

Abstract
Bromodomains (BRDs) are epigenetic interaction domains currently recognized as emerging drug targets for development of anticancer or anti-inflammatory agents. In this study, development of a selective ligand of the fifth BRD of polybromo protein-1 (PB1(5)) related to switch/sucrose nonfermenting (SWI/SNF) chromatin remodeling complexes is presented. A compound collection was evaluated by consensus virtual screening and a hit was identified. The biophysical study of protein–ligand interactions was performed using X-ray crystallography and isothermal titration calorimetry. Collective data supported the hypothesis that affinity improvement could be achieved by enhancing interactions of the complex with the solvent. The derived SAR along with free energy calculations and a consensus hydration analysis using WaterMap and SZmap algorithms guided rational design of a set of novel analogues. The most potent analogue demonstrated high affinity of 3.3 μM and an excellent selectivity profile, thus comprising a promising lead for the development of chemical probes targeting PB1(5).
Introduction
Medical interventions directly targeting epigenetic functionalities are among the most promising novel approaches for treatment of serious pathological states such as cancer, metabolic and neurological diseases, inflammation, and viral infections.1−5 The first marketed epigenetic drug (Vorinostat) provided in 2006 the “proof-of-concept” for targeting epigenetic machinery by small molecules in a therapeutic manner. Since then, a multitude of epigenetic proteins have been suggested as potential targets for pharmacotherapy, including families of enzymes like the histone acetyltransferases (HATs), the DNA methyltransferases (DNMTs), and the histone deacetylases and sirtuins (HDACs and SIRTs). Bromodomains (BRDs) comprise a family of 61 epigenetic modules implicated in recognition of acetylated lysine (Kac) residues mainly on histones.6 The BRDs can be found as components of at least 46 multidomain proteins of the human genome.7,8 The discovery of a small molecule selectively inhibiting BRDs in bromo- and extra-terminal (BET) proteins (BRD2, BRD3, BRD4, BRDT) has provided solid evidence for BRD druggability.9 The fused triazole-thienodiazepine scaffold-based 1 ((+)-JQ1) has shown antiproliferative effects in BRD4-dependent human NUT midline carcinoma (NMC) cells in vivo.10 An increasing number of studies provides a compelling rationale for using 1 as well as a structurally related triazolobenzodiazepine-based BET inhibitor (I-BET762, see Figure 1A), mainly as antiproliferative or immunomodulatory agents.11−15 The first BRD inhibitors have already entered clinical trials as chemotherapeutic agents.16
Figure 1.

(A) Structures of known BRD inhibitors along with their specificity profile within the various BRD subfamilies. (B) Chemical structures for selected compounds evaluated as BRD inhibitors in this study.
The establishment of the BET group of BRDs as valid therapeutic targets for the aforementioned aggressive form of cancer, along with the increasing amount of data showing that BRD modules critically affect several cellular functions, have led to the emergence of BRDs as appealing targets for development of highly specific inhibitors. As all BRD are not equally druggable, the family can be subdivided into nine subfamilies (I to IX) according to structural features that define druggability of each member.17 At present, an increasing number of compounds selectively inhibiting BRDs outside the BET group (subfamily II) are described. Among the available molecules are I-CBP112 and CBP30 that target subfamily III c-AMP response element-binding protein binding protein (CREBBP) and E1A binding protein p300 (EP300), compounds LP99, I-BRD9, and BI9564 inhibiting subfamily IV BRD7 and BRD9, compounds NI57, OF-1, and PFI-4 targeting subfamily IV bromodomain and PHD finger containing-1, -2, and -3 (BRPF1, BRPF2, and BRPF3), compounds GSK2801 and BAZ2ICR targeting subfamily V bromodomain adjacent to zinc finger containing-domain 2A and 2B (BAZ2B/A), and 2 (PFI-3) targeting subfamily VIII SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily A, member 2 and 4 (SMARCA2/4) and PB1(5), while a number of molecules targeting sets of BRDs have been developed as well (compound structures are shown in Figure 1A,B; for a more complete overview please see review refs (29, 30)).18−30 Such compounds with single or group-wise BRD-selective inhibitory properties can significantly advance the elucidation of biological roles of targeted BRDs and may thus serve as invaluable chemical biology probes for mechanistic studies or as possible drug candidates.31
The BRDs of subfamily VIII have been characterized as proteins of intermediate or difficult druggability, in contrast to highly druggable subfamilies such as I or II (the BETs) or individual members of subfamilies III, IV, and VII.17 Subfamily VIII is comprised by BRDs that are mostly components of the chromatin remodeling complexes of SWI/SNF family. Indeed, all SWI/SNF complexes contain a central helicase with a bromodomain (SMARCA2/4) while the polybromo-associated BRG1 or hBRM-associated factor (PBAF) complex, a subtype of the SWI/SNF complex (or SWI/SNF-B), contains also the polybromo protein PB1 (or BAF180) which comprises six individual BRD modules.32 Remodeling complexes of the SWI/SNF family play a central role in development, particularly of the cardiac cells.33−35 Mutations in components of these chromatin modulators have been tightly linked to development of numerous cancer types including epithelioid sarcoma and malignant rhabdoid tumors.36,37 Especially in the case of renal cell carcinoma and pancreatic cancer, normal expression of the PBRM1 gene encoding for PB1 protein is considered as a critical factor of tumor progression.38−41 Moreover, truncating mutations of PB1 which have been found in breast cancer cells suggest a possible role for PB1 as a tumor suppressor. Although the precise mechanism for its tumor repressor activity is not yet clearly understood, it is thought that this functionality might be partially mediated through the ability of PB1 to regulate p21 induction.42−44 Moreover, the tumor-suppressing role of PB1 has been shown to be related to promotion of centromere cohesion and thus preservation of genomic stability in a manner independent from its involvement in transcriptional regulation.45 An additional aspect of its tumor suppressing role is highlighted by its activity as a repressor of transcription in response to double-strand breaks of DNA.46,47 Finally, the occurrence of six distinct BRDs on PB1 has raised a number of questions regarding their concerted or complementary function or even their redundancy.32 Two of PB1 BRDs lack the canonical asparagine that coordinates the Kac carbonyl (or equivalent moieties in inhibitors).6 It is likely that these BRDs bind therefore much weaker to inhibitors that typically hydrogen bond directly with this asparagine residue. As PB1(5) showed strongest interaction to acetyl-lysine containing histone peptide arrays, it is therefore likely the most relevant BRD anchoring PB1 (and the SWI/SNF complex) to chromatin in an acetyl-lysine dependent way. As a result, discovery of highly specific chemotypes with inhibitory properties toward BRDs of subfamily VIII such as PB1 and more specifically PB1(5) can be regarded as a challenging project, which would advance the existing knowledge concerning their underlying biological roles of these domains in health and disease. This notion is further highlighted by the finding that, while the ATPase module seemingly surpasses SMARCA2/4 as a drug target for synthetic-lethal interventions in a number of SWI/SNF-related cancer types, targeting PB1(5) and SMARCA2/4 does not result in toxicity but in differentiation phenotypes and thus simultaneous inhibition of several SWI/SNF BRDs can possibly provide alternative routes for therapeutically modulating chromatin remodeling.28,29,48
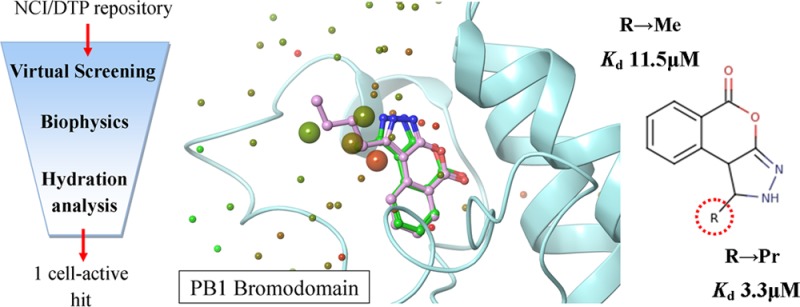
In this study, the utilization of three virtual screening (VS) methodologies of high orthogonality and their combination in a simple consensus scheme is presented. The National Cancer Institute/Developmental Therapeutics Program (NCI/DTP) Repository was systematically evaluated against a group of underexplored BRDs with moderate druggability and a pyrazoloisocoumarin hit showing low micromolar affinity, and a promising selectivity profile toward the fifth BRD of PB1 was identified. The attempt to optimize this ligand was based on the integration of thermodynamic and structural data obtained by X-ray crystallography and isothermal titration calorimetry (ITC) analysis with theoretical calculations. Hydration mapping was performed by implementation of the two most robust algorithms, namely SZmap and WaterMap, which afforded a high degree of convergence. Subsequent synthesis of rationally designed analogues where specific water molecules would be targeted by gradual expansion of the exocyclic methyl group resulted in derivatives demonstrating improved binding affinity and high selectivity for PB1(5) over related BRDs, displacing the protein from chromatin in cells and reducing viability of human fibroblasts.
Results and Discussion
In Silico Screening Protocol and Consensus Ranking
A screening initiative was undertaken as a means to discover compounds that selectively bind BRDs of subfamily VIII, thus providing novel and tractable scaffolds for sustaining an inhibitor development project. The NCI/DTP compound repository comprising approximately 260000 entries was selected due to its open-access policy. It was reasoned that an in silico computational evaluation of the collection prior to experimental screening would increase the anticipated hit recovery rate and additionally enhance the overall rational character of the study by providing structural insight from the initial steps of the process. Therefore, three distinct yet complementary VS methodologies were implemented in a stepwise protocol aiming at the optimal exploitation of sampling efficacy in a time-efficient manner. These methods would directly account for binding to the protein cavity (docking and scoring calculations featuring protein–ligand energy terms and implicit solvent contributions to binding) as well as conformational likeliness of the screened compounds to a known active compound (three-dimensional similarity involving pharmacophoric sites and shape in real space) or for the existence of hidden or nonobvious molecular motifs (two-dimensional similarity of topological fingerprints representing structural features and chemical functionality) commonly occurring between the screened collection entries and a known active template.
The overall workflow of the implemented integrated VS protocol is depicted in Figure 2. In the first step, a ligand-based approach would be used to evaluate the total NCI/DTP collection and find compounds that were similar to 1. For this purpose, two-dimensional similarity screening was performed using Canvas (Schrödinger Inc.).49−51 Canvas was additionally used in this step to create a potentially enriched subset of the initial library containing 5000 diverse compounds (∼2% of the total). In the second step, results of the ligand-based diversity evaluation would be redirected as input to a structure-based screening approach by implementing docking-scoring calculations of the enriched subset toward the bromodomain Kac-binding pocket using Glide SP (Schrödinger Inc.).52−55 In parallel, a separate ligand-based three-dimensional similarity screening of the global library would be performed using the ROCS algorithm (OpenEye Inc.).56,57 Finally, results of the different approaches were combined by a consensus scoring scheme and the top-ranked compounds would be selected for in vitro assays. The high degree of orthogonality between the two different screening pathways was thought as a means to effectively enhance sampling robustness and thus accuracy of the VS protocol, while their combination in a mixed serial-parallel manner was expected to optimize screening speed with the most time-consuming structure-based method being preceded by a ligand-based, fast prescreening of 2D similarity.
Figure 2.
