

Abstract
Background
Smith-Lemli-Opitz syndrome (SLOS) is a multiple malformation/cognitive impairment syndrome characterized by the accumulation of 7-dehydrocholesterol (7DHC), a precursor sterol of cholesterol. Simvastatin, an HMG-CoA reductase inhibitor that crosses the blood-brain-barrier, has been proposed for treatment of SLOS based on in vitro and in vivo studies suggesting that simvastatin increases expression of hypomorphic DHCR7 alleles.
Methods
Safety and efficacy of simvastatin therapy in 23 mild to typical SLOS patients was evaluated in a randomized, double-blind, placebo-controlled trial. The cross-over trial consisted of two 12 month treatment phases separated by a 2 month wash-out period.
Results
No safety issues were identified in this study. Plasma dehydrocholesterol levels decreased significantly 8.9 ± 8.4% on placebo to 6.1 ± 5.5% on simvastatin (p<0.005) and we observed a trend toward decreased cerebral spinal fluid dehydrocholesterol levels. A significant improvement (p=0.017, paired t-test) was observed in the Aberrant Behavior Checklist-C Irritability when subjects were on simvastatin.
Conclusions
This paper reports the first randomized, placebo-controlled trial designed to test the safety and efficacy of simvastatin therapy in SLOS. Simvastatin appears to be relatively safe in SLOS patients, improves the serum dehydrocholesterol/total sterol ratio, and significantly improves irritability symptoms in mild to classical SLOS patients.
Introduction
Smith-Lemli-Opitz syndrome (SLOS, OMIM #270400) is an autosomal recessive, multiple malformation syndrome first described by Smith, Lemli, and Opitz in 1964 [1]. Nearly 30 years would pass before Irons and Tint [2] determined the biochemical nature of the disorder. Biochemically, SLOS is characterized by the abnormal accumulation of 7-dehydrocholesterol (7DHC). The gene 7-dehydrocholesterol reductase (DHCR7) encodes the enzyme that reduces 7-dehydrocholesterol to cholesterol. DHCR7 was independently cloned by three groups in 1998, and mutations within the gene were shown to be the molecular basis of SLOS [3–5]. Impaired DHCR7 activity results in increased levels of 7DHC and its isomer 8-dehydrocholesterol (8DHC). Cholesterol levels are frequently low in SLOS patients, but can be in the normal range. The incidence of SLOS has been estimated to be between 1 in 25,000 to 60,000 [6–9].
The SLOS phenotype varies broadly. At the severe end of the phenotypic spectrum, SLOS is a lethal disorder due to multiple major congenital anomalies. In contrast, at the mild end of the phenotypic spectrum, SLOS patients manifest fewer and minor physical abnormalities in combination with lesser functional cognitive and behavioral deficits. Congenital anomalies are variable but can include second + third toe syndactyly, microcephaly, neonatal or postnatal cataracts, micrognathia, cleft palate, postaxial polydactyly, cardiac defects, pyloric stenosis, and genital malformations [10, 11].
Currently, most SLOS patients are treated with dietary cholesterol supplementation. Although cholesterol therapy reduces serum 7DHC levels to a degree, significant levels of 7DHC persist even after years of therapy [12, 13]. Anecdotal case studies and case series support the idea that cholesterol supplementation benefits the overall well-being of SLOS patients [10, 14–18]; however, effects of dietary cholesterol supplementation on cognitive or behavioral aspects of this disorder have not been reported by others [19, 20] nor substantiated in a limited controlled trial [21]. Efficacy of dietary cholesterol supplementation is likely limited by the inability of dietary cholesterol to cross the blood-brain-barrier [22]. Moreover, increased levels of 7DHC or 7DHC derived oxysterol levels could have toxic effects [23–26]. In mild to classical SLOS patients specialists have hypothesized that many aspects of the abnormal behavioral and cognitive phenotype could be the result of altered sterol composition in the central nervous system. Thus, interventions that ameliorate the central nervous system biochemical disturbances in SLOS are critical to understanding the pathological processes that underlie this inborn error of cholesterol synthesis and to development of effective therapies for the neurological deficits.
The use of HMG-CoA reductase inhibitors as an adjunctive therapy to dietary cholesterol supplementation was considered soon after the biochemical defect was determined [27] and several case reports with divergent outcomes were published. For example, Starck et al. [18] reported adverse events in two severely affected SLOS patients treated with simvastatin, whereas Jira et al. [12, 28] reported that simvastatin therapy reduced serum and cerebral spinal fluid (CSF) levels of 7DHC and paradoxically increased plasma and CSF cholesterol levels in two SLOS patients. Although the studies were confounded by concurrent dietary cholesterol supplementation, an increase in serum cholesterol in SLOS patients treated with simvastatin was also reported in two additional case reports [29, 30].
Expression of DHCR7 is regulated by SREBP2 which, when activated by low levels of cholesterol in the endoplasmic reticulum, increases the transcription of most genes of the cholesterol synthetic pathway. Having shown that DHCR7 expression is increased in SLOS fibroblasts treated with simvastatin [31], we hypothesized that the paradoxical increase in serum cholesterol could be due to increased expression of a DHCR7 allele with residual enzymatic function, and we demonstrated that many DHCR7 alleles encode an enzyme with residual activity [31]. Furthermore, both in vitro experiments with human fibroblasts [31] and in vivo experiments using hypomorphic Dhcr7T93M/delta mice [32] support the hypothesis that increased expression of DHCR7 alleles with residual enzymatic activity can significantly improve plasma and tissue sterol levels. Because residual DHCR7 activity varies among SLOS patients, this hypothesis could explain the paradoxical increase in cholesterol in some patients and the adverse reactions observed in others.
