

Summary
Induction of broadly neutralizing antibodies (bnAbs) capable of inhibiting infection with diverse variants of human immunodeficiency virus type 1 (HIV‐1) is a key, as‐yet‐unachieved goal of prophylactic HIV‐1 vaccine strategies. However, some HIV‐infected individuals develop bnAbs after approximately 2‐4 years of infection, enabling analysis of features of these antibodies and the immunological environment that enables their induction. Distinct subsets of CD4+ T cells play opposing roles in the regulation of humoral responses: T follicular helper (Tfh) cells support germinal center formation and provide help for affinity maturation and the development of memory B cells and plasma cells, while regulatory CD4+ (Treg) cells including T follicular regulatory (Tfr) cells inhibit the germinal center reaction to limit autoantibody production. BnAbs exhibit high somatic mutation frequencies, long third heavy‐chain complementarity determining regions, and/or autoreactivity, suggesting that bnAb generation is likely to be highly dependent on the activity of CD4+ Tfh cells, and may be constrained by host tolerance controls. This review discusses what is known about the immunological environment during HIV‐1 infection, in particular alterations in CD4+ Tfh, Treg, and Tfr populations and autoantibody generation, and how this is related to bnAb development, and considers the implications for HIV‐1 vaccine design.
Keywords: autoantibody, broadly neutralizing antibody, CD4+ T follicular helper cell, CD4+ T follicular regulatory cell, human immunodeficiency virus, regulatory T cell
This article is part of a series of reviews covering B cells and Immunity to HIV appearing in Volume 275 of Immunological Reviews.
1. Introduction
Human immunodeficiency virus type 1 (HIV‐1) infection remains a persistent global health threat, and approximately two million new adult infections occur each year.1 Education, identification and treatment of infected persons with antiretroviral drugs, male circumcision, condoms, and needle exchange programs have been effective at curtailing the epidemic, but declines in the rate of new infections have plateaued, and it appears unlikely that the goal of <500 000 new adult infections per year will be achieved by 2020. An effective HIV‐1 vaccine could contribute to further reductions of infections as part of a coordinated prevention strategy,2 but to date, testing of candidate vaccines in efficacy trials has been disappointing 3, 4, 5, 6, 7 with only one trial showing any degree of vaccine efficacy.8
A number of antibody‐mediated functions against HIV‐1 have been studied, including virus capture and phagocytosis,9, 10 antibody‐dependent cell‐mediated virus inhibition,11, 12 antibody‐dependent cell‐mediated cytotoxicity (ADCC),13, 14 chemokine secretion of monocytes stimulated by antibodies,15 and virus neutralization.16, 17, 18 Neutralization has been shown to exert immune pressure on HIV‐1,19, 20 while the role of other antibody‐mediated functions in exerting immune pressure is unclear, primarily because antibodies that mediate ADCC and other activities often also neutralize.21 Regardless, multiple studies have demonstrated that virus neutralization is a driver of both virus and antibody diversity,22, 23, 24, 25, 26 although neutralization of autologous viruses does not always cause the extinction of susceptible virus populations in an infected individual.25
Because of their ability to neutralize many different circulating strains of HIV‐1, broadly neutralizing antibodies (bnAbs) are attractive targets for vaccine development.27 Passive infusion studies in animals have shown that bnAbs can prevent infection by intravenous28 and mucosal28, 29, 30, 31, 32, 33 challenge, suggesting that a vaccine that elicits robust and durable levels of bnAbs could be protective. Vaccine development strategies that leverage our understanding of antibody‐virus coevolution34 and that use knowledge of antibody‐antigen structure relationships27 are currently being tested in animal models35, 36 and human studies are planned. However, to date, no vaccine has reliably elicited bnAbs, and one possibility is that in addition to optimizing antigen structure, it may be necessary to recreate the immunological environment in which bnAbs develop. Understanding the conditions in which bnAbs have developed is a critical first step toward recreating those conditions.
2. Development of broadly neutralizing antibodies in HIV‐1 infected individuals
2.1. The antibody response in HIV‐1 infection
During acute HIV‐1 infection, there is a vigorous immune response that is unable to contain virus replication or the establishment of latency.37 In many cases, a single transmitted/founder virus establishes infection and then evolves within the host resulting in a diverse virus population.38 The earliest changes to the virus population are driven by the CD8+ T‐cell response that is induced as viremia increases39 and places strong selection pressure on the virus, resulting in complete turnover of the virus pool within the first few weeks of infection.40 The development of antibody responses follows a pattern, with antiviral antibodies being detected first as immune complexes41 followed by free antibody directed at the HIV‐1 envelope (Env) glycoprotein 41 (gp41) subunit,41, 42 and then by the development of Env glycoprotein 120 (gp120)‐binding antibodies.41 These early antibody responses do not neutralize or place selective pressure on virus evolution41; antibodies capable of neutralizing autologous viruses are not detectable until weeks to months after infection is established19, 20 and have little to no activity against heterologous HIV‐1 strains.43 The initial gp41‐directed antibody response is polyreactive and the antibodies are highly mutated,42 and evidence indicates that at least some early responding B cells are primed prior to infection by non‐HIV‐1 antigens such as proteins contained in intestinal microbiota.44
Acutely HIV‐1‐infected individuals do not make bnAbs,41, 43 but during chronic HIV‐1 infection, neutralization breadth develops to a greater or lesser degree in each person.45, 46, 47 Breadth of plasma neutralizing activity develops incrementally,48 and evidence suggests that protracted exposure to HIV‐1 Env is required.22, 23, 24, 47, 48, 49, 50 Neutralization breadth is not an all‐or‐nothing phenomenon—in one study, about half of a cohort of 205 chronically infected persons were capable of neutralizing about half of a panel of 219 hard‐to‐neutralize (tier 2) HIV‐1 isolates.45 When bnAbs arise, neutralization breadth is thought to be mediated by 1‐2 specificities per individual,51, 52 although serum mapping studies53, 54, 55, 56 and algorithms designed to deconvolute neutralization data57 suggest that in some individuals breadth may be mediated by three or more specificities of antibodies. In addition, the acquisition of additional neutralization breadth can continue during ongoing HIV‐1 infection. For example, in one infected individual (CH505), a CD4‐binding site‐directed bnAb developed24 that was aided in its evolution by a cooperating antibody lineage also directed at the CD4 binding site but that did not initially have neutralization breadth.23 During ongoing infection, this cooperating antibody lineage further evolved, developing bnAb activity.22 The latter example suggests that the development of neutralization breadth is a dynamic process and that conditions favorable to the evolution of bnAbs may persist during chronic infection in some individuals.
Despite the fact that B cells are not a primary target of HIV‐1 infection, virus replication and immune activation in HIV‐1 infection are associated with profound dysregulation of the B‐cell compartment. It is possible that trafficking of the negative regulatory factor (Nef) protein from HIV‐1‐infected macrophages to B cells may alter class switching and germinal center (GC) responses,58 but dysfunction is also likely driven by the early cytokine storm59 and ongoing immune dysfunction caused by HIV‐1 infection of T cells. B‐cell dysregulation is evidenced by the delayed antibody response in acute HIV‐1 infection,41 an increase in the proportion of activated memory B cells and exhausted B cells, non‐specific plasmablast activation (leading to polyclonal immunoglobulin production), and a decline in the frequency of long‐lived plasma cells.42, 60, 61, 62, 63 In addition, Env‐specific B cells are found in the activated and exhausted B‐cell subsets in viremic individuals.64 However, the B‐cell dysfunction in HIV‐1 infection does not prevent the generation of bnAbs, and the extent of dysregulation of circulating B‐cell subsets during chronic infection shows no correlation with neutralization breadth.65, 66 At this time, it is not known whether B‐cell dysfunction is necessary for the development of breadth in HIV‐1‐infected individuals.
2.2. Clinical characteristics of individuals making bnAbs
Longitudinal studies of HIV‐1 infection have allowed researchers to map the development of bnAbs.22, 23, 24, 26, 48 During acute/early HIV‐1 infection, high viral load and an early decline in circulating CD4+ T cells were associated with the development of neutralization breadth,48, 50 whereas individuals who had low or undetectable viral loads, such as long‐term non‐progressors, were found to have less neutralization breadth.49 Time since infection also correlated with the development of breadth,56 although whether this was due to the need for protracted Env exposure, persistent immune perturbation caused by HIV‐1 infection, or both is not clear.
The development of neutralization breadth does not appear to impact the progression of HIV‐1 disease48, 67; for example, many individuals in these longitudinal cohorts went on antiretroviral therapy as part of standard of care during the course of study,48, 50 including those with high degrees of breadth.48 Examination of viruses and antibodies taken from the same HIV‐1‐infected individuals has consistently shown that circulating bnAbs cause extinction of susceptible virus populations despite ongoing viremia,23, 24, 25 although viruses susceptible to less potent neutralizing antibodies continue to circulate.25 Whether individuals who have developed bnAbs exhibit a reduced susceptibility to HIV‐1 super‐infection has not been addressed, although as bnAbs constitute only part of the overall HIV‐specific antibody response they may not reach sufficient titers to do so. The lack of clinical impact of bnAbs is consistent with early bnAb infusion studies in animal models68 and humans69 that did not show a lasting impact on HIV‐1 viral loads, although newer, more potent bnAbs appear to have a more persistent impact on viral load70, 71, 72 and subsequent antibody development.73 At this time, ongoing clinical trials are being conducted to determine if bnAb infusion can augment other anti‐HIV‐1 therapies.
The duration and magnitude of Env exposure that is permissive for bnAb development is not completely known. Longitudinal studies suggest that bnAbs do not develop until at least 2 years after HIV‐1 infection, but a study of two HIV‐1‐infected individuals who made Env gp41 membrane proximal external region (MPER) bnAbs showed that activity developed at about 1 year of infection while they were not on antiretroviral therapy.26 A second study compared individuals taking and not taking antiretroviral therapy and determined that the frequency of individuals making bnAbs was similar between the groups,74 although not all individuals taking antiretrovirals were completely suppressed. Thus, while prolonged Env exposure appears to be highly correlated with the development of neutralization breadth, there does appear to be a pathway to bnAb development in some individuals that lack prolonged or high‐level Env exposure.
Common demographic characteristics do not appear to influence bnAb development. In a large longitudinal study of African HIV‐1‐infected individuals, there was no correlation between neutralization breadth and geographical origin, age, or gender, and no correlation with reported mode of transmission or risk factor.50 In this same study, there was a correlation with subtype C infection and with the presence of human leukocyte antigen (HLA)‐A*03,50 although these two correlations were not observed in a different study.47 In this second study, exome sequencing performed on matched sets of individuals with and without bnAbs did not reveal any specific gene that correlated with breadth, although some candidate variants were found.47 To date, the data suggest that multiple factors contribute to the development of breadth in HIV‐1‐infected individuals.
2.3. The association between autoimmunity and HIV‐1 Infection
Early during the HIV‐1 epidemic, clinicians noted that some HIV‐1‐infected individuals would go on to develop autoimmune phenomena,75, 76 usually in the setting of uncontrolled infection. In addition, investigators noted that there was an underreporting of coincident HIV‐1 infection and systemic lupus erythematosus (SLE),77, 78, 79, 80 recognizing that lack of reporting does not mean a lack of coincident disease. It was suggested that SLE and other autoimmune diseases might provide protection against infection,75 but to date no study has isolated the protective factor. One candidate antibody class, antiphospholipid antibodies that can be found in autoimmune disease, was shown to block infection in vitro but testing of patient samples gave inconclusive results.15 Thus, whether SLE or other autoimmune diseases are protective against HIV‐1 infection remains an open question.
However, many isolated bnAbs have been shown to have autoreactivity when tested in assays used for the diagnosis of autoimmune disease22, 24, 81, 82 or when tested on arrays of human proteins.83, 84 While there are bnAbs that do not react with human antigens,83 most do. For some bnAbs where maturation has been studied from the initial B‐cell rearrangement to breadth, development of breadth was correlated with acquisition of autoreactivity.24 In one case, bnAb activity and autoreactivity were directly correlated, when a bnAb that also had reactivity with double stranded DNA was isolated from a person with coincident HIV‐1 infection and SLE,81 indicating that in that person, bnAb activity derived from an autoreactive B‐cell pool. In addition to autoreactivity, bnAbs usually have a high degree of somatic mutation and/or long heavy‐chain complementarity determining region 3 (HCDR3) loops,85 and no reported bnAb lacks all three characteristics. Autoreactivity, long HCDR3 loops, and high levels of somatic hypermutation are also associated with antibodies made by B cells that are deleted by central and peripheral tolerance controls.86, 87 Furthermore, studies of antibody knock‐in mice (engineered to have B cells with the potential to express immunoglobulins (Igs) composed of the variable region heavy (VH) and variable region light (VL) chains of mature bnAbs or their germline precursors) have demonstrated that B cells expressing many different bnAbs or bnAb precursors are under strong tolerance control,88, 89, 90, 91, 92, 93 although this does not hold for all bnAbs studied.89
Thus, while it may not be necessary for a HIV‐1 neutralizing antibody to exhibit characteristics of autoantibodies to have breadth, the two appear to be highly correlated. Because of this apparent correlation, we recently completed a study examining the relationship between autoantibodies and bnAb activity.47 In two different cohorts, we found that HIV‐1‐infected persons making bnAbs were more likely to have serum autoantibodies as well as perturbations of their circulating T‐cell populations, thus suggesting relaxed tolerance controls. T‐cell populations involved in regulation of B‐cell responses and their relationship to bnAb induction are discussed in the following sections.