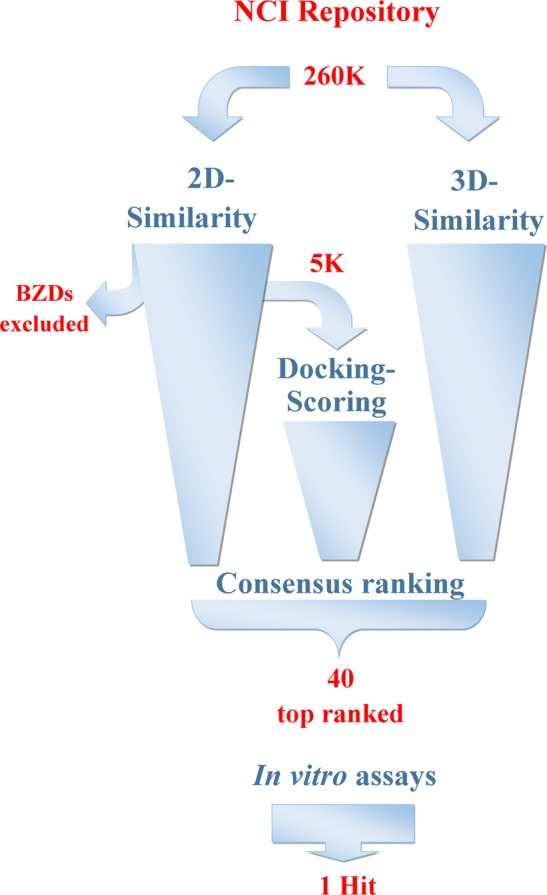
Virtual screening protocol utilized for the evaluation of the NCI/DTP Repository. Compounds were subjected to three different in silico screening methodologies. Two-dimensional similarity (toward 1, Canvas software, left-side funnel) was performed after discarding benzodiazepine-containing structures (BZDs). The top 2% diverse NCI/DTP molecules were additionally docked to the BRD (Glide software, middle funnel). In parallel, three-dimensional similarity was undertaken for the total collection (toward 1, ROCS software, right-side funnel). Finally, rank-ordered results originating from each of the three distinct filters were combined by a consensus ranking approach and the top 40 compounds were assayed experimentally for their BRD-binding affinity.
The NCI/DTP repository was independently prepared in terms of correct protonation state, tautomerization, and stereoisomerization for docking calculations and 3D similarity searches using LigPrep (Schrödinger, Inc.) and Quacpac (OpenEye, Inc.), respectively. In the case of 3D similarity screening, Filter and Omega (OpenEye Inc.) were additionally utilized for drug-likeness filtering and conformer ensemble generation. No special preparation was needed for 2D similarity searches. The query molecule used in both ligand-based approaches was 1, the only high-potency BRD inhibitor known at the time of the study. The compound does not show strong binding to any BET group BRD. However, on the basis of the highly conserved fold and high homology between BRD4 and PB1 (sequence similarity 61.6%) as well as in the absence of any other template, it was thought that the overall molecular geometry of 1 would adequately outline the fundamental structural requisites for competing Kac binding needed for hit recovery. It should also be noted that all NCI repository entries containing the benzodiazepine (BZD) core were filtered out and excluded from the screening study as a means to aid the discovery of novel scaffolds with BRD binding properties.
In the 2D screening approach, the structures of all NCI/DTP compounds were converted to fingerprint (FP) strings by utilizing a variety of different algorithms for FP derivation. Then, the structural similarity of each NCI/DTP compound with respect to the query molecule was determined by comparing the Tanimoto distance of its FPs with the corresponding FPs calculated for 1. Setup of the 2D screening calculations was not straightforward, as the selection of the most appropriate combination among the wide array of FP derivation methods and similarity measuring metrics available in Canvas is not always trivial.51 For this reason, a minimal training set was prepared with 20 fragments of known binding affinities for BRD4, CREBBP, and BAZ2B (SGC unpublished data) seeded into a set of 6000 randomly chosen entries from the NCI/DTP repository regarded as inactive decoys. Then, a total of 60 screens were performed using several recommended combinations of the Canvas 2D-screening parameters and the efficiency of each screen to top-rank the 20 active fragments among the 6000 decoys was quantified.50 Of these 60 screens, 11 afforded high recovery rates of the known actives, with enrichment factor at 20% of screened library (EF20%) reaching 2.2–2.5 for the 11 selected FP combinations while it was 0–1.2 for the remaining (EFmax20% is 5). These FP combinations were further selected for screening the global NCI/DTP repository (concerning FP derivation, scaling, and atom-typing methods; see Supporting Information, Table S1).
In the structure-based approach, the 5000 molecules of the enriched NCI/DTP subset derived from 2D-prescreening were sequentially docked to the BRD Kac-binding pocket and their theoretical affinity for the protein was evaluated using the GlideScore empirical scoring function as implemented in Glide. Concerning the 3D-sceening methodology, similarity searches were performed for 200-conformer ensembles of each NCI/DTP entry against 1 at its bioactive conformation. The 3D similarity was determined in terms of overall molecular shape overlap (shape similarity) as well as spatial similarity of pharmacophoric sites such as positive and negative atoms, hydrogen bond donors and acceptors, hydrophobes, and rings (color similarity).
Each of the implemented screening approaches (2D similarity-assisted docking and scoring screen, independent 2D and 3D similarity screens) afforded a distinct list of compounds ranked according to their potential to bind the BRD pocket and compete with acetylated histone binding. The lists were combined by a simple consensus scoring approach accounting for the frequency of appearance of each compound within a given percentage threshold of the top of the individual lists. By using this consensus scoring scheme, the 40 top-ranked compounds were selected for assessing their BRD binding properties in vitro using a differential scanning fluorimetry (DSF) assay.
In Vitro Affinity Assessment of Top-Ranked Compounds
The 40 compounds that scored higher in the consensus scheme were assayed in vitro using the thermal shift DSF screening assay (Supporting Information, Table S2). This method is based on measurement of the thermal stabilization of a protein in the presence of a small-molecule binder, as determined by the increase of the protein–ligand complex melting temperature (Tm) compared to that of the apoprotein.58 A screening panel of six BRDs was utilized for obtaining insight into the specificity profile of the potential hits. The DSF assay was implemented with high protein and ligand concentrations and resulted in the identification of seven compounds (3–9, Figure 1B) with detectable temperature shifts (Supporting Information, Table S3 for Tm curves and screening method of selection). Out of those, five failed to bind PB1(5) but showed very weak binding toward BRD4(2), with corresponding Tm values ranging between 0.8 and 2.0 °C. These results were in accordance with the well-established, favorable druggability of this BRD (and the BET group in general).17 Only two compounds afforded a Tm value larger than 3 °C. Compound 8 (NSC76484)59 resulted in a 3.2 °C stabilization toward BRD4(2). This particular scaffold demonstrated a rather obvious structural similarity with known BRD4 inhibitors, as its tricyclic ring system contained a triazole moiety reminiscent of the corresponding pharmacophore of 1. However, the most interesting finding was compound 9 (NSC356476),59 which showed a marked stabilization of 4.1 °C toward PB1(5) and at the same time only weak binding to BRD4(2) (Tm of 1.9 °C) at 100 μM compound concentration. The results of the thermal shift assay were considered as a good indication that 9, a pyrazoloisocoumarin derivative of relatively low molecular weight (200 Da), could constitute a novel BRD-binding scaffold and at the same time a promising hit with a highly preferable specificity profile toward BRD related to the SWI/SNF chromatin remodeling complexes.
X-ray Crystallography of Ligand–BRD Complex
To better understand the interaction between 9 and PB1(5), the cocrystal structure of the protein–ligand complex was determined at a resolution of 2.02 Å and the binding mode of 9 was elucidated (Supporting Information, Table S4). The ligand bound with the benzopyrone system buried deeply in the Kac pocket, while the N3 of the pyrazole ring accepted a hydrogen bond from the amide nitrogen of the conserved N739 (Figure 3B–D). With the exception of this hydrogen bond, the ligand was stabilized mainly through hydrophobic interactions. The most important contacts of 9 with the bromodomain are accommodated by L687 and L693 of the ZA loop, by I745 of αC-helix as well as by I683 and F684, two residues that constitute along with A682 the WPF shelf of the Kac cavity in PB1(5). A weak edge-to-face π–π stacking interaction could be observed between the aromatic system of the ligand and Y738. Notably, the overall conformation of the ZA loop in the complex of 7 and PB1(5) did not induce structural changes when compared to the apoprotein, suggesting that the unusual loop conformation observed for PB1(5) in a previously released structure (PDB code: 3G0J) is not induced as a result of ligand binding.
Figure 3.

Binding of 9 to PB1(5) leads to displacement of water molecules. (A) Apo-structure of PB1(5) (PDB code: 3G0J) with the canonical water network. Water molecules are shown as transparent spheres and are labeled W1–W5. The main secondary structure elements as well as acetyl-lysine binding site residues are shown in stick representation. (B) Co-crystal structure of 9 with PB1(5). The inset shows a 2FoFc electron density map contoured at 2σ around the ligand. (C) Overlay of 9 with the apo-structure. The main residues and water molecules present in the apo-structure are shown. (D) Surface representation showing that 9 displaces four of five structural waters present in the acetyl-lysine binding site.
Interestingly, compound 9 displaced four of the five of the conserved buried water molecules that are observed in the majority of the available BRD–ligand complexes (Figure 3A–D). These waters form an extensive hydrogen bond network in the Kac-binding cavity which is usually retained in most of the released bromodomain–inhibitor cocrystal structures (B-values of waters between 28 and 40, similar to main chain Calpha positions in the protein backbone). In the case of 9, however, the only water molecule that was not displaced seems to be stably bound through a multidirectional system of four hydrogen bonds to the side chain hydroxyl of Y696, the side chain amide of N734, and the backbone carbonyls of M704 and M731 (Supporting Information, Figure S1). This water was the most deeply buried of the aforementioned network of conserved waters, and it was located within hydrogen-bonding distance to both the exocyclic carbonyl of 9 and the side chain of the conserved Y696 of ZA loop. This water molecule is highly conserved and forms a hydrogen bond with the hydroxyl group of Y696 in acetyl-lysine containing peptides and acetyl-lysine mimetic inhibitor complexes. An additional water that participates in ligand binding was found in the first hydration shell on the solvent-accessible side of the Kac cavity, forming a hydrogen bond bridge between N2 of 9 and the side chain carbonyl of N739. Finally, the good complementarity of the ligand to the acetyl-lysine binding cavity was indicated by the solvent-accessible surface area that was buried upon binding which was 172 Å2 for the ligand (135 Å2 apolar) and 188 Å2 for the protein (177 Å2 apolar).
A binding mode displacing four of the five structural water molecules similar to 9 has also been described for 2 and salicylic acid-containing fragments.27 It was therefore explored whether compounds containing phenolic moieties similar to 2 and salicylic acid or pyrone-related structures similar to the isocoumarin system of 9 could induce the same water displacement motif with 9 and hence demonstrate binding affinity for PB1(5). Moreover, due to increased stability of compounds bearing closed rings, such as 9, against hydrolysis when compared to 2, identifying new scaffolds interacting with SWI/SNF bromodomains using this unique water displacing binding mode was of high interest to us. Screening of a small in-house library of natural products identified 5,3′,4′-trihydroxyflavone (14) and luteolin (15) as binders of BRDs with strongest temperature shifts observed for PB1(5) (Figure 4A,B). Subsequent cocrystallization studies indeed confirmed the hypothesized water displacing acetyl-lysine binding modes for both flavones, maintaining only water W5 in the binding pocket (Figure 4C–F). The critical role this buried solvent molecule seems to have in complex stabilization should certainly be considered when planning chemical modifications targeting hit optimization. Yet, its net energetic contribution to binding should be carefully assessed because there might be a considerable penalty component due to its restrained positioning. The affinity of 15 was determined using isothermal titration calorimetry, resulting in a Kd of 12.9 μM.