Subsequent to initiation of our trial, additional reports describing the use of simvastatin in SLOS were published, although none were placebo-controlled trials. Chan et al. [33] reported a decrease in plasma 7DHC but no increase in the cholesterol level in three subjects treated with up to 0.4 mg/kg/d of simvastatin and a high cholesterol diet. Szabó et al. [30] reported decreased 7DHC and increased cholesterol levels after 2 years’ of a single subject given 0.2 mg/kg/d simvastatin and dietary cholesterol supplementation (150–250 mg/kg/d). They also reported improved behavior and gross motor function. Roullet et al. [34] observed no change in the dehydrocholesterol (7DHC + 8DCH)/cholesterol ratio in 9 SLOS subjects treated with an average simvastatin dose of 0.23 mg/kg/d. Hass et al. [35] published a retrolective study of simvastatin treatment (1 mg/kg/d) of 15 SLOS subjects. They found a decrease in the dehydrocholesterol to cholesterol ratio, but no positive effects on behavior. Six of their subjects taking this dose of simvastatin had significant adverse effects (sleep disturbance, increased serum transaminase levels, autoagression) that resulted in reduction or discontinuation of simvastatin therapy. None of these studies included a control group, none evaluated cerebrospinal fluid sterols or included prespecified clinical outcome measures. In this manuscript we report the findings of a placebo-controlled, crossover trial of simvastatin therapy in SLOS in which we demonstrate a significant decrease in the plasma dehydrocholesterol/total sterol ratio, a trend toward decreased CSF 7DHC levels, and a significant decrease in subject irritability.
Materials and Methods
Human Subjects Research Protections
This study was approved by the NICHD Institutional Review Board. Neurocognitive testing performed at Kennedy Krieger Institute in Baltimore was also approved by the Johns Hopkins Institutional Review Board. This study was registered on ClinicalTrials.gov (NCT00064792). Informed consent was obtained from parents or guardians. Due to the cognitive impairment associated with this disorder, patient assent was not feasible. This study was monitored by the NICHD Data Safety Monitoring Committee.
Subjects
The study was limited to individuals 4 to 18 years of age at enrollment with a biochemical diagnosis (increased level of 7DHC) of SLOS. For both safety reasons and because our hypothesis assumes the presence of a DHCR7 allele with residual enzymatic function, enrollment was limited to subjects with clinically mild to typical SLOS with an SLOS severity score [10] ≤ 30, a diagnostic dehydrocholesterol(DHC)/cholesterol ratio ≤ 1.0, and demonstration of residual cholesterol synthesis in the subjects fibroblasts of ≥ 10% of that observed in control lines [31]. Subjects were excluded if they had acute liver disease or serum transaminase levels > 3-times normal, history of myopathy or serum creatine phosphokinase (CPK) > 3-times normal, or a weight less than 10 kg. Accrual limit was 25 subjects with the goal of having at least 18 subjects complete the study.
Simvastatin, placebo and cholesterol preparations
Simvastatin was obtained from Xenos Bioresources, and compounded by the NIH Clinical Center Pharmacy Development Service (PDS) at a concentration of (5 mg/mL) in Ora-Plus© 0.75 mL and cherry flavor 0.25 mL. The PDS also prepared a placebo preparation that was indistinguishable from the active product in appearance and taste. Use of simvastatin in this research study was covered by IND#67,321. Simvastatin was administered as a single daily dose of 0.5 mg/kg/day for the first six weeks of the trial and 1.0 mg/kg/day for the remaining 46 weeks of the trial. The maximum dose was 40 mg/d. The dose selected for this study was based upon that used by Jira et al. [12]. Dose adjustments for weight were made at 6, 14, and 20 months. The placebo dose matched the corresponding simvastatin volume. All subjects were maintained on dietary cholesterol supplementation (150 mg/kg/d) utilizing cholesterol suspension (150 mg/mL) in Ora Plus prepared by PDS.
Trial Design
The study was designed as a randomized, double-blind, placebo-controlled, crossover trial consisting of two 12-month treatment and placebo arms separated by a 2-month washout period. Randomization was performed by the PDS in blocks of four. Neither the participants nor evaluating physicians knew the assignments. Study participants were clinically evaluated, and behavioral assessments were obtained, at baseline, 6, 12, 20 and 26 months during the study. Behavioral assessment was also obtained at 14 months. Anthropomorphic measurements were obtained at each admission. The Side Effects Profile for Exceptional Children modified with additional questions related to SLOS and simvastatin (Supplemental Information) was used to screen participants for adverse effects. Serum/plasma was collected for sterol, biochemical, or safety testing at 0, 0.5, 1, 3, 9, 14, 14.5,15, 17, 23 and 26 months. CSF was collected at 0, 12 and 26 months. Parents and guardians could elect to enroll the study participants in an open-label extension after completing the crossover trial. Data regarding adverse events observed during the open-label extension were also collected.
Laboratory Testing
Clinical laboratory testing was performed by the NIH Clinical Center Department of Laboratory Medicine. Sterol levels were measured by the Kennedy Krieger Institute Biochemical Genetics Laboratory. Residual cholesterol synthesis in subject fibroblasts was determined as previously reported [31].
Statistical Evaluation
The primary outcome measure for this study was the plasma DHC/total sterol ratio. Secondary outcome measures were a change in the severity of the irritability subscale of the Aberrant Behavior Checklist-community [36] completed by the parent or primary caretaker and the assessment of overall behavioral plus cognitive improvement using the Clinician’s Global Impression-Improvement scale (CGI-I) [37] completed by the behavioral clinician (ET) at each follow-up visit.