3. T follicular helper cells and their role in bnAb induction
3.1. T follicular helper cells
T follicular helper (Tfh) cells are a subset of CD4+ T cells specialized for provision of help to B cells. Help is provided in a cognate fashion, i.e. antigen bound to B‐cell surface Ig is internalized and presented with MHC class II, and Tfh cells with specificity for the peptides presented interact with and provide help to the B cell.94 The first cognate interactions between B cells and CD4+ T cells occur at the interface between the T‐ and B‐cell zones in lymphoid tissues.95 T‐cell‐mediated ligation of CD40 on the B cell triggers B‐cell clonal expansion and differentiation,96 and cytokines produced by the T cell including interleukin (IL)‐21 and IL‐4 promote growth and differentiation and direct Ig class switching.97, 98 Provision of help is dependent on an active two‐way interaction between the B and T cell, with inducible T‐cell costimulatory (ICOS) triggering being important for IL‐21 production99 and IL‐4 production being dependent on signaling lymphocytic activation molecule (SLAM).100 Following activation, the B cells may differentiate into short‐lived extrafollicular plasma cells that have very few mutations in their Ig variable (V)‐region genes and secrete low‐affinity Ig, develop into early memory B cells, or undergo proliferation within the follicle, giving rise to GCs (reviewed in 101). GCs are comprised of B cells and Tfh cells (which migrate into GCs following initial interaction with B cells at the margin of the B‐cell follicle), together with antigen‐bearing follicular dendritic cells (FDCs), macrophages, and stromal cells. B cells undergo further cognate interactions with Tfh cells in the light zones of the GC, in which help is again mediated via two‐way receptor‐ligand interactions and production of cytokines including IL‐21.102, 103, 104 This enables Tfh cells to control the affinity of B cells entering the GC reaction.105 B cells then migrate into the dark zones where they proliferate and undergo somatic hypermutation, resulting in antibody diversification (reviewed in 106). The number of divisions B cells undergo in the dark zone and the speed of cycling are determined by the help provided by Tfh cells.107, 108 B cells then exit into the light zones, where those cells expressing surface Ig of sufficiently high affinity to enable them to efficiently acquire antigen from FDCs undergo further interactions with Tfh cells and receive signals, in particular via CD40, that enable continued survival. Tfh cells thus, select high‐affinity B cells following affinity maturation.109, 110 These B cells may then re‐enter the dark zone and undergo further round(s) of proliferation and somatic hypermutation, or alternatively become long‐lived memory B cells or differentiate into long‐lived plasma cells that typically migrate out of the lymph node (LN) to reside in the bone marrow or gut.106 The help provided by Tfh cells to B cells evolves over the course of the GC response: Tfh cells initially produce IL‐21 and select high‐affinity B cells, then subsequently shift to IL‐4 production and high CD154 (CD40 ligand) expression and promote B‐cell differentiation into plasma cells.111
3.2. T follicular helper cell differentiation
Tfh cell differentiation is a complex multi‐step process that is influenced by numerous heterogeneous signals (reviewed in 112). The first step occurs when naive CD4+ T cells are primed by dendritic cells (DCs) in LNs: an initial Tfh‐lineage fate decision is made during the first few rounds of cell division.113 Factors that influence CD4+ cell Tfh‐lineage differentiation include the cytokine environment, ICOS ligation, and signaling via the T‐cell receptor (TCR). In mice, IL‐6 plays an important role in promoting Tfh cell generation and induces upregulation of the transcription factor B‐cell lymphoma 6 (Bcl6), which is a central regulator of Tfh differentiation.114, 115, 116 However, although there is evidence that IL‐6 can also promote Tfh differentiation in humans,117 it is a not a good inducer of Bcl6 expression in human CD4+ T cells.118, 119 By contrast, human (but not murine) Tfh cell differentiation is potently induced by activin A120 and can also be driven by transforming growth factor β (TGFβ), particularly in combination with IL‐12 or IL‐23.121 Tfh differentiation is negatively regulated by other cytokines, including IL‐2122, 123 and IL‐7.124, 125
Tfh cell differentiation also requires ligation of ICOS by its ligand ICOSL.126 Studies in mice have shown that CD8α(−) DCs at the interface of the follicle and T‐cell zone play a prominent role in Tfh cell induction by upregulating expression of ICOSL and also OX40L.127 The G‐protein‐coupled receptor EBI‐2 positions T cells in this location, where Tfh cell differentiation is further enhanced by DC‐mediated production of soluble and membrane‐bound CD25 (the IL‐2 receptor α chain), which binds to and quenches IL‐2.128 Signals received via the TCR during DC‐mediated antigen presentation also influence CD4+ differentiation into early Tfh cells, although there is not a simple relationship between TCR signal strength and Tfh cell differentiation.129 Instead, the duration of signaling appears more important, with Tfh cell induction being associated with prolonged antigen presentation by DC.130
As CD4+ T cells differentiate into pre‐Tfh cells they downregulate expression of C‐C chemokine receptor type 7 (CCR7) and P‐selectin glycoprotein ligand 1 (PSGL‐1), which promote localization to and retention within the T‐cell zone, and upregulate expression of the chemokine receptor characteristic of Tfh cells, C‐X‐C chemokine receptor type 5 (CXCR5), which binds C‐X‐C motif chemokine ligand 13 (CXCL13), a chemokine produced in B‐cell follicles.131, 132 This enables pre‐Tfh cells to migrate to the margin of the B‐cell follicle,133 where they interact with B cells and the next step in Tfh differentiation takes place. B cells present antigen to pre‐Tfh cells, enabling further cell division. ICOSL expressed by the B cells also binds ICOS on the pre‐Tfh cell, which maintains Bcl6 upregulation, reinforcing the Tfh differentiation program and inducing directional migration of the early Tfh cells into the B‐cell follicle.134
As early Tfh cells undergo further interactions with B cells, GC formation occurs and they complete their differentiation into mature GC Tfh cells. Most GC Tfh cells have a CXCR5hi, CCR7lo, PSGL‐1lo, programmed cell death protein 1 (PD‐1)hi phenotype and express high levels of Bcl6, Maf, and SLAM‐associated protein (SAP). SAP expression is essential for GC formation,135 preventing inhibitory signaling through SLAM family member 6 (SLAMF6) to enable Tfh‐B cell adhesion,136 which is crucial for Tfh cell retention in GCs and provision of help to B cells. Bcl6 plays a central role in Tfh cell differentiation,115, 137, 138 but other transcription factors that act upstream and downstream of Bcl6 are also required (reviewed in 139). These include lymphoid enhancer‐binding factor 1 (LEF‐1) and T‐cell‐specific transcription factor 1 (TCF‐1), which act upstream of Bcl6 to promote Tfh cell differentiation140, 141, 142; basic leucine zipper transcription factor ATF‐like (Batf), which positively regulates Bcl6 and is important for IL‐4 expression143; interferon regulatory factor 4 (IRF4), which drives differentiation of IL‐12‐stimulated CD4 T cells to Tfh rather than Th1 cells144; achaete‐scute homolog 2 (Ascl2), which induces CXCR5 expression145; c‐Maf, which enhances expression of CXCR5, IL‐21, and IL‐4146; signal transducer and activator of transcription (STAT)s, STAT3 and 4 being particularly important for human Tfh cell differentiation and regulation of IL‐21 expression by human CD4+ T cells121, 147; and forkhead box protein O1 (FOXO1), which can both promote and inhibit Tfh cell differentiation.148 In addition, Tfh cell differentiation is regulated by microRNAs including members of the miRNA‐17‐92 family.149, 150 Bcl6 is a transcriptional repressor and orchestrates the expression of genes involved in Tfh cell migration, differentiation, and function as well as repressing genes involved in alternative fate differentiation.151 Importantly, it represses B lymphocyte‐induced maturation protein 1 (Blimp 1), an antagonist of Tfh cell differentiation,137 as well as repressing expression of receptors for cytokines that promote CD4+ T‐cell differentiation into T‐helper (Th)1, Th2, Th17, and regulatory (Treg) cells. Nonetheless Tfh cells can be polarized, and subsets of Th1, Th2, or Th17‐like Tfh cells develop in certain infections/diseases.
Unlike GC B cells, which usually remain in a single GC, Tfh cells can move from one GC to another or exit the follicle altogether.152, 153 As Bcl6 expression requires ongoing induction for its maintenance, its expression is downregulated when GC Tfh cells leave the follicle, and they gradually transition to a resting memory state.152, 153 Early Tfh cells can also differentiate into memory Tfh cells without becoming GC Tfh cells. Memory Tfh cells are generated in both mice113, 154, 155, 156, 157 and humans99, 158, 159, 160 and can be long‐lived. They have a central memory (Tcm) phenotype and reside in spleen, lymph nodes, and bone marrow as well as recirculating in blood.
3.3. Circulating Tfh populations
Human peripheral blood memory Tfh cells are heterogeneous in phenotype (reviewed in 161). They are found within the CXCR5+ subset of circulating CD4+ T cells,99, 159 although CXCR5+ can also be expressed on non‐Tfh cells. Precise definition of circulating memory Tfh cells is challenging, as resting memory Tfh cells in blood do not express Bcl6 or the high levels of PD‐1 and ICOS characteristic of GC Tfh cells and require re‐stimulation in order to mediate B‐cell helper activity.99, 159, 160, 162 A subpopulation of blood CXCR5+ CD4+ T cells expresses PD‐1, although at lower levels than GC Tfh cells. Some of the PD‐1+ cells co‐express ICOS; this delineates a more activated Tfh cell population. PD‐1+ CXCR5+ CD4+ Tcm cells have a gene expression profile more similar to that of GC Tfh cells than PD‐1− CXCR5+ CD4+ or CXCR5− CD4+ Tcm cells and mediate superior help for B cells in in vitro culture assays.160, 163 However, memory Tfh cells are not exclusively found in this population, as PD‐1− CXCR5+ CD4+ cells can upregulate PD‐1 expression and provide help for B cells if suitably re‐stimulated. CXCR5+ CD4+ Tcm cells can also be subdivided on the basis of differential chemokine receptor expression into CXCR3+ (Th1‐like), CXCR3− CCR6+ (Th17‐like), and CXCR3− CCR6− (Th2‐like) subpopulations. The CXCR3− (Th2 and Th17‐like) CXCR5+ CD4+ subpopulations provide help for B cells in vitro, whereas the CXCR3+ CXCR5+ CD4+ population produces little IL‐21 and has a very poor B‐cell helper capacity99, 158, 160, 163; furthermore the blood CXCR3− PD1+ CXCR5+ CD4+ subset has a transcriptional profile most similar to that of GC Tfh cells.160 Nonetheless, although the CXCR3− PD1+ subset of resting memory CXCR5+ CD4+ cells have been deemed to constitute resting memory Tfh cells,160 this does not provide an exclusive or comprehensive definition. Notably, CXCR3+ CXR5+ memory Tfh cells are generated under some conditions, e.g. the seasonal influenza vaccine elicits ICOS+ PD‐1+ CXCR3+ CXCR5+ Tfh cells, and the circulating frequency of this population in the periphery correlates with the quantity and avidity of the influenza‐specific antibody response.158, 164
3.4. Tfh cells in HIV‐1/simian immunodeficiency virus (SIV) infection
HIV‐specific CD4+ T‐cell responses are primed as viremia increases in acute HIV‐1 infection, but following a transient initial expansion typically decline to relatively low frequencies in the absence of antiretroviral therapy,165, 166 due in part to the preferential susceptibility of virus‐specific CD4+ T cells to infection by HIV.167 The dynamics of Tfh cell induction in acute HIV‐1 infection have not been precisely determined, but LN GCs containing proliferating Tfh cells are present in macaques by day 14 post‐SIV infection,168, 169 suggesting rapid induction of a Tfh cell response following infection.
In vitro, GC Tfh cells are more susceptible to infection with HIV‐1 than non‐GC Tfh cells or CXCR5− extrafollicular CD4+ T cells,170, 171 and in vivo they have been shown to constitute the major CD4 T‐cell compartment for virus replication in both HIV‐1 172, 173 and SIV174, 175 infections. Furthermore, Tfh cell populations in both LNs and blood constitute a major (although not the only) site of SIV/HIV latency in macaques/humans receiving antiretroviral therapy.171, 176, 177, 178, 179 GC Tfh cells are not only highly activated cells that are good targets for HIV‐1 infection but are also located in close proximity to FDCs, which are an important reservoir of infectious virus and can readily transmit infection to Tfh cells.180, 181, 182, 183 Virus replication in GC Tfh cells may also be facilitated by the limited ability of antiviral CD8+ T cells to enter B‐cell follicles,168, 171, 175, 179, 184 although recent studies in the lymphocytic choriomeningitis virus (LCMV) mouse model show that persisting virus can be cleared from Tfh cells and B cells by a CXCR5+ CD8+ T‐cell population that is able to enter B‐cell follicles,185, 186, 187 raising the intriguing prospect that if an analogous antiviral CD8+ T‐cell population expressing sufficiently high levels of CXCR5 could be induced in humans, this may enable targeting of Tfh cells that harbor persistent HIV‐1 such as the latent pool.
