

Abstract
Aminoacyl tRNA synthetases (AARSs) are best known for their essential role in translation in the cytoplasm. The concept that AARSs also exist in the nucleus started to draw attention around the turn of the new millennium, when aminoacylated tRNAs were first found in the nuclei of Xenopus oocytes. It is now expected that all cytoplasmic AARSs are present in the nucleus. In addition to tRNA aminoacylation, nuclear AARSs were found to regulate a spectrum of biological processes and responses, with many AARSs functioning through regulation at the level of gene transcription. In this paper, we focus on describing methods that have been successfully implemented to study AARSs in transcriptional regulation. These include a cell fractionation assay to detect nuclear localization, an in vitro DNA-cellulose pull-down assay to determine DNA binding capacity, and a chromatin immunoprecipitation (ChIP)-DNA deep sequencing assay to identify DNA binding sites. Application of these methods would expand our understanding of AARS functions and reveal critical insights on the coordination of gene transcription and translation.
Keywords: Aminoacyl tRNA synthetase, Nuclear function, Transcriptional regulation Cell fractionation, Chromatin Immunoprecipitation
1. Introduction
Back in 1985, in an attempt to explore the regulatory role of cytoskeleton in protein synthesis, Mirande et al. unexpectedly detected PheRS in the nuclei of rapidly growing mammalian cells [1]. However, it was not until 1998, when tRNA aminoacylation was found in the nuclei of Xenopus oocytes and postulated to act as a functional proofreading step for newly made tRNAs, that nuclear AARSs started to draw attention [2, 3]. A systematic study later detected major tRNA species of at least 19 tRNA families in the aminoacylated form in the nuclei of yeast cells, suggesting the nuclear presence of almost all AARSs in eukaryotic cells [4].
As it turns out, the nuclear functions of AARSs go far beyond tRNA aminoacylation and are involved in regulating a broad range of biological processes including vascular development, inflammation, and stress response [5–8]. As early as 1993, Popenko et al. detected TrpRS in the nuclei of mammalian cells located with diffuse rather than condensed chromatin regions [9]. Recent studies revealed several AARSs act at the level of gene transcription to achieve their important regulatory functions. For example, allergen activated MAPK cascade in mast cells triggers phosphorylation of LysRS and the corresponding conformational change that allows the translocation of LysRS to the nucleus, where it interacts and activates transcriptional factor MITF to stimulate the expression of genes related to inflammation reactions [10, 11] (Fig. 1). Oxidative stress triggers TyrRS to translocate from the cytosol to the nucleus to sequestrate a transcriptional repressor TRIM28 to active transcription factor E2F1 and the expression of its target genes involved in DNA damage repair [12, 13] (Fig. 1). A small portion of SerRS (~10%) is constitutively located in the nuclei of vertebrate cells to counteract transcription factor c-Myc to achieve a balanced regulation in angiogenesis during vascular development [14–17] (Fig. 1). Transcriptional control has become a paradigm for regulation by nuclear AARSs.
Fig. 1.

Three key methods for studying the transcriptional regulatory functions of AARSs in cell nuclei. In addition to their cytoplasmic functions of charging tRNAs with cognate amino acids for ribosomal protein synthesis, many AARS family members in higher eukaryotes can enter nuclei, where they modulates the expression of genes involved in a broad range of biological processes. Three examples are illustrated: Nuclear TyrRS can compete with transcriptional factor E2F1 for binding to transcriptional repressor TRIM28 and activate transcription of DNA damage response (DDR) genes. Nuclear LysRS can activate transcriptional factor MITF during an allergic response by producing Ap4A to remove HINT1 from inhibiting MITF. SerRS can directly bind onto genomic DNA and repress the expression of genes involved in angiogenesis such as VEGFA. The the nuclear import of an AARS can be determined by cell fractionation assay. Once the nuclear localization is confirmed, the AARS can be further studied by DNA-cellulose pull-down assay in vitro and ChIP analysis in cells to understand its potential function related to gene transcription.