Figure 4.

Flavonoids 14 and 15 interact with the acetyl-lysine binding site of PB1(5) in a water displacing binding mode. (A) Temperature shift assays of 14 and 15 measured on diverse bromodomains at 10 μM and 100 μM inhibitor concentration. (B) Isothermal titration calorimetry revealed a dissociation constant of 12.9 μM for 15. Shown are raw titration heats as well as normalized binding heats (inset). (C) Binding mode of 14 shown in ball and stick representation with yellow carbon atoms in PB1(5) (inset: 2FoFc map). (D) Binding mode of 15 shown in ball and stick representation with yellow carbon atoms in PB1(5) (inset: 2FoFc map). (E) Overlay of apo-PB1(5) with the 15 complex. Water molecules present in apo-PB1(5) are shown as transparent spheres. Water molecules that are displaced in the 14 complex are highlighted by a red cross. (F) Overlay of apo-PB1(5) with the 15 complex. Water molecules present in apo-PB1(5) are shown as transparent spheres. Water molecules that are displaced in the 15 complex are highlighted by a red cross.
Isothermal Titration Calorimetry Analysis
To accurately determine the binding affinity of pyrazoloisocoumarin 9 for PB1(5), we measured the dissociation constant (Kd) in solution using ITC. This analysis revealed a Kd value of 11.5 μM, single site binding stoichiometry and a favorable binding enthalpy change (ΔH of −5.6 kcal/mol) were measured at 288 K, affording a fairly high ligand efficiency (LE) index of 0.46 for this hit (Table 1). The low micromolar affinity of 9 in combination with its high LE showed that the compound could indeed be considered as a promising starting point for rational development of analogues with improved binding affinity. The estimated ΔGbinding indicated that binding is dominated by enthalpic interactions, in accordance with the structural analysis, showing the formation of polar interactions. As mentioned before, the flavonoid hit 15 had a similar affinity (Kd of 12.9 μM) (Figure 4B). However, by considering the less pronounced originality and chemical tractability of the flavone system and, most importantly, the poor selectivity of 15 (Figure 4A) over other BRDs and other target families (e.g., kinases), the aforementioned scaffold was not prioritized at that stage and 9 was selected for further activity optimization.
Table 1. ITC Data along with the Predicted ΔΔG Values of the Novel Analogues 8, 9, and 10 Relative to 7 As Obtained by the FEP-REST Calculations (N-Values: 7, 1.0; 8, 1.2; 9, 0.8; 10, 1.1)a.
| FEP-REST | ITC |
||||
|---|---|---|---|---|---|
| compd | ΔΔGpredicted (kcal/mol) | Kd (μM) | ΔG (kcal/mol) | –TΔS (kcal/mol) | ΔH (kcal/mol) |
| 7 | 0.0 | 11.5 | –6.5 | –0.9 | –5.6 |
| 8 | –0.5 ± 0.1 | 3.4 | –7.2 | –0.6 | –6.7 |
| 9 | –0.9 ± 0.1 | 3.3 | –7.2 | –4.3 | –2.9 |
| 10 | –1.1 ± 0.1 | 5.1 | –7.0 | –0.6 | –6.4 |
The ΔΔG values predicted by FEP-REST for each mutation along with corresponding errors are calculated using the Bennett method as implemented in Desmond software, while the maximum error between predicted and experimental binding affinities is within 0.6 kcal/mol.
In an effort to devise a rational optimization strategy for the newly characterized pyrazoloisocoumarin hit to a possible lead compound, structural and thermodynamic data were combined and considered collectively. Inspection of the binding mode of 9 indicated three major extension vectors that could be utilized for hit optimization (shown in Figure 5A,B). Nevertheless, no obvious hydrogen bonding or electrostatic interaction partner was located within a reasonable radius around the bound ligand. Moreover, the apparent optimal fit of 9 and PB1(5) in combination with the known overall relative rigidity of the BRD fold set skepticism with respect to its capacity to explore more extensive models for hit expansion. It was hypothesized that affinity of 9 could be potentially enhanced through manipulation and fine-tuning of solvent effects that may affect the binding interaction. To explore this, the computational analysis of the protein hydration pattern was undertaken using the two currently available algorithms, SZmap (OpenEye Inc.) and WaterMap (Schrödinger, Inc.).60−63
Figure 5.

Comparison of the 9–PB1(5) complex hydration analysis results obtained using SZmap and Watermap. (A) Hydration sites predicted using SZmap algorithm are shown as yellow (negative ΔG, stable) and magenta (positive ΔG, unstable) spheres. (B) Water molecules predicted for 9–PB1(5) complex using Watermap algorithm are depicted as spheres colored according to their ΔG (green, low; red, high). The three most feasible extension vectors of the pyrazoloisocoumarin scaffold are depicted as red dashed arrows, and the related protein solvation sites are marked with their corresponding energy values in kcal/mol. The two methods afforded fairly comparable results showing highest convergence toward predicting unstable solvent molecules of ZA channel as the most promising hydration site that was therefore targeted by structural modifications of the hit.
Computational Hydration Analysis
Computational solvation mapping of a binding site (e.g., as performed by SZmap or WaterMap) can identify hydration sites that may confer ligand potency should that ligand for example efficiently displace or replace water molecules of appropriate energetics. As such, solvation mapping can suggest possible lead optimization modifications to optimize enthalpic protein–ligand interactions while simultaneously decreasing unfavorable entropy that results from localization of bulk water. The aforementioned approach can be of particular importance in the case of BRDs. Indeed, it has been shown that BRDs accommodate solvent-mediated interactions with their ligands that involve buried, conserved water molecules and are regarded as critical for binding. This highlights the considerable impact that the equilibrium between solvent dynamics and ligand binding can have to the druggability of each BRD.17
The hydration analysis performed for PB1(5) was focused to three separate regions of interest corresponding to the most promising extension vectors of the pyrazoloisocoumarin scaffold (Figure 5A,B). Those three regions were (i) the inner side of the Kac cavity, (ii) the entrance of the binding cavity, and (iii) the periphery of the ZA channel, a narrow saddle-shaped surface created by the ZA loop in the boundary of the BRD cavity in direct contact to the bound ligand. Those regions were extensively mapped utilizing the two above-mentioned computational approaches and inspected visually. Concerning the first region, SZmap predicted a hydration site partially overlapping with the buried water of the Kac cavity, a site thus needed to be efficiently replaced by a ligand polar group capable of forming equally extensive stabilizing interactions with the protein. For the second region, SZmap predicted a hydration site also corresponding to a highly stable water molecule that would consequently favor binding if merely being replaced by a polar group of the ligand. However, concerning the third region, SZmap predicted a cluster of unstable waters residing at the outer boundary of this hydration site. Those waters were seemingly trapped in a relatively hydrophobic environment yet in close proximity to the bulk solvent, thus they could be possibly targeted by nonpolar ligand groups (Figure 5A).
The utilization of WaterMap was also successful in identifying the aforementioned regions of interest concerning the protein hydration landscape (Figure 5B). Results obtained using WaterMap showed high convergence with the corresponding SZmap data with respect to the role of the buried water molecule (Supporting Information, Figure S1). Both approaches predicted that this was a highly stable water that would be rather difficult to increase binding affinity by replacing it by the ligand. However, WaterMap predicted that the hydration site located at the entrance of the binding site harbors a relatively unstable water, but these results were not confirmed by SZmap. This slight discrepancy was likely due to the actual position of these waters close to the first hydration shell of the protein and also to algorithmic differences implemented by each methodology. However, the prediction concerning the third region of interest was in good agreement between both methods. More specifically, at least two sites of unstable waters were predicted in the periphery of the ZA channel, in good overlap with the cluster of unstable waters predicted by SZmap. Moreover, another conserved water site was identified in fair proximity at the inner side of the ZA channel. Interestingly, prediction of the latter as well as one of the former sites was well supported by the actual presence of crystallographic water molecules in the protein–ligand complex, as the unstable water was visible only in one BRD molecule present in the asymmetric unit, while a water molecules predicted to be stable was present in both binding sites and interacted with three hydrogen bonds to the backbone carbonyls or amide nitrogens of residues A682, F684, L685, and R686.
In a consensus point of view, this third site at the periphery of the ZA channel was the most promising region in terms of structural interventions targeting affinity optimization through manipulation of the solvent component. Both approaches used for hydration analysis suggested that this site, as occupied by unstable waters, can be regarded as moderately exposed to the solvent environment and, therefore, it is possibly suitable to be targeted by nonpolar ligand groups in a manner that could favor affinity by replacing those waters.
Design of Analogues and FEP-REST Calculations
In light of the structural, thermodynamic, and hydration analysis performed so far, inspection of the available extension vectors of 9 led to the selection of the 1-methyl group of the pyrazole ring as the most suitable for chemical elaborations. Most substitutions on the phenyl ring of 9 would possibly lead to steric clashes with the protein as was suggested by the optimal fit observed for 9 in the X-ray structure of the complex. Efficacious replacement of the buried structural water was deemed highly challenging given the enthalpic penalty predicted for the elimination of this water, a notion in agreement with several lead optimization studies available for BRDs (Figure 5 and Supporting Information, Figure S1). Likewise, efficient replacement of the stable water at the inner side of the ZA channel seemed unlike due to the pronounced high free energy penalty estimated by SZmap (−12.55 kcal/mol) and WaterMap (1.97 kcal/mol) for this particular water. Moreover, any possible modifications targeting the nitrogens of the pyrazole ring were avoided, as this part of the scaffold constitutes an essential element of the pharmacophore that anchors 9 to the protein, as shown in Figures 3B–D. Combined with the insufficient convergence between the two hydration mapping methodologies concerning the region corresponding to the binding site entrance, the unstable waters of the ZA channel were finally selected to be targeted. A conservative, proof-of-concept strategy was devised wherein the 1-methyl would be gradually extended to an alkyl chain of up to four carbon atoms (i.e., from 1-methyl to 1-n-butyl).
Indeed, in accordance with the predictions, the microenvironment at the outer boundary of the ZA channel toward where the introduced alkyl chain was anticipated to be directed and reside is highly hydrophobic and consists of residues I683, F684, L685, and P688. Moreover, the limited concavity and the overall shape of the region suggested that the unavoidable conformational entropy penalty expected by restriction of the alkyl chain upon binding could be kept minimal, thus outbalancing any unfavorable impact on ligand affinity. Yet, to check the appropriateness of the designed modifications, free energy perturbation-replica exchange solute tempering (FEP-REST) calculations were undertaken and the predicted changes in free energy of binding were determined. The ligand mutation module as implemented in program Desmond (D.E. Shaw Research) was used, and 5 ns simulations were performed for each transformation of the ligand either inside the protein environment or in pure water using an explicit solvent representation. The simulation results showed that the designed analogues demonstrated moderately higher predicted affinities compared to the original methyl analogue. Interestingly, the most promising modification was shown to be the introduction of the long butyl chain. While there is no accurate methodology based on FEP calculations for distinguishing between the enthalpic and entropic components of the predicted free energy release, visual inspection of the structures indicated that the displacement of the unstable waters by the ZA channel periphery by the extruding alkyl chain was realistic. Moreover, because of the limited nature of the designed structural modifications, FEP simulations were considered as reliable. Thus, the encouraging predictions of FEP prompted us to proceed with synthesis of the novel analogues.
