

Abstract
Background
The Human Microbiome has been variously associated with the immune-regulatory mechanisms involved in the prevention or development of many non-infectious human diseases such as autoimmunity, allergy and cancer. Integrative approaches which aim at associating the composition of the human microbiome with other available information, such as clinical covariates and environmental predictors, are paramount to develop a more complete understanding of the role of microbiome in disease development.
Results
In this manuscript, we propose a Bayesian Dirichlet-Multinomial regression model which uses spike-and-slab priors for the selection of significant associations between a set of available covariates and taxa from a microbiome abundance table. The approach allows straightforward incorporation of the covariates through a log-linear regression parametrization of the parameters of the Dirichlet-Multinomial likelihood. Inference is conducted through a Markov Chain Monte Carlo algorithm, and selection of the significant covariates is based upon the assessment of posterior probabilities of inclusions and the thresholding of the Bayesian false discovery rate. We design a simulation study to evaluate the performance of the proposed method, and then apply our model on a publicly available dataset obtained from the Human Microbiome Project which associates taxa abundances with KEGG orthology pathways. The method is implemented in specifically developed R code, which has been made publicly available.
Conclusions
Our method compares favorably in simulations to several recently proposed approaches for similarly structured data, in terms of increased accuracy and reduced false positive as well as false negative rates. In the application to the data from the Human Microbiome Project, a close evaluation of the biological significance of our findings confirms existing associations in the literature.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1516-0) contains supplementary material, which is available to authorized users.
Keywords: Bayesian hierarchical model, Data integration, Dirichlet-multinomial, Microbiome data, Variable selection
Background
The human microbiome is defined as the collection of microorganisms, including bacteria, viruses, and some unicellular eukaryotes, that live in and on our bodies [1]. Research on the microbiome has grown exponentially in the past few years and it has been argued that the microbiota can be regarded as a “second genome”[2, 3]. Indeed, just the human gut microbiome is estimated to be composed of approximately 1014 bacterial cells, i.e. ten times more than the total number of human cells in the body [4]. The contribution of the human microbiome on several health outcomes has been frequently reported in the literature. For example, microbial dysbiosis in the gut has been linked to irritable bowel syndrome and Crohn’s disease [5], type 2 diabetes [6], cardiovascular disease [7], and psychological conditions via the so-called “gut-brain axis” [8]. The composition of microbiota at other body sites have also been associated with conditions such as eczema [9] and pre-term labor [10]. This stream of research holds great potential for a better understanding of many mechanistic processes in the development of human diseases, especially with respect to immune regulation and barrier defense [11, 12].
Microbiome data is most commonly obtained by sequencing variable regions of the 16S rRNA gene, then grouping the transcripts into Operational Taxonomic Units (OTUs), based on their similarity to one another. The OTUs are then defined as a cluster of reads based on a similarity threshold (typically, 97%) set by the researcher. The membership count of each cluster is then used as a proxy for taxa abundances in the sample [13]. See [14] for a discussion of how the selection of the cutoff might impact the resulting OTUs, in particular for rare species. Many studies summarize the taxa abundances by constructing several indicators of community composition (e.g. alpha and beta diversity indexes, see, [15]). Alternatively, the full OTUs abundance table can be used to obtain more detailed information about existing associations between environment or phenotypes and microbes. Well-established statistical models for the analysis of count data (e.g., Poisson or Negative Binomial distribution) can be efficaciously employed for the analysis of taxonomic count data [16]. Although less common, other distributions (e.g., a two-parameter Weibull distribution) have also been shown to provide a good fit to the data for some communities (see, e.g. [17]). One distinctive characteristic of the microbiome data is their overdispersion: while some taxa (e.g., Bacteroides and Lactobacillus species) are common among samples, many other taxa are present at much lower abundances, and often never recorded in a sample, leading to zero-inflated distributions. Many of the existing tools for microbial community analysis (e.g., the QIIME platform, [18]) bypass those characteristics and rely on nonparametric tests to compare species across different conditions [19, 20]. Other approaches use ordination, e.g. multidimensional scaling, to summarize abundances, and are sometimes employed to link the microbiome data with available clinical covariates and phylogenetic information [21, 22]. In those approaches, the choice of the distance metric is often crucial. The interpretation of biological phenomena can also be challenging in low dimensional projections. Most importantly, distance-based methods do not explicitly quantify the relative importance of significant associations between taxa and covariates, and therefore are of limited use for clinical decisions.
In this manuscript, we consider an integrative Bayesian approach based on the use of Dirichlet-Multinomial (DM) distributions [23] for studying the association between taxa abundance data and available measurements on clinical, genetic and environmental covariates. Recently, La Rosa et al. [24] proposed the use of a DM model for hypothesis testing and power calculations in microbiome experiments. Holmes et. al [25] used a finite mixture of DM distributions to directly model the taxa counts. Neither method incorporate predictors to study the influence of external factors on the microbiome’s abundance. A penalized likelihood approach based on a DM regression model has been proposed instead by [26] to determine significant associations between the microbiome composition and a set of covariates which describe the individual dietary nutrients’ intakes. Similarly, [27] develop a structure constrained version of sparse canonical correlation analysis that integrates compositionalized microbiome data, phylogenetic information, and nutrient information. Furthermore, [28] propose penalized regression models to associate the multivariate compositionalized microbiome data with some univariate phenotype of interest, e.g. body mass index, as a response. However, the use of a constrained optimization approach does not allow to fully characterize the uncertainty in the selection of the significant associations, which is of particular importance, especially when dealing with high-dimensional and highly-correlated data.
Here, we propose a probabilistic modeling approach which both flexibly takes into account the typical features of microbiome count data and also allows for straightforward incorporation of available covariate information within a DM log-linear regression framework. With respect to modeling approaches as in [28], our framework allows the study of associations between multivariate microbiome data and multivariable predictors. By imposing sparsity inducing spike-and-slab priors on the regression coefficients, our model obtains a parsimonious summary of the effects of the associations and also allows an assessment of the uncertainty of the selection process. We evaluate the performance of our model first on simulated data, where we provide comparisons with methods developed for microbiome or similar type of data. We also illustrate our method on data obtained from the Human Microbiome Project [29], to investigate the association between taxonomic abundances and metabolic pathways inferred from whole genome shotgun sequencing reads. It is known that the combination of environmental and host genetic factors shape the composition of the gut microbiota, and these interactions appear to have a significant effect on several biological mechanisms, which may be related, for example, to the individual immunity and barrier defense, as well as metabolism and diet [30, 31]. The approach has been implemented in a user-friendly R code, which has been made publicly available (see the Licensing Section).