Here we present the detailed methods for studying nuclear functions of AARSs, with a special focus on methods related to transcriptional regulation. We omit gene expression analysis, as most laboratories are familiar with the method. We include three other key experiments, which are 1) cell fractionation study to detect the nuclear presence of an AARS; 2) in vitro pull-down assay with genomic DNA linked cellulose beads to determine the DNA binding capacity of an AARS and therefore evaluate whether the potential regulation would involve direct DNA binding; and 3) if DNA binding is detected, cell-based chromatin immunoprecipitation assay followed by DNA deep sequencing (ChIP-Seq) to determine the DNA binding sites of an AARS (Fig. 1).
2. Materials
2.1 Cell fractionation
0.25% Trypsin-EDTA (ThermoFisher Scientific, Cat. No. 25200056)
Swelling buffer (SB) [10 mM Tris-HCl (pH 7.4), 2 mM EDTA, proteinase inhibitor cocktail (added before use) (Roche, Cat. No. 11873580001)]
Plasma membrane lysis buffer (PMLB) [10 mM Tris-HCl (pH 7.1), 2 mM MgCl2, 1% Triton X-100]
Nuclear extraction buffer (NEB) [20 mM HEPES (pH 7.6), 300 mM NaCl, 2 mM EDTA, 1 mM 1,4-Dithiothreitol (DTT), 10% glycerol, 1% Triton X-100, protease inhibitor cocktail (added before use)]
Phosphate-buffered saline (PBS) [10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.2]
Branson digital sonifier 250 (Branson Ultrasonics Corporation)
2.2 DNA-cellulose pull-down assay
Calf thymus genomic DNA-cellulose (Sigma-Aldrich, D8515), Sigmacell cellulose-type 50 (Sigma-Aldrich, S5504)
Basic dilution buffer (BDB) [20 mM Tris-HCl (pH 7.5), 100 mM KCl, 10% glycerol, 1 mM EDTA, 1 mg/mL bovine serum albumin (BSA), filter and store at 4°C]
Binding buffer [Add 1 mM DTT, 0.1% Triton X-100, and protease inhibitor cocktail into BDB buffer.]
Washing buffer I [20 mM Tris-HCl (pH 7.5), 110 mM potassium acetate, 10% glycerol, 1 mM EDTA, 1 mg/mL BSA, 0.2% Triton X-100]
Washing buffer II [20 mM Tris-HCl (pH 7.5)]
Elution buffer [10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 500 mM NaCl, pH 7.4]
Micro Bio-spin column (Bio-Rad, Cat. No. 7326204)
2.3 Chromatin immunoprecipitation (ChIP)
Protein G magnetic beads (Active Motif, Cat. No. 53014)
ChIP grade antibody and negative control IgG from same species
Enzymatic Shearing Kit (with optimized digestion buffer) (Active Motif, Cat. No. 53035)
Fixation solution [Add 0.55 mL of 37% formaldehyde to 20 mL minimal cell culture medium and mix. The final formaldehyde concentration is 1%. Keep at room temperature.]
Stop-fix solution [PBS buffer with 125 mM glycine, keep at room temperature.]
100 mM Phenylmethylsulfonyl fluoride (PMSF, protease inhibitor)
100× protease inhibitor cocktail (Roche, Cat. No. 11873580001)
Cell lysis buffer [5 mM PIPES (pH 8.0), 85 mM KCl, 0.5% Nonidet P-40 (NP-40), Store at 4°C.]
