the years. Some of them target specific types of amino acid side chains, whereas others are more non-specific. Hydroxyl radical footprinting is one of the commonly used covalent labeling techniques.15,37,39 Hydroxyl radicals can label up to 16 of 20 amino acid side chains and offer excellent sequence coverage. The structural information obtained can be quite detailed and rich, but the approach is impractical for many investigators because special equipment for generation of radicals is usually required. Moreover, due to the extremely high reactivity of hydroxyl radicals, the approach requires a comprehensive search for multiple different labeling products, and the data processing and interpretation can be very time-consuming and challenging. While different covalent labeling strategies have been reported, fewer can be used in a simple and rapid manner. EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride)-mediated carboxyl group footprinting (CGF) represents a promising strategy that is well suited to investigating the protein conformation of biopharmaceuticals. The approach makes use of simple bench-top chemistry that introduces GEE tags on solvent-accessible side chains of carboxyl residues,32,35,36,40 and requires no special equipment and can be done in any analytical lab. The specificity of labeling on Asp and Glu residues makes data analysis and interpretation relatively simple and straightforward, assuming that there are sufficient D/E residues distributed throughout the primary sequence. In the case of mAbs, approximately 10% of the sequence is composed of D/E residues, so the CGF approach can provide reasonable coverage,32 and the subsequent trypsin peptide mapping used for detecting and quantitating the extent of labeling does not have to vary significantly from standard methodologies used for in-depth PTM characterization within the industry.
In this work, we aimed to develop a site-specific CGF methodology to provide localized conformational insights into mAbs and ADCs. Site-specific CGF was performed on an IgG1 molecule, herein referred to as mAb-A, and deglycosylated mAb-A to demonstrate that the site-specific CGF is capable of pinpointing subtle and highly localized conformational differences across mAb samples. Site-specific CGF was then applied to ADC characterization to further our understanding of the conformational impact of drug-conjugation on the antibodies. We believe the site-specific CGF methodology can be readily implemented in the industry and generally applied to mAbs and ADCs for localized conformational interrogation.
Results
mAb vs. deglycosylated mAb
To interrogate the conformational changes in a mAb induced by deglycosylation, CGF was performed on mAb-A and deglycosylated mAb-A in parallel as described in Materials and Methods. The labeled samples were then denatured and fully reduced with dithiothreitol (DTT). The labeling kinetics were first monitored at the subunit level by acquiring light chain (LC) and heavy chain (HC) mass spectra as a function of labeling time. As expected, the LC or HC of labeled samples displayed +85 Da adducts that are characteristic of GEE incorporation. No additional fragments were detected compared with unlabeled samples (data not shown). Fig. 1 illustrates the deconvoluted mass of HC for labeled deglycosylated mAb-A. After labeling for 2 min, the unlabeled HC with a mass of 49379 Da was still the predominant species (Fig. 1A), but satellite peaks with a mass shift of +85 Da are clearly apparent. As expected, continuing increases in labeling time yield an increase in the +85 Da satellite peaks in the HC and a corresponding decrease in the intensity of unlabeled HC (Fig. 1B and 1C). The results from monitoring GEE incorporation at the subunit level suggested that the labeling conditions were appropriate, generally mild and did not cause fragmentation or cross-linking to the mAb.
Figure 1.

Deconvoluted mass spectra of heavy chain (HC) for labeled deglycosylated mAb-A. (A) labeling time = 2 min, (B) labeling time = 5 min, (C) labeling time = 10 min.
In order to assess the labeling kinetics of each D/E residue distributed throughout the mAbs primary sequence, all labeled samples were subjected to trypsin peptide mapping as described in Materials and Methods. Fig. 2 shows a representation of the primary sequence of mAb-A superimposed above the recovered tryptic peptides, and the distribution of D/E residues throughout the sequence is indicated in red font. Coverage of ∼95% of the amino acid sequence in mAb-A was obtained in the tryptic peptide maps and all subsequent quantitative and qualitative comparisons were performed on the basis of these data sets. Many of the covalent labeling studies discussed in the literature report the level of modification seen at the peptide level instead of the level of modification seen at individual amino acid sites. It is especially common for hydroxyl radical-based labeling to be analyzed and presented at the peptide level.6,40,42 because the approach labels up to ∼16 of the 20 amino acids and usually results in multiple different modifications within a peptide. It is very challenging to identify and separate each individual modified species due to the extreme complexity of MS/MS data and coelution of peptide isomers. Residue level of data analysis has been reported in some cases where the studied proteins were considerably smaller than mAbs.38 Nevertheless, it remains difficult to achieve a residue level of modification assignment and separation when applying hydroxyl radical labeling to large proteins like mAbs.41 Since EDC-mediated GEE labeling is highly specific for D and E carboxylates, identifying labeling sites at the amino acid level is relatively straightforward. MAbs typically contain more than one D/E residue in tryptic peptides, thus quantitating CGF results at the peptide level (and not amino acid level) may result in an averaging out of subtle conformational differences in comparative studies. In this work, our goal was to achieve site-specific CGF results and provide highly localized conformational information for mAbs and ADCs. For individual labeled tryptic peptides, we primarily observed a mass shift of +85.0527 (predominant form), and very low levels of +57.0215 (label hydrolysis product) or +170.1055 Da (doubly modified, only for complementarity-determining region peptides). All these modifications were separated chromatographically and site-specifically assigned to particular D/E residues. For any peptide consisting of n labeling sites (D/E residues), fraction unlabeled, Fun (i), of a specific labeling site i was determined by Eq. (1) and Eq. (2). Labeling rate constant (RC), RC(i), of the specific site i was then determined by the dose-response plot Fun (i) as a function of labeling time.
Figure 2.