Despite the susceptibility of Tfh cells to infection with SIV/HIV, the frequency of GC Tfh cells begins to increase within a few months of infection188 and elevated Tfh cell numbers are routinely observed in LNs during chronic SIV and HIV infections,168, 173, 189, 190, 191 although Tfh cells are eventually depleted as progression to acquired immunodeficiency syndrome (AIDS) occurs.192 The expansion in Tfh cells is associated with an increase in GC B cells and plasma cells and elevated IgG production, suggesting that Tfh cells contribute to the dysregulation of B cells and antibody production that occurs during HIV‐1 infection.191 The magnitude and duration of antigenic stimulation are known to be important determinants of the GC Tfh cell response,193 and although GC Tfh cell frequencies do not generally show a direct correlation with viral load, the increase in GC Tfh cells during chronic SIV/HIV infection is dependent on ongoing antigenic stimulation, as GC Tfh cell frequencies decline when viral replication is contained by antiretroviral therapy.168, 189, 191 Sustained LCMV replication in mice is also associated with expansion of GC Tfh cells, and here IL‐6 production has been shown to play a key role in driving Tfh cell differentiation during chronic infection.194, 195 GC Tfh cell expansion in chronic HIV‐1 infection is also associated with elevated IL‐6 production,189 but as IL‐6 is not such a potent stimulus of human CD4+ differentiation into Tfh cells, it can be speculated that upregulation of other Tfh‐differentiating cytokines, in the context of the reduction in IL‐2 expression known to occur in chronic HIV infection,196 may play a more important role in enhancing Tfh cell differentiation during HIV infection. Reduced control of Tfh cell numbers by regulatory T‐cell populations likely also facilitates expansion of Tfh cells during SIV/HIV infection; this is discussed further below.
Although GC responses are rapidly induced in acute HIV‐1 infection and a pronounced expansion of GC Tfh cells occurs during chronic infection, T‐cell help for B‐cell responses may nonetheless be limited. LN GC Tfh cells from chronically HIV‐infected individuals were shown to have an impaired ability to provide help for B cells in in vitro co‐culture assays, with Tfh cell helper capacity correlating negatively with viral load.190 GC B cells from HIV‐1 infected individuals express elevated levels of programmed cell death ligand 1 (PD‐L1), binding of which to PD‐1 on Tfh cells reduced their activation, ICOS expression, and IL‐21 production in vitro, contributing to their poor helper capacity.190 The functional capacity of circulating Tfh cells was also found to be impaired: the proportion of cells producing cytokines including IL‐21 was reduced, and they again exhibited a reduced B‐cell helper capacity in vitro.163 These defects in Tfh cell function appear to develop very rapidly, during the acute phase of HIV‐1 infection.190, 197 Analysis of both GC Tfh cells in SIV‐infected macaques198 and circulating CXCR3− CXCR5+ cells in individuals chronically infected with HIV‐1199 suggests that Tfh cells become more Th1‐polarized by chronic infection. They also express higher levels of CD25 and exhibit enhanced IL‐2 signaling, and in vitro use of an anti‐IL‐2 antibody to block IL‐2 signaling improved the B‐cell helper capacity of circulating CXCR3− CXCR5+ Tfh cells from infected subjects.199
3.5. Relationship between Tfh cells and bnAb induction in HIV‐1 infection
As the majority of bnAbs show evidence of extensive somatic hypermutation, it is likely that Tfh cell activity plays an important role in bnAb induction. Analysis of the relationship between Tfh cell responses and bnAb induction during HIV‐1 infection is hampered by the limited availability of longitudinal samples from individuals who eventually develop bnAbs and the difficulty in studying LN Tfh cell responses in humans. Nonetheless individuals developing bnAbs have been shown to have a higher frequency of circulating memory Tfh cells (defined as PD1+ CXCR3− cells within CXCR5+ CD4+ T cells47, 160 or PD1+ CXCR5+ cells within CD4+ T cells200) both early (approximately 6 months) after infection and during chronic infection than individuals developing antibodies with little or no cross‐neutralizing breadth, an observation independent of the higher viral loads in subjects developing bnAbs.47, 160 Subjects developing bnAbs also have higher plasma levels of CXCL13, a biomarker of GC activity, at both early and chronic infection timepoints, than subjects who develop little/no antibody neutralization breadth.200, 201 Furthermore, in in vitro B‐cell co‐culture assays CXCR5+ CD4+ Tfh cells isolated during acute infection from subjects who subsequently developed good neutralization breadth showed induction of similar levels of antibody secretion, but higher levels of class‐switch antibodies than Tfh cells from subjects developing little/no neutralization breadth, suggesting that Tfh cell function may potentially be superior in those who make bnAbs.200 Although these studies only addressed total circulating Tfh frequencies and function, in macaques infected with a simian‐human immunodeficiency virus (SHIV) expressing the HIV‐1 AD8 Env (SHIVAD8), the frequency of Env‐specific CD154+ or IL‐4+ (but not IFNγ+) Tfh cells (LN PD1+ CXCR5+ CD4+ cells) at 44‐47 weeks post‐infection was shown to correlate positively with the concurrent frequency of IgG+ GC B cells and with antibody neutralization breadth at both this and later timepoints during infection.188 Transcriptional profiling of Env‐specific CD154+ Tfh cells (LN PD1+ CXCR5+ CD4+ cells) in SHIVAD8‐infected macaques also showed higher levels of Tfh‐related genes (Bcl6, MAF, MYB, CXCL13 and IL‐21) and the Th2 gene GATA3 (encoding GATA‐binding protein 3) and lower Foxp3 (encoding forkhead box protein 3) in animals developing greater nAb breadth.
Together, these observations support an important role for Tfh cells in the generation of HIV‐1 antibody neutralization breadth. Nonetheless, many important questions remain about the relationship between HIV‐specific Tfh cell responses and bnAb generation. First, does the epitope specificity of Tfh cells impact on bnAb induction? As Tfh cells mediate cognate interactions with B cells, Tfh cell epitopes must be physically linked to bnAb epitopes, although they need not necessarily be in Env, e.g. in macaques primed with group‐specific antigen (Gag) plus polymerase (Pol) (Gag‐Pol) immunogens and boosted with virus‐like particles containing Gag‐Pol and Env, Gag‐Pol‐specific CD4+ T cells were found to enhance Env‐specific antibody production.202 However, as antigens can undergo degradation in vivo, it may be advantageous for Tfh cell and bnAb epitopes to be in close proximity. Second, is Tfh cell avidity important? A recent study of the influenza‐virus‐specific CD4+ T‐cell response indicated that T cells responding to different epitopes exhibited distinct tendencies to develop into Tfh cells, with those exhibiting a higher functional avidity being more likely to become Tfh cells203; but whether Tfh cells of higher avidity also mediate superior help for B cells is not clear. No associations have been reported between HLA class II type and bnAb induction during HIV‐1 infection that may support a role for T‐cell responses of particular specificity favoring or disfavoring bnAb induction47; but as many CD4 T‐cell epitopes are promiscuously presented by multiple HLA class II alleles,204 this does not preclude a relationship between epitope recognition and help for bnAb induction. Finally, how does the functional capacity of Tfh cells impact on bnAb induction? If, as discussed above, Tfh cell function is impaired in HIV‐1‐infected individuals, does this hamper bnAb generation; and/or does preservation of certain aspects of Tfh cell function favor bnAb induction during chronic infection?
Although it has not yet proved possible to elicit bnAbs by vaccination, in the RV144 phase IIb vaccine trial, priming with a recombinant canarypox vector (ALVAC‐HIV vCP1521) and boosting with a recombinant gp120 subunit vaccine (AIDSVAX B/E) were found to exhibit a 31.2% efficacy in preventing infection in a low‐risk heterosexual Thai population.8, 205 A correlates analysis revealed that IgG responses to variable regions 1 and 2 (V1‐V2) of Env associated with a decreased infection risk, while IgA responses were associated with an increased risk of infection acquisition.206, 207 Interestingly, IgG responses to V1‐V2 were higher and were associated with a decreased risk of infection acquisition only in individuals with the HLA‐DPB1*13 class II allele, while Env‐specific IgA responses were associated with an enhanced infection risk only in individuals with HLA‐DQB1*06, two class II alleles that were both common (present at frequencies of >10%) in the RV144 vaccine trial participants.208 Env‐specific CD4+ T cells directed against V2 were the most common T‐cell response induced by the RV144 vaccine regimen209; furthermore, RV144 vaccinees exhibited higher frequencies of circulating HIV‐specific IL‐21‐producing CD4+ T cells than participants in other trials of non‐protective HIV vaccines.210 Together, these observations suggest an important role for qualitative features of the vaccine‐induced CD4+ T‐cell response in determining the protective capacity of the antibody response elicited—a relationship that may prove even more critical for bnAb induction.
4. Regulatory cell populations and their relationship to bnAb induction
4.1. Regulation of GC responses
GC responses need to be precisely controlled to enable generation of high‐affinity antibodies and prevent the production of autoantibodies and development of autoimmune disease and chronic inflammation (reviewed in 211). Although the magnitude of the GC response is diminished if Tfh cell numbers are markedly reduced, limiting the number of Tfh cells is important to promote competition among B cells for interaction with Tfh cells and enable stringent selection of high‐affinity B‐cell clones. The presence of high numbers of Tfh cells results in a reduction in the selection threshold and enables survival of lower affinity and self‐reactive B cells, e.g. in sanroque mice homozygous for a loss of function mutation in Roquin, which results in high ICOS expression on Tfh cells and excessive Tfh cell accumulation, there is spontaneous B‐cell activation, GC formation, and autoantibody production, and the animals develop a lupus‐like phenotype.212, 213 Likewise increased numbers of circulating CXCR5+ CD4+ T cells, frequencies of which correlate with autoantibody titers, are observed in patients with SLE and other autoimmune diseases including Sjogren's syndrome and myasthenia gravis.162, 214
As discussed above, Tfh cell differentiation is a multi‐step process initiated by the signals received by naive CD4+ T cells as they undergo priming by DCs in LN T‐cell zones and begin to expand, and reinforced by subsequent cross‐talk with B cells at margin of and within B‐cell follicles, and within GCs. The immunologic environment at the time of CD4+ T‐cell priming thus has an important impact on initial Tfh induction, and regulatory cell populations including FoxP3+ CD4+ regulatory T cells (Tregs),215, 216 IL‐10‐producing regulatory B cells,217, 218 and natural killer (NK) cells219 may all modulate the generation of Tfh cells and hence influence GC formation and the subsequent humoral response.
However, Tfh cell help for B cells is also regulated locally within GCs. Tfh cell regulation within the GC is mediated in part by inhibitory receptors, negative feedback loops, and the availability of antigen and growth factors, and it is also regulated by specialized populations of regulatory cells. The inhibitory receptor PD‐1 is expressed at high levels on Tfh cells, and engagement of PD‐1 by PD‐L1 on B cells prevents Tfh cell proliferation following antigen recognition on B cells.109, 220, 221 In addition to B cells, antigen‐specific plasma cells can also present antigen to Tfh cells. However, they do not stimulate Bcl6 expression in Tfh cells and promote their differentiation into non‐Tfh cells, providing a negative feedback mechanism for reducing the GC response when large numbers of antigen‐specific plasma cells have been generated.222 Furthermore, as antigenic stimulation is required to sustain Tfh cell responses, the GC response is down‐modulated as antigen is cleared193; conversely sustained antigen persistence may promote Tfh expansion in situations of chronic infection or autoimmunity. In addition, GC responses are also regulated by regulatory cell populations that are found within the GC, including CD4+ T follicular regulatory cells (Tfr)223 and major histocompatibility complex (MHC)‐E‐restricted regulatory CD8+ T cells.224
4.2. Control of GC responses by regulatory CD4+ T cells
CD4+ cells expressing the transcription factor FoxP3 and high levels of CD25 play an important role in the maintenance of self‐tolerance and immune homeostasis, and the absence of FoxP3 expression results in susceptibility to development of autoimmunity, immunopathology, and lymphoproliferative disease (reviewed in 216, 225). Naive CD4+ T cells expressing FoxP3 are produced by the thymus. These cells, which are termed natural CD4+ Treg cells (nTregs) (reviewed in 225), respond to antigenic stimulation by differentiating into effector Treg cells.226 There are nTreg cells with specificity for both self and foreign antigens, although the ratio of antigen‐specific FoxP3+ to FoxP3− cells is higher for the former, enabling more stringent control of responses to autoantigens.227 FoxP3 expression can also be induced in FoxP3− naive CD4+ T cells in the periphery in response to antigenic stimulation in the presence of IL‐2 and TGFβ,228, 229 resulting in the generation of induced (i)Treg cells. nTreg and iTreg cells can be differentiated by expression of the transcription factor Helios, which is only expressed in nTreg cells.230 FoxP3+ CD4+ Treg cells can mediate control of immune responses via multiple mechanisms. They reduce CD4+ T‐cell activation via cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4)‐mediated downregulation of CD80/86 on antigen‐presenting cells, which limits T‐cell costimulation via CD28; CD39/73‐mediated adenosine triphosphate (ATP) degradation, and CD25‐mediated IL‐2 sequestration; and can also downregulate immune responses by perforin and granzyme‐dependent lysis of antigen‐presenting cells and T cells and by production of immunosuppressive cytokines, such as IL‐10, TGFβ, and IL‐35 (reviewed in 231).