Chemistry
Compound 9 was synthesized along with a series of new analogues as depicted in Scheme 1. Yoon et al. synthesized 4-acetyl-1,3-isochromandione from homophthalic acid and acetic anhydride.64 Ozcan et al. later achieved the synthesis of the nor-pyrazoloisocoumarin 13 (Scheme 1) from homophthalic anhydride using a one-pot reaction consisting of Vilsmeier/1,3-cyclization.65 Taking advantage of the reactive methylene group in benzylic position (C4) of homophthalic anhydride, a combined strategy of the two above-mentioned literature data was developed for the generation of a first set of analogues. Introduction of carbonyl moieties with different carbon chains lengths in the C4-position of homophthalic acid is rendered possible by nucleophilic substitution in alkaline media. The reaction of homophthalic acid with thionyl chloride afforded homophthalic anhydride in excellent yields, in accordance with literature data. C4-deprotonation of homophthalic anhydride in mild conditions using anhydrous pyridine and followed by nucleophilic substitution of acetic, propionic, butyric, and valeric anhydride afforded the corresponding intermediates 9a–12a in low to moderate yields despite numbers of efforts to optimize the conditions (time, base, temperature), slightly improved only by the addition of anhydrous THF as solvent in the reaction media. The 4-acyl-isochroman-1,3-diones 9a–12a were subjected to pyrazole formation in the presence of hydrazine monohydrate in refluxing DMF. The corresponding isochromeno[3,4-c]pyrazol-5(2H)-ones 9–12 were obtained in low to moderate yields.
Scheme 1. Synthesis of Isochromeno[3,4-c]pyrazol-5(2H)-one Derivatives.

Structural Basis of Affinity Improvement and Selectivity Assessment of New Analogues
The affinity of the novel analogues for PB1(5) was determined by ITC, which demonstrated that the synthesized derivatives were indeed stronger binders than the original methyl hit (Figure 6B). The Kd values of 10 (ethyl), 11 (propyl), and 12 (butyl) derivatives were 3.4, 3.3, and 5.1 μM, respectively thus affording a 3.5-fold affinity improvement (Table 1). Moreover, the conservative structural modifications had a minor effect on LE, as the corresponding index was 0.48 for 10 and 0.45 for 11, while 12 afforded a slightly lower LE of 0.41. Notably, good agreement was obtained between ΔGbinding determined by ITC and the corresponding values predicted by FEP-REST calculations for the three novel analogues with errors within 0.6 kcal/mol (Table 1). To address the structural basis of the improved affinity, the crystallographic determination of the complexes of 10, 11, and 12 with PB1(5) was undertaken (Supporting Information, Table S4). As expected, the overall binding mode of the analogues carrying the extended alkyl chain on position 1 was highly similar to the corresponding mode of the original methyl hit, thus sustaining the basic protein–ligand interactions such as the hydrogen bond of the pyrazole to N739 and the overall hydrophobic packing of the ligand aromatic system inside the BRD cavity (Figure 7A,B). The alkyl chain of the novel analogues was positioned toward the periphery of the ZA channel where it resided, most notably the butyl moiety, in fully extended conformation stabilized by extensive contacts with P688 and the side chains of I683, L693, and I745. These crystallographic data were in good agreement with the computational hydration analysis, in terms of the anticipated effect on binding affinity of solvent reorganization and increase of buried nonpolar surface area of the ligand. Indeed, superposition of the alkyl chain binding geometries with the hydration sites predicted by WaterMap and SZmap showed that the alkyl groups overlapped with water molecules characterized by high free energy and thus predicted as unstable (Figure 5). Because of the lack of specific stabilizing interactions between those molecules and the BRD, their anticipated release to bulk solvent would offer a fair gain in binding affinity without serious perturbations on the overall enthalpy/entropy compensation landscape, as the ligand groups would substitute the solvent by forming comparable contacts with the protein. Furthermore, a binding geometry comparison between 12, 15, and 2 additionally showed that the butyl chain of 12 matches the interactions of the aromatic decorations of both 2 and the flavonoid 15 (Figure 7C,D). It is thus possible that ring B of flavonoids contributes to binding affinity by displacing unstable waters in a similar manner to that of the butyl chain of 12. In addition, another common SAR element between the new analogues and 15 was the presence of a pair of H-bond acceptors anchoring each ligand to N739 and water W5, respectively (Figure 7C,D).
Figure 6.
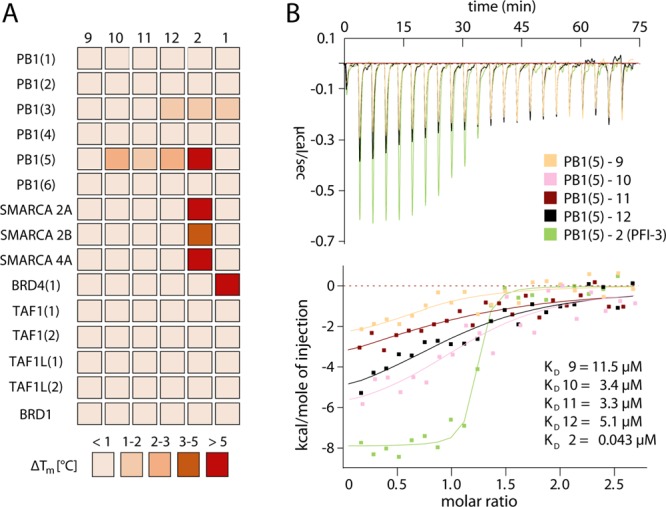
Potency and selectivity of synthesized analogues to compound 9. (A) Temperature shift assays of compounds 9–12 on subfamily VIII BRDs as well as selected other bromodomains. The inhibitors 1 and 2 were added for comparison. The heat map is color coded as described in the figure. (B) Isothermal titration calorimetry data for compounds 9–12 as well as 2. Shown are raw binding heats after baseline subtraction as well as normalized binding heats (lower panel). The solid lines represent nonlinear least-squares fits to the experimental data, and experimental binding heats are color coded as indicated in the figure. The Kd values determined for each experiments are also shown. All fitted and calculated thermodynamic data are shown in Supporting Information, Table S6.
Figure 7.

Binding mode comparison. (A) Binding mode of compound 10 in PB1(5). (B) Superimposition of the binding modes of inhibitors 10–12 showing a high degree of similarity. The remaining conserved water molecule (W5) is highlighted. Hydrogen bonds are indicated by dotted lines. The surface of the acetyl-lysine binding pocket is shown as a transparent sphere. (C) Comparison of binding modes of 12 and 14. (D) Comparison of binding modes of 12 and the PB1/SMARCA inhibitor 2.
Notably, systematic thermodynamic and structural studies of similar systems have shown that in many cases, increasing the length of an alkyl chain leads to gain in enthalpy that is, hence, fully offset by an unfavorable entropy change, thus affording a negative net effect on binding affinity.66 In this study, we demonstrated the use of designing constructive interactions with the water environment of a protein/ligand system on the basis of utilizing available solvation mapping algorithms. In a theoretical druggability assessment of BRDs based on the available X-ray crystallographic data, the degree by which different members of the family constitute tractable drug targets was determined. In this partition, BRDs of PB1 were clustered along with 57% of total studied BRDs that were classified as of either intermediate (PB1 BD2 and 5) or difficult (PB1 BD1, 3, 4, and 6) druggability. However, in that study, structural water molecules of the included BRDs were left intact and thus druggability determination was based on the assumption that those waters are not displaceable. The results presented herein show that such an approach could be challenged by the notion that the net contribution of solvent thermodynamics in binding cannot always be straightforwardly generalized across a whole family of proteins solely on the basis of structural similarity of related proteins.
To address the issue of selectivity and check whether the applied structural modifications and the observed affinity improvement had any unfavorable impact to the specificity profile of the scaffold, the affinity of 12 was determined toward a panel of 15 BRDs (Figure 6A and Supporting Information, Table S5) extended with a representative group of subfamily VIII members. Compound 12 showed selectivity toward PB1(5) over the closely related BRDs, with low binding (Tm not exceeding 2 °C) observed only toward PB1(3) (Supporting Information, Table S5). Notably, no binding was observed toward either BRD4 or, most importantly, the closely related SMARCA2, thus highlighting the possibility of designing a high affinity chemical probe targeting exclusively PB1(5). To further confirm these data, ITC was used, confirming that 12 did not interact with SMARCA BRDs (Supporting Information, Table S6).
Cell-Based Assays
To test the ability of 12 to bind to the bromodomain of PB1 and displace binding of PB1 from chromatin, a fluorescence recovery after photobleaching (FRAP) assay was performed. Chromatin was hyperacetylated using the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) to increase the assay window.67 Cells were transfected with a plasmid encoding GFP fused to either full length PB1 or to mutant PB1(2) and PB1(5), showing reduced ability to bind to chromatin. Cells were bleached with a laser and recovery time measured with or without compound treatment. Despite the fairly low potency in vitro 12 was able to displace PB1 from chromatin to levels of the control 2 and the mutant protein and showed similar half recovery times of ∼2 s (Figure 8).28 The Kd of 2 is 48 nM for PB1(5) measured by ITC as compared to 5.1 μM for 12. This is therefore in line with the effects observed in FRAP, where an effect on PB1(5) is seen for 2 at 1 μM and for 12 at 20 μM. FRAP experiments suggest that, at least in part, the complex can be displaced. Yet, as this experiment has been performed using overexpressed proteins, it cannot be excluded that the endogenous SWI/SNF complex remains bound to chromatin in the presence of PB1(5) inhibitors.
Figure 8.

Compound 12 reduces PB1 association with chromatin in cells. (A) FRAP half recovery times of GFP-PB1 are significantly decreased when treated with compound 12 at 20 μM as indicated. Cells expressing mutants of the bromodomain PB1(2) (N263A) or PB1(5) (N754Y) reducing the binding to chromatin were analyzed as comparison as was the compound 2. Significant differences to cells treated with SAHA of p < 0.05 are shown by *. (B) Time dependence of fluorescence recovery in the bleached area of cells expressing wt or mutant GFP-PB1 with the corresponding treatment as in (A).