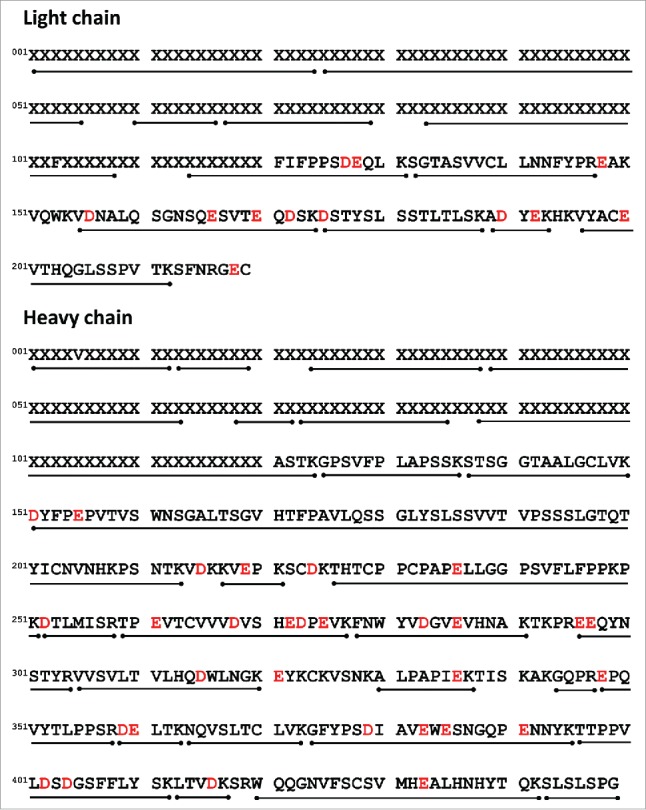
Primary sequence of mAb-A with amino acid position numbering. Each black bar under the sequence indicates the recovered tryptic peptide. Potential labeling sites (D/E residues) are highlighted in red color. Variable domain residues are represented with “X.”
The labeled HC peptide 258–280 (259TPEVTCVVVDVSHEDPEVK) represents one of the most complicated scenarios in that it contains 5 potential labeling sites (D/E residues), and is therefore a good example to illustrate the data analysis and RC determination process. Extracted ion chromatogram (EIC) of the unmodified peptide and the +85.0527 Da modified species in the labeled, deglycosylated mAb-A are shown in Fig. 3A and B, respectively. There are extremely low levels of +57.0215 Da modified species (which are not shown), but no detectable doubly labeled species found on this peptide. Based on the EIC shown in Fig. 3B, there are likely 5 unique modified species. MS/MS data from the 5 individual peaks was used to confidently assign each peak to a specific labeled site (shown in Fig. S1) and the identified labeling site was shown on the top of each peak. With the precise labeling localization and separation, we are able to determine the RC of each labeling site by generating a dose-response plot of each specific labeling site using Eq. (1)-(2). For comparison, first we determined the averaged RC of the peptide (Fig. 4A) by generating a dose-response plot of the peptide using Eq. (3). Comparing the averaged RC of the peptide in the glycosylated mAb (0.73/hr) and the deglycosylated mAb (0.84/hr), one could draw the conclusion that deglycosylation does not induce a significant change in side chain solvent accessibility in this peptide based on the error bar (Fig. 4A). At a residue level, however, the RCs of individual D/E amino acids in this peptide (Fig. 4 B-D) indicate that there are profound differences between these samples. For example, the RC for the Asp residue at position 268 increased over 20-fold after deglycosylation. This suggests that the side chain accessibility of D268 changed substantially after deglycosylation and highlights the advantages of site-specific RC comparison for pinpointing highly localized conformational changes in mAbs that would be missed by traditional peptide-level averaged RC comparisons.
Figure 3.
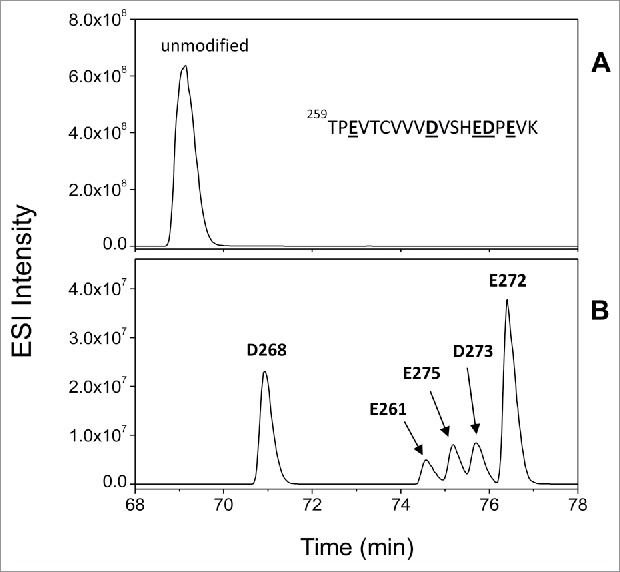
Extracted ion chromatogram (EIC) of the HC peptide 259–280 unmodified (A) and different +85.0527 Da modified species (B) from the labeled deglycosylated mAb-A. The carboxyl residue on each individual modified species denotes the identified labeling site based on MS/MS data shown in S1.
Figure 4.

Dose response plots of the whole peptide 259TPEVTCVVVDVSHEDPEVK (A), or individual residues E261 (B), D268 (C), E272 (D) in intact or deglycosylated mAb-A. Black color represents the intact mAb and red color represents deglycosylated mAb-A and each data point is an average of three experiments. The solid lines show the best fit to first-order exponential equation on which the RC of the peptide or individual residues was determined and shown on the right.