FoxP3+ CD4+ Treg cells play an important role in the regulation of humoral immune responses, as both mice and humans lacking FoxP3 have elevated levels of circulating antibodies.232, 233 By regulating initial CD4+ T‐cell activation and differentiation FoxP3+ CD4+ Treg cells can reduce the initial generation of Tfh cells, although they may also enhance CD4+ T‐cell differentiation into Tfh cells by sequestering IL‐2.234 However, regulatory CD4+ T cells are also found within GCs,235 and recent studies have shown that a subset of CXCR5+ FoxP3+ CD4+ Treg cells, termed T follicular regulatory (Tfr) cells, are generated as immune responses are induced and localize to the GC to control the GC response.236, 237, 238 In mice, Tfr cells have been shown to reduce antibody production and the number of GC B cells, antigen‐specific B cells, and plasma cells.236, 237, 238, 239, 240, 241 Multiple stages of the B‐cell differentiation process are inhibited, from initial B‐cell activation to the generation of class‐switched B cells and plasma cells. Tfr cells also inhibit Tfh cell expansion, differentiation, and production of cytokines such as IL‐21 and IL‐4.239, 240, 242 Human Tfr cells (defined as CD127− CD25+ CXCR5+ CD4+ cells) have likewise been shown to reduce antibody production in in vitro Tfh:B cell co‐culture assays.243 Importantly, although Tfr cells reduce the overall magnitude of the humoral response, the antibody produced is of higher affinity than that generated in the absence of Tfr cells.240, 244
CD4+ Tfr cells develop from thymically derived Tregs that co‐opt a Tfh‐like differentiation pathway following activation and can also be generated from FoxP3− precursors.245 Like Tfh cells, Tfr cell differentiation is initiated early after antigen priming by DCs. Although Tfr cells express FoxP3 and proteins characteristic of CD4+ Tregs including Blimp‐1, CTLA‐4, and CD39, they acquire expression of Tfh cell markers including CXCR5, Bcl6, PD‐1, and ICOS (although they do not express proteins involved in mediating help for B cells such as CD40L, IL‐21, and IL‐4).236, 237, 238 Their differentiation involves some of the same pathways as those used by Tfh cells, although there are differences, e.g. CXCR5 expression on FoxP3+ T cells is regulated by nuclear factor of activated T cells (NFAT) rather than Ascl2.246 Following their initial generation, Tfr cells may either leave the LN and enter the circulation to become memory Tfr cells242 or migrate into the B‐cell follicles where they interact with B cells and undergo full differentiation into effector Tfr cells.239 Tfr cell differentiation following interaction with both DCs and B cells requires co‐stimulation though CD28 and ICOS, and SAP‐dependent stimulation by B cells.236, 237, 239, 247, 248 CTLA‐4 expression on Tfr cells inhibits their differentiation and maintenance via interaction with CD80/86 on DCs and B cells240, 249; and PD‐1 ligation inhibits both Tfr cell differentiation and their subsequent function.239
It is not known whether Tfr cells regulate Tfh‐B cell interactions at the border of the follicle and modulate GC formation; but they act within the GC to suppress GC Tfh cells and B cells. The mechanisms by which Tfr cells mediate suppression of GC Tfh cells and B cells are not well defined. CTLA‐4 has been shown to play an important role in Tfr cell function,240, 249 but the importance of other receptor‐ligand interactions and suppressive mechanisms such as production of IL‐10 and IL‐35, perforin/granzyme‐dependent lysis of GC Tfh and B cells, or interference with Tfh cell interaction with B cells remain to be elucidated. Antibody responses are determined by the ratio of Tfr:Tfh cells.239, 240, 250 In the resting state, there is a 1:1 ratio of Tfr:Tfh cells within CXCR5+ CD4+ T cells in LNs (although lower ratios are present in the spleen and Peyer's patches). During an immune response, Tfh cells differentiate more rapidly than Tfr cells, and as GC formation occurs, Tfr:Tfh ratios decrease, typically to 1:4‐5. This decrease precedes the increase in GC B cells and antibody production. It has been speculated that as full Tfr cell differentiation is dependent on interaction with GC B cells, the increase in GC B cells may enhance Tfr cell differentiation, resulting in a feedback loop that controls the GC response.223 In autoimmune BXD2 mice, high levels of IL‐21 enhance Tfh cell generation and drive a decline in the Tfr:Tfh cell ratio, promoting autoantibody production.251
4.3. Control of GC responses by regulatory CD8+ T cells
Studies in mice have identified a subset of CXCR5+ CD8+ T cells in LN GCs that also contribute to the regulation of humoral responses (reviewed in 224). These cells have been shown to suppress T‐cell‐dependent B‐cell responses in a H‐2 Qa‐1‐dependent manner and play an important role in maintenance of self‐tolerance.252 Qa‐1 and its human homolog HLA‐E are non‐classical MHC class Ib molecules that predominantly present a peptide derived from the signal sequence of classical MHC class Ia molecules, recognition of which by the inhibitory NK cell receptor NKG2A/CD94 contributes to inhibition of triggering of NK cell effector activity on contact with cells that express normal levels of MHC 1a molecules.253 However, MHC‐E can also present some peptides derived from autoantigens and pathogens, which are recognized by CD8+ T cells. Qa‐1 is expressed at high levels on GC CXCR5+ CD4+ cells, while CXCR5− CD4+ T cells express very low levels of Qa‐1; hence, Qa‐1‐restricted CXCR5+ CD8+ T cells specifically target CXCR5+ Tfh cells.254 By mediating perforin‐dependent lysis of GC Tfh cells, they reduce the GC response and help to prevent autoantibody development.252, 254, 255 Although Qa‐1‐restricted CD8+ Treg cells express CXCR5, they do not express ICOS or PD‐1 or markers characteristic of CD4+ Treg cells such as FoxP3. However, they do express ICOSL and CD122 (the IL‐2 receptor β chain, which also forms part of the IL‐15 receptor).224 The mechanisms involved in their differentiation are not fully understood, although Helios‐dependent STAT5 activation has been shown to be important to enable their survival and prevent terminal differentiation.256 Whether similar CXCR5+ CD8+ Treg cells contribute to regulation of GC responses in humans remains unclear, although HLA‐E‐restricted CD8+ Treg cells have been suggested to be involved in the control of type 1 diabetes in humans.257
4.4. CD4+ Treg cells and CD4+ Tfr cells in HIV‐1/SIV infection
FoxP3+ CD4+ Treg cells have potential to mediate both beneficial and detrimental effects during HIV‐1 infection, by suppressing the generalized T‐cell activation and inflammation that contributes to ongoing virus replication and disease progression and/or impairing HIV‐specific immune responses. However, despite considerable investigation, their roles remain incompletely understood (reviewed in 258). Analysis of regulatory CD4+ T cells in HIV‐1 and SIV infections is complicated by the difficulty in distinguishing CD25+ FoxP3+ CD4+ Treg cells from activated conventional CD4+ T cells, as the latter also express CD25 and FoxP3 transiently following activation.259
Despite the fact that CD4+ Treg cells can be infected with HIV‐1 and the absolute number of both total CD4+ T cells and CD4+ Treg cells is reduced in HIV‐1‐infected individuals, multiple reports indicate that the frequency of CD4+ Treg cells within circulating CD4+ T cells is increased.260, 261, 262, 263, 264, 265, 266, 267, 268 Enhanced generation of nTreg cells from the thymus,269 increased iTreg induction in the periphery,270, 271 and enhanced CD4+ Treg cell survival272 have all been suggested to contribute to this increase. Furthermore, as HIV‐specific Tregs are induced during infection,273, 274 the increase in CD4+ Treg frequencies may be, in part, driven by antigenic stimulation. CD4+ Treg cells have also been found to accumulate in tissues during HIV‐1 infection.
The in vitro suppressive capacity of CD4+ Treg cells from HIV‐1 infected individuals is reported to be comparable to that of HIV‐seronegative donors, and CD4+ Treg cells from subjects acutely or chronically infected with HIV‐1 have also been shown to suppress HIV‐specific CD4+ T‐cell proliferation and cytokine production in vitro.274, 275 However, the circulating frequency of CD4+ Treg cells has not been found to show any correlation with the magnitude of HIV‐specific T‐cell responses,276, 277, 278 leaving the contribution of CD4+ Treg cells to regulation of virus‐specific T‐cell responses in HIV‐infected individuals unclear. Their role in suppressing immune activation is also uncertain. Many studies report a positive correlation between CD4+ Treg cell frequencies and levels of T‐cell activation260, 262, 263, 279, 280, 281, and CD4+ Treg cell frequencies also correlated inversely with the circulating CD4 T‐cell count. However, this may reflect enhanced CD4+ Treg generation in the context of immune activation (and potentially also enhanced virus replication, although CD4+ Treg cell frequencies are not always reported to correlate with plasma viral load).
Relatively few studies have addressed Tfr cell frequencies during HIV‐1 or SIV infection. The frequency of Tfr cells within total LN CD4+ T cells does not appear to be increased during chronic SIV282, 283 or HIV‐1 infection,284 although the absolute number of Tfr cells is increased. One report suggests that the Tfr cell frequency in the spleen may be increased during HIV‐1 infection, however.285 Two studies in which the LN Tfr:Tfh cell ratio was analyzed in macaques chronically infected with SIV report conflicting results, with one finding an increase284 and the other a decrease.283 In the latter study, the decrease in the Tfr:Tfh cell ratio correlated with an increase in the percentage of GC B cells, suggesting that Tfr cells suppress GC B‐cell formation. LN Tfr:Tfh cell ratios have not been investigated during HIV‐1 infection, but the ratio of Tfr:Tfh (i.e. CD25+ FoxP3+:CD25− FoxP3−) cells within the circulating CXCR5+ CD4+ T‐cell pool in a cohort of chronically infected individuals was found to be similar to that of uninfected subjects.47 Whether there are alterations in Tfr cell function during HIV‐1/SIV infections remains to be investigated; however, in experiments in which tonsil cells were infected with HIV‐1 in vitro, Tfr cells were found to express increased levels of CTLA‐4, lymphocyte activation gene 3 (LAG‐3), IL‐10, and TGF‐β and retained the capacity to impair Tfh cell proliferation and production of IL‐21 and IL‐4.284
4.5. Relationship between CD4+ Treg cells/CD4+ Tfr cells and bnAb induction in HIV‐1/SIV infection
Regulatory CD4+ T‐cell populations, in particular Tfr cells, control the overall magnitude of the GC response and regulate the stringency of B‐cell selection within GCs, limiting the production of lower‐affinity antibodies and auto‐reactive antibodies. They may, therefore, constrain the production of HIV‐1 bnAbs, which, as discussed earlier, commonly exhibit extensive somatic hypermutation and show evidence of autoreactivity, suggesting a need for strong GC Tfh cell activity and relaxed selection controls for their generation. One study in SIV‐infected macaques found that circulating CD4+ Treg numbers correlated positively with autoantibody titers and the spectrum of autoantigens recognized, which does not support a relationship between relaxation of host tolerance controls and generation of antibodies with greater autoreactivity286; however, LN Tfr cell numbers and the Tfr:Tfh cell ratio were not addressed. Conversely in another SIV study, the LN Tfr cell frequency (% FoxP3+ CD25+ CXCR5+ CD4+ T cells) was found to show an inverse correlation with the frequency of LN Tfh cells and the avidity of antibodies recognizing the SIV gp120 envelope protein in plasma, indicating a role for Tfr cells in constraining the maturation of the envelope‐reactive antibody response.282
We recently analyzed circulating CD4+ Treg and CD4+ Tfr cell populations in subjects chronically infected with HIV‐1 who had generated bnAbs and matched individuals who had developed little or no antibody neutralization breadth.47 The frequency of CD4+ Treg cells within lymphocytes was significantly lower in the bnAb group, although there was no difference between groups in the percentage of CD4+ Treg cells within CD4+ T cells, the circulating frequency of Tfr cells, or the Tfr:Tfh ratio (ratio of CD25+ FoxP3+:CD25− FoxP3− cells within circulating CXCR5+ CD4+ T cells). Interestingly, however, PD‐1 was expressed at significantly higher levels on both CD4+ Treg and CD4+ Tfr cells in the subjects who had generated bnAbs. As PD‐1 inhibits the function of CD4+ Treg and CD4+ Tfr cells,239 this suggested that the suppressive capacity of CD4+ Treg/Tfr cells in the bnAb‐producing subjects may be impaired, an observation supported by the finding that the PD‐1hi subset of CD4+ Treg cells from some HIV‐seronegative donors exhibited an impaired capacity to inhibit the proliferation of conventional CD4+ T cells in vitro.47 As discussed above, autoantibodies were also detected in a higher proportion of the group of subjects who produced bnAbs than those who did not. Together, these findings suggest that bnAb development during HIV‐1 infection may be favored by a relaxation in regulatory CD4+ T‐cell control of antibody production that enables a strong GC response and permits some degree of autoreactive antibody production.47
5. Implications for design of bnAb‐inducing vaccines
A major focus of efforts to develop bnAb‐inducing vaccines has been on antigen design.287, 288, 289 Dissection of the pathways by which bnAbs evolve in HIV‐1‐infected individuals has led to recognition of the need for antigens that are able to trigger B cells expressing the unmutated common ancestors of mature bnAbs, drive the selection of daughter cells expressing antibodies with potential for further maturation along bnAb lineages, and ultimately enable bnAb evolution.34 However, in addition to design of appropriate vaccine antigens, immunization strategies need to be devised that will elicit an environment supportive of an appropriate B‐cell response to them. Here too, insights can be gained from analysis of HIV‐1 infected individuals who develop bnAbs. Factors shown to correlate with bnAb induction in HIV‐1 infection are summarized in Figure 1. Together they suggest that a strong CD4+ Tfh cell response, reduction in host tolerance controls, potentially mediated by a reduction in regulatory CD4+ T‐cell suppression of GC responses, and strong, sustained antigenic stimulation are all of importance to enable a potent GC response and drive B cells to undergo multiple rounds of somatic hypermutation, in the context of a relaxation of host constraints on autoreactive antibody production.