Methods
We describe our Bayesian variable selection approach for the analysis of microbiome data and their association with a set of available covariates in the context of DM log-linear regression models.
Dirichlet-multinomial regression with variable selection
Let y i=(y i1,…,y iJ) indicate the vector of counts representing the taxonomic abundance table obtained from the ith patient, with y ij denoting the frequency of the jth microbial taxon, for j=1,…,J and i=1,…,n. Furthermore, let X=(x 1,…,x P) indicate a n×P matrix of measurements on P covariates. We start by modeling the taxonomic count data with a Multinomial distribution
| 1 |
with the summation of all counts in the vector, and where the parameters ϕ’s are defined on the J dimensional simplex
We further impose a conjugate Dirichlet prior on ϕ, that is ϕ∼Dirichlet(γ), where γ=(γ 1,…,γ J) indicates a J-dimensional vector of strictly positive parameters. An advantage of our hierarchical formulation is that conjugacy can be exploited to integrate ϕ out, obtaining the Dirichlet–Multinomial model, y i∼DM(γ), with probability mass function
where . First described in [23] as the compound multinomial, the DM(γ) allows more flexibility than the Multinomial when encountering overdispersion in multivariate count data, as it induces an increase in the variance by a factor of (y ++γ +)/(1+γ +).
Next, we incorporate the covariates into the modeling via a log-linear regression framework where the DM parameters depend on the available covariates X’s. More specifically, we define ζ j= log(γ j) and assume
| 2 |
i=1,…,n; j=1,…,J. In this formulation, the intercept term α j corresponds to the log baseline parameter for the taxon j, whereas the regression parameter β pj captures the effect of the pth covariate on the abundance for that taxon.
Identifying the significant associations between taxa and covariates in model (1)–(2) is equivalent to determining the non-zero β pj parameters. One way to address this issue is through variable selection and the use of spike-and-slab mixture priors [32, 33]. First, we introduce latent binary indicator vectors ξ j=(ξ 1j,ξ 2j,…,ξ pj), such that ξ pj=1 if the pth covariate influences the abundance of the jth taxa and ξ pj=0 otherwise. Then, we write the prior on the β pj’s as
| 3 |
where δ 0 denotes a Dirac-delta at 0 and is some suitably large value [34, 35]. It is common to choose relatively large values for . Such a choice suggest a flat prior distribution on the location of the coefficients {β pj∣ξ pj=1} and therefore encourages the selection of relatively large effects. In the Results Section, we discuss the results of a sensitivity analysis to assist with the choice of this parameter. We place Bernoulli priors on the selection indicators ξ pj, that is
| 4 |
We also specify Beta hyperpriors on the hyperparameters p pj, i.e., p pj∼Beta(ab), as this has been shown to provide an automatic adjustment for multiplicity [36]. This is equivalent to placing a Beta mixed Binomial distribution on ξ pj,
with λ=(a,b). As a practical suggestion, the hyper-parameters a and b should be chosen so to induce a relatively weakly specification of the prior as a “flat” Beta distribution. This can be obtained by setting a and b so that a+b=2, and the prior expected mean value m=a/(a+b). For most cases, a value of m=0.01, which corresponds to assuming a priori that 1% of the P covariates will be selected, provides an adequate balance between false positives and false negative counts, as we further illustrate in a sensitivity analysis in the Results Section. Finally, we assume normal priors on the α j’s, i.e. . Large values for encode a diffuse prior, to describe non-informative or objective prior beliefs. However, results are typically quite robust to prior choices on the intercept parameters, and is usually assumed as a default specification in Bayesian regression when dealing with standardized variables. Figure 1 provides an overview of the proposed integrative modeling approach, with reference to the application to the Human Microbiome Project data we describe later.
Fig. 1.
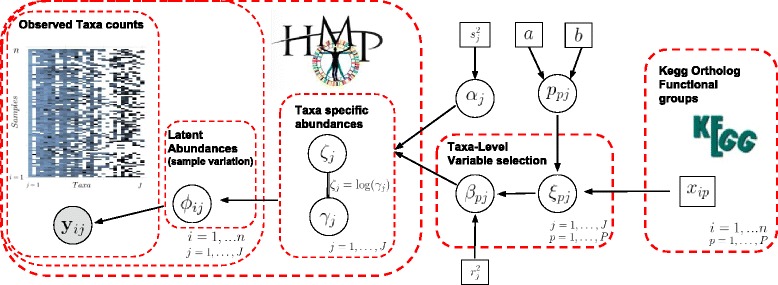
Schematic overview of the proposed integrative Bayesian approach for the application to data from the Human Microbiome Project. The observed data counts (right) are regressed on the available covariates (left), through a variable selection approach, which informs the (unknown) population abundance of each taxon
MCMC algorithm
We implement a stochastic search Markov Chain Monte Carlo (MCMC) algorithm for posterior inference that employs a Gibbs scan to sample the non-zero regression coefficients [37]. We encourage an efficient sampling by employing a component-wise adaptive Metropolis algorithm [38] as described below. A generic iteration of the MCMC algorithm comprises the following steps:
Update of α : This is a Metropolis-Hastings step with a symmetric random walk proposal , for j=1,…,J.
- Joint update of ( ξ , β ): We sample these parameters jointly via a Gibbs scan that employs a Metropolis acceptance step. For each j=1,…,J and p=1,…,P:
- if ξ pj=1: propose and .
-
if ξ pj=0: propose and then propose following an adaptive Metropolis-Hasting schemewhere is the current estimate of the variance of the target distribution. The value of is updated using a recursive formula as in [39] on all the previous draws for β pj.
- Accept () with probability
where , and .
For posterior inference, we are interested in identifying the relevant associations between taxa and covariates as captured by the selection indicators ξ pj’s and the corresponding regression coefficients β pj’s. Estimates of the marginal posterior probabilities of inclusion (PPIs) of the latent indicators ξ pj can be calculated by counting the number of times that each taxa/covariate association is included across the MCMC iterations. A selection of the significant associations can then be made by choosing those elements that have marginal PPIs greater than a specific value, for example greater than 0.5 for the median probability model of [40]. Another choice for the threshold which controls for multiplicity [41] relies on an estimated pre-specified Bayesian false discovery rate α calculated as
where 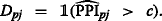 . An optimal threshold c
′ can be found for error rate α by choosing c
′ such that . Estimates of the non-zero regression coefficients β
pj can also be calculated by averaging over the sampled MCMC values.