As shown in Fig. 2, the recovered tryptic peptides of mAb-A include 59 D/E residues that will act as structural probes for the conformational interrogation of the mAb in the carboxyl labeling strategy. Fig. 5A shows the site-specific CGF of mAb-A (top) and deglycosylated mAb-A (bottom). The labeling kinetics of the 59 D/E residues that are distributed across the mAb primary structure are quite different and the RCs differ by over three orders of magnitude. This result suggests that there are profound differences in side-chain accessibility, which is an indicator for different side chain conformations. Note that some bars are absent in the plots depicted in Fig. 5A, this is because either the corresponding D/E residues are not labeled or their RCs are less than 0.002 hr−1. It was generally established that the EDC-mediated carboxyl group labeling in a protein solution is a pseudo first-order reaction.32,35,36,40 To ensure the structural integrity of proteins in the labeling experiments, the extent of modification has been well controlled and optimized to ensure that deviation from first-order reaction kinetics, which would indicate chemical modification-induced protein unfolding,18,32 does not occur. The RCs of some D/E residues vary from 0.01 hr−1 to as high as 1.1 hr−1 (shown in Fig. S2); nevertheless, the dose response plots fit well to a first-order exponential equation. These data confirm that structural perturbation induced by the chemical labeling in our experiments is negligible.18,32
Figure 5.

Site-specific rate constant (RC) comparison. (A) Mirror plot of RC for individual carboxyl residues in mAb-A (top) versus deglycosylated mAb-A (bottom). X-axis corresponds to individual carboxyl residues. The primary sequence and numerical position are defined in Fig. 2. The Y-axis is experimental RC of individual carboxyl residues and each data point is an average of three experiments. (B) RC difference plot of individual carboxyl residues for mAb-A and deglycosylated mAb-A. The ΔRC% was calculated from the data shown in (A) using Eq.(4). The dotted lines at the y-axis value of ± 100% represent the threshold for identifying significantly different RCs between two compared samples. RCs of carboxyl residues with star in the x-axis are absent in (A) because these residues are either not labeled or their RCs are less than 0.002 hr−1 in both samples and the ΔRC% of these residues is defined as zero in (B).
To facilitate the comparison of RCs between samples, a RC difference (ΔRC %) of individual carboxyl residues between mAb-A and deglycosylated mAb-A was plotted (Fig. 5B). The ΔRC% of each carboxyl residue was calculated using Eq. (4). Based on multiple independent experimental replicates, the experimental error or variance associated with RC determination was 5∼20% for most carboxyl residues. However, we found that when RC is less than 0.01 hr−1, the associated error tends to increase and could be as high as 35%, which is consistent with previous reports.32 Based on these observations, we consider the variance of RC determination for a carboxyl residue to be ± 35%. We then used ∼3 times the max variance as the threshold for defining significant differences. Thus, we suggested that a threshold of ± 100% for ΔRC% be adopted for the purpose of identifying significantly different RCs for the labeling of carboxyl residues between two comparator samples. From Fig. 5B, it is apparent that most D/E residues have similar RCs in the mAb and deglycosylated mAb samples. However, two residues (D268 and E297) stand out from the others as having RCs that change significantly upon deglycosylation. The ΔRC% for these residues are much greater than 100%, indicating the side chains of D268 and E297 have very different (solvent) accessibility in the two samples, and, that they become much more accessible after deglycosylation.
To better contextualize the carboxyl labeling result, we interrogated the crystal structure of a model Fc domain (PDB: 3AVE), which has the same primary sequence and glycan molecules as mAb-A. Fig. 6 shows a zoomed-in view of the model structure of the Fc domain (PDB: 3AVE). The residues of interest, D268 and E297 are highlighted in magenta sticks and green dotted lines represent stable H-bonds identified by Swiss PDB Viewer with default values,42 i.e., a donor−acceptor distance between 2.195 and 3.3 Å, respectively, and a minimum angle of 90°C. The crystal structure indicates that the side chains of D268 and K249 form stable H-bonds with the glycan molecules. The D268 is buried inside and highly protected by the glycan molecules. The absence the N-glycans eliminates the H-bond between the glycan molecules and the side-chain of D268, which explains why D268 become so much more accessible for carboxyl labeling after deglycosylation. As for D297, the increased labeling at this site is likely due to its location, proximal to D268 and N300. In general, the case study illustrates the capability of site-specific CGF for pinpointing subtle and highly localized conformational differences between mAb samples.
Figure 6.

Zoomed-in view of the crystal structure of a model Fc domain (PDB: 3AVE). The backbone amide hydrogen is depicted in blue and the carbonyl oxygen is depicted in red. Glycans (Man3GlcNAc4Fuc1) are depicted as yellow sticks. The glycosylation site N300 is highlighted with light blue sticks, residues D268 and E297 are highlighted with magenta sticks and K249 is highlighted with red sticks. Green dotted lines represent stable H-bonds identified by Swiss PDB Viewer with default values. Note that Protein Data Bank file 3AVE has a different amino acid numbering. Subtracting 3 from the number used here corresponds to the numbering in 3AVE.
mAb vs. ADC
The interchain cysteine-linked ADC was manufactured by conjugating drug-linkers (vcMMAE) with the partially reduced mAb-A.27,43 Site-specific CGF comparison of mAb-A and the corresponding ADC was performed in the same manner as the comparison of mAb-A and deglycosylated mAb-A. Briefly, mAb-A and ADC were labeled side-by-side in native condition (sodium phosphate buffer, pH 7.0), followed by reduced LC/MS analysis and trypsin peptide map. RCs of individual carboxyl residues were determined by their dose-response plots and the ΔRC% of each carboxyl residue is calculated by Eq.(5) and plotted in Fig. 7. All 59 carboxyl residues displayed similar RCs in the mAb and ADC, suggesting that there are no significant differences in the solvent accessibility of these side chains in the two samples. Since the 59 D/E residues are well distributed throughout the entire primary sequence, our results suggest that the mAb and ADC share similar side chain conformation under native conditions. It should be noted that the kinetics of the carboxyl labeling chemistry that we used here are relatively slow. Since the structural integrity of proteins is preserved in the labeling experiments, the labeling kinetics of a carboxyl group will only reflect the dominant or averaged conformation of the side chain at the particular condition. We would not expect the data to reflect conformational dynamics or stability of side chains. The data interpretation is thus different than a classical HDX study where the labeling kinetics of any amide hydrogen depends on both solvent accessibility and conformational fluctuations of the amide hydrogen-bonding network.12,14 To understand the conformational impacts of drug conjugation from orthogonal assays, circular dichroism (CD) spectra comparison and DSC thermogram comparison of the mAb and ADC are also provided (shown in Fig. S3 and S4), and discussed in the next section.