The study was powered based on the primary outcome measure. Prior data on plasma DHC/total sterol ratios from our Natural History study gave a standard deviation of 0.13 for the between-subject variability. Serial data from four subjects gave a correlation of 0.95 supporting feasibility of a cross-over design, and yielded an expected same subject standard deviation of approximately 0.04. A power calculation indicated that, in a cross-over design, we could detect a difference of 0.03 between treatment and control means at p=0.05 with 80% power in 18 subjects. Statistical analysis was performed using Prism 6 (GraphPad Software, La Jolla, CA). Two sided paired t-tests were used when data were available from both phases; otherwise, unpaired t-tests were used. An F-test was performed to compare variances. A p-value of less than 0.05 was considered to be significant, and values between 0.05 and 0.10 were considered to indicate a trend. No adjustment was made for multiple comparisons on the prespecified primary and secondary outcome measures. Data is expressed as mean ± SD unless otherwise noted.
Results
Study subjects
Of 23 SLOS subjects initially enrolled in this study, 22 were randomized, and 18 completed the study (Fig. 1). Demographics, SLOS severity, DHCR7 genotype, SLOS severity score, residual cholesterol synthesis and baseline sterol values for all participants are provided in Table 1. Of the 22 randomized subjects, 13 (59%) were male and 9 were female. Age at enrollment ranged from 4.0 to 17.5 years with a mean and median of 8.2 and 7.3 years, respectively. The SLOS Severity Score ranged from 6 to 28, with a mean and median of 13.2 and 11 respectively. Fibroblast residual fractional cholesterol synthesis ranged from 0.11 to 0.76 with a mean and median of 0.37 and 0.30, respectively. Baseline DHC/sterol ratio for plasma and CSF were 9.1 ± 7.4% and 6.7 ± 4.0% respectively. The plasma and CSF DHC/sterol ratios were highly correlated (r2 = 0.83, p<0.0001).
Figure 1. Study design.

Subjects were randomized to either placebo or simvastin in the first phase of this crossover trial. After 12 months in phase 1 and a 2-month washout period, subjects were switched to the alternative therapy for 12 months. One subject was excluded and 4 subjects were withdrawn.
Table 1.
| Baseline Plasma Sterols | Baseline CSF Sterols | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study Identifier |
NIH SLOS Subject Identifier |
Gender | Age at Enrollment |
DHCR7 Genotype | SLOS Severity Score |
Fibroblast Residual Cholesterol Synthesis |
Cholesterol (mcg/ml) |
7DHC (mcg/ml) |
8DHC (mcg/ml) |
Percent DHC/total sterols |
Cholesterol (mcg/ml) |
7DHC (mcg/ml) |
8DHC (mcg/ml) |
Percent DHC/total sterols |
| S01a | SLOS-025 | F | 10.0 | R242C/R450L | 11 | 0.19 | 1168 | 40 | 20 | 4.9 | NSOc | NSO | NSO | NSO |
| S02 | SLOS-055 | F | 4.4 | C380Y/c.964-1 G>C | 11 | 0.61 | 1287 | 22 | 28 | 3.7 | 1.94 | 0.035 | 0.083 | 5.7 |
| S03 | SLOS-004 | M | 8.8 | c.964-1 G>C/T93M | 22 | 0.21 | 649 | 95 | 85 | 21.7 | 1.64 | 0.065 | 0.140 | 11.1 |
| S04b | SLOS-026 | M | 13.3 | R242C/R450L | 11 | 0.24 | 783 | 33 | 44 | 9.0 | 2.02 | 0.028 | 0.084 | 5.3 |
| S05 | SLOS-012 | M | 8.9 | c.964-1 G>C/T289I | 11 | 0.47 | 1061 | 28 | 33 | 5.4 | 1.66 | 0.024 | 0.077 | 5.7 |
| S06 | SLOS-013 | M | 10.6 | c.964-1 G>C/T289I | 6 | 0.49 | 1102 | 20 | 29 | 4.3 | 2.68 | 0.043 | 0.132 | 6.1 |
| S07d | SLOS-001 | M | 7.1 | c.964-1 G>C/T93M | 11 | 0.31 | 858 | 93 | 66 | 15.6 | 1.69 | 0.088 | 0.155 | 12.6 |
| S08 | SLOS-002 | M | 9.8 | W151X/T93M | 28 | 0.19 | 788 | 71 | 63 | 14.5 | 2.00 | 0.081 | 0.151 | 10.4 |
| S09 | SLOS-024 | F | 7.6 | T154M/R443H | 22 | 0.51 | 1085 | 59 | 54 | 9.4 | 1.98 | 0.029 | 0.080 | 5.2 |
| S10 | SLOS-018 | F | 12.0 | R242C/W177R | 17 | 0.48 | 819 | 40 | 42 | 9.1 | 1.94 | 0.033 | 0.073 | 5.2 |
| S11 | SLOS-033 | M | 5.6 | c.964-1 G>C/A247V | 6 | 0.49 | 1008 | 63 | 62 | 11.0 | 1.73 | 0.041 | 0.079 | 6.5 |
| S12 | SLOS-005 | F | 17.5 | c.964-1 G>C/R352W | 17 | 0.11 | 849 | 139 | 98 | 21.8 | 1.22 | 0.086 | 0.101 | 13.3 |
| S13 | SLOS-062 | F | 10.3 | P51S/W151X | 6 | 0.27 | 942 | 17 | 28 | 4.6 | 1.66 | 0.020 | 0.047 | 3.9 |
| S14b | SLOS-034 | M | 5.7 | c.964-1 G>C/V326L | 33 | 0.33 | ||||||||
| S15 | SLOS-060 | M | 4.8 | T289I/c.964-1 G>C | 6 | 0.35 | 1495 | 29 | 49 | 5.0 | 1.90 | 0.036 | 0.102 | 6.8 |
| S16 | SLOS-063 | F | 5.4 | T93M/R443C | 22 | 0.21 | 738 | 168 | 121 | 28.1 | 0.96 | 0.057 | 0.094 | 13.6 |
| S17 | SLOS-061 | M | 6.3 | S169L/c.964-1 G>C | 11 | 0.20 | 1082 | 2 | 8 | 0.9 | 2.99 | 0.003 | 0.028 | 1.0 |
| S18c | SLOS-064 | M | 4.0 | c.964-1 G>C/V466M | 11 | 0.76 | 467 | 4.2 | 3.7 | 1.7 | 1.22 | 0.010 | 0.038 | 3.8 |
| S19 | SLOS-066 | M | 4.1 | M1V/Q98X | 17 | 0.25 | 1620 | 0.8 | 2.6 | 0.2 | 2.18 | 0.000 | 0.006 | 0.3 |
| S20 | SLOS-073 | M | 6.9 | M1V/R446Q | 6 | 0.37 | 1311 | 0.4 | 1.4 | 0.1 | 3.66 | 0.002 | 0.009 | 0.3 |
| S21 | SLOS-068 | F | 4.1 | c.964-1 G>C/Y462H | 11 | 0.66 | 1053 | 45 | 35 | 7.1 | 1.85 | 0.049 | 0.074 | 6.2 |