0.5 M EDTA
5 M NaCl
RNase A, 10 mg/mL (ThermoFisher Scientific, Cat. No. EN0531)
Protease K, 20 mg/mL (ThermoFisher Scientific, Cat. No. EO0491)
Phenol/Chloroform (1:1, TE saturated at pH 8.0) (Sigma-Aldrich, Cat. No. P2069)
ChIP dilution buffer [20 mM Tris-HCl (pH 8.0), 167 mM NaCl, 1.2 mM EDTA, 0.01% (wt/vol) SDS, 1.1% Triton X-100]
Low salt washing buffer [20 mM Tris (pH 8.0), 150 mM NaCl, 0.1% (wt/vol) SDS, 1% Triton X-100, 2 mM EDTA]
High salt washing buffer [20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 2 mM EDTA, 0.1% (wt/vol) SDS, 1% Triton X-100]
LiCl washing buffer (0.25 M LiCl; 1% NP-40; 1% Sodium Deoxycholate, 1 mM EDTA; 10 mM Tris pH 8.0)
TE buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA]
Elution buffer [1% (wt/vol) SDS, 0.1 M NaHCO3]
QIAquick PCR purification kit (QIAGEN, Cat. No. 28104)
Magnetic stand (ThermoFisher Scientific, Cat. No. 12320D)
3. Cell fractionation protocol
This chapter describes the procedure to perform cell fractionation to study the nuclear localization of AARSs. To ensure the cell fractionation experiment is performed properly, it is necessary to monitor a cytoplasmic and a nuclear marker (e.g., α-tubulin and lamin A/C, respectively), alongside with the AARS (Fig. 2). Nuclear localization of an AARS may be regulated by environmental cues [11, 13]. To study the effect of an inducing agent, cells need to be treated prior to the procedures below. For example, in order to test if the nuclear localization of SerRS is responsive to DNA damage stimuli to potentially regulate DNA damage response, we treated cells with UV irradiation before the cell fractionation assay (Fig. 2). In this protocol, we use 0.5–1×107 HEK293 cells cultured in one 10-cm diameter dish with ~80–90% confluency.
Figure 2.

Nuclear import of SerRS tested by cell fractionation assay. HEK293 cells (0.5–1×107) were treated with UV (6 mJ/cm2) irradiation or not treated before the whole cell lysates (WCL) were prepared and the nuclear (N) and cytoplasmic (C) fractions were extracted. The WCL, N and C fractions were further analyzed by Western blot. WCL was blotted to evaluate the effect of the treatment on the expression of the AARS. Blotting cytoplasmic specific protein α-tubulin and nuclear specific protein lamin A/C was used to evaluate the purities of the fractions. In this experiment, there were small amounts of cytoplasmic contamination in the nuclear fractions as indicated by the presence of α-tubulin. The contamination can be removed by additional wash of the nuclear pellets with PBS buffer after the cytoplasmic fractions is removed.
3.1 Cell preparation
Harvest cells with 0.25% trypsin-EDTA and transfer the cells to a 1.5 mL microcentrifuge tube. Centrifuge at 500 ×g for 5 minutes at 4°C. Remove the supernatant. From now on, keep cells and cell extracts on ice.
Wash cells by gently suspending the cell pellet with 1 mL of ice-cold PBS by pipetting. Take 100 μL suspended cells for preparation of whole cell lysate sample and the rest 900 μL suspended cells will be used to prepare cytoplasmic and nuclear fraction. Spin down the cells at 500 ×g for 5 minutes at 4°C. Carefully remove the supernatant, leaving the cell pellet as dry as possible. The pellet volume of 0.5–1×107 HEK293 cells is around 50–100 μL.
3.2 Cell fractionation
Add ice-cold SB buffer to the cell pellet. The volume of SB is tenfold of cell pellet volume. For an estimated 50 μL of cell pellet, add 500 μL of SB buffer. Immediately suspend the cell pellet by pipetting and then vortex the tube at the highest speed for 15 seconds. Incubate the tube on ice for 10 minutes.
Add ice-cold PMLB buffer and immediately vortex the tube at the highest speed for 5 seconds. The volume of PMLB is 1/20 of the cell suspension volume made in last step. For 50 μL of cell pellet suspended in 500 μL of SB buffer, add 27.5 μL of PMLB buffer. Incubate the tube on ice for 1 minute1. Vortex the tube at the highest speed for 5 seconds again to fully release the nuclei. Spin down the cell nuclei at 16,000 ×g for 5 minutes at 4°C.
Immediately transfer the supernatant (i.e. cytoplasmic fraction) to a clean tube and keep it on ice until use.
