

Abstract
Water contamination is generally considered to be detrimental to the performance of aprotic lithium–air batteries, whereas this view is challenged by recent contrasting observations. This has provoked a range of discussions on the role of water and its impact on batteries. In this work, a distinct battery chemistry that prevails in water-contaminated aprotic lithium–oxygen batteries is revealed. Both lithium ions and protons are found to be involved in the oxygen reduction and evolution reactions, and lithium hydroperoxide and lithium hydroxide are identified as predominant discharge products. The crystallographic and spectroscopic characteristics of lithium hydroperoxide monohydrate are scrutinized both experimentally and theoretically. Intriguingly, the reaction of lithium hydroperoxide with triiodide exhibits a faster kinetics, which enables a considerably lower overpotential during the charging process. The battery chemistry unveiled in this mechanistic study could provide important insights into the understanding of nominally aprotic lithium–oxygen batteries and help to tackle the critical issues confronted.
Water is believed to undermine the performance of aprotic lithium–air batteries. However, the authors here disclose different battery chemistry, showing that both lithium ions and protons are involved in the battery reactions in the presence of water, leading to an unprecedented dynamic product.
The pursuit of high-energy power sources going beyond the state-of-the-art Li-ion batteries has evoked a surge of intensive studies of the lithium–air battery, as it has the potential of achieving nearly the same level of energy density as that of gasoline1. Although profound studies have been carried out, several technical challenges severely hinder the development of lithium–air batteries for practical application. Taking the most studied aprotic lithium–oxygen (Li-O2) system as an example, the formation of insoluble and insulating lithium peroxide (Li2O2) during the discharge process leads to surface passivation and pore clogging of the cathode, which results in low round-trip energy efficiency and limited capacity2,3,4. Advances in electrocatalysts so far seem to have achieved only limited success in addressing the above issues. It remains a significant challenge that in a Li-air battery the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) take place electrocatalytically at the ‘solid–solid' interface, which is intrinsically less favourable than those at the ‘liquid–solid' interface in other metal-air batteries (or fuel cells)5,6,7,8,9. As such, soluble redox catalysts have recently been extensively investigated to transform the solid-state electrode reaction into a solution phase reaction10,11,12,13,14,15,16,17,18,19. Among the soluble OER catalyst, iodide received the most attention owing to its relatively good stability. Another critical issue for the aprotic Li-O2 battery is that it is in essence an open system nominally, for which not only oxygen is fed into the battery upon operation; other species in air such as moisture are also inevitably introduced into the system. The presence of water in the electrolyte is generally considered to be detrimental as it attacks lithium metal at the anode and it may become involved in the ORR reaction at the cathode. For instance, water and protons were found in one study to significantly influence the crystal growth of Li2O2 (refs 20, 21). In other studies, lithium hydroxide (LiOH) was however identified as the main discharge product in the presence of moisture15,16, whereas disputes persist on the oxidation of LiOH by triiodide (I3−) during charging process22,23,24,25,26,27. Moreover, water was believed to catalyse the ORR reaction in aprotic Li-O2 battery resulting in the formation of LiOH28, and good cycling performance was achieved in humid O2 (ref. 29). Therefore, owing to the complexity of the reaction, the battery chemistry of water-contaminated aprotic Li-O2 cell remains to be elucidated30.
Here we carefully investigate the influence of water on the battery chemistry of aprotic Li-O2 cells when LiI is used as the OER redox catalyst. With the help of a Li+-conducting ceramic membrane, we safely exclude any side-effects that may incur by the reactions of water and redox mediators with the lithium anode. One finding is that along with LiOH, lithium hydroperoxide (LiOOH) is detected to be one of the predominant discharge products, heralding a distinct battery chemistry for water-contaminated Li-O2 batteries. As a rarely reported lithium compound, we study the crystallographic and spectroscopic characteristics of LiOOH both experimentally and theoretically, and find LiOOH presents much faster reaction kinetics towards I3− as compared with Li2O2 and LiOH. A two-stage charging process is proposed in terms of the detailed studies to elucidate the mechanism of Li-O2 cells involving water in the battery reactions.
Results
Identification and characterization of LiOOH
Our study started off from the assessment of the reactivity of various discharge products of Li-O2 batteries towards oxidation by I3−. Titrations of Li2O2 and LiOH with I3− were firstly conducted in dimethoxyethane (DME) solutions. Despite the slightly more positive potential I3− stays intact in Li2O2 suspension even after stirring for 12 h (Supplementary Fig. 1), which can be explained by the sluggish reaction between the two species in accordance with our previous computational and experimental observation17. A similar phenomenon was observed here for LiOH, suggesting that in vigorously dried aprotic solution I3− cannot be removed by Li2O2 and LiOH within the timescale of the titration. After adding H2O into the above Li2O2 suspension, the colour of I3− was quickly bleached as a result of rapid reduction of I3−. In contrast, the presence of H2O in LiOH solution did not cause an appreciable change, whereas the bleaching happened instantaneously when H2O2 was added into the LiOH solution, although H2O2 itself was found to be stable with I3− (Supplementary Fig. 1). To understand the above phenomena, we noticed the following two reactions for Li2O2 and LiOH have been reported, respectively31,32:
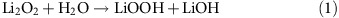 |
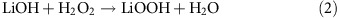 |
In the presence of surplus water, the solid products of both reactions are expected to be in the hydrated form, LiOOH·H2O and LiOH·H2O33. Interestingly, if not coincidentally, both reactions point to the same product—LiOOH, implying that the bleaching of I3− might be induced by this compound. Though its crystal structure is not known, various authors suggest it to be the intermediate when producing Li2O2 by reactions of H2O2 with Li alcoholates in the corresponding alcohol (that is, methanol, ethanol and references therein)34.