Analogue 12 was additionally subjected to a viability assay in 1BR-hTERT human fibroblast cells. The drug very slightly reduced viability of the cells above 5 μM (Supporting Information, Figure S2A), while 2 also reduced viability to a similar level (Supporting Information, Figure S2B). It was next determined whether 12 reduces the ability of BAF180 to bind to chromatin using chromatin fractionation and Western blotting. Consistent with the FRAP results, treatment of 1BR-hTERT cells with 12 slightly reduced the levels of chromatin-bound PB1 (Supporting Information, Figure S2C). Loss of PB1 sensitizes cells to DNA damaging agents including mitomycin C (MMC).45 While U2OS PB1 knockout cells were more sensitive to MMC than control cells, 12 did not result in any significant sensitization to this drug and this was true for experiments using 2 as well (Supporting Information, Figure S2D). This suggests that the remaining level of chromatin-bound PB1 is sufficient to perform the function of PB1 in MMC-induced DNA repair. Finally, the effect of 12 on cell proliferation was studied using WST-1 and the multiple myeloma (MM) lines L363, H929, and JJN3. In agreement with earlier reports, exposure of cells to 12 did not lead to significant inhibition of proliferation on JJN3 cells.
Conclusion
Precise estimation of the energetics accompanying solvent dynamics and reorganization upon ligand binding is an aspect of structure-based drug design that demands attention, as it concerns one of the less comprehensible events addressed by currently available theoretical approaches in response to the increasing demand for high-quality free energy predictions. Especially with BRDs, the available structural data seem to converge toward identifying an ensemble of four to five buried water molecules that are considered as an almost integral part of the Kac binding cavity. However, whether those waters should (or could) be effectively displaced upon compound binding, aiming at an affinity gain in a selective fashion over the various BRD members is still debatable. Discovery of the low micromolar pyrazoloisocoumarin ligand of PB1(5) through a consensus virtual screening approach provided a valuable starting point for probing the importance of solvent effects in BRD hit optimization. Although relatively low (1 molecule active toward PB1(5) out of 40 top-ranked compounds assayed in vitro), the hit rate achieved in this virtual screen was considered satisfactory, given the serendipitous fact that the discovered hit demonstrated a highly original binding mode involving displacement of waters in a manner not seen in most BRD–ligand binding events including the interaction of the template JQ1 with its target BRD4. Rational elaboration of the scaffold was pursued by combining experimental results obtained by ITC and X-ray crystallography with theoretical data derived by predictive algorithms modeling hydration of the target protein. The effort was not focused on structural waters occupying the BRD Kac cavity but rather on solvent trapped on the macromolecule surface. By doing so, it successfully indicated a means to affinity gain by chemical modifications targeting secondary contributors as an indirect and alternative route to binding affinity optimization.
Naïvely, it would seem that growing an alkyl chain into solvent where it cannot sustain considerable hydrophobic interactions with the protein, would not contribute favorably to ligand binding potency in any system.66 However, in this study, it was shown that by considering the impact of solvent dynamics in binding, one can achieve optimization of a hit through alternative and possibly more efficient routes than those solely based on inspection of protein–ligand interactions. To facilitate this, one should go beyond utilization of structure-based techniques like protein X-ray crystallography or docking and also look to solvent mapping or other methods which take into consideration solvent dynamics. The combination of different approaches demonstrating considerable orthogonality with each other but with the potential to be reasonably integrated affords a highly reliable and constructive representation of complex systems such as those of protein/ligand/solvent regularly studied in drug discovery. More specifically, by using innovative computational tools for solvation mapping such as SZmap and Watermap, independently and in a consensus fashion, one can distinguish between stable (structural) and unstable (trapped) waters and thus devise modifications targeting each category appropriately. This work shows that by releasing unstable waters into bulk solvent, one may achieve a favorable, although small, improvement in Kd, in contrast to the expected view that growing an alkyl chain toward water would result to derivatives of lower affinity.
The advances in the field of epigenetics, and more specifically the discovery of compounds interfering with BRDs in a medically relevant manner, underline the necessity for focused screening efforts targeting proteins of the underexplored BRD subfamilies and thus enabling elucidation of their functionalities and involvement in different pathological states. On the other hand, rational approaches for hit identification or lead optimization can substantially facilitate the discovery process. The integral approach presented in this study can enable the most efficient exploitation of available structural, biophysical, and biochemical data toward rational development of hit compounds into promising leads for probe or drug discovery. The characterization of cell-permeable pyrazoloisocoumarin derivatives showing single-digit micromolar affinity toward the fifth BRD and high selectivity over other subfamily members such as SMARCA is anticipated to facilitate development of a chemical probe for exploring the biological role of this protein interaction module in the epigenetic mechanisms mediated by SWI/SNF complexes.
Experimental Section
Library Preparation and Virtual Screening
Prior to calculations, the NCI/DTP Repository compounds were enumerated and prepared in terms of correct protonation states, tautomerism, and stereoisomerism using the LigPrep routine (Schrödinger Inc.). The NCI/DTP collection was prepared independently for ROCS screening using the enumeration and conformer generation tools provided by OpenEye Inc. (Filter, Quacpac, Omega) with default settings. The ligand-based approach was based on the ROCS algorithm (OpenEye Inc.) with the Tanimoto-combo scoring scheme and the implicit Mills–Dean force field, while 1 was utilized as the query molecule. With respect to the structure-based approach, rigid docking was performed using the Glide v. 5.7 SP sampling algorithm and the corresponding GScore SP5 scoring scheme (Schrödinger Inc.) with the van der Waals atom radii scaling set to 0.9 for the protein grid and to 0.8 for the screened ligands. Protein preparation was performed by the corresponding routine as implemented in Schrödinger 2014 suite. The three screening methodologies were combined by a simple consensus statistical scheme on the basis of frequencies of appearance for each compound at the top of a given percentage threshold in each of the computationally ranked lists using Microsoft Excel 2010. For FEP-REST calculations, the ligand mutation module as implemented in Desmond software (D.E. Shaw Research) was used with default settings and 5 ns simulations were performed for each transformation of the ligand either inside the protein environment or in pure water using an explicit solvent representation.
Synthesis and Characterization of the Compounds
All chemicals were purchased from Aldrich Chemical Co. Microwave-assisted reactions were performed in a single mode CEM apparatus. NMR spectra were recorded on Bruker DRX 400 and Bruker Avance 600 spectrometers (1H 400 and 600 MHz); chemical shifts are expressed in ppm downfield from TMS. HRMS spectra were determined on a MSQ Orbitrap Thermofinnigan spectrometer. Columns chromatography were conducted using flash silica gel 60 (40–63 μm) from Merck. Purity of the compounds has been determined by HPLC and was above 95%.
HPLC Conditions
HPLC chromatograms have been recorded on a Thermo Finnigan chain (pump P4000, controller SN4000, autosampler AS3000, UV detector UV6000LP) equipped with a Supelco Discovery HS C18 5 μm column (25 cm × 4.6 mm). All the solvents were degassed in an ultrasound bath for 15 min and then filtered (nylon membrane, 0.45 μm) prior to use. Samples were diluted in HPLC grade methanol (1 mg/mL) and filtered (nylon membrane, 0.45 μm). Retention times (tR) are given in minutes. Two methods were used. Method A: H2O + 0.1% CH3COOH/MeOH, 15/85 for 45 min. Method B: H2O/CH3CN, 10/90 for 30 min.
Synthesis of Homophthalic Anhydride
Homophthalic acid (3g, 0.01665 mol) was dissolved in anhydrous CH2Cl2. Thionyl chloride was then added (4.83 mL, 0.067 mol) dropwise. The mixture was then warmed at reflux for 12 h. After completion of the reaction, solvent was evaporated and the crude residue dried under high vacuum to remove remaining traces of thionyl chloride. Homophthalic was obtained with 98% yield and used without purification. 1H NMR (400 MHz, CDCl3, δ in ppm, J in Hz): 8.25 (d, J = 7.56, 1H, H-1), 7.37 (d, J = 7.6, 1H, H-4), 7.54 (t, J = 7.56, 1H, H-2), 7.72 (t, J = 7.56, 1H, H-3), 4.17 (s, 2H, H-5).
General Procedure for the Synthesis of 4-Acyl-1,3-isochromandione 9a–12a
Homophthalic anhydride (1 equiv) was dissolved in anhydrous pyridine (2 equiv). After 15 min, the corresponding anhydride (4 equiv) was added dropwise. The mixture was then stirred for 5 h at room temperature. Et2O was then added and the precipitate filtered and washed with an extra amount of Et2O to afford the desired 4-acyl-1,3-isochromandione as a white solid. The product was then used for the next step without further purification.
General Procedure for the Synthesis of Isochromeno[3,4-c]pyrazol-5(2H)-one 9–12
The desired 4-acyl-1,3-isochromandione 9a–12a (1 equiv) was dissolved in anhydrous DMF and stirred for 15 min. Hydrazine monohydrate was then added dropwise (4.2 equiv) and the mixture warmed to reflux for 12 h. After completion of the reaction, the mixture was cooled and water was added. The precipitate is then filtered, washed with water, and dried, affording the corresponding isochromeno[3,4-c]pyrazol-5(2H)-one 9–12.
Data for 1-Methylisochromeno[3,4-c]pyrazol-5(2H)-one 9
Yield: 40%. 1H NMR (600 MHz, DMSO, δ in ppm, J in Hz): 12.39 (s, 1H, H-2), 8.20 (d, J = 8.00, 1H, H-6), 7.85 (m, 2H, H-8 and H-9), 7.49 (t, J = 8.00, 1H, H-7), 2.63 (s, 3H, 1′-CH3). 13C NMR (150 MHz, DMSO, δ in ppm): 161.39 (C-5), 156.97 (C-3a), 135.78 (C-5a), 135.47 (C-8), 133.58 (C-9a), 130.94 (C-6), 126.12 (C-7), 122.01 (C-9), 118.07 (C-1), 96.55 (C-9b), 11.68 (1′-CH3). HRMS (ESI+) m/z = 201.0659 ([M + H]+) (calculated mass: 200.19). HPLC: method A tR = 3.17 min; method B tR = 2.83 min.
Data for 1-Ethylisochromeno[3,4-c]pyrazol-5(2H)-one 10
Yield: 30%. 1H NMR (600 MHz, DMSO, δ in ppm, J in Hz): 12.95 (s, 1H, H-2), 8.21 (d, J = 7.9, 1H, H-6), 7.87 (t, J = 7.5, 1H, H-8), 7.82 (d, J = 7.5, 1H, H-9), 7.49 (t, J = 7.90, 1H, H-7), 3.04 (q, J = 7.55, 2H, H-2′), 1.32 (t, J = 7.55, 3H, 1′-CH3). 13C NMR (150 MHz, DMSO, δ in ppm) 161.35 (C-5), 156.94 (C-3a), 141.43 (C-5a), 135.59 (C-8), 133.42 (C-9a), 131.02 (C-6), 126.13 (C-7), 122.10 (C-9), 118.16 (C-1), 95.71 (C-9b), 19.24 (C-1′), 12.61 (C-2′). HRMS (ESI-) m/z = 213.0672 ([M – H]+) (calculated mass: 214.22). HPLC: method A tR = 3.40 min; method B tR = 2.91 min.
Data for 1-Propylisochromeno[3,4-c]pyrazol-5(2H)-one 11
1H NMR (600 MHz, DMSO, δ in ppm, J in Hz): 12.93 (s, 1H, H-2), 8.17 (d, J = 7.91 Hz, 1H, H-6), 7.81 (m, 2H, H-8 and H-9), 7.45 (t, J = 7.91 Hz, 1H, H-7), 2.96 (t, J = 7.41 Hz, 2H, H-1′), 1.7 (h, J = 7.31 Hz, 2H, H-2′), 0.95 (t, J = 7.31 Hz, 3H, H-3′). 13C NMR (150 MHz, DMSO, δ in ppm) 161.34 (C-5), 156.89 (C-3a), 140.04 (C-5a), 135.59 (C-8), 133.44 (C-9a), 131.03 (C-6), 126.15 (C-7), 122.09 (C-9), 118.20 (C-1), 96.01 (C-9b), 27.47 (C-1′), 21.30 (C-2′), 13.43 (C-3′). HRMS (ESI−) m/z = 227.1287 [M – H]+ (calculated mass: 228.25). HPLC: method A tR = 3.72 min; method B tR = 3.01 min.