Figure 1.
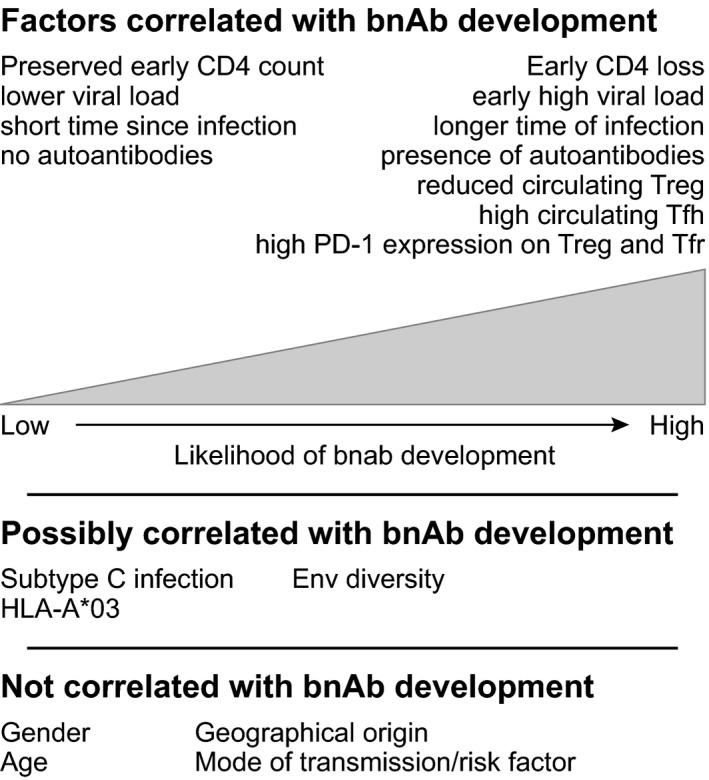
Factors associated with bnAb development in HIV‐1‐infected individuals. HIV‐1‐infected individuals who make bnAbs have been found to have perturbations of their immune system. In contrast, demographic characteristics have not been found to correlate with bnAb development
Vaccine development efforts are, thus, likely to benefit from consideration of these three inter‐related factors. First, a strong antigen‐specific CD4+ Tfh cell response needs to be elicited. As discussed above, it is currently unclear whether this should ideally target particular CD4+ epitopes within the vaccine antigen, although it can be speculated that targeting of high‐affinity epitopes in close proximity to bnAb epitopes may be beneficial. However, the use of immunization regimes (e.g. vectors and adjuvants) that promotes CD4+ T‐cell differentiation into Tfh cells will be of great importance. For example, recent studies in murine models demonstrated that adenoviral vectors elicited a strong Tfh cell response to encoded vaccine antigens,290 and that adenoviral priming enabled induction of strong humoral responses to a subsequent protein boost delivered with a relatively weak adjuvant291; and adenoviral vectors have also been shown to elicit strong Tfh cell responses to encoded HIV antigens in macaques.292 Second, immunization platforms need to be developed that will enable high level and prolonged antigen availability. This is of importance to sustain the Tfh cell response and the GC reaction and enable B cells to undergo multiple rounds of somatic hypermutation. Repeat immunizations, vectors that drive prolonged antigen expression, and/or platforms enabling slow antigen release over time (e.g. nanoparticle delivery systems293) may all be helpful. Third, it may be necessary to transiently modulate host constraints on autoreactive antibody development at the time of bnAb induction to enable the evolution of bnAbs whose development is constrained by host tolerance controls. For example, this could be achieved by the use of immune modulatory strategies such as transient CTLA‐4 blockade,294 although this type of approach will need to be carefully safety‐tested in animal models prior to study in human trials.
In summary, study of the mechanisms that enable bnAb induction in some HIV‐infected individuals has given important insight into how bnAb induction may be achieved by vaccination. The benefit to rational vaccine design strategies is such that achievement of bnAb induction by vaccination now seems very likely.
Conflicts of interest
The authors have no conflicts of interest.
AcknowledgEments
The authors thank Prof Andrew McMichael and Dr Isabela Pedroza‐Pacheco for their helpful comments on this review. Support for this work was provided by the NIH, NIAID, Division of AIDS, UM‐1 grant for the Duke Center for HIV/AIDS Vaccine Immunology‐Immunogen Discovery AI100645; and MRC Programme grant MR/K012037. P. B. is a Jenner Institute Investigator.
References
- 1. UNAIDS . Prevention Gap Report. Geneva: World Health Organisation, 2016. [Google Scholar]
- 2. Fauci AS, Marston HD. Ending AIDS—Is an HIV vaccine necessary? N Engl J Med. 2014;370:495–498. [DOI] [PubMed] [Google Scholar]
- 3. Buchbinder SP, Mehrotra DV, Duerr A, et al. Efficacy assessment of a cell‐mediated immunity HIV‐1 vaccine (the Step Study): A double‐blind, randomised, placebo‐controlled, test‐of‐concept trial. Lancet. 2008;372:1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilbert PB, Peterson ML, Follmann D, et al. Correlation between immunologic responses to a recombinant glycoprotein 120 vaccine and incidence of HIV‐1 infection in a phase 3 HIV‐1 preventive vaccine trial. J Infect Dis. 2005;191:666–677. [DOI] [PubMed] [Google Scholar]
- 5. Gray GE, Allen M, Moodie Z, et al. Safety and efficacy of the HVTN 503/Phambili study of a clade‐B‐based HIV‐1 vaccine in South Africa: A double‐blind, randomised, placebo‐controlled test‐of‐concept phase 2b study. Lancet Infect Dis. 2011;11:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hammer SM, Sobieszczyk ME, Janes H, et al. Efficacy trial of a DNA/rAd5 HIV‐1 preventive vaccine. N Engl J Med. 2013;369:2083–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pitisuttithum P, Gilbert P, Gurwith M, et al. Randomized, double‐blind, placebo‐controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV‐1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194:1661–1671. [DOI] [PubMed] [Google Scholar]
- 8. Rerks‐Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV‐1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. [DOI] [PubMed] [Google Scholar]
- 9. Liu P, Williams LD, Shen X, et al. Capacity for infectious HIV‐1 virion capture differs by envelope antibody specificity. J Virol. 2014;88:5165–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu P, Yates NL, Shen X, et al. Infectious virion capture by HIV‐1 gp120‐specific IgG from RV144 vaccinees. J Virol. 2013;87:7828–7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forthal DN, Landucci G, Daar ES. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural‐killer effector cells. J Virol. 2001;75:6953–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hessell AJ, Hangartner L, Hunter M, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. [DOI] [PubMed] [Google Scholar]
- 13. Lyerly HK, Reed DL, Matthews TJ, et al. Anti‐GP 120 antibodies from HIV seropositive individuals mediate broadly reactive anti‐HIV ADCC. AIDS Res Hum Retroviruses. 1987;3:409–422. [DOI] [PubMed] [Google Scholar]
- 14. Tyler DS, Lyerly HK, Weinhold KJ. Anti‐HIV‐1 ADCC. AIDS Res Hum Retroviruses. 1989;5:557–563. [DOI] [PubMed] [Google Scholar]
- 15. Moody MA, Liao HX, Alam SM, et al. Anti‐phospholipid human monoclonal antibodies inhibit CCR5‐tropic HIV‐1 and induce beta‐chemokines. J Exp Med. 2010;207:763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr Protoc Immunol. 2005;12:12.11. [DOI] [PubMed] [Google Scholar]
- 17. Montefiori DC. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol. 2009;485:395–405. [DOI] [PubMed] [Google Scholar]
- 18. Zhou JY, Montefiori DC. Antibody‐mediated neutralization of primary isolates of human immunodeficiency virus type 1 in peripheral blood mononuclear cells is not affected by the initial activation state of the cells. J Virol. 1997;71:2512–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richman DD, Wrin T, Little SJ, Petropoulos CJ. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci USA. 2003;100:4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei X, Decker JM, Wang S, et al. Antibody neutralization and escape by HIV‐1. Nature. 2003;422:307–312. [DOI] [PubMed] [Google Scholar]
- 21. Pollara J, Bonsignori M, Moody MA, Pazgier M, Haynes BF, Ferrari G. Epitope specificity of human immunodeficiency virus‐1 antibody dependent cellular cytotoxicity [ADCC] responses. Curr HIV Res. 2013;11:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonsignori M, Zhou T, Sheng Z, et al. Maturation pathway from germline to broad HIV‐1 neutralizer of a CD4‐mimic antibody. Cell. 2016;165:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao F, Bonsignori M, Liao HX, et al. Cooperation of B cell lineages in induction of HIV‐1‐broadly neutralizing antibodies. Cell. 2014;158:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liao HX, Lynch R, Zhou T, et al. Co‐evolution of a broadly neutralizing HIV‐1 antibody and founder virus. Nature. 2013;496:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moody MA, Gao F, Gurley TC, et al. Strain‐specific V3 and CD4 binding site autologous HIV‐1 neutralizing antibodies select neutralization‐resistant viruses. Cell Host Microbe. 2015;18:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sather DN, Carbonetti S, Malherbe DC, et al. Emergence of broadly neutralizing antibodies and viral coevolution in two subjects during the early stages of infection with human immunodeficiency virus type 1. J Virol. 2014;88:12968–12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burton DR, Mascola JR. Antibody responses to envelope glycoproteins in HIV‐1 infection. Nat Immunol. 2015;16:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baba TW, Liska V, Hofmann‐Lehmann R, et al. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian‐human immunodeficiency virus infection. Nat Med. 2000;6:200–206. [DOI] [PubMed] [Google Scholar]
- 29. Hessell AJ, Rakasz EG, Poignard P, et al. Broadly neutralizing human anti‐HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009;5:e1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mascola JR, Stiegler G, VanCott TC, et al. Protection of macaques against vaginal transmission of a pathogenic HIV‐1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. 2000;6:207–210. [DOI] [PubMed] [Google Scholar]
- 31. Moldt B, Rakasz EG, Schultz N, et al. Highly potent HIV‐specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc Natl Acad Sci USA. 2012;109:18921–18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parren PW, Marx PA, Hessell AJ, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virol. 2001;75:8340–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pegu A, Yang ZY, Boyington JC, et al. Neutralizing antibodies to HIV‐1 envelope protect more effectively in vivo than those to the CD4 receptor. Sci Transl Med. 2014;6:243ra288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B‐cell‐lineage immunogen design in vaccine development with HIV‐1 as a case study. Nat Biotechnol. 2012;30:423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bradley T, Fera D, Bhiman J, et al. Structural constraints of vaccine‐induced tier‐2 autologous HIV neutralizing antibodies targeting the receptor‐binding site. Cell Rep. 2016;14:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanders RW, van Gils MJ, Derking R, et al. HIV‐1 VACCINES. HIV‐1 neutralizing antibodies induced by native‐like envelope trimers. Science. 2015;349:aac4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen MS, Shaw GM, McMichael AJ, Haynes BF. Acute HIV‐1 infection. N Engl J Med. 2011;364:1943–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keele BF, Giorgi EE, Salazar‐Gonzalez JF, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV‐1 infection. Proc Natl Acad Sci USA. 2008;105:7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ndhlovu ZM, Kamya P, Mewalal N, et al. Magnitude and kinetics of CD8+ T cell activation during hyperacute HIV infection impact viral set point. Immunity. 2015;43:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goonetilleke N, Liu MK, Salazar‐Gonzalez JF, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV‐1 infection. J Exp Med. 2009;206:1253–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tomaras GD, Yates NL, Liu P, et al. Initial B‐cell responses to transmitted human immunodeficiency virus type 1: Virion‐binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti‐gp41 antibodies with ineffective control of initial viremia. J Virol. 2008;82:12449–12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liao HX, Chen X, Munshaw S, et al. Initial antibodies binding to HIV‐1 gp41 in acutely infected subjects are polyreactive and highly mutated. J Exp Med. 2011;208:2237–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Derdeyn CA, Moore PL, Morris L. Development of broadly neutralizing antibodies from autologous neutralizing antibody responses in HIV infection. Curr Opin HIV AIDS. 2014;9:210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trama AM, Moody MA, Alam SM, et al. HIV‐1 envelope gp41 antibodies can originate from terminal ileum B cells that share cross‐reactivity with commensal bacteria. Cell Host Microbe. 2014;16:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hraber P, Seaman MS, Bailer RT, Mascola JR, Montefiori DC, Korber BT. Prevalence of broadly neutralizing antibody responses during chronic HIV‐1 infection. AIDS. 2014;28:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mikell I, Sather DN, Kalams SA, Altfeld M, Alter G, Stamatatos L. Characteristics of the earliest cross‐neutralizing antibody response to HIV‐1. PLoS Pathog. 2011;7:e1001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moody MA, Pedroza‐Pacheco I, Vandergrift NA, et al. Immune perturbations in HIV‐1—infected individuals who make broadly neutralizing antibodies. Sci Immunol. 2016;1:aag0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gray ES, Madiga MC, Hermanus T, et al. The neutralization breadth of HIV‐1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J Virol. 2011;85:4828–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doria‐Rose NA, Klein RM, Daniels MG, et al. Breadth of human immunodeficiency virus‐specific neutralizing activity in sera: Clustering analysis and association with clinical variables. J Virol. 2010;84:1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Landais E, Huang X, Havenar‐Daughton C, et al. Broadly neutralizing antibody responses in a large longitudinal sub‐Saharan HIV primary infection cohort. PLoS Pathog. 2016;12:e1005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bonsignori M, Montefiori DC, Wu X, et al. Two distinct broadly neutralizing antibody specificities of different clonal lineages in a single HIV‐1‐infected donor: Implications for vaccine design. J Virol. 2012;86:4688–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walker LM, Simek MD, Priddy F, et al. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV‐1 infected individuals. PLoS Pathog. 2010;6:e1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dhillon AK, Donners H, Pantophlet R, et al. Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1‐infected donors. J Virol. 2007;81:6548–6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gray ES, Taylor N, Wycuff D, et al. Antibody specificities associated with neutralization breadth in plasma from human immunodeficiency virus type 1 subtype C‐infected blood donors. J Virol. 2009;83:8925–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li Y, Svehla K, Louder MK, et al. Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1‐infected individuals. J Virol. 2009;83:1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sather DN, Armann J, Ching LK, et al. Factors associated with the development of cross‐reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J Virol. 2009;83:757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Georgiev IS, Doria‐Rose NA, Zhou T, et al. Delineating antibody recognition in polyclonal sera from patterns of HIV‐1 isolate neutralization. Science. 2013;340:751–756. [DOI] [PubMed] [Google Scholar]