Figure 7.

Site-specific rate constant (RC) comparison of mAb-A and ADC under native conditions. The ΔRC% was calculated using Eq.(5) and the dotted lines at the y-axis value of ± 100% represent the threshold for identifying significantly different RCs between two comparator samples.
Discussion
MAbs, and increasingly ADCs, are among the most rapidly expanding protein therapeutics in the biopharmaceutical development pipeline. Characterizing the domain-specific conformation of mAbs and ADCs is challenging due to their structural heterogeneity and large size. HDX MS has demonstrated great utility as a technique for probing the conformation and dynamics of mAbs and ADCs, and has been increasingly adopted by researchers in the biopharmaceutical industry. Classical HDX MS utilizes the isotope exchange kinetics of amide hydrogens on a protein to provide conformational insights into the protein backbone. However, the rapid back-exchange limits the application of HDX for some protein samples. Also, this approach does not directly probe amino acid side chain conformation, which plays an important role in protein-protein interactions and protein-ligand binding.44,45 For a given protein amino acid, conformational changes at the side chain (e.g., dihedral angle rotation, solvent-exposed or buried, free or H-bonded with other molecules) may or may not necessarily occur concomitantly with conformational changes in the backbone amide and thus be detectable by HDX. In this work, we highlighted an orthogonal methodology based on covalent labeling on amino acid side chains to provide complementary structural information for complex biologics mAbs and ADCs.
Site-specific CGF results from our investigation of conformational changes in mAb-A induced by deglycosylation pinpointed two residues, D268 and E297 (N-glycan attached at N300) in the Fc domain, that were more solvent accessible in the mAb after deglycosylation. The change observed in D268 was especially pronounced as its RC was found to increase 25-fold after deglycosylation. These findings are supported by a crystal structure of a model Fc domain with glycan molecules, as discussed above. Houde et al previously used HDX MS to study the effect of deglycosylation on IgG1 mAbs20,21 Their HDX comparison at a peptide level found two regions in Fc domain (residues “FLFPPKPKDTLM” and “PREEAYNSTYRVVSVLT,” which correspond to HC 244–254 and HC 294–307 in our mAb-A) have altered isotope exchange upon deglycosylation.21 Since the second region contains the glycosylation site N300, it is difficult to directly interpret the HDX data at the peptide level because its comparator is the glycosylated peptide. The more recent study22 narrowed down the first region to residues “LFPPK” (HC 245–249 in our mAb-A) and found that these residues had increased deuterium uptake after deglycosylation. In contrast, our CGF data indicated that the side chains of D268 and E297 in Fc domain have significantly enhanced accessibility after deglycosylation. These results illustrate that the two approaches (HDX and CGF) are complementary. In principle, CGF is sensitive to changes in the solvent accessibility of D/E side chains while classical HDX MS reveals information about the conformation and dynamics of the backbone amide H-bonding network. Regardless of the principle behind these methods, it is not surprising that conformational changes in the Fc region 245LFPPK induced by deglycosylation were not detected by CGF since this region lacks D/E residues, which would be labeled in this approach. The change in side chain accessibility of E297 and the changed HDX kinetics of HC294–307 after deglycosylation may be linked in some manner to fundamental structural changes wrought by deglycosylation. Likely E297 has been shielded to some extent by the presence of the glycan molecules and become more exposed and susceptible to labeling as a consequence of glycan removal. The region HC294–307 probably also becomes more dynamic or solvent-exposed after glycan removal. In previous HDX studies,20,21 HDX MS did not detect local conformational changes close to D268. This may be because the deglycosylation results in significantly increased side chain accessibility of D268, but does not concomitantly induce significant conformational changes in the backbone amide. It is also quite possible that the backbone conformational differences were not detected because the HDX MS data are essentially a deuterium uptake readout that is averaged across all amino acid amides in a peptide. It is also possible that small changes were lost due to back-exchange during chromatographic separation, which is typically 10∼25% in an HDX experiment. Overall, this case study highlighted that site-specific CGF is well suited for detecting subtle and highly localized conformational differences across comparator mAb samples and it is complementary to classic HDX MS.
Variations on the site-specific CGF that we used here have been applied to different types of protein conformation studies but, to our knowledge, CGF methodology has never been used to assess the downstream effects of drug conjugation on mAb conformation. A side-by-side comparison of mAb-A and the corresponding interchain cysteine linked ADC was performed under native conditions. All the 59 carboxyl residues that are well-distributed on the primary sequence displayed very similar RCs in the mAb and ADC, indicating there are no significant changes in solvent accessibility of these side chains after drug conjugation. It suggests that the mAb and ADC have a high degree of similarity in side chain conformation at native conditions. But it does not necessarily mean the ADC and parent mAb share the same conformation at any local area, since the CGF approach can only directly monitor the side chain accessibility of D/E resides and there are uncovered regions such as the 222SCDK. Nevertheless the CGF result is consistent with the high degree of similarity observed in circular dichroism (CD) spectra of the mAb and ADC at native conditions (shown in Fig. S3), and is generally consistent with findings obtained by other groups using orthogonal approaches.7,11,46
Spectroscopic analyses performed on similar interchain cysteine-linked ADCs composed of the same drug-linker (vcMMAE) and lgG1 mAbs indicated that the ADC and parent mAb share similar secondary and tertiary structures.7 Native ion mobility MS of this class of ADCs revealed that differentially drug-loaded subpopulations of the ADC have a gas phase conformation that is similar to the unconjugated mAb.46 However, DSC studies from different groups indicated that interchain cysteine-linked ADCs have lower thermal stability relative to parental mAbs.7,11 Our DSC comparison of mAb-A and the corresponding ADC also indicated that the ADC has lower melting temperature in the CH2 domain than mAb-A (shown in Fig. S4), but this does not mean that the DSC and CGF data are inconsistent. Slow covalent labeling approaches such as the carboxyl group labeling used here provides an assessment of the dominant or averaged conformation of side chains on a protein in a particular solution environment, but it does not include information on the conformational stability of the protein itself. Caution is thus warranted when comparing the CGF with stability related assays such as DSC. To investigate the conformational stability of side chains on mAb/ADCs, carboxyl group labeling experiments can be performed on stressed samples and, under these conditions, ADCs and their parental mAbs may display different side chain accessibility on some specific carboxyl residues.