Data for 1-Butylisochromeno[3,4-c]pyrazol-5(2H)-one 12
1H NMR (600 MHz DMSO, δ in ppm, J in Hz) 12.95 (s, 1H, H-2), 8.21 (d, J = 7.99 Hz, 1H, H-6), 7.87 (t, J = 7.5 Hz, 1H, H-8), 7.81 (d, J = 7.5 Hz, 1H, H-9), 7.48 (t, J = 7.99 Hz, 1H, H-7), 3.01 (t, J = 7.50 Hz, 2H, H-1′), 1.69 (quintuplet, J = 7.52 Hz, 2H, H-2′) 1.40 (h, J = 7.52 Hz, 2H, H-3′), 0.94 (t, J = 7.39 Hz, 3H, H-4′). 13C NMR (150 MHz, DMSO, δ in ppm) 161.33 (C-5), 156.90 (C-3a), 140.21 (C-5a), 135.59 (C-8), 133.44 (C-9a), 131.04 (C-6), 126.13 (C-7), 122.05 (C-9), 118.20 (C-1), 95.94 (C-9b), 30.01 (C-1′), 25.35 (C-2′), 21.65 (C-3′), 13.59 (C-4′). HRMS (ESI−) m/z = 241.0976 (M – H). HPLC: tR = [M – H]+ (calculated mass: 242.28). HPLC: method A tR = 4.29 min; method B tR = 3.21 min.
The activity of the compounds was validated by X-ray crystallography of all protein–compound complexes and by various different binding affinity measuring methodologies such as ITC and DSF as well as cell-based assays. All active compounds are therefore considered as not PAINS.
Protein Expression and Purification
cDNA encoding human the fifth BRD of PB1 (NCBI Gene ID: 55193) was obtained from the SGC clone collection and was used as template to amplify the kinase domain region of the protein, using the polymerase chain reaction (PCR) in the presence of Platinum Pfx DNA polymerase (Invitrogen, UK). PCR products were purified (QIAquick PCR Purification Kit, Qiagen Ltd. UK) and further subcloned into a pMCSG7 derived expression vector (pNIC28-Bsa4), using ligation independent cloning. This vector includes sites for ligation-independent cloning and a Tobacco Etch Virus (TEV)-cleavable N-terminal His6-tag (extension MHHHHHHSSGVDLGTENLYFQ*SM−). After digestion with TEV protease, the protein retains an additional serine and methionine on the N-terminus. The constructs were transformed into competent Mach1 cells (Invitrogen, UK) to yield the final plasmid DNA.
Colonies from freshly transformed plasmid DNA in competent Escherichia coli BL21(DE3)-R3-pRARE2 cells (phage-resistant derivative of BL21(DE3) cell (Invitrogen), with a pRARE plasmid encoding rare codon tRNAs) were grown overnight at 37 °C in 30 mL of 2× Luria–Bertani medium (LB-broth, Amresco) with 50 μg/mL kanamycin and 34 μg/mL chloramphenicol (startup culture). The startup culture was diluted 1:100 in terrific broth (TB, Merck), and cell growth was allowed at 37 °C to an optical density of about 1.5 (OD600) before the temperature was decreased to 18 °C. When the system equilibrated at 18 °C, the optical density was about 3.0 (OD600) and protein expression was induced overnight at 18 °C with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The bacterial cells were harvested by centrifugation (8700g for 15 min at 4 °C, FIBERLITE F9-6x1000 LEX rotor, on a Thermo Scientific SORVALL LYNX 6000 centrifuge) and were frozen at −20 °C as pellets for storage. Cells expressing His6 tagged protein were resuspended in lysis buffer (50 mM HEPES, pH 7.5 at 25 °C, 500 mM NaCl, 20 mM imidazole, 5% glycerol, 1 mM tris(2-carboxyethyl)phosphine (TCEP)) and lysed using a SONICS Vibra cell sonicator on ice. The lysate was cleared by centrifugation (23000 rpm for 30 min at 4 °C, T29-8x50 rotor, on a Thermo Scientific SORVALL LYNX 6000 centrifuge) and was applied to a cobalt-nitrilothiacetic acid agarose column (IMAC Sepharose 6 Fast Flow, GE Healthcare charged with Co2+, 5 mL, equilibrated with 20 mL of lysis buffer). The column was washed with 30 mL of lysis buffer at gravity flow, and the protein was eluted using a step elution of imidazole in lysis buffer (50, 100, 2 × 300 mM imidazole in 50 mM HEPES, pH 7.5 at 25 °C, 500 mM NaCl, 1 mM TCEP). All fractions were collected and monitored by SDS-polyacrylamide gel electrophoresis (Bio-Rad Criterion Precast Gels, 10–20% Tris-HCl 1.0 mm, from Bio-Rad, CA; gel run conditions, 200 V, 20–30 mA, 50 min in SDS buffer). The eluted protein was further purified with size exclusion chromatography on a Superdex 75/200 16/60 HiLoad gel filtration column (GE/Amersham Biosciences) on an ÄktaPrime plus system (GE/Amersham Biosciences). Samples were monitored by SDS-polyacrylamide gel electrophoresis and concentrated to ∼10 mg/mL in the gel-filtration buffer, 10 mM Hepes pH 7.5, 150 mM NaCl, 5% glycerol, 0.5 mM TCEP using a 10 kDa cutoff concentrator and were used for DSF assay and isothermal titration calorimetry. Protein handling was carried out on ice or in a cold room in all the above steps.
Protein Crystallization
Aliquots of the purified PB1 domain 5 (Uniprot identifier as PB1_HUMAN Q86U86-1 fragment 645–766) were set up for crystallization using a mosquito crystallization robot (TTP Labtech, Royston UK). Coarse screens were typically setup onto Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (200 + 100 nL, 150 + 150 nL, and 100 + 200 nL). Initial hits were optimized further scaling up the drop sizes. All crystallizations were carried out using the sitting drop vapor diffusion method at 277.15 K. Crystals with compound 9 were grown by mixing 150 nL of the protein (12.65 mg/mL and 5 mM final ligand concentration) with an equal volume of reservoir solution containing 18% PEG3350, 0.15 M sodium malonate pH 7.0, 10% ethylene glycol, and 0.1 M bis-tris-propane pH 8.1. Diffraction quality crystals grew within a few days. Crystals in complex with the compound 10 (5 mM final concentration) were obtained by mixing 150 nL of the protein (6.5 mg/mL) and 150 nL crystallization buffer (0.1 M trisodium citrate dihydrate pH 5.6, 20% 2-propanol, 20% PEG 4K); PB1 bromodomain 5 crystals in complex with the compound 11 (5 mM final concentration) were grown by mixing 200 nL of the protein (6.45 mg/mL) and 100 nL of crystallization buffer (0.2 M ammonium sulfate, 0.1 M MES pH 6.5, 30% PEGMME 5K); PB1 bromodomain 5 crystals in complex of the compound 12 (5 mM final concentration) were grown by mixing 200 nL of the protein (6.45 mg/mL) and 100 nL of crystallization buffer (0.1 M lithium sulfate monohydrate, 0.1 M Tris pH 8.5, 30% PEG4K). Complex crystals were cryoprotected using the well solution supplemented with additional 20% ethylene glycol and was flash frozen in liquid nitrogen.
Data Collection and Structure Solution
Data for the PB1(5) complex with 9 were collected in-house on a Rigaku FRE rotating anode system equipped with a RAXIS-IV detector at 1.52 Å. All other diffraction data were collected at Diamond Light Source 03 and I04-1 beamlines at a single wavelength of 0.9763 and 0.9207 Å, respectively. Data for the PB1(5) complex with 9 were indexed and integrated using MOSFLM, and scaling was performed with SCALA.68,69 Other data sets were indexed and integrated using XDS,70 and scaling was performed with AIMLESS.71 Initial phases were calculated by molecular replacement with PHASER using the apo template structure 3G0J.pdb.72 Unique and initial solutions were improved in a total of 50 cycles of automated protein chain tracing starting from existing model and computed using ARP/wARP,73 further manual building with COOT, and refinement against maximum likelihood target using REFMAC5.74,75 Thermal motions were analyzed using TLSMD, and hydrogen atoms were included in late refinement cycles.76 All model validations were carried out using MolProbity.77 Data collection and refinement statistics are compiled in Supporting Information, Table S2. The models and structure factors have been deposited with PDB accession codes: 5II1 (compound 9), 5HRV (compound 10), 5HRW (compound 11), 5HRX (compound 12), 5IID (compound 14), and 5II2 (compound 15).
DSF Assay and Isothermal Titration Calorimetry
The proteins at 2.5 μM (20 μM in preliminary high-throughput screening mode) were mixed with ligands at 100 μM (200 μM in preliminary high-throughput screening mode), and the assays and data evaluation for melting temperatures were performed using a RT-PCR Mx3005p machine (Stratagene) as previously described.58 All calorimetric titration experiments were carried out in a VP-ITC calorimeter at 15 °C. The buffer condition used was 20 mM HEPES pH 7.5, 150 mM NaCl, and 0.5 mM TCEP. Titration was performed by injecting the proteins (200 μM) into a reaction cell containing the inhibitors (15 μM). Data analysis was performed with TA NanoAnalyze. Corrected data were fitted to a single binding site model using a nonlinear least-squares minimization algorithm and binding parameters including reaction enthalpy changes (ΔH), reaction entropy changes (ΔS), equilibrium dissociation constants (Kd), and stoichiometry were calculated. In all experiments, the N-value was almost equal to 1.
Fluorescence Recovery After Photobleaching (FRAP)
Full length cDNA for the isoform 8 of human polybromo-1 protein (PB1, Q86U86-8) was amplified by PCR from an IMAGE clone (IMAGE: 40082629) and cloned into pDONR-221 vector using Gateway BP reaction producing Gateway entry clones. Mutations which impair binding to a second (N263A) and fifth (N754Y) BRDs were introduced into full length PB1 Gateway entry clones using 15 cycles QuikChange II PCR protocol (Agilent Technologies). Mammalian expression constructs encoding N-terminal EGFP tag were constructed by Gateway LR recombination reaction between Vivid Colors pcDNA6.2/N-EmGFP-DEST (Invitrogen, catalogue no. V356-20) and a wild-type or mutated PB1 Gateway entry clone.