. An optimal threshold c
′ can be found for error rate α by choosing c
′ such that . Estimates of the non-zero regression coefficients β
pj can also be calculated by averaging over the sampled MCMC values.
In order to compare selection performance of different methods, we calculate accuracy, false positive rate (FPR), false negative rate (FNR) and Matthews correlation coefficient (MCC), across 30 replicated datasets. We define accuracy as ACC = (TP + TN)/(P + N), with TP the number of true positives out of P selected and TN the number of true negatives out of N not selected. The false negative rate is calculated as FNR = FN/(FN + TP), the false positive rate as FPR = FP/(FP + TN), and the Matthews correlation coefficient as
with N=TN+TP+FN+FP, and [42]. Since the MCC balances TP and FP counts, and can be used even if the classes are of very different sizes, it is generally regarded as one of the most appropriate measures of classification accuracy. We further computed receiving operating curves (ROC) to compare the performance of the selection procedure across the different methods.
Comparison study on simulated data
We carry out a simulation study to assess the performance of our model and compare results to alternative methods. More specifically, we consider two methods which have been specifically employed for the integrative analysis of microbiome data: the penalized approach of Chen and Li [26], and the false discovery rate-corrected pair-wise correlation tests considered in [19]. In addition, we consider the factorized maximum a posteriori (MAP) Bayesian lasso of [43], a recently proposed general statistical method for conducting variable selection in multivariate count-response regression. When fitting the Bayesian Gamma Lasso method of [43], model selection was done using the minimum AIC, while for Chen and Li’s approach the minimum BIC was calculated with the group penalty set to 20%. We also fit the method of Chen and Li to the untransformed data. The false discovery rate threshold for the Spearman’s correlation tests was set to 0.05.
In simulating data, we set n=100, P=50 and J=50, and chose P r=9 and J r=5 to obtain a total number of relevant taxa/covariate associations equal to 25. We simulated the covariate matrix X according to a Multivariate-Normal (0,Σ) with Σ i,j=ρ |i−j| and ρ=0.4. We drew each vector y i of counts from a Dirichlet-Multinomial distribution as follows. For i=1,…,n, , with the row sum N i∼DiscreteUnif[1,0000;2,000], and . For , we set , j=1,…,J, with γ j= exp{α j+X β j}, and ψ∈ [ 0,1] an overdispersion parameter. When ψ→0, the simulated values approximate a Multinomial (π) distribution, while for large ψ, the sampled values are more disperse. Here, we set ψ=0.01. We sampled the non-zero β pj’s from the intervals ±[ 0.5,1.0] and the intercept parameters from a Uniform(–2.3, 2.3). Below we report performance results as averages over 30 replicated simulated datasets.
When running the MCMC, we used a vague prior for the intercept by setting the variance parameter to . Similarly, we set , to provide sufficiently vague prior information on the non-zero log-linear regression coefficients. Finally, we set m=0.01 (or a=0.02 and b=1.98), resulting in a sparse prior mean on selected associations of 1% of the total. We provide comments on the sensitivity of the selection results to the choice of these hyperparameters in the Section below. We ran the MCMC algorithm for 10,000 iterations and thinned to every fifth iteration. On a single dataset, the C code took approximately 31.5 min to run on an Intel Xeon E5-2630 2.30 GHz processor. We assessed convergence visually and via the Geweke diagnostic [44] as implemented in the R package coda. Convergence was checked for a) the number of active variables in each iteration and b) the samples from each of the selected β pj. The five number summary of the 25 Geweke z-scores was (–3.43, –1.06, –0.63, 0.71, 1.98).
Inferring associations between taxonomic abundances and metabolic pathways
We demonstrate our approach on publicly available data obtained from the Human Microbiome Project (HMP) website [29] from which we use 79 samples from healthy individuals. The Y matrix in our model contains 16S rRNA microbial counts from stool samples at the genus taxonomic level. As common in microbiome studies, the genera abundances (Bacteroides, Prevotella, etc.) were filtered by requiring each genus to be present in at least 5% of the samples. This procedure removes extremely low-abundance genera leaving 80 genera for the analysis. From the same 79 individuals, we obtained KEGG orthology group abundances which are used as the matrix of covariates X of our model. The KEGG orthology groups were reconstructed from metagenomic shotgun sequencing (WGS) using the HMP Unified Metabolic Analysis Network (HUMAnN) pipeline [45] and were also provided on the HMP website. These values represent inferred abundances of biochemical functional groups and metabolic pathways present due to the shotgun sequenced reads of bacterial and non-bacterial genes in the sample. To reduce correlation among the covariates we used average linkage clustering on the correlation matrix of the KEGG groups and chose one representative from each cluster, according to its relevance to microbiome research, leaving 76 columns in X. Finally, the columns in X were mean centered and scaled to unit variance. Though the HMP sampled 300 individuals for several timepoints and over many sites, there were relatively few samples that included the WGS used to obtain the KEGG orthology data. Thus, when joining the samples from the 16S rRNA data and the KEGG orthology data, a total of 79 matched samples remained.
We used the same hyperparameter settings as in the simulation study, that is and and set m=0.01, resulting in a sparse mean selection prior of 1% of the total 6,080 possible associations. The MCMC algorithm described in “MCMC algorithm” section above was run for 500,000 iterations and thinned to every 100th draw. We assessed convergence visually and via the Geweke diagnostic [44] as implemented in the R package coda. The five number summary of the Geweke z-scores for the 26 β pj’s was (–3.83, –1.19, 0.15, 1.46, 3.38).
Results
Simulation study
In Fig. 2 we show the plot of the marginal PPIs of the P×J elements ξ pj, obtained by computing the proportion of times that ξ pj=1 across all iterations, after burn-in. The selected median model, corresponding to a threshold of 0.5 on the PPIs, results in a false positive rate of 0.0004 and a false negative rate of 0.04. The value of the AUC for this replicate was 0.99.
Fig. 2.