| S22d | SLOS-070 | M | 13.3 | W151X/V466M | 22 | 0.29 | 776 | 71 | 68 | 15.2 | 1.81 | 0.097 | 0.165 | 12.6 |
| S23 | SLOS-056 | F | 6.1 | S192F/Y462H | 6 | 0.51 | 962 | 34 | 29 | 6.1 | 1.58 | 0.026 | 0.063 | 5.3 |
Shaded rows indicate phase 1 placebo and phase 2 simvastatin
Excluded, Severity Score >30
No sample obtained
Subjects were randomized in blocks of four with 10 and 12 subjects starting on simvastatin and placebo, respectively, for the first 12-month phase. Simvastatin therapy was initiated at 0.5 mg/kg/d and increased to 1.0 mg/kg/d at six weeks. The placebo was increased by a corresponding volume. Two subjects on placebo were withdrawn during the first phase. S04 reported symptoms consistent with muscular pain and had increasing CPK levels and was withdrawn at 1 month. S18 was withdrawn at 6 months due to noncompliance. Twenty subjects, equally distributed between simvastatin and placebo groups, were crossed over to the alternate study arm after a two-month washout period. Two subjects (S07 and S22) were withdrawn at 14 and 20 months into the second phase for noncompliance. Although both of these subjects were on simvastatin, there were no indications that noncompliance was due to adverse drug effects. A total of 18 subjects completed both phases of the trial. There were no significant differences in gender, age, SLOS Severity Score, fibroblast residual cholesterol synthesis or baseline sterol ratios between subjects who were initially randomized to either simvastatin or placebo or the 4 subjects withdrawn from the study (Supplemental Table 1). Because we did not observe an effect of phase order, the data were analyzed as simvastatin treated versus placebo.
Safety and adverse events
Twenty-seven adverse events occurred in 15 subjects during the controlled part of the trial (Supplemental Table 2). Seven additional adverse events were noted during the open-label extension. Five adverse events were classified as serious. Only one serious adverse event, a post-LP headache requiring extended hospitalization, occurred while on simvastatin. No serious adverse events were considered to be drug related. Thirteen (48%) of the adverse events occurred while on simvastatin and 10 of the 27 (37%) had an infectious etiology. Eight episodes in seven subjects involved gastrointestinal symptoms (gastroenteritis, diarrhea, vomiting, gas) with three of these (38%) occurring when on simvastatin. Increased aggression and self-injurious behavior was reported clinically in one subject during the open-label extension phase. Safety laboratory testing was performed at 0, 0.5, 1, 3, 6, 9, 12, 14, 14.5, 15, 17, 20, 23, and 26 months. Values remained within normal ranges, and there were no significant changes in serum transaminase, alkaline phosphatase or total bilirubin levels (Supplemental Fig. 1) between placebo and simvastatin phases. Serum CPK levels remained within the normal range and did not significantly differ between placebo and simvastatin phases. The trend toward increased serum AST and CPK levels during the simvastatin phase was not considered clinically significant. Subject S04 was noted to have increasing CPK levels after initiation of the study. CPK levels in subject S04 increased from 199 U/L (normal range 52–386 U/L) at baseline to 220 U/L and 347 U/L at 2 and 4 weeks respectively. Although the levels were still within the normal range, because the subject reported thigh pain and exhibited signs of weakness, fatigue and worsening behavior, the investigational product was discontinued and the subject was withdrawn from the study. CPK levels were followed and returned to baseline after the treatment was stopped. After completion of the study, it was determined that the subject had been assigned to placebo. No abnormalities in serum electrolytes, creatinine, glucose, inorganic phosphate, magnesium, ionized calcium were observed. Hemoglobin and hematocrit as well as white blood cell and platelet counts were normal. These data suggest that patients with mild to typical SLOS are not at increased risk of liver or muscle toxicity when treated with simvastatin.
Anthropomorphic parameters
Weight and height were measured at each admission. Since subjects were not seen at 14 months the values obtained at the 12-month visit were used as a baseline for phase 2. Only two subjects lost weight during the trial. Both S12 and S16 each lost 1.2 kg during the placebo phase. Mean monthly weight gain during the placebo and simvastatin treatment periods were 0.20 ± 0.16 and 0.21 ± 0.13 kg, respectively (S Fig. 2A, n = 18, p=0.76, paired t-test). Complete data sets for height were available for 15 subjects. For two subjects, the monthly height gain could only be determined for either the simvastatin or placebo phase. Mean monthly height gain during the placebo and simvastatin treatment periods were 0.49 ± 0.23 and 0.42 ± 0.20 cm, respectively (S Fig. 2B, n=16, p=0.42, unpaired t-test). Our data do not support a significant change in growth parameters in mild to typical SLOS subjects treated with simvastatin.
Figure 2. Plasma sterol levels.