FRAP studies were performed essentially as described.62 In brief, U2OS cells were transfected (Fugene HD; Roche) with mammalian overexpression constructs encoding GFP fused to the N-terminus of full length wild-type or mutant PB1. The imaging system consisted of a Zeiss LSM 710 laser-scanning and control system (Zeiss) coupled to an inverted Zeiss Axio Observer Z1 microscope equipped with a high numerical aperture (N. A. 1.3) 40× oil immersion objective (Zeiss). Samples were placed in an incubator chamber in order to maintaining temperature and humidity. FRAP and GFP fluorescence imaging were both carried out with an argon-ion laser (488 nm) and with a PMT detector set to detect fluorescence between 500 and 550 nm. Once an initial scan had been taken, a region of interest corresponding to approximately 50% of the entire GFP positive nucleus was empirically selected for bleaching. A time lapse series was then taken to record GFP recovery using 1% of the power used for bleaching. The image data sets and fluorescence recovery data were exported from ZEN 2009, the microscope control software, into Origin to determine the average half-time for full recovery for 10–20 cells per treatment point. Data were analyzed using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test.
Cell Culture
1BR-hTERT and U2OS cells were cultured at 37 °C in a 5% CO2 incubator in DMEM (Gibco) supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine.
Viability Assays
First, 5000 1BR-hTERT cells were seeded into 96-well plates in triplicate format. Then after 24 h, cells were incubated with DMSO or 10 μM 12 with or without mitomycin C. Viability was analyzed 4 days following drug treatment using CellTiter-Glo Reagent (Promega).
Chromatin Fractionation
First, 6 ×106 1BR-hTERT cells were plated into 10 cm dishes and grown for 24 h. DMSO or 10 μM 12 was then added for 48 h, and the cells were harvested, washed in PBS, and pelleted. Cell pellets were resuspended in low salt lysis buffer (50 mM Tris pH 8.0, 2 mM EDTA, 2 mM EGTA, 150 mM NaCl, 0.2% Triton-X100, 0.3% NP40) and incubated on ice for 5 min. Cell lysates were centrifuged at 3000 rpm for 4 min at 4 °C and the supernatant removed as the soluble fraction. The chromatin pellet was washed three times in ice-cold PBS before incubation with Benzonase (Sigma) in nuclease buffer (50 mM Tris pH 8.0, 20 mM NaCl, 2 mM MgCl2) for 30 min on ice. An equal volume of twice the high salt/Triton buffer (1.86 M NaCl, 0.4% Triton-X100) was added before sonication in a water bath and centrifuged at 14000 rpm for 5 min at 4 °C. The supernatant was retained as the chromatin fraction. Then 50 μg of protein from the soluble or chromatin fractions were run on polyacrylamide gels using SDS-PAGE before Western blotting using antibodies against BAF180 (Bethyl, A301-591A), α-tubulin (Abcam, ab7291), and CENPA (Abcam, ab13939).
Cell Proliferation Assays
Cell proliferation of MM lines was measured by the colorimetric WST-1 assay (Clontech, Mountain View, USA) according to the manufacturer’s instructions. The method measures the metabolic activity of viable cells based on the enzymatic cleavage of the tetrazolium salt WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium) to formazan dye by cellular mitochondrial dehydrogenases, present in viable cells. As cells proliferate more WST-1 is converted to the formazan dye product, which can be quantified by measuring the absorbance at 450 nm in a multiwell DAS plate reader. A number of 5 × 104 cells (1 × 106 cells/mL) were seeded into each of 96-well microtiter plates (Corning). The plates were incubated in a humidified incubator in 5% CO2 at 37 °C for 24 h, and 2–3 h prior to analysis, 10 μL of WST-1 solution was added to each well.
Acknowledgments
This study was supported by EU-COST action TD0905 “Epigenetics: bench to bedside” and EU-FP7REGPOT-2011 project INsPiRE 284460 (V.M.). The screened compounds were provided free of charge from the National Cancer Institute (NCI)/Division of Cancer Treatment and Diagnosis (DCTD)/Developmental Therapeutics Program (DTP) repository (https://dtp.cancer.gov). S.K. is grateful for support by the SGC, a registered charity (no. 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, Merck, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust. M.W. is grateful for support by the Research Training Group Translational Research Innovation—Pharma (TRIP), supported by the Else Kröner-Fresenius Foundation (EKFS).
Glossary
Abbreviations Used
- BRD
bromodomain
- BET
bromodomain and extra terminal
- PB1
polybromo protein 1
- SWI/SNF
switch-sucrose nonfermentable
- NCI
National Cancer Institute
- FEP-REST
free energy perturbation-replica exchange solute tempering
- HATs
histone acetyltransferases
- DNMTs
DNA-methyltransferases
- HDAC
histone deacetylases
- SIRTs
sirtuins
- Kac
acetyl-lysine
- NMC
NUT midline carcinoma
- CREBBP
c-AMP response element-binding protein binding protein
- EP300
E1A binding protein p300
- BRPF
bromodomain and PHD finger containing
- BAZ2B/A
bromodomain adjacent to zinc finger containing domain 2A and 2B
- SMARCA2/4
SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily A, member 2 and 4
- PBAF
polybromo-associated BRG1 or hbrm-associated factor
- VS
virtual screening
- NCI/DTP
National Cancer Institute/Developmental Therapeutics Program
- BZDs
benzodiazepines
- FP
fingerprint
- DSF
differential scanning fluorimetry
- ITC
isothermal titration calorimetry
- 534tOHF
5,3′,4′-trihydroxyflavone
- FRAP
fluorescence recovery after photobleaching
- MM
multiple myeloma
- ANOVA
analysis of variance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00355.
X-ray crystallographic data collection and refinement statistics, fingerprint derivation combinations used for virtual screening, structures and details of compounds evaluated using DSF and ITC, and details on assays with fibroblast cells and chromatin fractionation (PDF)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates of the PB1(5) bound to compounds 7 (5II1), 8 (5HRV), 9 (5HRW), 10 (5HRX), 12 (5IID), and 13 (5II2) have been deposited with the PDB. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
Supplementary Material
References
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Covre A.; Coral S.; Di Giacomo A. M.; Taverna P.; Azab M.; Maio M. Epigenetics meets immune checkpoints. Semin. Oncol. 2015, 42, 506–513. 10.1053/j.seminoncol.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Szyf M. Prospects for the development of epigenetic drugs for CNS conditions. Nat. Rev. Drug Discovery 2015, 14, 461–474. 10.1038/nrd4580. [DOI] [PubMed] [Google Scholar]
- Hamm C. A.; Costa F. F. Epigenomes as therapeutic targets. Pharmacol. Ther. 2015, 151, 72–86. 10.1016/j.pharmthera.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Juo Y. Y.; Gong X. J.; Mishra A.; Cui X.; Baylin S. B.; Azad N. S.; Ahuja N. Epigenetic therapy for solid tumors: from bench science to clinical trials. Epigenomics 2015, 7, 215–235. 10.2217/epi.14.73. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S.; Zeng L.; Zhou M. M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J. S.; Mercan F.; Rivera K.; Pappin D. J.; Vakoc C. R. BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell 2015, 58, 1028–1039. 10.1016/j.molcel.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia P. L.; Miller A.L.; Kreitzburg K. M.; Council L. N.; Gamblin T. L.; Christein J. D.; Heslin M. J.; Arnoletti J. P.; Richardson J. H.; Chen D.; Hanna C. A.; Cramer S. L.; Yang E. S.; Qi J.; Bradner J. E.; Yoon K. J. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. 10.1038/onc.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Peng R.; Phillips J. E.; Deguzman J.; Ren Y.; Apparsundaram S.; Luo Q.; Bauer C. M.; Fuentes M. E.; DeMartino J. A.; Tyagi G.; Garrido R.; Hogaboam C. M.; Denton C. P.; Holmes A. M.; Kitson C.; Stevenson C. S.; Budd D. C. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am. J. Pathol. 2013, 183, 470–479. 10.1016/j.ajpath.2013.04.020. [DOI] [PubMed] [Google Scholar]
- Mazur P. K.; Herner A.; Mello S. S.; Wirth M.; Hausmann S.; Sánchez-Rivera F. J.; Lofgren S. M.; Kuschma T.; Hahn S. A.; Vangala D.; Trajkovic-Arsic M.; Gupta A.; Heid I.; Noël P. B.; Braren R.; Erkan M.; Kleeff J.; Sipos B.; Sayles L. C.; Heikenwalder M.; Heßmann E.; Ellenrieder V.; Esposito I.; Jacks T.; Bradner J. E.; Khatri P.; Sweet-Cordero E. A.; Attardi L. D.; Schmid R. M.; Schneider G.; Sage J.; Siveke J. T. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. 10.1038/nm.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Wang Z.; Zhang Z.; Ren X.; Lu X.; Ding K. Small-molecule BET inhibitors in clinical and preclinical development and their therapeutic potential. Curr. Top. Med. Chem. 2015, 15, 776–794. 10.2174/1568026615666150302110135. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Brown N.; Knapp S.; Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55, 7346–7359. 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammitzsch A.; Tallant C.; Fedorov O.; O’Mahony A.; Brennan P. E.; Hay D. A.; Martinez F. O.; Al-Mossawi M. H.; de Wit J.; Vecellio M.; Wells C.; Wordsworth P.; Müller S.; Knapp S.; Bowness P. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10768–10773. 10.1073/pnas.1501956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J.; Fedorov O.; Tallant C.; Monteiro O.; Meier J.; Gamble V.; Savitsky P.; Nunez-Alonso G. A.; Haendler B.; Rogers C.; Brennan P. E.; Müller S.; Knapp S. Discovery of a chemical tool inhibitor targeting the bromodomains of TRIM24 and BRPF. J. Med. Chem. 2016, 59, 1642–1647. 10.1021/acs.jmedchem.5b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P. G.; Vieira L. C.; Tallant C.; Fedorov O.; Singleton D. C.; Rogers C. M.; Monteiro O. P.; Bennett J. M.; Baronio R.; Müller S.; Daniels D. L.; Méndez J.; Knapp S.; Brennan P. E.; Dixon D. J. LP99: discovery and synthesis of the first selective BRD7/9 bromodomain inhibitor. Angew. Chem., Int. Ed. 2015, 54, 6217–6221. 10.1002/anie.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Bamborough P.; Bannister A. J.; Becher; Bit R. A.; Che K. H.; Chung C. W.; Dittmann A.; Drewes G.; Drewry D. H.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Michon A. M.; Molnar J.; Robson S. C.; Tomkinson N. C.; Kouzarides T.; Prinjha R. K.; Humphreys P. G. Discovery of I-BRD9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J. Med. Chem. 2016, 59, 1425–1439. 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin L.; McGrath S.; Vidler L. R.; Chaikuad A.; Monteiro O.; Tallant C.; Philpott M.; Rogers C.; Fedorov O.; Liu M.; Akhtar W.; Hayes A.; Raynaud F.; Müller S.; Knapp S.; Hoelder S. Structure enabled design of BAZ2-ICR, a chemical probe targeting the bromodomains of BAZ2A and BAZ2B. J. Med. Chem. 2015, 58, 2553–2559. 