Simulated data: Marginal posterior probabilities of inclusion (PPI) for each coefficient β jp, j=1,…,50, p=1,…,50, describing the association between each taxa and each covariate. Each PPI is obtained by averaging the number of times that each taxa/covariate association is included across the MCMC iterations, after burn-in. The true associations are indicated as red dots
Figure 3 illustrates the selection performance of the proposed method, by plotting the average ROC curves over the 30 replicated datasets (ψ=0.01) for each of the methods included in the comparison. The Figure shows that our proposed model outperforms the competing methods in terms of achieved average true and false positive rates.
Fig. 3.
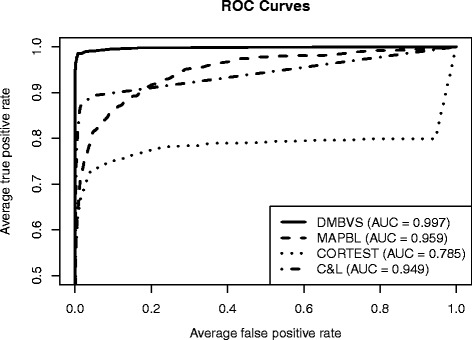
Simulated data: Comparison results of selection performances (ROC curves). DMBVS: Dirichlet–Multinomial Bayesian Variable Selection (our method), MAPBL : Maximum A Posteriori Bayesian Lasso, CORTEST: Multiplicity Corrected Correlation Tests as in Wu et al. (2010), C&L: composite penalty from Chen and Li (2013)
As an additional comparison, together with the total number of correctly identified regression parameters, which we term “overall recovery”, we also looked at the “taxa-wise recovery”, which we defined as the correct recovery of any element from one of the J taxa. Thus, recovery for overall selection occurs for P×J elements while taxa-wise selection occurs for J elements. Table 1 reports average values for accuracy, FPR, FNR and MCC, averaged across the 30 replicated datasets, for both overall and taxa-wise recovery. These results show that our method in particular outperforms competing methods for taxa-wise recovery. In the same Table we report results for a more challenging simulated scenario, obtained with a higher value of the overdispersion parameter (ψ=0.1). As expected, the increase in overdispersion makes the selection task more difficult for all methods. However, our method still outperforms or is commensurate with the competing methods, even in the presence of considerable overdispersion.
Table 1.
Simulated data: performance assessment for two different scenarios, characterized by different values of the dispersion parameter ψ
| Overall | Taxa | |||||||
|---|---|---|---|---|---|---|---|---|
| DMBVS | MAPBL | C&L | CORTEST | DMBVS | MAPBL | C&L | CORTEST | |
| ψ=0.01 | ||||||||
| MCC | 0.93 | 0.64 | 0.67 | 0.73 | 0.89 | 0.66 | 0.50 | 0.85 |
| FNR | 0.05 | 0.10 | 0.12 | 0.31 | 0.00 | 0.46 | 0.43 | 0.02 |
| FPR | 0.00 | 0.01 | 0.01 | 0.00 | 0.05 | 0.01 | 0.09 | 0.06 |
| Accuracy | 1.00 | 0.99 | 0.99 | 1.00 | 0.96 | 0.91 | 0.85 | 0.95 |
| ψ=0.1 | ||||||||
| MCC | 0.72 | 0.42 | 0.54 | 0.56 | 0.73 | 0.40 | 0.38 | 0.70 |
| FNR | 0.39 | 0.58 | 0.28 | 0.63 | 0.24 | 0.73 | 0.52 | 0.37 |
| FPR | 0.00 | 0.01 | 0.01 | 0.00 | 0.05 | 0.01 | 0.12 | 0.02 |
| Accuracy | 1.00 | 0.99 | 0.99 | 1.00 | 0.92 | 0.86 | 0.81 | 0.92 |
Values are rounded averages over thirty replicates. Results for Matthews’ Correlation Coefficient, Falso Positive Rate, False Negative Rate, and Accuracy, are based on the median probability model. DMBVS: Dirichlet–Multinomial Bayesian Variable Selection (our method), MAPBL: Maximum A Posteriori Bayesian Lasso, C&L: composite penalty from Chen and Li (2013), CORTEST: Multiplicity Corrected Correlation Tests as in Wu et al. (2010)
Sensitivity analysis
Since our proposal requires the choice of a number of hyperparameters, it is important to investigate how sensitive the results are to varying parameter sets. Therefore, we conclude our simulation study by briefly discussing the sensitivity of the results to the prior specifications. In general, we found that results were robust to the prior choices on the intercept parameters, α j, while, as expected, some sensitivity was observed with respect to the variance hyperparameters of the spike-and-slab prior (3) on the regression coefficients, β pj, and the hyperparameters of the Beta priors on p pj. In Table 2 we report results obtained by considering a full grid of values for the prior expected value of p pj, i.e. m∈{0.005,0.01,0.05}, and the slab variance, . In the Additional file 1, we further report the corresponding ROC curves. With only 25 truly non-zero β pj’s, out of 2,500 parameters, small increases in false positive rates can drastically decrease the Matthews correlation coefficient. Thus imposing some sparsity by using a smaller value for m improves overall performance while larger values of m allow for more false positives. The results appear to suggest that assuming moderate sparsity a priori (e.g., m=0.01) generally leads to good operating characteristics. Similarly, when the slab variance is small, e.g. , there is more prior density close to zero, which allows small but insignificant variables to be selected. Conversely, when the slab variance is large, e.g. , false positives are less likely but false negatives increase, since the prior density is spread more evenly over the support. Therefore, an intermediate value, e.g. , provides a reasonable compromise, which favors relatively large effect sizes and a small number of false positives. In the Additional file 1, we also report the performance of our method for varying values of the over-dispersion parameter ψ and the sample size n. As expected, the results show that the performances improve for larger sample sizes and decreasing overdispersion.
Table 2.