We can further contextualize the current CGF results by comparing the data to a previous HDX MS study on the same system (mAb-A and ADC).27 In the HDX study,27 it was found that ∼90% of the primary sequence of the ADC displayed the same HDX kinetics as the parent mAb-A under native conditions, which indicated that the ADC and mAb-A share very similar backbone conformation. Combining the results from the two complementary methods, HDX MS and site-specific CGF, as well as other orthogonal assays, it can generally be concluded that mAb-A and the ADC share similar conformation at native conditions. It is worth noting, however, that HDX MS results pointed to two regions (244FLFPPKPKDTLM and 337KTISKAKGQPREPQV) in the Fc domain that show increased HDX kinetics in the ADC.27 The data indicated that the backbone amide H-bonding network of the two regions become more conformationally dynamic or more solvent-accessible in the ADC, but no conclusions can be drawn regarding the side chain conformation of the two regions. Based on the HDX MS data, one might expect the CGF data to show differences in the side chain solvent-accessibility of D252 and E348 between mAb-A and the corresponding ADC; however, no such difference was detected in these experiments. Based on the HDX and CGF data, it is very likely that these two regions become more conformationally dynamic in the backbone as a consequence of drug conjugation, but nevertheless have similar solvent accessibility at native conditions. As mentioned earlier, HDX MS primarily monitors the conformation and dynamics of the protein backbone, while site-specific CGF provides an assessment on the dominant conformation of side chains. Since these approaches are measuring different aspects of conformation, it should not be assumed that result from one approach will mirror the result from the other even though the results from different approaches are likely correlated. In fact, this case study demonstrates that combining the two complementary methods for the analysis of a given system can provide a more complete picture of the conformational feature of proteins than either technique alone.
It is worth noting that while HDX and CGF are complementary approaches, the experimental execution of the two assays is quite different. The execution of HDX MS remains somewhat challenging. Pepsin and other acidic proteases used for HDX MS can result in unpredictable cleavage patterns and a large number of overlapping peptides that may require special software or instrumentation for identification and data processing. Rapid back-exchange of deuterium to hydrogen during the post-labeling sample handling limits the applications of HDX for some protein samples. A common challenge is the difficulty of generating and recovering enough peptic peptides across the entire sequence of a protein during the very short time at quench conditions (0°C and pH 2.5) that are required to minimize the back-exchange.17 However, HDX is generally considered to be “structurally benign.” Conformational changes induced by the isotope labeling are negligible. There is thus no need to intentionally control and optimize the isotope labeling level to ensure the structural integrity of proteins in the labeling experiments.
In contrast to HDX, we have found that EDC-mediated carboxyl group footprinting is essentially a simple bench chemistry experiment which can be executed in any analytical lab that routinely carries out peptide map experiments. More importantly, the stable label from the CGF approach makes sample handling much more robust and flexible. Unlike typical HDX MS approaches, back-exchange or scrambling of covalently attached labels during LC/MS/MS is not a concern. Site-specific CGF can be readily performed on protein therapeutics in complicated biological matrices that require intensive post-labeling sample preparation. These types of sample conditions are quite challenging to analyze using traditional HDX MS experiments. We have performed site-specific CGF on mAbs and ADCs in a variety of conditions and the experimental execution was not limited by complicated formulation buffers that are not compatible with downstream MS detection. Extensive post-labeling protein purification can be incorporated prior to enzymatic treatment and much longer chromatographic separations at higher temperatures can be used during LC/MS/MS analyses, and this can dramatically improve the CGF readout by peptide map. Also, different proteases can be used besides trypsin to recover all the peptides of interest if it is necessary. The introduction of covalent modifications in the CGF approach can potentially result in conformational changes of proteins, and thus the labeling experiments should be carefully controlled. Assuring that first-order labeling kinetics is maintained throughout the labeling time-course is an easy and well-accepted way to prevent artificial results.
For site-specific covalent labeling strategies like CGF, the common drawback is limited sequence coverage. Regardless of the targeted residue, it is difficult to cover more than 13% of mAb sequence based on typical amino acid composition of mAbs. Non-specific labeling strategies such as hydroxyl radical footprinting can offer excellent sequence coverage, but it requires special equipment and complex data processing workflows. In an ideal scenario, different labeling strategies (e.g., HDX, CGF, hydroxyl radical labeling) would be implemented in a complementary manner to provide a detailed and complete conformational assessment of mAbs and ADCs. This ideal, however, may not be easy to achieve for a given lab or investigator due to constraints in resources or expertise. Given the relative simplicity of the CGF labeling procedure, the extent of sequence coverage (∼10% of well-distributed sequence) and straightforward data analysis, we believe that the site-specific CGF approach is technically quite tractable, and can be readily and routinely implemented for localized conformational assessment of mAbs and ADCs. It could be performed in a stand-alone manner or complementarily with other techniques depending on different needs and available resources. The approach can be very useful for performing site-specific conformational comparability studies of mAbs and ADCs during process development and manufacturing.
Materials and methods
Materials
Glycine ethyl ester (GEE), sodium phosphate, sodium chloride, 8 M urea, formic acid, DTT, iodoacetic acid (IAA), 8 M guanidine hydrochloride (guanidine HCl), 1 M Tris buffer (pH = 8) were purchased from Sigma (St. Louis, MO). NAP-5 columns were obtained from GE Healthcare. HPLC grade water and acetonitrile, 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) were obtained from Thermo Fisher Scientific (Rockford, IL). Trypsin (V5111, lot: 0000190933) was obtained from Promega (Madison, WI) and PNGase F (P0704L, lot: 0421507) was obtained from New England Biolabs (Ipswich, MA). All chemicals were used as received.