Wash the nuclei pellet by suspending the pellet in 1 mL of ice-cold PBS and vortex at the highest speed for 5 seconds2. Spin down the nuclei pellet at 16,000 ×g for 5 minutes at 4°C. Remove the supernatant.
Suspend the nuclei pellet in ice-cold NEB buffer. The volume of NEB is 1/2 of the SB volume3. For 50 μL of nuclei pellet suspended in 500 μL of SB buffer, add 250 μL of NEB buffer. Sonicate the nuclei suspension on ice for 2 × 30 seconds, with a 30-second pause in between4. Use Branson digital sonifier with the following pulse settings: cycle 0.5, 10–15% power.
Centrifuge the tube at 16,000 ×g for 10 minutes at 4°C. Transfer the supernatant (i.e., nuclear fraction) to a clean tube and keep it on ice.
Western blot analysis to detect AARS, α-tubulin, and lamin A/C in each fraction.
4. DNA-cellulose pull-down assay protocol
Several AARSs were reported to bind to DNA [16, 18, 19]. In this section, we introduce the use of genomic DNA linked cellulose beads to determine the DNA binding capacity of an AARS. This method was used to detect the direct interaction of human SerRS with DNA, when GlyRS gave no interaction (Fig. 3) [16]. Therefore SerRS and GlyRS can serve as the positive and negative controls, respectively, for this experiment.
Figure 3.
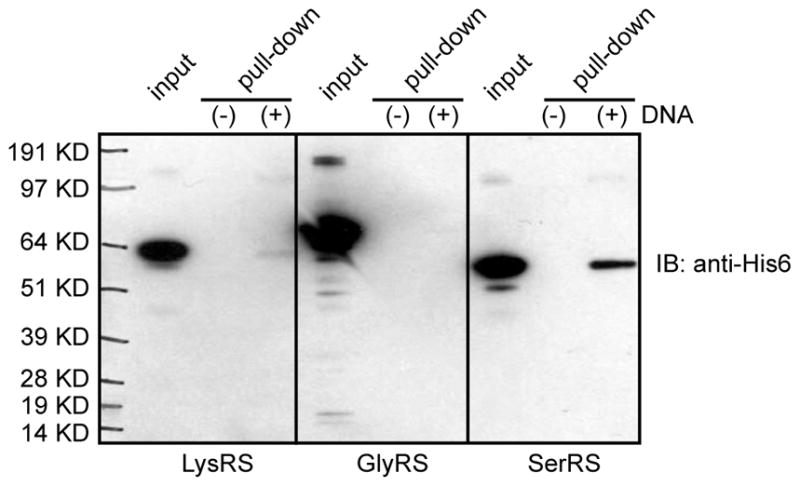
Detecting direct AARS-DNA interaction by DNA-cellulose pull down assay. Recombinant His6-tagged human LysRS, GlyRS, and SerRS were incubated with calf thymus genomic DNA linked cellulose or unconjugated cellulose beads for 1 hour at 4 °C. After wash and subsequent elution, the elutes were analyzed by Western blot analysis using anti-His6 tag antibody. The figure is adapted from a previous publication [16].
4.1. DNA-cellulose preparation
For each AARS to be tested, transfer 5 mg of Calf thymus genomic DNA-cellulose or 5 mg control cellulose (without conjugated DNA) into 0.5 mL of BDB buffer and incubate on ice for 30 minutes.
Wash DNA-cellulose or control cellulose 4 times with 0.5 mL of BDB buffer. Spin down the cellulose at 2000 ×g for 1 minute and discard the supernatant.
Add 0.5 mL of BDB buffer supplemented with 1% BSA to the cellulose and incubate at 25°C for 1 hour. Rotate the tube during incubation.
Spin down the cellulose at 2000 ×g for 1 minute and suspend the cellulose in 0.5 mL of binding buffer.
4.2. Pull-down assay
Transfer 400 μL of DNA-cellulose or control cellulose to 1.5 mL tube. Add AARS to a final concentration of ~50 nM (~1 μg of SerRS) into the celluloses and incubate at 4°C for 1 hour with rotation5.