Simulated data: sensitivity analysis for varying values of the prior expected value of p pj, m, and the slab variance , and for two different scenarios, characterized by different values of the dispersion parameter ψ
| m=0.005 | m=0.01 | m=0.05 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ψ=0.01 | |||||||||
| MCC | 0.69 | 0.93 | 0.96 | 0.69 | 0.93 | 0.95 | 0.69 | 0.93 | 0.95 |
| FPR | 0.01 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 |
| FNR | 0.02 | 0.04 | 0.08 | 0.02 | 0.05 | 0.09 | 0.02 | 0.05 | 0.08 |
| Accuracy | 0.99 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 |
| AUC | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| ψ=0.1 | |||||||||
| MCC | 0.53 | 0.73 | 0.72 | 0.53 | 0.73 | 0.71 | 0.52 | 0.73 | 0.72 |
| FPR | 0.01 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 |
| FNR | 0.26 | 0.37 | 0.46 | 0.26 | 0.37 | 0.47 | 0.26 | 0.37 | 0.47 |
| Accuracy | 0.99 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 |
| AUC | 0.96 | 0.96 | 0.93 | 0.96 | 0.96 | 0.93 | 0.95 | 0.95 | 0.93 |
Values are averages over 30 replicates
Data analysis
Figure 4 shows the traceplot of the number of included taxa/covariate associations and the plot of the marginal PPIs of the P×J elements ξ pj, obtained by computing the proportion of times that ξ pj=1 across all iterations, after burn-in. Here the median model, corresponding to a threshold of 0.5 on the PPIs, selects 92 associations. Among those, 26 have a marginal PPI greater than 0.98, which corresponds to a Bayesian FDR of 0.1. These 26 associations are listed in Table 3, together with the corresponding estimated regression coefficients, and depicted in Figs. 5 and 6, for positive and negative associations, respectively. In these Figures, the magnitude of the association, as captured by the estimated β pj’s, is proportional to the width of the edges, with red lines indicating negative associations and blue lines positive associations. As a comparison, the method by Chen and Li [26] identified 120 associations, whereas the Bayesian Lasso of [43] and the correlation test-based method of [19] identified, respectively, 220 and 711 associations. Those results appear to confirm the sparser selection achieved by our method, consistently with the results of the simulation study.
Fig. 4.
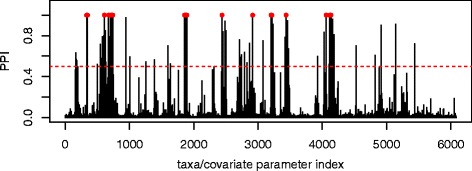
Real data: Marginal posterior probabilities of inclusion (PPI) for each coefficient β jp, in Eq. (2), describing the association between each taxa and each covariate. Each PPI is obtained by averaging the number of times that each taxa/covariate association is included across the MCMC iterations, after burn-in. Here, the median model, corresponding to a threshold of 0.5 on the PPIs, selects 92 associations. Among those, 26 have a marginal PPI greater than 0.98, which corresponds to a Bayesian FDR of 0.1. These 26 associations are indicated as red dots
Table 3.
Real data: selection results using a BFDR of 0.1
| KEGG ID | Pathway | Taxa | MPPI | β pj |
|---|---|---|---|---|
| ko04660 | T cell receptor signaling pathway | g.Subdoligranulum | 1.00 | 0.40 |
| ko04512 | ECM-receptor interaction | g.Subdoligranulum | 1.00 | 0.44 |
| ko00680 | Methane metabolism | g.Sutterella | 1.00 | -1.47 |
| ko05200 | Pathways in cancer | o.Bacteroidales | 1.00 | -1.04 |
| ko04540 | Gap junction | o.Bacteroidales | 1.00 | -0.92 |
| ko00534 | Glycosaminoglycan biosynthesis | g.Ruminococcus | 1.00 | -0.56 |
| ko00053 | Ascorbate and aldarate metabolism | g.Ruminococcus | 1.00 | -0.46 |
| ko00650 | Butanoate metabolism | g.Ruminococcus | 1.00 | -0.55 |
| ko00513 | Various types of N-glycan biosynthesis | g.Parabacteroides | 1.00 | 0.54 |
| ko00500 | Starch and sucrose metabolism | g.Parabacteroides | 1.00 | -0.61 |
| ko00904 | Diterpenoid biosynthesis | g.Dialister | 1.00 | -1.21 |
| ko00360 | Phenylalanine metabolism | g.Dialister | 1.00 | -2.18 |
| ko00626 | Naphthalene degradation | g.Bacteroides | 1.00 | 0.39 |
| ko00010 | Glycolysis / Gluconeogenesis | g.Bacteroides | 1.00 | -0.57 |
| ko00513 | Various types of N-glycan biosynthesis | g.Prevotella | 1.00 | 0.77 |
| ko00512 | Mucin type O-Glycan biosynthesis | g.Prevotella | 1.00 | -1.06 |
| ko00620 | Pyruvate metabolism | g.Prevotella | 1.00 | -1.76 |
| ko04340 | Hedgehog signaling pathway | g.Ruminococcus | 1.00 | 0.45 |
| ko04660 | T cell receptor signaling pathway | g.Roseburia | 1.00 | 0.46 |
| ko00983 | Drug metabolism | g.Sutterella | 1.00 | -1.20 |
| ko00260 | Glycine, serine and threonine metabolism | o.Bacteroidales | 1.00 | 0.88 |
| ko00330 | Arginine and proline metabolism | g.Ruminococcus | 0.99 | 0.39 |
| ko01057 | Biosynthesis of type II polyketide products | o.Bacteroidales | 0.99 | 0.76 |
| ko00620 | Pyruvate metabolism | g.Ruminococcus | 0.99 | 0.56 |
| ko00920 | Sulfur metabolism | f.Prevotellaceae | 0.99 | 1.36 |
| ko00010 | Glycolysis/Gluconeogenesis | o.Clostridiales | 0.98 | 0.54 |
The text in the KEGG column is hyperlinked to the KEGG orthology database for a more complete description of the selected pathways. Taxa names start with “g.”, “f.” or “o.” which stand for genus, family, or order, respectively, and correspond to the lowest taxonomic classification available
Fig. 5.

Real data: Selected positive taxa-by-covariate associations. The magnitude of the association, as captured by the median of the MCMC draws for each β pj, is proportional to the width of the edges
Fig. 6.