MAb-A, a humanized IgG1 kappa monoclonal antibody, was expressed in Chinese hamster ovary cells. mAb-A was conjugated with valine-citrulline-monomethyl auristatin E (vcMMAE) to form interchain cysteine-linked ADCs according to procedures established at Seattle Genetics.43 The averaged drug-antibody ratio of the ADC was determined to be 4.1 by hydrophobic interaction chromatography.27 Deglycosylated mAb-A was prepared by adding 2 µL of PNGase F per 100 µg of mAb-A and incubating at 37°C for 5 hours.
Labeling and trypsin peptide mapping
Prior to labeling, all samples (mAb or ADC) were buffer-exchanged into the labeling buffer (5 mM sodium phosphate, 100 mM sodium chloride, pH 7.0) using NAP-5 columns. Stock solutions of GEE (1 M) and EDC (120 mM) were prepared in the labeling buffer. The carboxyl labeling reactions were performed at room temperature by adding 35 µL of GEE and 15 µL of EDC stock solution to 100 µL of mAb or ADC (∼1.5 mg/mL) and incubating for 2, 5, or 10 minutes. The labeling reaction was quenched by adding 6 uL of 5% formic acid. The labeled samples were then buffer exchanged to 6 M guanidine/HCl (0.1 mM Tris, pH = 8), and fully reduced with 10 mM DTT at 45°C for 30 min. A portion of the fully reduced samples was set aside for LC/MS interrogation of labeling extent on the HC and LC subunits. The remainder was alkylated with 25 mM IAA for 25 min at room temperature (in the dark). The reduced and alkylated samples were then buffer exchanged to digestion buffer (50 mM Tris, pH 7.5). The resulting samples (except for the labeled, previously deglycosylated mAb-A sample) were subsequently deglycosylated by adding 2 µL of PNGase F per 100 µg of mAb or ADC and incubating at 37°C for 1.5 hours. Following deglycosylation, the samples were then digested with trypsin (enzyme: substrate ratio = 1:10) at 37°C for 4 hours and analyzed by LC/MS.
Reduced LC/MS analysis
Labeled samples (mAb or ADC) were denatured and reduced with DTT as described above. The resulting light chain (LC) and heavy chain (HC) subdomains of the mAb or ADC were separated with a 2.1×150 mm PLRP-S reversed-phase column (Agilent, Santa Clara, CA)) at 65°C. The column eluent was delivered to an Agilent 6510 QTOF MS (Agilent) operating in positive electrospray ionization (ESI) mode. The deconvoluted mass of the LC or HC was obtained using a maximum entropy deconvolution algorithm within the MassHunter workstation software version B.06.00.
LC/MS/MS analysis
Tryptic peptides of the labeled mAb or ADC were separated with a Waters UPLC BEH C18 column (2.1×150 mm, 1.7 µm) operated at 60 °C. Solvent A consisted of water with 0.1% formic acid and solvent B was acetonitrile with 0.08% formic acid. Peptides were eluted using a gradient of 0% to 34% solvent B over 114 min, and 34% to 70% solvent B over 10 min. The eluted peptides were analyzed with a Thermo Q-Exactive mass spectrometer with an ESI interface (San Jose, CA). Peptide mass spectra were acquired in positive ion mode at a resolution of 70,000 for MS1 scans and 17,500 for data-dependent MS/MS scans.
The MS data was analyzed with Byonic and Byologic software developed by Protein Metrics Inc. (San Carlos, CA). The expected tryptic peptides and labeled peptides were searched with precursor mass tolerance of 10 ppm for MS1 scans and fragment mass tolerance of 20 ppm for MS/MS scans with variable modifications of +85.0527 and +57.0215 Da on glutamic acid and aspartic acid (D/E) residues corresponding to the expected mass shift for GEE incorporation and its hydrolysis product, respectively. The labeled peptides were verified by manually comparing the MS/MS scans to the MS/MS from the corresponding unlabeled peptides. The extent of D/E labeling was determined by comparing the EIC area of the labeled and unlabeled peptides from the MS1 scan. Labeling RC of individual carboxyl residues in a given peptide was determined on the basis of EIC peak areas.
Site-specific rate constant (RC) determination
For a peptide consisting of N labeling sites (D/E residues), the labeled fraction Flabel (i) of a specific labeling site i was determined by
| (1) |
Aun represents EIC peak area of the unmodified peptide; Alabel (i) represents EIC peak areas of individual modified species that were labeled at the site i either exclusively or in combination with other labeling sites.
Fraction unlabeled Fun (i) of a specific labeling site i can be calculated as
| (2) |
RC (i) of a specific site i can be obtained from the dose-response plot Fun (i) as a function of labeling time fit to a first-order exponential equation.