Spin down the cellulose at 2000 ×g for 1 minute and discard the supernatant. Wash pellets twice with washing buffer I at 4°C.
Suspend the cellulose pellet in 0.7 mL of washing buffer I and transfer to a spin column. Wash the spin column five times with 0.7 mL of washing buffer I.
Wash the spin columns three times with 0.7 mL of washing buffer II.
Add 70 μL of elution buffer. Spin the columns at 2000 ×g for 1 minute to collect the elutes.
5. ChIP protocol
For AARSs that show DNA binding capacity in the above in vitro assay, we can subsequently identify their binding sites on the genomic DNA. In this section, we will introduce a well established ChIP method to capture AARS-interacting DNA fragments in human cells, which can be further analyzed by DNA deep sequencing or by quantitative real-time PCR (qPCR) (Fig. 4A). We suggest using the enzymatic digestion method to shear chromatin DNA. Compared to the traditional sonication method, the enzymatic approach is easier to control and be optimized. The enzymatic approach produces small DNA fragments (100–200 bp), which are suitable for the DNA deep sequencing platform and for qPCR. We also suggest using protein G-coated magnetic beads to pull out antibodies. Compared to the traditional agarose beads, the magnetic beads need fewer wash steps and provide cleaner results, therefore can ensure the sample-to-sample consistency.
Figure 4.

ChIP assay to identify AARS binding loci on genomic DNA. (A) Schematic representation of the ChIP procedure. (B) Agarose gel analysis to monitor enzymatic shearing results at various time points. Optimal shearing should give bands between 150 bp to 500 bp with a predominant band at 150 bp. In this experiment with HEK 293 cells, 10–12 minutes of enzymatic shearing was optimal. (C) Enrichment of VEGFA promoter DNA from HEK 293 cells overexpressing Flag-tagged SerRS versus the empty vector by ChIP assay using a Flag antibody. Quantitative real-time PCR (qPCR) was performed using DNA purified from the ChIP reactions and a primer pair specific for the VEGFA gene.
5.1. Cross-linking and chromatin shearing
When cells grow to 80–90% confluency in one 15-cm dish (for HEK293 cells, the estimated number of cells is 2–5×107)6, discard the medium and add 20 mL fixation solution7. Incubate on a shaker for 10 minutes at room temperature.
Discard the fixation solution. Stop fixation by adding 10 mL of Stop-fix solution and incubate on a shaker for 5 minutes at room temperature.
Discard the Stop-fix solution, wash the cells twice with 10 mL ice-cold PBS.
Discard the PBS washing solution, Add 5 mL of ice-cold PBS (supplemented with 30 μL of 100 mM PMSF and 50 μL of 100× protease inhibitor cocktail immediately before use) to the dish and scrape cells with a rubber policeman. Transfer the cells to a 15 mL tube on ice.
Pellet the cells at 800 ×g for 10 minutes at 4°C. Discard the supernatant. The pellet can be stored at −80°C after the addition of 1 μL of 100 mM PMSF and 1 μL of 100× protease inhibitor cocktail.
Resuspend cell pellet in 1 mL ice-cold cell lysis buffer (supplemented with 5 μL of 100 mM PMSF and 10 μL of 100× protease inhibitor cocktail) by gentle pipetting and brief vortex. Incubate on ice for 30 minutes.
Transfer the cells to an ice-cold dounce homogenizer. Dounce on ice with 50 strokes to fully release the nuclei8.
Pellet the cell nuclei at 2,500 ×g in a microcentrifuge for 10 minutes at 4°C. Discard the supernatant.
Resuspend the nuclei pellet in 350 μL digestion buffer (provided with the Enzymatic Shearing Kit) supplemented with 1.75 μL of 100 mM PMSF and 3.5 μL of 100× protease inhibitor cocktail. Incubate at 37°C for 5 minutes.