Real data: Selected negative taxa-by-covariate associations. The magnitude of the association, as captured by the median of the MCMC draws for each β pj, is proportional to the width of the edges
Discussion
A close investigation of the biological significance of the associations identified by our model reveals several interesting characteristics and affirms the relevance of these associations. Commensal microbiota that inhabit the human gut are proficient at scavenging glycans and polysaccharides, including those in plants, such as starches or cellulose, animal-derived tissues (glycosaminoglycans and N-linked glycans), and glycans from host mucus (O-linked glycans) [46]. Ruminococcus spp. are known to participate in both resistant starch and glycosaminoglycan degradation [46, 47]. It has been reported that long-term consumption of diets rich in protein and animal fat were associated with an enterotype primarily containing increased Bacteroides and Ruminococcus species [19]. Additionally, Ruminococcus torques and Ruminococcus gnavus have been shown to degrade mucins [48]. Thus, it is logical that Ruminococcus, which is one of the noteworthy genera involved in glycosaminoglycan degradation, would be negatively associated to glycosaminoglycan biosynthesis (ko00534) (Table 3). Similarly, Parabacteroides which is also negatively associated with N-glycan biosynthesis (ko00513), is involved in deglycosylation and utilization of N-glycans [49]. Also, among the associations identified for the glycan pathways, Prevotella was negatively associated with mucin type O-glycan biosynthesis (ko00512). In the literature, Prevotella has implications for mucosal homeostasis, as some Prevotella spp. express a unique mucin-desulfating glycosidase that can hydrolyze GlcNAc residues on mucin-type O-glycans, and thus is important for mucin degradation [50]. Other associations affirmed through the literature included that of Bacteroides with naphthalene degradation (ko00626). It has been reported that Bacteroidetes possess the capability to degrade polycyclic aromatic hydrocarbons such as naphthalene [51]. Associations of Ruminococcus with pyruvate metabolism (ko00620) are also supported, as phosphoenolpyruvate carboxykinase was previously reported to be associated with Ruminococcus flavefaciens in the rumen [52]. Another supported association is that of Prevotellaceae with sulfur metabolism. L-cysteine desulfhydrase enzymes have been characterized in Prevotella intermedia [53]. Additionally, glycosulfatase enzymes have been described in Prevotella [54]. Equally interesting is the selection of pathways that are expected to be omnipresent among many bacteria, such as glycolysis/gluconeogenesis (ko00010), as glycolysis occurs, with variations, in nearly all organisms, both aerobic and anaerobic. Thus, it is not surprising that taxa like Clostridiales are positively associated with glycolysis/gluconeogenesis as they are abundant taxa within the gut microbiome.
Given the complexity of metabolic pathways and the process of mapping specific genes to pathways, some of the selected associations are unexpected, and might be due to the 16S abundances that were made available at the HMP site and the mapping of metagenomic sequences to specific KEGG orthology groups by HUMAnN. For example, several species of Ruminococcus are known to participate in butanoate (butyrate) metabolism [55], Dialister spp. have phenylalanine arylamidase activities [56], and Prevotella spp. are known to participate in pyruvate metabolism [57, 58]. Since those associations should be driven exclusively by bacterial genes, it is interesting that we find significant associations between the abundance of certain bacterial taxa and KEGG pathways that are primarily reported among eukaryotic species (i.e., T-cell receptor signaling, hedgehog signaling, pathways in cancer, etc.). Indeed, although precautionary steps are performed, the HMP consortium reported that human contaminants are found in 50–90% of the sequences [15]. This might also explain the negative association exhibited by Bacteriodes and glycolysis/gluconeogenesis. These unexpected findings suggest the need for further investigations and validation.
Conclusion
Herein, we have developed a Bayesian approach to the Dirichlet-Multinomial regression models that allows for the selection of significant associations between covariates and taxa from a microbiome abundance table by imposing spike-and-slab priors on the log-linear regression coefficients of the model. We have applied our model to simulated data and compared performances with methods developed for similar applications. We have illustrated the performance of our method using publicly available data on taxonomic abundances and metabolic pathways inferred from whole genome shotgun sequencing reads, which we obtained from the Human Microbiome Project website. Our results have revealed interesting links between specific taxa (i.e. genera) and particular metabolic pathways, which we have validated via existing literature.
Several extensions of our model are possible. Because some habitats, e.g. the gut, are thought to have highly variable dynamics, longitudinal sampling may be preferred to cross-sectional sampling since it may give a better sense of long-term trends [59]. Thus, incorporating repeated samples with specified correlation structures in the linear predictor could produce additional insights. Another important aspect of microbiome data, which is receiving attention from researchers, is the heterogeneity in community structure across samples, as this can be an indication of the existence of “enterotypes” [60, 61]. This can be addressed within our modeling framework by employing Bayesian nonparametric models that would allow to cluster selected associations across partitions of the samples. These extensions are currently under investigation.
Acknowledgements
RA acknowledges the support received by CNR-IMATI (Center of National Research - The Institute for Applied Mathematics and Information Technologies “Enrico Magenes”, Milano, Italy) to conduct this research.
Funding
WDW is supported by NIH Grant NCI T32 CA096520 at Rice University. Jessica Galloway-Peña is supported by the Odyssey Program and CFP Foundation at The University of Texas MD Anderson Cancer Center. MG, JPG and SAS are been partially supported by the NIH/NCI award P30CA016672, MG used the Biostatistics Shared Resource.
Availability of data and materials
The datasets used in the study are included as Additional files 2 and 3, respectively. The code developed for this manuscript is publicly available on GitHub at https://github.com/duncanwadsworth/dmbvs, at http://www.micheleguindani.info and on http://www.stat.rice.edu/~marina/. An R package is under development and will be announced on those sites.
Authors’ contributions
WDW and RA have collaborated in the development of the algorithm and performed the statistical analyses, JGP and SAS have contributed to the interpretation of the biological findings, MG and MV have conceived the study and supervised the development of the algorithm and the statistical analyses. All authors have contributed to the writing of the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AIC
Akaike information criterion
- ACC
True positive
- AUC
Area under the curve
- BIC
Bayesian information criterion
- DM
Dirichlet-Multinomial
- FDR
False discovery rate
- FN
False negative
- FNR
False negative rate
- FP
False positive
- FPR
False positive rate
- N
Negative
- HMP
Human Microbiome Project
- KEGG
Kyoto encyclopedia of genes and genomes
- MAP
Maximum a posteriori
- MCC
Matthews correlation coefficient
- MCMC
Markov Chain Monte Carlo
- OTU
Operational taxonomic unit
- P
Positive
- PPI
Posterior probability of inclusion
- ROC
Receiving operating curve
- TN
True negative
- WGS
Whole genome shotgun sequencing
Additional files
Comparison with other methods and sensitivity analysis in the simulation study. (PDF 142 kb)
The dataset comprising the 16S rRNA microbial counts. (ZIP 1280 kb)
The dataset used for the KEGG orthology groups. (ZIP 718 kb)
Contributor Information
W. Duncan Wadsworth, Email: w.duncan.wadsworth@rice.edu.
Raffaele Argiento, Email: raffaele@mi.imati.cnr.it.