- 58. Xu W, Santini PA, Sullivan JS, et al. HIV‐1 evades virus‐specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long‐range intercellular conduits. Nat Immunol. 2009;10:1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stacey AR, Norris PJ, Qin L, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Buckner CM, Moir S, Ho J, et al. Characterization of plasmablasts in the blood of HIV‐infected viremic individuals: Evidence for nonspecific immune activation. J Virol. 2013;87:5800–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moir S, Ho J, Malaspina A, et al. Evidence for HIV‐associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV‐infected viremic individuals. J Exp Med. 2008;205:1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moir S, Malaspina A, Ogwaro KM, et al. HIV‐1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proc Natl Acad Sci USA. 2001;98:10362–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Levesque MC, Moody MA, Hwang KK, et al. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV‐1 infection. PLoS Med. 2009;6:e1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kardava L, Moir S, Shah N, et al. Abnormal B cell memory subsets dominate HIV‐specific responses in infected individuals. J Clin Invest. 2014;124:3252–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boliar S, Murphy MK, Tran TC, et al. B‐lymphocyte dysfunction in chronic HIV‐1 infection does not prevent cross‐clade neutralization breadth. J Virol. 2012;86:8031–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Doria‐Rose NA, Klein RM, Manion MM, et al. Frequency and phenotype of human immunodeficiency virus envelope‐specific B cells from patients with broadly cross‐neutralizing antibodies. J Virol. 2009;83:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Euler Z, van Gils MJ, Bunnik EM, et al. Cross‐reactive neutralizing humoral immunity does not protect from HIV type 1 disease progression. J Infect Dis. 2010;201:1045–1053. [DOI] [PubMed] [Google Scholar]
- 68. Poignard P, Sabbe R, Picchio GR, et al. Neutralizing antibodies have limited effects on the control of established HIV‐1 infection in vivo. Immunity. 1999;10:431–438. [DOI] [PubMed] [Google Scholar]
- 69. Trkola A, Kuster H, Rusert P, et al. Delay of HIV‐1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med. 2005;11:615–622. [DOI] [PubMed] [Google Scholar]
- 70. Lynch RM, Boritz E, Coates EE, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV‐1 infection. Sci Transl Med. 2015;7:319ra206. [DOI] [PubMed] [Google Scholar]
- 71. Scheid JF, Horwitz JA, Bar‐On Y, et al. HIV‐1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shingai M, Nishimura Y, Klein F, et al. Antibody‐mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature. 2013;503:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schoofs T, Klein F, Braunschweig M, et al. HIV‐1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV‐1. Science. 2016;352:997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Medina‐Ramirez M, Sanchez‐Merino V, Sanchez‐Palomino S, et al. Broadly cross‐neutralizing antibodies in HIV‐1 patients with undetectable viremia. J Virol. 2011;85:5804–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kaye BR. Rheumatologic manifestations of infection with human immunodeficiency virus (HIV). Ann Intern Med. 1989;111:158–167. [DOI] [PubMed] [Google Scholar]
- 76. Kopelman RG, Zolla‐Pazner S. Association of human immunodeficiency virus infection and autoimmune phenomena. Am J Med. 1988;84:82–88. [DOI] [PubMed] [Google Scholar]
- 77. Barthel HR, Wallace DJ. False‐positive human immunodeficiency virus testing in patients with lupus erythematosus. Semin Arthritis Rheum. 1993;23:1–7. [DOI] [PubMed] [Google Scholar]
- 78. Calza L, Manfredi R, Colangeli V, D'Antuono A, Passarini B, Chiodo F. Systemic and discoid lupus erythematosus in HIV‐infected patients treated with highly active antiretroviral therapy. Int J STD AIDS. 2003;14:356–359. [DOI] [PubMed] [Google Scholar]
- 79. Palacios R, Santos J. Human immunodeficiency virus infection and systemic lupus erythematosus. Int J STD AIDS. 2004;15:277–278. [DOI] [PubMed] [Google Scholar]
- 80. Palacios R, Santos J, Valdivielso P, Marquez M. Human immunodeficiency virus infection and systemic lupus erythematosus. An unusual case and a review of the literature. Lupus. 2002;11:60–63. [DOI] [PubMed] [Google Scholar]
- 81. Bonsignori M, Wiehe K, Grimm SK, et al. An autoreactive antibody from an SLE/HIV‐1 individual broadly neutralizes HIV‐1. J Clin Invest. 2014;124:1835–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Haynes BF, Fleming J, St Clair EW, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV‐1 antibodies. Science. 2005;308:1906–1908. [DOI] [PubMed] [Google Scholar]
- 83. Liu M, Yang G, Wiehe K, et al. Polyreactivity and autoreactivity among HIV‐1 antibodies. J Virol. 2015;89:784–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yang G, Holl TM, Liu Y, et al. Identification of autoantigens recognized by the 2F5 and 4E10 broadly neutralizing HIV‐1 antibodies. J Exp Med. 2013;210:241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Haynes BF, Moody MA, Alam M, et al. Progress in HIV‐1 vaccine development. J Allergy Clin Immunol. 2014;134:3–10; quiz 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Meffre E, Milili M, Blanco‐Betancourt C, Antunes H, Nussenzweig MC, Schiff C. Immunoglobulin heavy chain expression shapes the B cell receptor repertoire in human B cell development. J Clin Invest. 2001;108:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Meffre E, Wardemann H. B‐cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008;20:632–638. [DOI] [PubMed] [Google Scholar]
- 88. Doyle‐Cooper C, Hudson KE, Cooper AB, et al. Immune tolerance negatively regulates B cells in knock‐in mice expressing broadly neutralizing HIV antibody 4E10. J Immunol. 2013;191:3186–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ota T, Doyle‐Cooper C, Cooper AB, et al. B cells from knock‐in mice expressing broadly neutralizing HIV antibody b12 carry an innocuous B cell receptor responsive to HIV vaccine candidates. J Immunol. 2013;191:3179–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Verkoczy L, Chen Y, Bouton‐Verville H, et al. Rescue of HIV‐1 broad neutralizing antibody‐expressing B cells in 2F5 VH x VL knockin mice reveals multiple tolerance controls. J Immunol. 2011;187:3785–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Verkoczy L, Chen Y, Zhang J, et al. Induction of HIV‐1 broad neutralizing antibodies in 2F5 knock‐in mice: Selection against membrane proximal external region‐associated autoreactivity limits T‐dependent responses. J Immunol. 2013;191:2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Verkoczy L, Diaz M, Holl TM, et al. Autoreactivity in an HIV‐1 broadly reactive neutralizing antibody variable region heavy chain induces immunologic tolerance. Proc Natl Acad Sci USA. 2010;107:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Verkoczy L, Moody MA, Holl TM, et al. Functional, non‐clonal IgMa‐restricted B cell receptor interactions with the HIV‐1 envelope gp41 membrane proximal external region. PLoS ONE. 2009;4:e7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lanzavecchia A. Antigen‐specific interaction between T and B cells. Nature. 1985;314:537–539. [DOI] [PubMed] [Google Scholar]
- 95. Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. [DOI] [PubMed] [Google Scholar]
- 96. Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39‐kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA. 1992;89:6550–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bryant VL, Ma CS, Avery DT, et al. Cytokine‐mediated regulation of human B cell differentiation into Ig‐secreting cells: Predominant role of IL‐21 produced by CXCR5+ T follicular helper cells. J Immunol. 2007;179:8180–8190. [DOI] [PubMed] [Google Scholar]
- 98. Avery DT, Bryant VL, Ma CS, de Waal Malefyt R, Tangye SG. IL‐21‐induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL‐4. J Immunol. 2008;181:1767–1779. [DOI] [PubMed] [Google Scholar]
- 99. Morita R, Schmitt N, Bentebibel SE, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yusuf I, Kageyama R, Monticelli L, et al. Germinal center T follicular helper cell IL‐4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol. 2010;185:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nutt SL, Tarlinton DM. Germinal center B and follicular helper T cells: Siblings, cousins or just good friends? Nat Immunol. 2011;12:472–477. [DOI] [PubMed] [Google Scholar]
- 102. Linterman MA, Beaton L, Yu D, et al. IL‐21 acts directly on B cells to regulate Bcl‐6 expression and germinal center responses. J Exp Med. 2010;207:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zotos D, Coquet JM, Zhang Y, et al. IL‐21 regulates germinal center B cell differentiation and proliferation through a B cell‐intrinsic mechanism. J Exp Med. 2010;207:365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Reinhardt RL, Liang HE, Locksley RM. Cytokine‐secreting follicular T cells shape the antibody repertoire. Nat Immunol. 2009;10:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schwickert TA, Victora GD, Fooksman DR, et al. A dynamic T cell‐limited checkpoint regulates affinity‐dependent B cell entry into the germinal center. J Exp Med. 2011;208:1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247:52–63. [DOI] [PubMed] [Google Scholar]
- 107. Gitlin AD, Mayer CT, Oliveira TY, et al. T cell help controls the speed of the cell cycle in germinal center B cells. Science. 2015;349:643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. 2014;509:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Good‐Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD‐1 regulates germinal center B cell survival and the formation and affinity of long‐lived plasma cells. Nat Immunol. 2010;11:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Victora GD, Schwickert TA, Fooksman DR, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Weinstein JS, Herman EI, Lainez B, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. 2016;17:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Choi YS, Yang JA, Yusuf I, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. 2013;190:4014–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Choi YS, Yang JA, Crotty S. Dynamic regulation of Bcl6 in follicular helper CD4 T (Tfh) cells. Curr Opin Immunol. 2013;25:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nurieva RI, Chung Y, Martinez GJ, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin‐2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Chavele KM, Merry E, Ehrenstein MR. Cutting edge: Circulating plasmablasts induce the differentiation of human T follicular helper cells via IL‐6 production. J Immunol. 2015;194:2482–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ma CS, Suryani S, Avery DT, et al. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL‐12. Immunol Cell Biol. 2009;87:590–600. [DOI] [PubMed] [Google Scholar]
- 119. Schmitt N, Morita R, Bourdery L, et al. Human dendritic cells induce the differentiation of interleukin‐21‐producing T follicular helper‐like cells through interleukin‐12. Immunity. 2009;31:158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Locci M, Wu JE, Arumemi F, et al. Activin A programs the differentiation of human TFH cells. Nat Immunol. 2016;17:976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schmitt N, Liu Y, Bentebibel SE, et al. The cytokine TGF‐beta co‐opts signaling via STAT3‐STAT4 to promote the differentiation of human TFH cells. Nat Immunol. 2014;15:856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ballesteros‐Tato A, Leon B, Graf BA, et al. Interleukin‐2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu X, Lu H, Chen T, et al. Genome‐wide analysis identifies Bcl6‐controlled regulatory networks during T follicular helper cell differentiation. Cell Rep. 2016;14:1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. McDonald PW, Read KA, Baker CE, et al. IL‐7 signalling represses Bcl‐6 and the TFH gene program. Nat Commun. 2016;7:10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Choi YS, Kageyama R, Eto D, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shin C, Han JA, Koh H, et al. CD8alpha(‐) dendritic cells induce antigen‐specific T follicular helper cells generating efficient humoral immune responses. Cell Rep. 2015;11:1929–1940. [DOI] [PubMed] [Google Scholar]
- 128. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL‐2‐quenching dendritic cells. Nature. 2016;533:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tubo NJ, Pagan AJ, Taylor JJ, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell. 2013;153:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Benson RA, MacLeod MK, Hale BG, Patakas A, Garside P, Brewer JM. Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife. 2015;4:000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kim CH, Rott LS, Clark‐Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center‐localized subset of CXCR5+ T cells. J Exp Med. 2001;193:1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene‐1high germinal center‐associated subpopulation. J Immunol. 2007;179:5099–5108. [DOI] [PubMed] [Google Scholar]
- 134. Xu H, Li X, Liu D, et al. Follicular T‐helper cell recruitment governed by bystander B cells and ICOS‐driven motility. Nature. 2013;496:523–527. [DOI] [PubMed] [Google Scholar]
- 135. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP‐controlled T‐B cell interactions underlie germinal centre formation. Nature. 2008;455:764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kageyama R, Cannons JL, Zhao F, et al. The receptor Ly108 functions as a SAP adaptor‐dependent on‐off switch for T cell help to B cells and NKT cell development. Immunity. 2012;36:986–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Johnston RJ, Poholek AC, DiToro D, et al. Bcl6 and Blimp‐1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yu D, Rao S, Tsai LM, et al. The transcriptional repressor Bcl‐6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. [DOI] [PubMed] [Google Scholar]
- 139. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol. 2016;34:335–368. [DOI] [PubMed] [Google Scholar]
- 140. Choi YS, Gullicksrud JA, Xing S, et al. LEF‐1 and TCF‐1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. 2015;16:980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wu T, Shin HM, Moseman EA, et al. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep. 2015;12:2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Xu L, Cao Y, Xie Z, et al. The transcription factor TCF‐1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol. 2015;16:991–999. [DOI] [PubMed] [Google Scholar]