Add 17 μL of the working solution of 200 U/mL Enzymatic Shearing Cocktail (made by 1:100 dilution of stock Enzyme Shearing Cocktail [2×104 U/mL] with 50% glycerol in distilled water) to the pre-warmed nuclei and vortex to mix. Incubate at 37°C for an optimized time period. (For HEK293 cells, the optimal digestion time is 10–12 minutes (Fig. 4B)). During the incubation, vortex the tube briefly every 2 minutes to increase the shearing efficiency.
Stop the digestion by the addition of 7 μL ice-cold 500 mM EDTA and incubate on ice for 10 minutes.
Centrifuge the sheared samples at 16,000 ×g for 10 minutes at 4°C. Transfer the supernatant to clean tubes. DNA concentration in the supernatant needs to be measured first (5.2) before proceeding to the immunoprecipitation step (5.3).
5.2. Measuring the concentration of sheared DNA and assessing shearing efficiency
Add 150 μL of distilled water and 10 μL of 5 M NaCl to 50 μL of sheared chromatin sample and mix. Incubate at 65°C overnight to reverse the cross-link.
Remove RNAs by adding 2 μL RNase A (10 mg/mL) and incubate at 37°C for 15 minutes.
Remove proteins by adding 10 μL of Protease K and incubate at 42°C for 1.5 hours.
Add 200 μL Phenol/Chloroform (1:1, TE saturated at pH 8.0) to the samples, vortex at highest speed for 15 seconds and centrifuge at 16,000 ×g for 5 minutes. Transfer the aqueous phase to a clean tube, add 20 μL of 3 M sodium acetate (pH 5.2) and 500 μL of ethanol. Vortex and store the samples in −80°C for at least 1 hour to precipitate DNA fragments.
Centrifuge at 16,000 ×g for 5 minutes at 4°C. Discard the supernatant.
Add 500 μL of 70% ice-cold ethanol to wash the pellet. Centrifuge at 16,000 ×g for 5 minutes at 4°C. Discard the supernatant and allow the pellet to air-dry.
Dissolve the pellet in 50 μL distilled water. Measure the absorbance at 260 nm with a spectrophotometer to determine the DNA concentration. This is the concentration of DNA in the sheared chromatin samples.
Load the DNA fragments with loading buffer on 1.5% agarose gel and run the gel at 100V for 45 minutes. Enzymatic digestion prefers open regions that are easily accessible, such as the regions in between nucleosomes. Therefore, the optimal enzymatic shearing should result in predominately ~150 bp fragments with small amounts of larger fragments (i.e., 300 bp, 450 bp, and etc.) on the agarose gel.
5.3. Immunoprecipitation
Based on the DNA concentration measured during step 5.2, take 10–50 μg of sheared chromatin samples from step 5.1 to set up the ChIP reactions by adding the components following the order shown in Table 1.
Take same amount of sheared DNA as that for immunoprecipitation and add in 5 M NaCl to 0.3 M, 2 μL of 10 mg/mL RNase A, 10 μL of 0.5 M EDTA, 20 μL of 1M Tris-HCl (pH 6.5) and Elution buffer to 100 uL. This is the ‘input DNA sample’ to be used as a reference in the subsequent DNA deep sequencing or qPCR experiment.
Incubate the ChIP reactions on a rotator overnight at 4°C.
Spin the tube briefly to collect liquid from the cap. Place the tube on magnetic stand to pellet beads on the tube side. Discard the supernatant.
Table 1.
ChIP reaction
| Adding order | Reagent | Volume |
|---|---|---|
| 1 | Protein G magnetic beads | 25 μL |
| 2 | ChIP dilution buffer | 10 μL |
| 3 | Sheared chromatin sample (10–50 μg) | ≤ 60 μL* |
| 4 | Protease inhibitor cocktail (100×) | 1 μL |
| 5 | Distilled water | Add to make the final reaction volume be 100 μL. |
| 6 | ChIP grade antibody Or appropriate IgG control |
3 μg |
| Total volume | 100 μL |
Note: If the sheared chromatin concentration is very low, the reaction volume can be expanded to 200 μL with double volume of ChIP dilution buffer and protease inhibitor cocktail.
5.4. Wash, Elution and DNA purification
Wash beads one time with 1mL of ChIP dilution buffer.