Cholesterol and dehydrocholesterol (7DHC + 8DHC) levels were measured at baseline (B), washout (W, 14 mo) as well as at 1, 3, 6, 9 and 12 months in both the placebo and simvastatin treatment phase. Plasma cholesterol levels (A, B) and DHC (C, D) decreased significantly during the simvastatin phase compared to the placebo phase. The plasma DHC/Total Sterol ratio (E, F), which was the primary outcome measure of this study, also decreased significantly. Data expressed as mean ± SEM.
Plasma and Cerebral Spinal Fluid Sterol Biochemistry
Cholesterol and cholesterol precursor levels were measured at 0 (Baseline), 1, 3, 6, 9, 12, 14 (Washout), 15, 17, 20, 23, and 26 months. Plasma sterol data is presented in Figure 2 and Supplemental Table 3. No significant differences were noted between plasma cholesterol levels (p=0.07), dehydrocholesterol levels (p=0.54) or the DHC/total sterol ratio (p=0.69) at baseline (time 0) or at 14 months after a 2 month washout. Because changes in the plasma sterol levels were evident by 1 month of treatment (Fig. 2A,C,E), data from all time points in each phase were combined. In contrast to prior case reports, our data do not support a paradoxical increase in plasma cholesterol levels with simvastatin therapy when subjects are evaluated either as a group (Fig. 2B) or as individuals. When the subjects were evaluated as a group, we observed a significant (p<0.005, paired t-test) decrease in mean plasma cholesterol levels while on simvastatin (105 ± 16 mg/dl) compared to placebo (120 ± 31 mg/dl). On an individual basis (Supplemental Table 3), a trend toward increased plasma cholesterol was only observed in one subject (S06, p=0.05). In contrast, a significant decrease in plasma cholesterol was observed in 7 subjects (S04, S10, S12, S14, S15, S16, and S18). Plasma DHC levels were decreased in SLOS subjects treated with simvastatin compared to placebo (Fig. 2C, D). Mean DHC levels decreased from 106 ± 91 mcg/ml on placebo to 66 ± 58 mcg/ml on simvastatin (p<0.001, paired t-test).
The primary outcome measure of this study was the plasma DHC/total sterol ratio. We observed a decrease in the plasma DHC/total sterol ratio in subjects treated with simvastatin. The mean fraction of dehydrocholesterol decreased from 8.9 ± 8.4% on placebo to 6.1 ± 5.5% on simvastatin (p<0.005, paired t-test). On an individual basis, the mean fraction of dehydrocholesterol decreased in 17/18 subjects (Supplemental Table 3). The decrease was significant (p<0.05) in 12 subjects, and two additional subjects demonstrated a trend toward (p<0.10) significance.
CSF sterols were evaluated as an exploratory outcome measure (Fig. 3, Supplemental Table 3). We observed a trend (p=0.07, paired t-test) toward decreased CSF DHC levels when subjects were treated with simvastatin compared to placebo. No significant difference was observed for either the CSF cholesterol level or the DHC/total sterol ratio in paired samples.
Figure 3. Cerebral spinal fluid sterol levels.

Neither cholesterol (A) nor the DHC/Total Sterol ratio (B) were decreased in CSF from subjects when they were treated with simvastatin compared to when they were treated with placebo. A trend toward decreased DHC (C) levels was observed during simvastatin therapy compared to placebo. Data expressed as mean ± SEM.
Clinical outcome measures
In addition to determining if simvastatin had an effect on sterol levels, we assessed whether treatment would provide a clinical benefit by improving behavior. Our secondary outcome measures were the Irritability subscale of the ABC-C and the CGI-I completed by the behavioral clinician at each follow up visit. No significant differences were observed with the CGI. For the ABC-C, paired data was available for both baseline (0 mo) and washout (14 mo) for 13 subjects. We did not observe any significant difference (p=0.89, paired t-test) in the ABC-C Irritability subscale between these two control time points (Fig. 4A). However, paired comparison of the 14 subjects for whom we had data from both the placebo and simvastatin treatment arms demonstrated a significant (p=0.017) decrease (clinical improvement) in the ABC-C Irritability subscale (Fig. 4B).
Figure 4. Simvastin therapy reduces irritability in SLOS subjects.
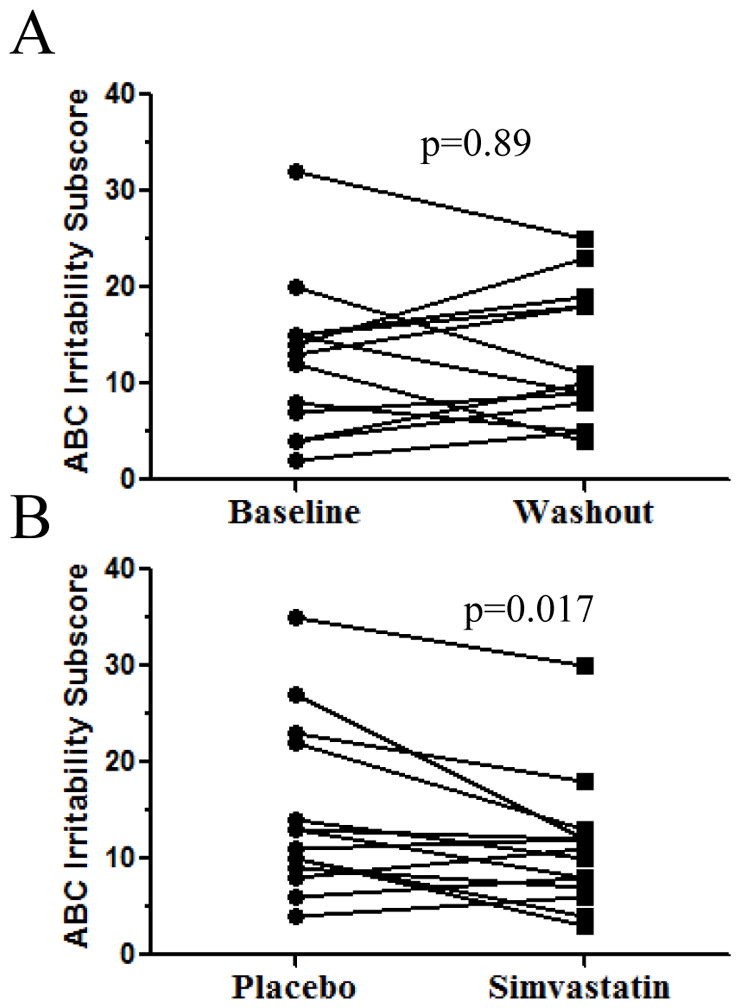
A) Comparison of ABC Irritability subscores from baseline (0 mo) and washout (14 mo). No significant differences were noted. B) Paired evaluation of data from the 14 subjects for whom we had data from both the placebo and simvastatin treatment arms demonstrated a significant improvement in the ABC-C Irritability subscale (p=0.017, paired t-test).