Unlike the site-specific RC determination, averaged RC of a peptide was determined by the dose-response plot Fun (peptide) as a function of labeling time, where Fun (peptide) represents fraction unlabeled of the peptide and was defined as previously reported15,36,39,47
| (3) |
Eq. (1)∼(3) are based on the commonly made assumption that differences in the ionization efficiency between unmodified peptides and the corresponding modified forms are negligible.15,36,39,47
The RC difference ΔRC % of individual labeling sites between the mAb, deglycosylated mAb and ADC was determined as:
| (4) |
Or
| (5) |
where RC mAb, RC degly, and RC ADC are the RC of individual labeling sites in the mAb, deglycosylated mAb or ADC, respectively.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs 2015; 7:9-14; PMID:25529996; http://dx.doi.org/ 10.4161/19420862.2015.989042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med 2012; 64:15-29; PMID:23043493; http://dx.doi.org/ 10.1146/annurev-med-050311-201823 [DOI] [PubMed] [Google Scholar]
- 3.Adair JR, Howard PW, Hartley JA, Williams DG, Chester KA. Antibody-drug conjugates - a perfect synergy. Expert Opin Biol Ther 2012; 12:1191-206; PMID:22650648; http://dx.doi.org/ 10.1517/14712598.2012.693473 [DOI] [PubMed] [Google Scholar]
- 4.Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, Benjamin DR. The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther 2009; 330:932-8; PMID:19498104; http://dx.doi.org/ 10.1124/jpet.109.155549 [DOI] [PubMed] [Google Scholar]
- 5.Ikeda H, Hideshima T, Fulciniti M, Lutz RJ, Yasui H, Okawa Y, Kiziltepe T, Vallet S, Pozzi S, Santo L, et al.. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin Cancer Res 2009; 15:4028-37; PMID:19509164; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2867 [DOI] [PubMed] [Google Scholar]
- 6.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011; 3:161-72; PMID:21441786; http://dx.doi.org/ 10.4161/mabs.3.2.14960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo J, Kumar S, Prashad A, Starkey J, Singh SK. Assessment of physical stability of an antibody drug conjugate by higher order structure analysis: Impact of thiol- maleimide chemistry. Pharm Res 2014; 31:1710-23; PMID:24464270; http://dx.doi.org/ 10.1007/s11095-013-1274-2 [DOI] [PubMed] [Google Scholar]
- 8.Hawe A, Kasper JC, Friess W, Jiskoot W. Structural properties of monoclonal antibody aggregates induced by freeze-thawing and thermal stress. Eur J Pharm Sci 2009; 38:79-87; PMID:19540340; http://dx.doi.org/ 10.1016/j.ejps.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 9.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci 2008; 97:1414-26; PMID:17721938; http://dx.doi.org/ 10.1002/jps.21104 [DOI] [PubMed] [Google Scholar]
- 10.Wakankar AA, Feeney MB, Rivera J, Chen Y, Kim M, Sharma VK, Wang YJ. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: changes due to modification and conjugation processes. Bioconjug Chem 2010; 21:1588-95; PMID:20698491; http://dx.doi.org/ 10.1021/bc900434c [DOI] [PubMed] [Google Scholar]
- 11.Acchione M, Kwon H, Jochheim CM, Atkins WM. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs 2012; 4:362-72; PMID:22531451; http://dx.doi.org/ 10.4161/mabs.19449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom 2006; 17:1481-89; PMID:16876429; http://dx.doi.org/ 10.1016/j.jasms.2006.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirrone GF, Iacob RE, Engen JR. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal Chem 2015; 87:99-118; PMID:25398026; http://dx.doi.org/ 10.1021/ac5040242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konermann L, Tong X, Pan Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J Mass Spectrom 2008; 43:1021-36; PMID:18523973; http://dx.doi.org/ 10.1002/jms.1435 [DOI] [PubMed] [Google Scholar]
- 15.Konermann L, Stocks BB, Pan Y, Tong X. Mass spectrometry combined with oxidative labeling for exploring protein structure and folding. Mass Spectrom Rev 2010; 29:651-67; PMID:19672951; http://dx.doi.org/ 10.1002/mas.20256 [DOI] [PubMed] [Google Scholar]
- 16.Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev 2009; 28:785-815; PMID:19016300; http://dx.doi.org/ 10.1002/mas.20203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Y, Konermann L. Membrane protein structural insights from chemical labeling and mass spectrometry. Analyst 2010; 135:1191-200; PMID:20498872; http://dx.doi.org/ 10.1039/b924805f [DOI] [PubMed] [Google Scholar]
- 18.Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the strucuture of macromolecular complexes. Annu Rev Biophys Biomol Struct 2006; 35:251-76; PMID:16689636; http://dx.doi.org/ 10.1146/annurev.biophys.35.040405.102050 [DOI] [PubMed] [Google Scholar]
- 19.Zhu MM, Rempel DL, Du Z, Gross ML. Quantification of protein-ligand interactions by mass spectrometry, titration, and H/D Exchange: PLIMSTEX. J Am Chem Soc 2003; 125:5252-53; PMID:12720418; http://dx.doi.org/ 10.1021/ja029460d [DOI] [PubMed] [Google Scholar]
- 20.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal Chem 2009; 81:2644-51; PMID:19265386; http://dx.doi.org/ 10.1021/ac802575y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics 2010; 9:1716-28; PMID:20103567; http://dx.doi.org/ 10.1074/mcp.M900540-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang L, Sundaram S, Zhang J, Carlson P, Matathia A, Parekh B, Zhou Q, Hsieh MC. Conformational characterization of the charge variants of a human IgG1 monoclonal antibody using H/D exchange mass spectrometry. MAbs 2012; 5:114-25; PMID:23222183; http://dx.doi.org/ 10.4161/mabs.22695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan J, Zhang S, Parker CE, Borchers CH. Subzero temperature chromatography and top-down mass spectrometry for protein higher-order structure characterization: method validation and application to therapeutic antibodies. J Am Chem Soc 2014; 136:13065-71; PMID:25152011; http://dx.doi.org/ 10.1021/ja507880w [DOI] [PubMed] [Google Scholar]
- 24.Fang J, Richardson J, Du Z, Zhang Z. Effect of Fc-glycan structure on the conformational stability of IgG revealed by hydrogen/deuterium exchange and limited proteolysis. Biochemistry 2016; 55:860-68; PMID:26812426; http://dx.doi.org/ 10.1021/acs.biochem.5b01323 [DOI] [PubMed] [Google Scholar]