- 143. Betz BC, Jordan‐Williams KL, Wang C, et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med. 2010;207:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bollig N, Brustle A, Kellner K, et al. Transcription factor IRF4 determines germinal center formation through follicular T‐helper cell differentiation. Proc Natl Acad Sci USA. 2012;109:8664–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Liu X, Chen X, Zhong B, et al. Transcription factor achaete‐scute homologue 2 initiates follicular T‐helper‐cell development. Nature. 2014;507:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bauquet AT, Jin H, Paterson AM, et al. The costimulatory molecule ICOS regulates the expression of c‐Maf and IL‐21 in the development of follicular T helper cells and TH‐17 cells. Nat Immunol. 2009;10:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ma CS, Avery DT, Chan A, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 2012;119:3997–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Stone EL, Pepper M, Katayama CD, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity. 2015;42:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Baumjohann D, Kageyama R, Clingan JM, et al. The microRNA cluster miR‐17 approximately 92 promotes TFH cell differentiation and represses subset‐inappropriate gene expression. Nat Immunol. 2013;14:840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kang SG, Liu WH, Lu P, et al. MicroRNAs of the miR‐17 approximately 92 family are critical regulators of T(FH) differentiation. Nat Immunol. 2013;14:849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hatzi K, Nance JP, Kroenke MA, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med. 2015;212:539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kitano M, Moriyama S, Ando Y, et al. Bcl6 protein expression shapes pre‐germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34:961–972. [DOI] [PubMed] [Google Scholar]
- 153. Shulman Z, Moriyama S, Ando Y, et al. T follicular helper cell dynamics in germinal centers. Science. 2013;341:673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Hale JS, Youngblood B, Latner DR, et al. Distinct memory CD4+ T cells with commitment to T follicular helper‐ and T helper 1‐cell lineages are generated after acute viral infection. Immunity. 2013;38:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Liu X, Yan X, Zhong B, et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J Exp Med. 2012;209:1841–1852, S1841‐1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Luthje K, Kallies A, Shimohakamada Y, et al. The development and fate of follicular helper T cells defined by an IL‐21 reporter mouse. Nat Immunol. 2012;13:491–498. [DOI] [PubMed] [Google Scholar]
- 157. Weber JP, Fuhrmann F, Hutloff A. T‐follicular helper cells survive as long‐term memory cells. Eur J Immunol. 2012;42:1981–1988. [DOI] [PubMed] [Google Scholar]
- 158. Bentebibel SE, Lopez S, Obermoser G, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5:176ra132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chevalier N, Jarrossay D, Ho E, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol. 2011;186:5556–5568. [DOI] [PubMed] [Google Scholar]
- 160. Locci M, Havenar‐Daughton C, Landais E, et al. Human circulating PD‐1+CXCR3‐CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Schmitt N, Bentebibel SE, Ueno H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol. 2014;35:436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. He J, Tsai LM, Leong YA, et al. Circulating precursor CCR7(lo)PD‐1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013;39:770–781. [DOI] [PubMed] [Google Scholar]
- 163. Boswell KL, Paris R, Boritz E, et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog. 2014;10:e1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Bentebibel SE, Khurana S, Schmitt N, et al. ICOS(+)PD‐1(+)CXCR3(+) T follicular helper cells contribute to the generation of high‐avidity antibodies following influenza vaccination. Sci Rep. 2016;6:26494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Oxenius A, Fidler S, Brady M, et al. Variable fate of virus‐specific CD4(+) T cells during primary HIV‐1 infection. Eur J Immunol. 2001;31:3782–3788. [DOI] [PubMed] [Google Scholar]
- 166. Rosenberg ES, Billingsley JM, Caliendo AM, et al. Vigorous HIV‐1‐specific CD4+ T cell responses associated with control of viremia. Science. 1997;278:1447–1450. [DOI] [PubMed] [Google Scholar]
- 167. Douek DC, Brenchley JM, Betts MR, et al. HIV preferentially infects HIV‐specific CD4+ T cells. Nature. 2002;417:95–98. [DOI] [PubMed] [Google Scholar]
- 168. Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Spatial alterations between CD4(+) T follicular helper, B, and CD8(+) T cells during simian immunodeficiency virus infection: T/B cell homeostasis, activation, and potential mechanism for viral escape. J Immunol. 2012;188:3247–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Hong JJ, Amancha PK, Rogers KA, et al. Early lymphoid responses and germinal center formation correlate with lower viral load set points and better prognosis of simian immunodeficiency virus infection. J Immunol. 2014;193:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kohler SL, Pham MN, Folkvord JM, et al. Germinal center T follicular helper cells are highly permissive to HIV‐1 and alter their phenotype during virus replication. J Immunol. 2016;196:2711–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Pallikkuth S, Sharkey M, Babic DZ, et al. Peripheral T follicular helper cells are the major HIV reservoir within central memory CD4 T cells in peripheral blood from chronically HIV‐infected individuals on combination antiretroviral therapy. J Virol. 2016;90:2718–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hufert FT, van Lunzen J, Janossy G, et al. Germinal centre CD4+ T cells are an important site of HIV replication in vivo. AIDS. 1997;11:849–857. [DOI] [PubMed] [Google Scholar]
- 173. Perreau M, Savoye AL, De Crignis E, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV‐1 infection, replication, and production. J Exp Med. 2013;210:143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Brenchley JM, Vinton C, Tabb B, et al. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood. 2012;120:4172–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Connick E, Folkvord JM, Lind KT, et al. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of rhesus macaques is linked to disease stage and inversely related to localization of virus‐specific CTL. J Immunol. 2014;193:5613–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Banga R, Procopio FA, Noto A, et al. PD‐1(+) and follicular helper T cells are responsible for persistent HIV‐1 transcription in treated aviremic individuals. Nat Med. 2016;22:754–761. [DOI] [PubMed] [Google Scholar]
- 177. Baxter AE, Niessl J, Fromentin R, et al. Single‐cell characterization of viral translation‐competent reservoirs in HIV‐infected individuals. Cell Host Microbe. 2016;20:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Boritz EA, Darko S, Swaszek L, et al. Multiple origins of virus persistence during natural control of HIV infection. Cell. 2016;166:1004–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Fukazawa Y, Lum R, Okoye AA, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. 2015;21:132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Haase AT, Henry K, Zupancic M, et al. Quantitative image analysis of HIV‐1 infection in lymphoid tissue. Science. 1996;274:985–989. [DOI] [PubMed] [Google Scholar]
- 181. Heath SL, Tew JG, Tew JG, Szakal AK, Burton GF. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature. 1995;377:740–744. [DOI] [PubMed] [Google Scholar]
- 182. Spiegel H, Herbst H, Niedobitek G, Foss HD, Stein H. Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T‐helper cells. Am J Pathol. 1992;140:15–22. [PMC free article] [PubMed] [Google Scholar]
- 183. Thacker TC, Zhou X, Estes JD, et al. Follicular dendritic cells and human immunodeficiency virus type 1 transcription in CD4+ T cells. J Virol. 2009;83:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Connick E, Mattila T, Folkvord JM, et al. CTL fail to accumulate at sites of HIV‐1 replication in lymphoid tissue. J Immunol. 2007;178:6975–6983. [DOI] [PubMed] [Google Scholar]
- 185. He R, Hou S, Liu C, et al. Follicular CXCR5‐expressing CD8+ T cells curtail chronic viral infection. Nature. 2016;537:412–428. [DOI] [PubMed] [Google Scholar]
- 186. Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature. 2016;537:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Leong YA, Chen Y, Ong HS, et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. 2016;17:1187–1196. [DOI] [PubMed] [Google Scholar]
- 188. Yamamoto T, Lynch RM, Gautam R, et al. Quality and quantity of TFH cells are critical for broad antibody development in SHIVAD8 infection. Sci Transl Med. 2015;7:298ra120. [DOI] [PubMed] [Google Scholar]
- 189. Petrovas C, Yamamoto T, Gerner MY, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. 2012;122:3281–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Cubas RA, Mudd JC, Savoye AL, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19:494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lindqvist M, van Lunzen J, Soghoian DZ, et al. Expansion of HIV‐specific T follicular helper cells in chronic HIV infection. J Clin Invest. 2012;122:3271–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xu H, Wang X, Malam N, Lackner AA, Veazey RS. Persistent simian immunodeficiency virus infection causes ultimate depletion of follicular Th cells in AIDS. J Immunol. 2015;195:4351–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Baumjohann D, Preite S, Reboldi A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. [DOI] [PubMed] [Google Scholar]
- 194. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. 2011;208:987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin‐6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. 2011;334:825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Fan J, Bass HZ, Fahey JL. Elevated IFN‐gamma and decreased IL‐2 gene expression are associated with HIV infection. J Immunol. 1993;151:5031–5040. [PubMed] [Google Scholar]
- 197. Muir R, Metcalf T, Tardif V, et al. Altered memory circulating T follicular helper‐B cell interaction in early acute HIV infection. PLoS Pathog. 2016;12:e1005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Velu V, Mylvaganam GH, Gangadhara S, et al. Induction of Th1‐biased T follicular helper (Tfh) cells in lymphoid tissues during chronic simian immunodeficiency virus infection defines functionally distinct germinal center Tfh cells. J Immunol. 2016;197:1832–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Cubas R, van Grevenynghe J, Wills S, et al. Reversible reprogramming of circulating memory T follicular helper cell function during chronic HIV infection. J Immunol. 2015;195:5625–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Cohen K, Altfeld M, Alter G, Stamatatos L. Early preservation of CXCR5+ PD‐1+ helper T cells and B cell activation predict the breadth of neutralizing antibody responses in chronic HIV‐1 infection. J Virol. 2014;88:13310–13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Havenar‐Daughton C, Lindqvist M, Heit A, et al. CXCL13 is a plasma biomarker of germinal center activity. Proc Natl Acad Sci USA. 2016;113:2702–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Nabi G, Genannt Bonsmann MS, Tenbusch M, et al. GagPol‐specific CD4(+) T‐cells increase the antibody response to Env by intrastructural help. Retrovirology. 2013;10:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Knowlden ZA, Sant AJ. CD4 T cell epitope specificity determines follicular versus non‐follicular helper differentiation in the polyclonal response to influenza infection or vaccination. Sci Rep. 2016;6:28287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ranasinghe S, Cutler S, Davis I, et al. Association of HLA‐DRB1‐restricted CD4(+) T cell responses with HIV immune control. Nat Med. 2013;19:930–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Robb ML, Rerks‐Ngarm S, Nitayaphan S, et al. Risk behaviour and time as covariates for efficacy of the HIV vaccine regimen ALVAC‐HIV (vCP1521) and AIDSVAX B/E: A post‐hoc analysis of the Thai phase 3 efficacy trial RV 144. Lancet Infect Dis. 2012;12:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Haynes BF, Gilbert PB, McElrath MJ, et al. Immune‐correlates analysis of an HIV‐1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yates NL, Liao HX, Fong Y, et al. Vaccine‐induced Env V1‐V2 IgG3 correlates with lower HIV‐1 infection risk and declines soon after vaccination. Sci Transl Med. 2014;6:228ra239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Prentice HA, Tomaras GD, Geraghty DE, et al. HLA class II genes modulate vaccine‐induced antibody responses to affect HIV‐1 acquisition. Sci Transl Med. 2015;7:296ra112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. de Souza MS, Ratto‐Kim S, Chuenarom W, et al. The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV‐1 envelope. J Immunol. 2012;188:5166–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Schultz BT, Teigler JE, Pissani F, et al. Circulating HIV‐specific interleukin‐21(+)CD4(+) T cells represent peripheral Tfh cells with antigen‐dependent helper functions. Immunity. 2016;44:167–178. [DOI] [PubMed] [Google Scholar]
- 211. Pratama A, Vinuesa CG. Control of TFH cell numbers: Why and how? Immunol Cell Biol. 2014;92:40–48. [DOI] [PubMed] [Google Scholar]
- 212. Linterman MA, Rigby RJ, Wong RK, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206:561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Vinuesa CG, Cook MC, Angelucci C, et al. A RING‐type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. [DOI] [PubMed] [Google Scholar]
- 214. Simpson N, Gatenby PA, Wilson A, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62:234–244. [DOI] [PubMed] [Google Scholar]
- 215. Wing JB, Sakaguchi S. Foxp3(+) T(reg) cells in humoral immunity. Int Immunol. 2014;26:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Miyara M, Sakaguchi S. Human FoxP3(+)CD4(+) regulatory T cells: Their knowns and unknowns. Immunol Cell Biol. 2011;89:346–351. [DOI] [PubMed] [Google Scholar]
- 217. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241. [DOI] [PubMed] [Google Scholar]
- 218. Rosser EC, Mauri C. Regulatory B cells: Origin, phenotype, and function. Immunity. 2015;42:607–612. [DOI] [PubMed] [Google Scholar]