Wash beads twice with 1 mL of low salt washing buffer.
Wash beads twice with 1 mL of high salt washing buffer.
Wash beads twice with 1 mL of LiCl washing buffer.
Wash beads one time with 1 mL of TE buffer.
Resuspend the beads with 50 μL of Elution buffer and incubate 15 minutes at room temperature on a rotator. Spin tubes briefly to collect liquid from caps. Transfer the supernatant to a new tube. Repeat once more and combine both the elution.
To reverse the cross-links, add 6.5 μL of 5 M NaCl (to reach the final concentration 0.3 M) and 2 μL of RNase A to the eluted DNA samples. Incubate the ChIP eluted DNA and the ‘input DNA samples’ prepared above at 65°C overnight in a thermocycler.
Return samples to room temperature and add 2 μL of 20 mg/mL protease K. Mix well and incubate at 37°C for 1 hour. Centrifuge the samples at 16,000 ×g for 5 minutes at room temperature. Transfer the supernatant to new tubes.
Use QIAquick PCR purification kit to purify the DNA. Elute DNA with 50 μL of TE buffer for future qPCR or DNA sequencing analysis.
6. Conclusion
We have provided detailed protocols for studying the nuclear localization of AARSs and their potential roles in the nucleus including the regulation of gene transcription by direct interaction with genomic DNA. These methods will contribute to deepen our understanding of the broad biological functions of AARSs beyond translation and potentially reveal critical insights on the coordination of gene transcription and translation.
Highlights.
Methods for studying nuclear function of AARS are proposed.
Cell fractionation used for determining the nuclear localization of AARS is described.
DNA-cellulose pull-down assay for analyzing the direct interaction of AARS with DNA is provided.
Chromatin immunoprecipitation assay for identifying the AARS binding loci in the genome is described in detail.
Acknowledgments
The work is supported by a R01 grant from the US National Institutes of Health NS085092.
Footnotes
Prolong incubation time after the addition of PMLB can cause the lysis of the nuclear membrane and therefore contamination of the cytoplasmic fraction with nuclear proteins.
Given the relatively low amount of AARSs in cell nuclei comparing to their amount in the cytoplasm, it is necessary to wash the nuclei pellet once to completely remove cytoplasmic AARS contamination.
Considering that the nuclear-cytoplasmic volume ratio of many cell lines (such as HEK293 cells) is around 1:2, using the same volume ratio between NEB and SB can approximately maintain the original concentration ratio of nuclear and cytoplasmic proteins.
Comparing with vortex, sonication can more efficiently break nuclear membranes and release the nuclear proteins. For AARSs that can strongly bind to genomic DNA, sonication can fragmentize and solubilize DNA, increasing the recovery of DNA bound AARSs.
Although recombinant mammalian AARSs purified from E. coli are commonly used for the assay, we should keep in mind that these proteins may lack certain posttranslational modifications that are unique to the mammalian system and may influence DNA binding.
To pull down endogenous proteins or proteins with less efficient antibodies, more cells may be needed.
To study the effect of a particular treatment on AARS-DNA interaction, cells should be treated before the fixation step.