10.1021/jm501963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosmini R.; Nguyen V. L.; Toum J.; Simon C.; Brusq J. M.; Krysa G.; Mirguet O.; Riou-Eymard A. M.; Boursier E. V.; Trottet L.; Bamborough P.; Clark H.; Chung C. W.; Cutler L.; Demont E. H.; Kaur R.; Lewis A. J.; Schilling M. B.; Soden P. E.; Taylor S.; Walker A. L.; Walker M. D.; Prinjha R. K.; Nicodème E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem. 2014, 57, 8111–8131. 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- Hay D. A.; Fedorov O.; Martin S.; Singleton D. C.; Tallant C.; Wells C.; Picaud S.; Philpott M.; Monteiro O. P.; Rogers C. M.; Conway S. J.; Rooney T. P. C.; Tumber A.; Yapp C.; Filippakopoulos P.; Bunnage M. E.; Müller S.; Knapp S.; Schofield S. J.; Brennan P. E. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Chaikuad A.; Bamborough P.; Bantscheff M.; Bountra C.; Chung C.; Fedorov O.; Grandi P.; Jung D.; Lesniak R.; Lindon M.; Müller S.; Philpott M.; Prinjha R.; Rogers C.; Selenski C.; Tallant C.; Werner T.; Willson T. M.; Knapp S.; Drewry D. H. Discovery and characterization of GSK2801, a selective chemical probe for the bromodomains BAZ2A and BAZ2B. J. Med. Chem. 2016, 59, 1410–1424. 10.1021/acs.jmedchem.5b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah J. C.; Mujtaba S.; Karakikes I.; Zeng L.; Muller M.; Patel J.; Moshkina N.; Morohashi K.; Zhang W.; Gerona-Navarro G.; Hajjar R. J.; Zhou M. M. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 2011, 18, 531–541. 10.1016/j.chembiol.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Fedorov O.; Thanasopoulou A.; Leonards K.; Jones K.; Meier J.; Olzscha H.; Monteiro O.; Martin S.; Philpott M.; Tumber A.; Filippakopoulos P.; Yapp C.; Wells C.; Che K. H.; Bannister A.; Robson S.; Kumar U.; Parr N.; Lee K.; Lugo D.; Jeffrey P.; Taylor S.; Vecellio M. L.; Bountra C.; Brennan P. E.; O’Mahony A.; Velichko S.; Müller S.; Hay D.; Daniels D. L.; Urh M.; La Thangue N. B.; Kouzarides T.; Prinjha R.; Schwaller J.; Knapp S. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 2015, 75, 5106–5119. 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O.; Castex J.; Tallant C.; Owen D. R.; Martin S.; Aldeghi M.; Monteiro O.; Filippakopoulos P.; Picaud S.; Trzupek J. D.; Gerstenberger B. S.; Bountra C.; Willmann D.; Wells C.; Philpott M.; Rogers C.; Biggin P. C.; Brennan P. E.; Bunnage M. E.; Schüle R.; Günther T.; Knapp S.; Müller S. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 2015, 1 (10), e1500723. 10.1126/sciadv.1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Côté A.; Magnuson S. Disrupting acetyl-lysine recognition: Progress in the development of bromodomain inhibitors. J. Med. Chem. 2016, 59, 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Smith S. G.; Zhou M. M. Discovery of chemical inhibitors of human bromodomains. Chem. Rev. 2015, 115, 11625–11668. 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- Bunnage M. E.; Piatnitski Chekler E. L.; Jones L. H. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Brownlee P. M.; Chambers A. L.; Oliver A. W.; Downs J. A. Cancer and the bromodomains of BAF180. Biochem. Soc. Trans. 2012, 40, 364–369. 10.1042/BST20110754. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zhai W.; Richardson J. A.; Olson E. N.; Meneses J. J.; Firpo M. T.; Kang C.; Skarnes W. C.; Tjian R. Polybromo protein BAF180 functions in mammalian cardiac chamber maturation. Genes Dev. 2004, 18, 3106–3116. 10.1101/gad.1238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Gao X.; Diaz-Trelles R.; Ruiz-Lozano P.; Wang Z. Coronary development is regulated by ATP-dependent SWI/SNF chromatin remodeling component BAF180. Dev. Biol. 2008, 319, 258–266. 10.1016/j.ydbio.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Mehrotra A.; Joe B.; de la Serna I. L. SWI/SNF chromatin remodeling enzymes are associated with cardiac hypertrophy in a genetic rat model of hypertension. J. Cell. Physiol. 2013, 228, 2337–2342. 10.1002/jcp.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Q.; Xia Q. Y.; Wang Z. Y.; Li L.; Shen Q.; Shi S. S.; Wang X.; Liu B.; Wang Y. F.; Shi Q. L.; Ma H. H.; Lu Z. F.; He Y.; Zhang R. S.; Yu B.; Zhou X. J. Frequent co-inactivation of the SWI/SNF subunits SMARCB1, SMARCA2 and PBRM1 in malignant rhabdoid tumours. Histopathology 2015, 67, 121–129. 10.1111/his.12632. [DOI] [PubMed] [Google Scholar]
- Cristofaro M. F. D.; Betz B. L.; Rorie C. J.; Reisman D. N.; Wang W.; Weissman B. E. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell. Physiol. 2001, 186, 136–145. 10.1002/1097-4652(200101)186:1<136::AID-JCP1010>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Duns G.; Hofstra R. M.; Sietzema J. G.; Hollema H.; van Duivenbode I.; Kuik A.; Giezen C.; Jan O.; Bergsma J. J.; Bijnen H.; van der Vlies P.; van den Berg E.; Kok K. Targeted exome sequencing in clear cell renal cell carcinoma tumors suggests aberrant chromatin regulation as a crucial step in ccRCC development. Hum. Mutat. 2012, 33, 1059–1062. 10.1002/humu.22090. [DOI] [PubMed] [Google Scholar]
- Piva F.; Santoni M.; Matrana M. R.; Satti S.; Giulietti M.; Occhipinti G.; Massari F.; Cheng L.; Lopez-Beltran A.; Scarpelli M.; Principato G.; Cascinu S.; Montironi R. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert Rev. Mol. Diagn. 2015, 15, 1201–1210. 10.1586/14737159.2015.1068122. [DOI] [PubMed] [Google Scholar]
- Macher-Goeppinger S.; Keith M.; Tagscherer K. E.; Singer S.; Winkler J.; Hofmann T. G.; Pahernik S.; Duensing S.; Hohenfellner M.; Kopitz J.; Schirmacher P.; Roth W. PBRM1 (BAF180) protein is functionally regulated by p53-induced protein degradation in renal cell carcinomas. J. Pathol. 2015, 237, 460–471. 10.1002/path.4592. [DOI] [PubMed] [Google Scholar]
- Numata M.; Morinaga S.; Watanabe T.; Tamagawa H.; Yamamoto N.; Shiozawa M.; Nakamura Y.; Kameda Y.; Ohkawa S.; Rino Y.; Akaike M.; Masuda M.; Miyagi Y. The clinical significance of SWI/SNF complex in pancreatic cancer. Int. J. Oncol. 2013, 42, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W.; Nagase S.; Montia A. G.; Kalachikov S. M.; Keniry M.; Su T.; Memeo L.; Hibshoosh H.; Parsons R. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008, 68, 1667–1674. 10.1158/0008-5472.CAN-07-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine I.; Sato M.; Sunaga N.; Toyooka S.; Peyton M.; Parsons R.; Wang W.; Gazdar A. F.; Minna J. D. The 3p21 candidate tumor suppressor gene BAF180 is normally expressed in human lung cancer. Oncogene 2005, 24, 2735–2738. 10.1038/sj.onc.1207694. [DOI] [PubMed] [Google Scholar]
- Burrows A. E.; Smogorzewska A.; Elledge S. J. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 14280–14285. 10.1073/pnas.1009559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee P. M.; Chambers A. L.; Cloney R.; Bianchi A.; Downs J. A. BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep. 2014, 6, 973–981. 10.1016/j.celrep.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarougkas A.; Ismail A.; Chambers A. L.; Riballo E.; Herbert A. D.; Künzel J.; Löbrich M.; Jeggo P. A.; Downs J. A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. 10.1016/j.molcel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A.; Chambers A. L.; Downs J. A.; Lehmann A. R. A role for chromatin remodellers in replication of damaged DNA. Nucleic Acids Res. 2012, 40, 7393–7403. 10.1093/nar/gks453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangamudi B.; Paul T. A.; Shah P. K.; Kost-Alimova M.; Nottebaum L.; Shi X.; Zhan Y.; Leo E.; Mahadeshwar H. S.; Protopopov A.; Futreal A.; Tieu T. N.; Peoples M.; Heffernan T. P.; Marszalek J. R.; Toniatti C.; Petrocchi A.; Verhelle D.; Owen D. R.; Draetta G.; Jones P.; Palmer W. S.; Sharma S.; Andersen J. N. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: Insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 2015, 75, 3865–3878. 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2015-1: Canvas, Version 2.5; Schrödinger, LLC: New York, 2015.
- Duan J.; Dixon S. L.; Lowrie J. F.; Sherman W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graphics Modell. 2010, 29, 157–170. 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Sastry M.; Lowrie J. F.; Dixon S. L.; Sherman W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- Small-Molecule Drug Discovery Suite 2015-1: Glide, Version 6.6; Schrödinger, LLC: New York, 2015.
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shaw D. E.; Shelley M.; Perry J. K.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- ROCS 3.2.1.4; OpenEye Scientific Software: Santa Fe, NM, 2015; http://www.eyesopen.com.
- Hawkins P. C. D.; Skillman A. G.; Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute (NCI)/Division of Cancer Treatment and Diagnosis (DCTD)/Developmental Therapeutics Program (DTP), http://dtp.cancer.gov.
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the active-site solvent in the thermodynamics of Factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2015-1: WaterMap, Version 2.2; Schrödinger, LLC: New York, 2015.
- SZMAP 1.2.1.4; OpenEye Scientific Software: Santa Fe, NM, 2015; http://www.eyesopen.com.
- Yoon T.; De Lombaert S.; Brodbeck R.; Gulianello M.; Chandrasekhar J.; Horvath R. F.; Ge P.; Kershaw M. T.; Krause J. E.; Kehne J.; Hoffman D.; Doller D.; Hodgetts K. J. The design, synthesis and structure-activity relationships of 1-aryl-4-aminoalkylisoquinolines: a novel series of CRF-1 receptor antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 891–896. 10.1016/j.bmcl.2007.12.050. [DOI] [PubMed] [Google Scholar]
- Ozcan S.; Dengiz C.; Deliömeroglu M. K.; Sahin E.; Balci M. A novel one-pot, three-component reaction for the synthesis of isocoumarin-condensed pyrazoles. Tetrahedron Lett. 2011, 52, 1495–1497. 10.1016/j.tetlet.2011.01.114. [DOI] [Google Scholar]
- Myslinski J. M.; Clements J. H.; DeLorbe J. H.; Martin S. F. Protein-ligand interactions: Thermodynamic effects associated with increasing the length of an alkyl chain. ACS Med. Chem. Lett. 2013, 4, 1048–1053. 10.1021/ml400211q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott M.; Rogers C. M.; Yapp C.; Wells C.; Lambert J. P.; Strain-Damerell C.; Burgess-Brown N. A.; Gingras A. C.; Knapp S.; Müller S. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenet. Chromatin 2014, 7, 14. 10.1186/1756-8935-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H.. MOSFLM 7.01; MRC Laboratory of Molecular Biology: Cambridge, 2007.
- Evans P.SCALA—Scale Together Multiple Observations of Reflections 3.3.0; MRC Laboratory of Molecular Biology: Cambridge, 2007.
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J.. Phaser Crystallographic Software, 2007. [DOI] [PMC free article] [PubMed]
- Langer G.; Cohen S. X.; Lamzin V. S.; Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Painter J.; Merritt E. A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 439–450. 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.