Discussion
This study reports the first randomized, placebo-controlled trial of simvastatin therapy in SLOS. A crossover design was used. Although crossover trials have limitations, this design facilitates the conduct of controlled trials in rare disorders where the number of participants is limited and clinical heterogeneity is great. Our study design necessarily had several limitations. Because dietary cholesterol therapy has become standard of care in SLOS, participants were maintained on 150 mg/kg/d of cholesterol supplementation for the duration of the study. Thus, an evaluation of simvastatin therapy independent of dietary cholesterol supplementation was precluded. Only SLOS subjects with a mild to classical phenotype were included; severely affected subjects being excluded due to safety concerns. Although apparently random, missing data may have introduced an unrecognized bias. Because of the rarity of SLOS, the necessarily small study sample size limited the number of outcome measures that could be evaluated. Also, the lack of generally accepted outcome measures for characterization of abnormal behavior in SLOS necessitated the selection of clinical outcome measures without significant prior data available to validate their use.
Although difficult to conduct in rare disorders, controlled trials are essential for understanding both the safety and potential efficacy of experimental therapies. Repurposing of existing drugs can expedite the development of therapies for rare and neglected diseases. However, off-label use and case reports are not a substitute for controlled trials, and the conflicting literature regarding the safety and efficacy of simvastatin therapy in SLOS highlights the limitations of case reports and uncontrolled studies.
The underlying hypothesis for this study was that treatment with simvastatin would increase DHCR7 activity by increasing expression of DHCR7 alleles with residual enzymatic function. Increased DHCR7 activity would then decrease DHC levels relative to cholesterol and thus result in a lower DHC/total sterol ratio. This mechanism could also potentially explain the prior paradoxical increases in cholesterol that have been reported [12, 28]. This potential mechanism was supported by preclinical in vitro and in vivo studies [31, 32]. We further hypothesized that the behavioral and cognitive aspects of SLOS result from both developmental and functional defects due to altered neuronal cholesterol composition. Although brain developmental defects are irreversible, functional defects due to altered sterol composition or neurosteroid metabolism should be amendable to therapeutic interventions that correct the biochemical defect.
Consistent with our first hypothesis, we observed a significant decrease (Fig. 2) in the plasma DHC/total sterol ratio. This decrease was due to a greater decrease in DHC relative to that of cholesterol. This is consistent with the data reported by Haas et al [35] in their retrospective study. Our data do not support a concomitant increase in plasma cholesterol levels. For CSF sterol levels which were an exploratory outcome measure in this trial, we observed a trend toward decreased DHC levels in CSF (Fig. 3), although this was not reflected in the CSF DHC/total Sterol ratio. CSF sterol levels were measured under the assumption that they might reflect synaptic cholesterol levels, but the validity of this assumption is not known. Nonetheless, this result is consistent with our observation of a disproportionate decrease in plasma 7DHC levels with simvastatin therapy. It is plausible that a decrease in 7DHC levels could have a beneficial effect on central nervous system function in SLOS. In vitro and in vivo preclinical studies supported the idea that 7DHC or 7DHC metabolites are toxic. 7DHC alters the physiochemical properties, protein composition and signaling functions of cellular membranes (reviewed in [25]). More recently, Francis et al. [26] have shown that 7DHC disrupts Wnt/β-catenin signaling, and Xu et al. [23, 24] have shown that 7DHC-derived oxysterols, in particular 3β,5α-dihydroxycholest-7-en-6-one (DHCEO) accumulate in SLOS and likely have toxic effects. Thus, reduction of 7DHC or 7DHC metabolites levels could ameliorate abnormal SLOS behaviors.
In this study we also evaluated the potential of simvastatin to alter specific aspects of the SLOS behavioral phenotype. Our secondary outcome measures were the CGI-I and ABC-C Irritability scores. While we observed no significant effect on the CGI-I, we did observe significant improvement in the ABC-C Irritability score (Fig. 4). This paper therefore represents the first controlled study to demonstrate improved behavior in SLOS subjects in response to a therapeutic intervention. The CGI-I is a physician assessment which seeks to ascertain their impression of global improvement in both cognition and behavior. The ABC-C was developed as an outcome measure for pharmacological trials in people with developmental disabilities. The ABC-C is completed by the parent or guardian. The 15-item Irritability subscale includes questions about aggression, self-injury, tantrums, agitation, and unstable mood on a scale of 0 to 45. Although we cannot directly demonstrate improved synaptic sterol composition, we were able to demonstrate changes in plasma and CSF 7DHC levels consistent with the hypothesis that therapies correcting CNS sterol composition might improve behavior in SLOS and thus have clinical benefit. Alternatively, the reduction in 7DHC levels could have led to a reduction in 7DHC-derived metabolites with adverse biological effects. The small size of this study precludes the statistical evaluation of multiple neurocognitive outcome measures and thus the evidence for clinical improvement is limited. However, the fact that we observed a significant improvement in the irritability subscale of the ABC provides a proof of principle for continued development of drugs that can increase DHCR7 activity and decrease 7DHC levels in individuals with SLOS. DHCR7 activity could be enhanced by drugs that either increase DHCR7 expression or stabilize mutant DHCR7 protein with residual enzymatic activity.