- 25.Iacob RE, Bou-Assaf GM, Makowski L, Engen JR, Berkowitz SA, Houde D. Investigating monoclonal antibody aggregation using a combination of H/DX-MS and other biophysical measurements. J Pharm Sci 2013; 102:4315-29; PMID:24136070; http://dx.doi.org/ 10.1002/jps.23754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan LY, Salas-Solano O, Valliere-Douglass JF. Antibody structural integrity of site-specific antibody-drug conjugates investigated by hydrogen/deuterium exchange mass spectrometry. Anal Chem 2015; 87:5669-76; PMID:25938577; http://dx.doi.org/ 10.1021/acs.analchem.5b00764 [DOI] [PubMed] [Google Scholar]
- 27.Pan LY, Salas-Solano O, Valliere-Douglass JF. Conformation and dynamics of interchain cysteine-linked antibody-drug conjugates as revealed by hydrogen/deuterium exchange mass spectrometry. Anal Chem 2014; 86:2657-64; PMID:24512515; http://dx.doi.org/ 10.1021/ac404003q [DOI] [PubMed] [Google Scholar]
- 28.Kaltashov IA, Bobst CE, Abzalimov RR, Berkowitz SA, Houde D. Conformation and dynamics of biopharmaceuticals: transition of mass spectrometry-based tools from academe to industry. J Am Soc Mass Spectrom 2009; 21:323-37; PMID:19963397; http://dx.doi.org/ 10.1016/j.jasms.2009.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang RY, Chen G. Higher order structure characterization of protein therapeutics by hydrogen/deuterium exchange mass spectrometry. Anal Bioanal Chem 2014; 406:6541-58; PMID:24948090; http://dx.doi.org/ 10.1007/s00216-014-7924-3 [DOI] [PubMed] [Google Scholar]
- 30.Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Volkin DB, Weis DD. Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half-life. MAbs 2015; 7:84-95; PMID:25524268; http://dx.doi.org/ 10.4161/19420862.2014.985494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wecksler AT, Kalo MS, Deperalta G. Mapping of Fab-1:VEGF interface using carboxyl group footprinting mass spectrometry. J Am Soc Mass Spectrom 2015; 26:2077-80; PMID:26419770; http://dx.doi.org/ 10.1007/s13361-015-1273-0 [DOI] [PubMed] [Google Scholar]
- 32.Kaur P, Tomechko SE, Kiselar J, Shi W, Deperalta G, Wecksler AT, Gokulrangan G, Ling V, Chance MR. Characterizing monoclonal antibody structure by carboxyl group footprinting. MAbs 2015; 7:540-52; PMID:25933350; http://dx.doi.org/ 10.1080/19420862.2015.1023683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Lilyestrom W, Li C, Scherer T, van Reis R, Zhang B. Revealing a positive charge patch on a recombinant monoclonal antibody by chemical labeling and mass spectrometry. Anal Chem 2011; 83:8501-8; PMID:22004540; http://dx.doi.org/ 10.1021/ac2016129 [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Shen W, Rempe D, Monsey J, Vidavsky I, Gross ML, Bose R. Carboxyl-group footprinting maps the dimerization interface and phosphorylation-induced conformational changes of a membrane-associated tyrosine kinase. Mol Cell Proteomics 2011; 10:1-16; PMID:21422241; http://dx.doi.org:19339500 10.1074/mcp.M110.005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen J, Zhang H, Gross ML, Blankenship RE. Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectromety-based footprinting. Proc Natl Acad Sci USA 2009; 106:6134-39; PMID:19339500; http://dx.doi.org/ 10.1073/pnas.0901691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Wen W, Huang RY-C, Blankenship RE, Gross ML. Mass spectrometry-based carboxyl footprinting of proteins: method evaluation. Int J Mass Spectrom 2012; 312:78-86; PMID:22408386; http://dx.doi.org/ 10.1016/j.ijms.2011.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom 2005; 16:2057-63; PMID:16263307; http://dx.doi.org/ 10.1016/j.jasms.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 38.Li KS, Rempel DL, Gross ML. Conformational-sensitive fast photochemical oxidation of proteins and mass spectrometry characterize amyloid beta 1–42 aggregation. J Am Chem Soc 2016; 138:12090-98; PMID:27568528; http://dx.doi.org/ 10.1021/jacs.6b07543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev 2007; 107:3514-43; PMID:17683160; http://dx.doi.org/ 10.1021/cr0682047 [DOI] [PubMed] [Google Scholar]
- 40.Hoare DG, Koshland DEJ. A method for the quantitative modification and estimation of carboxylic acid groups in proteins. J Biol Chem 1967; 242:2447-53; PMID:6026234 [PubMed] [Google Scholar]
- 41.Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, Ling V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. MAbs 2013; 5:86-101; PMID:23247543; http://dx.doi.org/ 10.4161/mabs.22964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997; 18:2714-23; PMID:9504803; http://dx.doi.org/ 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- 43.Sun MM, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, Handley FG, Ihle NC, Senter PD, Alley SC. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 2005; 16:1282-90; PMID:16173809; http://dx.doi.org/ 10.1021/bc050201y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruvinsky AM, Kirys T, Tuzikov AV, Vakser IA. Side-chain conformational changes upon protein–protein association. J Mol Biol 2011; 408:356-65; PMID:21354429; http://dx.doi.org/ 10.1016/j.jmb.2011.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirys T, Ruvinsky AM, Tuzikov AV, Vakser IA. Correlation analysis of the side-chains conformational distribution in bound and unbound proteins. BMC Bioinformatics 2012; 13:236-43; PMID:22984947; http://dx.doi.org/ 10.1186/1471-2105-13-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Debaene F, Boeuf A, Wagner-Rousset E, Colas O, Ayoub D, Corvaia N, Van Dorsselaer A, Beck A, Cianferani S. Innovative native MS methodologies for antibody drug conjugate characterization: High resolution native MS and IM-MS for average DAR and DAR distribution assessment. Anal Chem 2014; 86:10674-83; PMID:25270580; http://dx.doi.org/ 10.1021/ac502593n [DOI] [PubMed] [Google Scholar]
- 47.Yan Y, Chen G, Wei H, Huang RY, Mo J, Rempel DL, Tymiak AA, Gross ML. Fast photochemical oxidation of proteins (FPOP) maps the epitope of EGFR binding to adnectin. J Am Soc Mass Spectrom 2014; 25:2084-92; PMID:25267085; http://dx.doi.org/ 10.1007/s13361-014-0993-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.