Take 10 μL of the lysate to check under a phase contrast microscope to confirm the full release of cell nuclei. For HEK293 cells, 50 strokes were performed on our hands to completely release the nuclei.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mirande M, Le Corre D, Louvard D, Reggio H, Pailliez JP, Waller JP. Association of an aminoacyl-tRNA synthetase complex and of phenylalanyl-tRNA synthetase with the cytoskeletal framework fraction from mammalian cells. Exp Cell Res. 1985;156:91–102. doi: 10.1016/0014-4827(85)90264-2. [DOI] [PubMed] [Google Scholar]
- 2.Lund E, Dahlberg JE. Proofreading and aminoacylation of tRNAs before export from the nucleus. Science. 1998;282:2082–2085. doi: 10.1126/science.282.5396.2082. [DOI] [PubMed] [Google Scholar]
- 3.Schimmel P, Wang CC. Getting tRNA synthetases into the nucleus. Trends Biochem Sci. 1999;24:127–128. doi: 10.1016/s0968-0004(99)01369-9. [DOI] [PubMed] [Google Scholar]
- 4.Steiner-Mosonyi M, Mangroo D. The nuclear tRNA aminoacylation-dependent pathway may be the principal route used to export tRNA from the nucleus in Saccharomyces cerevisiae. Biochem J. 2004;378:809–816. doi: 10.1042/BJ20031306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo M, Schimmel P. Homeostatic mechanisms by alternative forms of tRNA synthetases. Trends Biochem Sci. 2012;37:401–403. doi: 10.1016/j.tibs.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo M, Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat Chem Biol. 2013;9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo M, Yang XL, Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat Rev Mol Cell Biol. 2010;11:668–674. doi: 10.1038/nrm2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko YG, Kang YS, Kim EK, Park SG, Kim S. Nucleolar localization of human methionyl-tRNA synthetase and its role in ribosomal RNA synthesis. J Cell Biol. 2000;149:567–574. doi: 10.1083/jcb.149.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popenko VI, Cherny NE, Beresten SF, Ivanova JL, Filonenko VV, Kisselev LL. Immunoelectron microscopic location of tryptophanyl-tRNA synthetase in mammalian, prokaryotic and archaebacterial cells. Eur J Cell Biol. 1993;62:248–258. [PubMed] [Google Scholar]
- 10.Ofir-Birin Y, Fang P, Bennett SP, Zhang HM, Wang J, Rachmin I, Shapiro R, Song J, Dagan A, Pozo J, Kim S, Marshall AG, Schimmel P, Yang XL, Nechushtan H, Razin E, Guo M. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol Cell. 2013;49:30–42. doi: 10.1016/j.molcel.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yannay-Cohen N, Carmi-Levy I, Kay G, Yang CM, Han JM, Kemeny DM, Kim S, Nechushtan H, Razin E. LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol Cell. 2009;34:603–611. doi: 10.1016/j.molcel.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 12.Fu G, Xu T, Shi Y, Wei N, Yang XL. tRNA-controlled nuclear import of a human tRNA synthetase. J Biol Chem. 2012;287:9330–9334. doi: 10.1074/jbc.C111.325902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei N, Shi Y, Truong LN, Fisch KM, Xu T, Gardiner E, Fu G, Hsu YS, Kishi S, Su AI, Wu X, Yang XL. Oxidative stress diverts tRNA synthetase to nucleus for protection against DNA damage. Mol Cell. 2014;56:323–332. doi: 10.1016/j.molcel.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukui H, Hanaoka R, Kawahara A. Noncanonical activity of seryl-tRNA synthetase is involved in vascular development. Circ Res. 2009;104:1253–1259. doi: 10.1161/CIRCRESAHA.108.191189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herzog W, Muller K, Huisken J, Stainier DY. Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circ Res. 2009;104:1260–1266. doi: 10.1161/CIRCRESAHA.108.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y, Xu X, Zhang Q, Fu G, Mo Z, Wang GS, Kishi S, Yang XL. tRNA synthetase counteracts c-Myc to develop functional vasculature. Elife. 2014;3:e02349. doi: 10.7554/eLife.02349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Shi Y, Zhang HM, Swindell EC, Marshall AG, Guo M, Kishi S, Yang XL. Unique domain appended to vertebrate tRNA synthetase is essential for vascular development. Nat Commun. 2012;3:681. doi: 10.1038/ncomms1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Putney SD, Schimmel P. An aminoacyl tRNA synthetase binds to a specific DNA sequence and regulates its gene transcription. Nature. 1981;291:632–635. doi: 10.1038/291632a0. [DOI] [PubMed] [Google Scholar]
- 19.Dou X, Limmer S, Kreutzer R. DNA-binding of phenylalanyl-tRNA synthetase is accompanied by loop formation of the double-stranded DNA. J Mol Biol. 2001;305:451–458. doi: 10.1006/jmbi.2000.4312. [DOI] [PubMed] [Google Scholar]