Although increased aggression and self-injurious behavior was reported in one subject during the open label extension phase of the study, we did not observe any consistent, treatment-limiting adverse effects or laboratory abnormalities in SLOS subjects treated with simvastatin, nor did we observe sleep disturbances, increased aggression, or self-injurious behavior during the controlled part of the study. We also did not observe any significant alteration in growth. This study supports the hypothesis that simvastatin therapy can be safely used in mild to classical SLOS patients. However, caution is still warranted, in regard to the long-term safety of simvastatin in these patients. We are aware of one subject (S23) who developed cataracts on off-label simvastatin therapy after this trial ended. Postnatal development of cataracts in SLOS is rare after infancy.
In summary, this study represents the first controlled trial of simvastatin therapy in SLOS and the first controlled trial demonstrating the potential of drug therapy to modulate sterol composition and to improve SLOS behavior. We have established that treatment with simvastatin is relatively safe, can decrease DHC levels, and improve at least one aspect of the behavioral phenotype. These data support continued efforts to identify and rigorously evaluate potential therapies that may have clinically meaningful benefits for patients with SLOS.
Supplementary Material
S. Table 3. Plasma and cerebral spinal fluid sterol values.
Side Effects Profile for Exceptional Children modified with additional questions related to SLOS and simvastatin
S. Figure 1. Serum chemistries. Serum transaminase (A–C), alkaline phosphatase (D), total bilirubin (E), and creatine phosphokinase (F) levels during placebo and simvastatin phases.
S. Figure 2. Anthropomorphic measures. A) Monthly weight change (p=0.76) and B) monthly height change (p=0.42) did not differ significantly between placebo and simvastatin phases.
S. Table 1. Randomization demographics.
S. Table 2. Adverse Events.
Acknowledgments
This study was supported by the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Autism Speaks, and by the Johns Hopkins Institute for Clinical and Translational Research (ICTR) which is funded in part by Grant Number UL1 TR 001079 from the National Center for Advancing Translational Sciences (NCATS) a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. The authors would like to acknowledge the clinical and supporting assistance of Halima Goodwin, Sandra Conley, Irena Bukelis, Geeta Sarphare and Diane Lanham as well as the laboratory assistance of Cynthia Toth. We also would like to thank Judith Starling and George Grimes from the NIH Clinical Center Pharmaceutical Development Service for their assistance in the preparation of the simvastatin and placebo suspensions. We offer our greatest thanks to the individuals with SLOS and their family members who participated in this clinical study.
Footnotes
Disclosures
The authors have no disclosures to make.
References
- 1.Smith DW, Lemli L, Opitz JM. A Newly Recognized Syndrome of Multiple Congenital Anomalies. J Pediatr. 1964;64:210–7. doi: 10.1016/s0022-3476(64)80264-x. [DOI] [PubMed] [Google Scholar]
- 2.Irons M, et al. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 1993;341(8857):1414. doi: 10.1016/0140-6736(93)90983-n. [DOI] [PubMed] [Google Scholar]
- 3.Fitzky BU, et al. Mutations in the Delta7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc Natl Acad Sci U S A. 1998;95(14):8181–6. doi: 10.1073/pnas.95.14.8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wassif CA, et al. Mutations in the human sterol delta7-reductase gene at 11q12–13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet. 1998;63(1):55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waterham HR, et al. Smith-Lemli-Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene. Am J Hum Genet. 1998;63(2):329–38. doi: 10.1086/301982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciara E, et al. SLOS carrier frequency in Poland as determined by screening for Trp151X and Val326Leu DHCR7 mutations. Eur J Med Genet. 2006;49(6):499–504. doi: 10.1016/j.ejmg.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Cross JL, et al. Determination of the allelic frequency in Smith-Lemli-Opitz syndrome by analysis of massively parallel sequencing data sets. Clin Genet. 2015;87(6):570–5. doi: 10.1111/cge.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelley RI. A new face for an old syndrome. Am J Med Genet. 1997;68(3):251–6. doi: 10.1002/(sici)1096-8628(19970131)68:3<251::aid-ajmg1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.Nowaczyk MJ, Waye JS, Douketis JD. DHCR7 mutation carrier rates and prevalence of the RSH/Smith-Lemli-Opitz syndrome: where are the patients? Am J Med Genet A. 2006;140(19):2057–62. doi: 10.1002/ajmg.a.31413. [DOI] [PubMed] [Google Scholar]
- 10.Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37(5):321–35. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter FD. Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2008;16(5):535–41. doi: 10.1038/ejhg.2008.10. [DOI] [PubMed] [Google Scholar]
- 12.Jira PE, et al. Simvastatin. A new therapeutic approach for Smith-Lemli-Opitz syndrome. J Lipid Res. 2000;41(8):1339–46. [PubMed] [Google Scholar]
- 13.Merkens LS, et al. Effects of dietary cholesterol on plasma lipoproteins in Smith-Lemli-Opitz syndrome. Pediatr Res. 2004;56(5):726–32. doi: 10.1203/01.PDR.0000141522.14177.4F. [DOI] [PubMed] [Google Scholar]