Michele Guindani, Email: micheleguindani@gmail.com.
Jessica Galloway-Pena, Email: jrgalloway@mdanderson.org.
Samuel A. Shelbourne, Email: sshelburne@mdanderson.org
Marina Vannucci, Email: marina@rice.edu.
References
- 1.Morgan XC, Huttenhower C. Chapter 12: Human microbiome analysis. PLoS Comput Biol. 2012;8(12):1002808. doi: 10.1371/journal.pcbi.1002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu B, Wang X, Li L. Human gut microbiome: The second genome of human body. Protein Cell. 2010;1(8):718–25. doi: 10.1007/s13238-010-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grice EA, Segre JA. The Human Microbiome: our second genome. Annu Rev Genomics Hum Genet. 2012;13:151–70. doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraher MH, O’Toole PW, Quigley EMM. Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev Gastroenterol Hepatol. 2012;9(6):312–22. doi: 10.1038/nrgastro.2012.44. [DOI] [PubMed] [Google Scholar]
- 5.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 7.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WHW, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–85. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cryan JF, O’Mahony SM. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil. 2011;23(3):187–92. doi: 10.1111/j.1365-2982.2010.01664.x. [DOI] [PubMed] [Google Scholar]
- 9.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Program NCS, Murray PR, Turner ML, Segre JA. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–9. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Bieda J, Chaemsaithong P, Miranda J, Chaiworapongsa T, Ravel J. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome. 2014;2(1):18. doi: 10.1186/2049-2618-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devaraj S, Hemarajata P, Versalovic J. The human gut Microbiome and body metabolism: implications for obesity and diabetes. Clin Chem. 2013;59(4):617–28. doi: 10.1373/clinchem.2012.187617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ash C, Mueller K. Manipulating the Microbiota. Science. 2016;352(6285):530–1. doi: 10.1126/science.352.6285.530. [DOI] [PubMed] [Google Scholar]
- 13.Tyler AD, Smith MI, Silverberg MS. Analyzing the human Microbiome: A “How To” guide for physicians. Am J Gastroenterol. 2014;109:983–93. doi: 10.1038/ajg.2014.73. [DOI] [PubMed] [Google Scholar]
- 14.Lange A, Jost S, Heider D, Bock C, Budeus B, Schilling E, Strittmatter A, Boenigk J, Hoffmann D. Ampliconduo: A split-sample filtering protocol for high-throughput amplicon sequencing of microbial communities. PLoS ONE. 2015;10(11):1–22. doi: 10.1371/journal.pone.0141590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The Human Microbiome Project et al. A framework for human microbiome research. Nature. 2012;486(7402):215–1. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4):1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossmann L, Jensen M, Heider D, Jost S, Glucksman E, Hartikainen H, Mahamdallie SS, Gardner M, Hoffmann D, Bass D, Boenigk J. Protistan community analysis: key findings of a large-scale molecular sampling. ISME J. 2016;10(9):2269–279. doi: 10.1038/ismej.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, Mcdonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing. Nature. 2010;7(5):335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–9. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youmans BP, Ajami NJ, Jiang Z-d, Campbell F, Wadsworth WD, Petrosino JF, Dupont HL, Highlander SK. Characterization of the human gut microbiome during travelers’ diarrhea. Gut Microbes. 2015;6(2):110–9. doi: 10.1080/19490976.2015.1019693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamady M, Lozupone CA, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4(1):17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuyama J, McMurdie PJ, Dethlefsen L, Relman DA, Holmes S. Comparisons of distance methods for combining covariates and abundances in microbiome studies. Pac Symp Biocomput. 2017;148:352–63. [PMC free article] [PubMed] [Google Scholar]
- 23.Mosimann JE. On the compound multinomial distribution, the multivariate β-distribution, and correlations among proportions. Biometrika. 1962;1(331):65–82. [Google Scholar]
- 24.la Rosa PS, Brooks JP, Deych E, Boone EL, Edwards DJ, Wang Q, Sodergren E, Weinstock G, Shannon WD. Hypothesis testing and power calculations for taxonomic-based human microbiome data. PLoS ONE. 2012;7(12):1–13. doi: 10.1371/journal.pone.0052078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: Generative Models for Microbial Metagenomics. PLoS ONE. 2012;7(2):30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Li H. Variable selection for sparse Dirichlet-multinomial regression with an application to microbiome data analysis. Ann Appl Stat. 2013;7(1):418–42. doi: 10.1214/12-AOAS592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Bushman FD, Lewis JD, Wu GD, Li H. Structure-constrained sparse canonical correlation analysis with an application to microbiome data analysis. Biostatistics. 2013;14(2):244–58. doi: 10.1093/biostatistics/kxs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin W, Shi P, Feng R, Li H. Variable selection in regression with compositional covariates. Biometrika. 2014;101(4):785–97. doi: 10.1093/biomet/asu031. [DOI] [Google Scholar]
- 29.The Human Microbiome Project et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. PNAS. 2010;107(44):18933–8. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. Cross-species comparisons of host genetic associations with the microbiome. Science. 2016;352(6285):29–32. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George EI, McCulloch RE. Approaches for Bayesian Variable Selection. Stat Sin. 1997;7:339–73. [Google Scholar]
- 33.Brown PJ, Vannucci M, Fearn T. Multivariate Bayesian variable selection and prediction. J R Stat Soc Ser B Stat Methodol. 1998;60(3):627–41. doi: 10.1111/1467-9868.00144. [DOI] [Google Scholar]
- 34.Smith M, Kohn R. Nonparametric regression using Bayesian variable selection. J Econ. 1996;75(2):317–43. doi: 10.1016/0304-4076(95)01763-1. [DOI] [Google Scholar]