- 219. Rydyznski CE, Waggoner SN. Boosting vaccine efficacy the natural (killer) way. Trends Immunol. 2015;36:536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Hams E, McCarron MJ, Amu S, et al. Blockade of B7‐H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011;186:5648–5655. [DOI] [PubMed] [Google Scholar]
- 221. Kawamoto S, Tran TH, Maruya M, et al. The inhibitory receptor PD‐1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–489. [DOI] [PubMed] [Google Scholar]
- 222. Pelletier N, McHeyzer‐Williams LJ, Wong KA, Urich E, Fazilleau N, McHeyzer‐Williams MG. Plasma cells negatively regulate the follicular helper T cell program. Nat Immunol. 2010;11:1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. 2016;271:246–259. [DOI] [PubMed] [Google Scholar]
- 224. Kim HJ, Cantor H. Regulation of self‐tolerance by Qa‐1‐restricted CD8(+) regulatory T cells. Semin Immunol. 2011;23:446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. [DOI] [PubMed] [Google Scholar]
- 226. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. [DOI] [PubMed] [Google Scholar]
- 227. Moon JJ, Dash P, Oguin TH 3rd, et al. Quantitative impact of thymic selection on Foxp3+ and Foxp3− subsets of self‐peptide/MHC class II‐specific CD4+ T cells. Proc Natl Acad Sci USA. 2011;108:14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T‐cell receptor stimulation is transforming growth factor‐beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL‐2 is essential for TGF‐beta to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. [DOI] [PubMed] [Google Scholar]
- 230. Thornton AM, Korty PE, Tran DQ, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic‐derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non‐inflammatory conditions. Semin Immunol. 2011;23:424–430. [DOI] [PubMed] [Google Scholar]
- 232. Godfrey VL, Wilkinson JE, Russell LB. X‐linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol. 1991;138:1379–1387. [PMC free article] [PubMed] [Google Scholar]
- 233. Wildin RS, Ramsdell F, Peake J, et al. X‐linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. [DOI] [PubMed] [Google Scholar]
- 234. Leon B, Bradley JE, Lund FE, Randall TD, Ballesteros‐Tato A. FoxP3+ regulatory T cells promote influenza‐specific Tfh responses by controlling IL‐2 availability. Nat Commun. 2014;5:3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Lim HW, Hillsamer P, Kim CH. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC‐Th cells and GC‐Th cell‐driven B cell responses. J Clin Invest. 2004;114:1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Chung Y, Tanaka S, Chu F, et al. Follicular regulatory T cells expressing Foxp3 and Bcl‐6 suppress germinal center reactions. Nat Med. 2011;17:983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Linterman MA, Pierson W, Lee SK, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Wollenberg I, Agua‐Doce A, Hernandez A, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol. 2011;187:4553–4560. [DOI] [PubMed] [Google Scholar]
- 239. Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD‐1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA‐4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41:1026–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Sage PT, Tan CL, Freeman GJ, Haigis M, Sharpe AH. Defective TFH cell function and increased TFR cells contribute to defective antibody production in aging. Cell Rep. 2015;12:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory‐like properties. J Clin Invest. 2014;124:5191–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Wallin EF, Jolly EC, Suchanek O, et al. Human T‐follicular helper and T‐follicular regulatory cell maintenance is independent of germinal centers. Blood. 2014;124:2666–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Wu H, Chen Y, Liu H, et al. Follicular regulatory T cells repress cytokine production by follicular helper T cells and optimize IgG responses in mice. Eur J Immunol. 2016;46:1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Aloulou M, Carr EJ, Gador M, et al. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun. 2016;7:10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Vaeth M, Muller G, Stauss D, et al. Follicular regulatory T cells control humoral autoimmunity via NFAT2‐regulated CXCR5 expression. J Exp Med. 2014;211:545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Good‐Jacobson KL, Song E, Anderson S, Sharpe AH, Shlomchik MJ. CD80 expression on B cells regulates murine T follicular helper development, germinal center B cell survival, and plasma cell generation. J Immunol. 2012;188:4217–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Linterman MA, Denton AE, Divekar DP, et al. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. Elife. 2014;3:e03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen‐specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA‐4. Immunity. 2014;41:1013–1025. [DOI] [PubMed] [Google Scholar]
- 250. Sage PT, Sharpe AH. In vitro assay to sensitively measure T(FR) suppressive capacity and T(FH) stimulation of B cell responses. Methods Mol Biol. 2015;1291:151–160. [DOI] [PubMed] [Google Scholar]
- 251. Ding Y, Li J, Yang P, et al. Interleukin‐21 promotes germinal center reaction by skewing the follicular regulatory T cell to follicular helper T cell balance in autoimmune BXD2 mice. Arthritis Rheumatol. 2014;66:2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T‐helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature. 2010;467:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Braud VM, Allan DS, O'Callaghan CA, et al. HLA‐E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. [DOI] [PubMed] [Google Scholar]
- 254. Kim HJ, Wang X, Radfar S, Sproule TJ, Roopenian DC, Cantor H. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6‐Yaa mice. Proc Natl Acad Sci USA. 2011;108:2010–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Leavenworth JW, Tang X, Kim HJ, Wang X, Cantor H. Amelioration of arthritis through mobilization of peptide‐specific CD8+ regulatory T cells. J Clin Invest. 2013;123:1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Kim HJ, Barnitz RA, Kreslavsky T, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science. 2015;350:334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Jiang H, Canfield SM, Gallagher MP, et al. HLA‐E‐restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest. 2010;120:3641–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Chevalier MF, Weiss L. The split personality of regulatory T cells in HIV infection. Blood. 2013;121:29–37. [DOI] [PubMed] [Google Scholar]
- 259. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. 2009;106:13439–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Angin M, Kwon DS, Streeck H, et al. Preserved function of regulatory T cells in chronic HIV‐1 infection despite decreased numbers in blood and tissue. J Infect Dis. 2012;205:1495–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Bi X, Suzuki Y, Gatanaga H, Oka S. High frequency and proliferation of CD4+ FOXP3+ Treg in HIV‐1‐infected patients with low CD4 counts. Eur J Immunol. 2009;39:301–309. [DOI] [PubMed] [Google Scholar]
- 262. Epple HJ, Loddenkemper C, Kunkel D, et al. Mucosal but not peripheral FOXP3+ regulatory T cells are highly increased in untreated HIV infection and normalize after suppressive HAART. Blood. 2006;108:3072–3078. [DOI] [PubMed] [Google Scholar]
- 263. Jiao Y, Fu J, Xing S, et al. The decrease of regulatory T cells correlates with excessive activation and apoptosis of CD8+ T cells in HIV‐1‐infected typical progressors, but not in long‐term non‐progressors. Immunology. 2009;128:e366–e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Lim A, Tan D, Price P, et al. Proportions of circulating T cells with a regulatory cell phenotype increase with HIV‐associated immune activation and remain high on antiretroviral therapy. AIDS. 2007;21:1525–1534. [DOI] [PubMed] [Google Scholar]
- 265. Montes M, Sanchez C, Lewis DE, et al. Normalization of FoxP3(+) regulatory T cells in response to effective antiretroviral therapy. J Infect Dis. 2011;203:496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Nikolova M, Carriere M, Jenabian MA, et al. CD39/adenosine pathway is involved in AIDS progression. PLoS Pathog. 2011;7:e1002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Tsunemi S, Iwasaki T, Imado T, et al. Relationship of CD4+CD25+ regulatory T cells to immune status in HIV‐infected patients. AIDS. 2005;19:879–886. [DOI] [PubMed] [Google Scholar]
- 268. Weiss L, Donkova‐Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus‐driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV‐specific CD4 T‐cell responses in HIV‐infected patients. Blood. 2004;104:3249–3256. [DOI] [PubMed] [Google Scholar]
- 269. Bandera A, Ferrario G, Saresella M, et al. CD4+ T cell depletion, immune activation and increased production of regulatory T cells in the thymus of HIV‐infected individuals. PLoS ONE. 2010;5:e10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Manches O, Munn D, Fallahi A, et al. HIV‐activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3‐dioxygenase‐dependent mechanism. J Clin Invest. 2008;118:3431–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Nilsson J, Boasso A, Velilla PA, et al. HIV‐1‐driven regulatory T‐cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood. 2006;108:3808–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Fritzsching B, Oberle N, Eberhardt N, et al. In contrast to effector T cells, CD4+CD25+FoxP3+ regulatory T cells are highly susceptible to CD95 ligand‐ but not to TCR‐mediated cell death. J Immunol. 2005;175:32–36. [DOI] [PubMed] [Google Scholar]
- 273. Angin M, King M, Altfeld M, Walker BD, Wucherpfennig KW, Addo MM. Identification of HIV‐1‐specific regulatory T‐cells using HLA class II tetramers. AIDS. 2012;26:2112–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Kared H, Lelievre JD, Donkova‐Petrini V, et al. HIV‐specific regulatory T cells are associated with higher CD4 cell counts in primary infection. AIDS. 2008;22:2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Kinter AL, Horak R, Sion M, et al. CD25+ regulatory T cells isolated from HIV‐infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV‐specific CD8+ T cells in vitro. AIDS Res Hum Retroviruses. 2007;23:438–450. [DOI] [PubMed] [Google Scholar]
- 276. Angin M, Streeck H, Wen F, et al. Regulatory T cell frequencies do not correlate with breadth or magnitude of HIV‐1‐specific T cell responses. AIDS Res Hum Retroviruses. 2012;28:749–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Eggena MP, Barugahare B, Jones N, et al. Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol. 2005;174:4407–4414. [DOI] [PubMed] [Google Scholar]
- 278. Owen RE, Heitman JW, Hirschkorn DF, et al. HIV+ elite controllers have low HIV‐specific T‐cell activation yet maintain strong, polyfunctional T‐cell responses. AIDS. 2010;24:1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Cao W, Jamieson BD, Hultin LE, Hultin PM, Detels R. Regulatory T cell expansion and immune activation during untreated HIV type 1 infection are associated with disease progression. AIDS Res Hum Retroviruses. 2009;25:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Del Pozo‐Balado Mdel M, Leal M, Mendez‐Lagares G, Pacheco YM. CD4(+)CD25(+/hi)CD127(lo) phenotype does not accurately identify regulatory T cells in all populations of HIV‐infected persons. J Infect Dis. 2010;201:331–335. [DOI] [PubMed] [Google Scholar]
- 281. Schulze Zur Wiesch J, Thomssen A, Hartjen P, et al. Comprehensive analysis of frequency and phenotype of T regulatory cells in HIV infection: CD39 expression of FoxP3+ T regulatory cells correlates with progressive disease. J Virol. 2011;85:1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Blackburn MJ, Zhong‐Min M, Caccuri F, et al. Regulatory and helper follicular T cells and antibody avidity to simian immunodeficiency virus glycoprotein 120. J Immunol. 2015;195:3227–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Chowdhury A, Del Rio Estrada PM, Tharp GK, et al. Decreased T follicular regulatory cell/T follicular helper cell (TFH) in simian immunodeficiency virus‐infected rhesus macaques may contribute to accumulation of TFH in chronic infection. J Immunol. 2015;195:3237–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Miles B, Miller SM, Folkvord JM, et al. Follicular regulatory T cells impair follicular T helper cells in HIV and SIV infection. Nat Commun. 2015;6:8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Colineau L, Rouers A, Yamamoto T, et al. HIV‐infected spleens present altered follicular helper T Cell (Tfh) subsets and skewed B cell maturation. PLoS ONE. 2015;10:e0140978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Ansari AA, Pereira LE, Mayne AE, et al. The role of disease stage, plasma viral load and regulatory T cells (Tregs) on autoantibody production in SIV‐infected non‐human primates. J Autoimmun. 2007;28:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Burton DR, Ahmed R, Barouch DH, et al. A blueprint for HIV vaccine discovery. Cell Host Microbe. 2012;12:396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Haynes BF. New approaches to HIV vaccine development. Curr Opin Immunol. 2015;35:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Kwong PD, Mascola JR, Nabel GJ. Rational design of vaccines to elicit broadly neutralizing antibodies to HIV‐1. Cold Spring Harb Perspect Med. 2011;1:a007278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Wang C, Hart M, Chui C, et al. Germinal center B cell and T follicular helper cell responses to viral vector and protein‐in‐adjuvant vaccines. J Immunol. 2016;197:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. de Cassan SC, Forbes EK, Douglas A, et al. The requirement for potent adjuvants to enhance the immunogenicity and protective efficacy of protein vaccines can be overcome by prior immunization with a recombinant adenovirus. J Immunol. 2011;187:2602–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Vargas‐Inchaustegui DA, Demers A, Shaw JM, et al. Vaccine induction of lymph node‐resident simian immunodeficiency virus Env‐specific T follicular helper cells in rhesus macaques. J Immunol. 2016;196:1700–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci USA. 2012;109:1080–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Keler T, Halk E, Vitale L, et al. Activity and safety of CTLA‐4 blockade combined with vaccines in cynomolgus macaques. J Immunol. 2003;171:6251–6259. [DOI] [PubMed] [Google Scholar]