- 14.Elias ER, et al. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS) Am J Med Genet. 1997;68(3):305–10. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 15.Irons M, et al. Treatment of Smith-Lemli-Opitz syndrome: results of a multicenter trial. Am J Med Genet. 1997;68(3):311–4. [PubMed] [Google Scholar]
- 16.Linck LM, et al. Cholesterol supplementation with egg yolk increases plasma cholesterol and decreases plasma 7-dehydrocholesterol in Smith-Lemli-Opitz syndrome. Am J Med Genet. 2000;93(5):360–5. doi: 10.1002/1096-8628(20000828)93:5<360::aid-ajmg4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 17.Nwokoro NA, Mulvihill JJ. Cholesterol and bile acid replacement therapy in children and adults with Smith-Lemli-Opitz (SLO/RSH) syndrome. Am J Med Genet. 1997;68(3):315–21. doi: 10.1002/(sici)1096-8628(19970131)68:3<315::aid-ajmg13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 18.Starck L, et al. Beneficial effects of dietary supplementation in a disorder with defective synthesis of cholesterol. A case report of a girl with Smith-Lemli-Opitz syndrome, polyneuropathy and precocious puberty. Acta Paediatr. 1999;88(7):729–33. doi: 10.1080/08035259950169008. [DOI] [PubMed] [Google Scholar]
- 19.Abuelo DN. Cholesterol supplementation in Smith-Lemli-Opitz syndrome. Am J Med Genet. 1998;78(4):378–80. [PubMed] [Google Scholar]
- 20.Sikora DM, et al. Cholesterol supplementation does not improve developmental progress in Smith-Lemli-Opitz syndrome. J Pediatr. 2004;144(6):783–91. doi: 10.1016/j.jpeds.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 21.Tierney E, et al. Analysis of short-term behavioral effects of dietary cholesterol supplementation in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2010;152A(1):91–5. doi: 10.1002/ajmg.a.33148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390(4):287–93. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu L, et al. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J Lipid Res. 2011;52(6):1222–33. doi: 10.1194/jlr.M014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L, et al. 7-Dehydrocholesterol-derived oxysterols and retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Biochim Biophys Acta. 2012;1821(6):877–83. doi: 10.1016/j.bbalip.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52(1):6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francis KRT, AN, Xin Y, O’Halloran PE, Wassif CA, Malik N, Williams IM, Cluzeau CV, Trivedi NS, Pavan WJ, Cho W, Westphal H, Porter FD. Modeling Smith-Lemli-Opitz syndrome with iPS cells reveals a causal role for Wnt/β-catenin defects in neuronal cholesterol synthesis phenotypes. Nature Medicine. doi: 10.1038/nm.4067. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irons M, et al. Abnormal cholesterol metabolism in the Smith-Lemli-Opitz syndrome: report of clinical and biochemical findings in four patients and treatment in one patient. Am J Med Genet. 1994;50(4):347–52. doi: 10.1002/ajmg.1320500409. [DOI] [PubMed] [Google Scholar]
- 28.Jira P, et al. New treatment strategy for Smith-Lemli-Opitz syndrome. Lancet. 1997;349(9060):1222. doi: 10.1016/S0140-6736(05)62415-4. [DOI] [PubMed] [Google Scholar]
- 29.Oberthur A, et al. Smith-Lemli-Opitz syndrome--case report, diagnostics and therapeutic options. Z Geburtshilfe Neonatol. 2009;213(5):210–4. doi: 10.1055/s-0029-1224190. [DOI] [PubMed] [Google Scholar]
- 30.Szabo GP, et al. A patient with Smith-Lemli-Opitz syndrome: novel mutation of the DHCR7 gene and effects of therapy with simvastatin and cholesterol supplement. Eur J Pediatr. 2010;169(1):121–3. doi: 10.1007/s00431-009-0987-z. [DOI] [PubMed] [Google Scholar]
- 31.Wassif CA, et al. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith-Lemli-Opitz syndrome fibroblasts. Mol Genet Metab. 2005;85(2):96–107. doi: 10.1016/j.ymgme.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 32.Correa-Cerro LS, et al. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet. 2006;15(6):839–51. doi: 10.1093/hmg/ddl003. [DOI] [PubMed] [Google Scholar]
- 33.Chan YM, et al. Effects of dietary cholesterol and simvastatin on cholesterol synthesis in Smith-Lemli-Opitz syndrome. Pediatr Res. 2009;65(6):681–5. doi: 10.1203/PDR.0b013e31819ea4eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roullet JB, et al. No evidence for mevalonate shunting in moderately affected children with Smith-Lemli-Opitz syndrome. J Inherit Metab Dis. 2012;35(5):859–69. doi: 10.1007/s10545-012-9453-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haas D, et al. Effects of cholesterol and simvastatin treatment in patients with Smith-Lemli-Opitz syndrome (SLOS) J Inherit Metab Dis. 2007;30(3):375–87. doi: 10.1007/s10545-007-0537-7. [DOI] [PubMed] [Google Scholar]
- 36.Marshburn EC, Aman MG. Factor validity and norms for the aberrant behavior checklist in a community sample of children with mental retardation. J Autism Dev Disord. 1992;22(3):357–73. doi: 10.1007/BF01048240. [DOI] [PubMed] [Google Scholar]
- 37.Guy W. D. NIMH, editor. ECDEU assessmetn manual for psychopharmacology. Rev. Rockville, MD: 1976. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S. Table 3. Plasma and cerebral spinal fluid sterol values.
Side Effects Profile for Exceptional Children modified with additional questions related to SLOS and simvastatin
S. Figure 1. Serum chemistries. Serum transaminase (A–C), alkaline phosphatase (D), total bilirubin (E), and creatine phosphokinase (F) levels during placebo and simvastatin phases.
S. Figure 2. Anthropomorphic measures. A) Monthly weight change (p=0.76) and B) monthly height change (p=0.42) did not differ significantly between placebo and simvastatin phases.
S. Table 1. Randomization demographics.
S. Table 2. Adverse Events.