- 35.Chipman H, George EI, Mcculloch RE. The Practical Implementation of Bayesian Model Selection. IMS Lect Notes - Monogr Ser. 2001;38:67–134. [Google Scholar]
- 36.Scott JG, Berger JO. Bayes and empirical-Bayes multiplicity adjustment in the variable-selection problem. Ann Stat. 2010;38(5):2587–619. doi: 10.1214/10-AOS792. [DOI] [Google Scholar]
- 37.Savitsky T, Vannucci M, Sha N. Variable selection for nonparametric gaussian process priors: models and computational strategies. Stat Sci. 2011;26(1):130–49. doi: 10.1214/11-STS354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts GO, Rosenthal JS. Examples of Adaptive MCMC. J Comput Graph Stat. 2009;18(2):349–67. doi: 10.1198/jcgs.2009.06134. [DOI] [Google Scholar]
- 39.Haario H, Saksman E, Tamminen J. Componentwise adaptation for high dimensional MCMC. Comput Stat. 2005;20(2):265–73. doi: 10.1007/BF02789703. [DOI] [Google Scholar]
- 40.Barbieri MM, Berger JO. Optimal predictive model selection. Ann Stat. 2004;32(3):870–97. doi: 10.1214/009053604000000238. [DOI] [Google Scholar]
- 41.Newton MA, Noueiry A, Sarkar D, Ahlquist P. Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics. 2004;5(2):155–76. doi: 10.1093/biostatistics/5.2.155. [DOI] [PubMed] [Google Scholar]
- 42.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405(2):442–51. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 43.Taddy MA. Multinomial inverse regression for text analysis (with discussion) J Am Stat Assoc. 2013;108(503):755–70. doi: 10.1080/01621459.2012.734168. [DOI] [Google Scholar]
- 44.Geweke J. Evaluating the accuracy of sampling-based approaches to the calculation of posterior moments. Bayesian Stat 4. 2012;8(6):169–93. [Google Scholar]
- 45.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methé B, Schloss PD, Gevers D, Mitreva M, Huttenhower C. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8(6):1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10(5):323–35. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, Louis P, McIntosh F, Johnstone AM, Lobley GE, Parkhill J, Flint HJ. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5(2):220–30. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crost EH, Tailford LE, Le Gall G, Fons M, Henrissat B, Juge N. Utilisation of Mucin Glycans by the Human Gut Symbiont Ruminococcus gnavus Is Strain-Dependent. PLoS ONE. 2013;8(10). doi:10.1371/journal.pone.0076341. [DOI] [PMC free article] [PubMed]
- 49.Cao Y, Rocha ER, Smith CJ. Efficient utilization of complex N-linked glycans is a selective advantage for Bacteroides fragilis in extraintestinal infections. PNAS. 2014;111(35):12901–6. doi: 10.1073/pnas.1407344111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rho JH, Wright DP, Christie DL, Clinch K, Furneaux RH, Roberton AM. A novel mechanism for desulfation of mucin: Identification and cloning of a mucin-desulfating glycosidase (sulfoglycosidase) from Prevotella strain RS2. J Bacteriol. 2005;187(5):1543–1551. doi: 10.1128/JB.187.5.1543-1551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hilyard EJ, Jones-Meehan JM, Spargo BJ, Hill RT. Enrichment, isolation, and phylogenetic identification of polycyclic aromatic hydrocarbon-degrading bacteria from Elizabeth River sediments. Appl Environ Microbiol. 2008;74(4):1176–82. doi: 10.1128/AEM.01518-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schöcke L, Weimer PJ. Purification and characterization of phosphoenolpyruvate carboxykinase from the anaerobic ruminal bacterium Ruminococcus flavefaciens. Arch Microbiol. 1997;167(5):289–94. doi: 10.1007/s002030050446. [DOI] [PubMed] [Google Scholar]
- 53.Yano T, Fukamachi H, Yamamoto M, Igarashi T. Characterization of L-cysteine desulfhydrase from Prevotella intermedia. Oral Microbiol Immunol. 2009;24(6):485–92. doi: 10.1111/j.1399-302X.2009.00546.x. [DOI] [PubMed] [Google Scholar]
- 54.Wright DP, Rosendale DI, Roberton AM. Prevotella enzymes involved in mucin oligosaccharide degradation and evidence for a small operon of genes expressed during growth on mucin. FEMS Microbiol Lett. 2000;190(1):73–9. doi: 10.1111/j.1574-6968.2000.tb09265.x. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi K, Nishida A, Fujimoto T, Fujii M, Shioya M, Imaeda H, Inatomi O, Bamba S, Andoh A, Sugimoto M. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion. 2016;93(1):59–65. doi: 10.1159/000441768. [DOI] [PubMed] [Google Scholar]
- 56.Jumas-Bilak E, Jean-Pierre H, Carlier JP, Teyssier C, Bernard K, Gay B, Campos J, Morio F, Marchandin H. Dialister micraerophilus sp nov and Dialister propionicifaciens sp nov., isolated from human clinical samples. Int J Syst Evol Microbiol. 2005;55(Pt 6):2471–478. doi: 10.1099/ijs.0.63715-0. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi N, Yamada T. Pathways for amino acid metabolism by Prevotella intermedia and Prevotella nigrescens. Oral Microbiol Immunol. 2000;15(2):96–102. doi: 10.1034/j.1399-302x.2000.150205.x. [DOI] [PubMed] [Google Scholar]
- 58.Ruan Y, Shen L, Zou Y, Qi Z, Yin J, Jiang J, Guo L, He L, Chen Z, Tang Z, Qin S. Comparative genome analysis of Prevotella intermedia strain isolated from infected root canal reveals features related to pathogenicity and adaptation. BMC Genomics. 2015;16(1):1–22. doi: 10.1186/1471-2164-16-S7-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, Rosenbaum M, Gordon JI. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A Guide to Enterotypes across the Human Body: Meta-Analysis of Microbial Community Structures in Human Microbiome Datasets. PLoS Comput Biol. 2013;9(1):1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Linnenbrink M, Künzel S, Fernandes R, Nadeau MJ, Rosenstiel P, Baines JF. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. PNAS. 2014;111:2703–10. doi: 10.1073/pnas.1402342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used in the study are included as Additional files 2 and 3, respectively. The code developed for this manuscript is publicly available on GitHub at https://github.com/duncanwadsworth/dmbvs, at http://www.micheleguindani.info and on http://www.stat.rice.edu/~marina/. An R package is under development and will be announced on those sites.