Hence, on the basis of the above titration tests, the reactivity of LiOOH is the highest and LiOH is the lowest towards oxidation by I3−. Whereas the comparison is arguably supported by the titrations, the formation of LiOOH, its reaction with I3−, and more importantly the existence of the compound in Li-O2 battery, need to be unambiguously characterized and identified.
To discern the characteristics of LiOOH from LiOH and Li2O2, the three compounds were concertedly characterized by synchrotron X-ray diffraction (XRD), Raman and Fourier transform infrared (FTIR) spectroscopy measurements with attenuated total reflection (ATR) mode, for which both LiOH and Li2O2 were used as received, whereas wet powder of LiOOH was prepared by a simple reaction between LiOH and H2O2 in water/DME following reaction (2).
The structures of four different Li compounds were characterized by powder XRD (Supplementary Fig. 2). The diffraction pattern of LiOOH is clearly discriminated from those of LiOH, Li2O2 and Li2O. In order to figure out the structure of the obtained LiOOH, high-resolution powder XRD was performed at 11-BM, Advanced Photon Source, Argonne National Lab (Fig. 1a). As seen from the LeBail fit results in Supplementary Fig. 3, the diffraction pattern of the LiOOH phase could be well indexed to a triclinic structure with P1 or 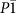 symmetry and lattice parameters: a=6.3688 Å, b=6.0878 Å, c=3.2074 Å, α=79.598°, β=101.832°, γ=102.311°, volume=117.69 Å3. The goodness of indexing, F(28), is as high as 795.1 with a zero-shift as small as −0.0009°, which together indicates that the fitting result is highly reliable. LeBail fits of an additional high-resolution XRD pattern for the 2θ range up to 22° (2θ angle) showed result very close to those derived from the lower 2θ angle range.
symmetry and lattice parameters: a=6.3688 Å, b=6.0878 Å, c=3.2074 Å, α=79.598°, β=101.832°, γ=102.311°, volume=117.69 Å3. The goodness of indexing, F(28), is as high as 795.1 with a zero-shift as small as −0.0009°, which together indicates that the fitting result is highly reliable. LeBail fits of an additional high-resolution XRD pattern for the 2θ range up to 22° (2θ angle) showed result very close to those derived from the lower 2θ angle range.
Figure 1. Characterizations of the various lithium compounds and the discharge product with or without water contamination.

(a) Rietveld refinement of the high-resolution X-ray diffraction pattern of the chemically synthesized LiOOH·H2O phase. The wavelength is λ=0.41423±0.00004 Å. The R factors of Rietveld refinement are Rp=6.10 %, Rwp=7.86%, χ2=2.93. (b) Raman spectra of Li2O2, LiOH, LiOOH·H2O and that of the discharge product collected from the cathode of a Li-O2 cell without water or containing 9.1 vol.% H2O in the electrolyte.
The crystal structure of LiOOH was solved from powder data starting from a comparison of the atomic arrangements of various compounds with related chemical compositions and reduced cells, noting that the close similarity of lattice parameters a and b as well as of β and γ suggests that the structure may be seen as a distorted variant of a monoclinic C2/m or even an orthorhombic body-centred structure as originally proposed for LiOOH·H2O by Cohen33. Rietveld refinements of several of these starting models converged to the structure shown in Fig. 2 with profile R and Chi2 values (Rwp=7.86%, Rp=6.10%, χ2=2.93) closely approximating those of the model-free LeBail fit (Supplementary Fig. 3) clarifying that for the available data quality no alternative structure model can yield a significantly closer match. Accordingly, the bond valence sums (when using soft BV parameters)35,36,37 of all atoms in the refined structure are close to the expectation value leading to a low global instability index of GII=0.077 underlying the plausibility of the structure model. Geometry optimization of the result from the Rietveld refinement by DFT confirmed that this structure is metastable. Details of the structure parameters resulting from both the Rietveld refinement and the DFT geometry optimization are given in the supplementary material (Supplementary Tables 1 and 2). The structure consists of chains of H-bridged OOH− ions as well as of Li+ ions arranged along the c-direction (O−H···O distances 1.12 and 1.46 Å from Rietveld refinement, or 1.07 and 1.46 Å from the DFT data), where each Li+ is tetrahedrally coordinated by O atoms of two water molecules and two HOO− anions. The atomic arrangement is closely related to the one reported earlier for monoclinic LiOH·H2O (Space group C2/m), as becomes more clearly visible when the primitive cell is used for that structure (see Fig. 2c). The structural similarity of LiOOH·H2O and LiOH·H2O also leads to similar stability. At 0 K the LiOOH·H2O should according to the DFT calculations be marginally stable against the decomposition into LiOH·H2O and ½ O2, whereas at ambient conditions, the entropically favoured decomposition of LiOOH·H2O proceeds easily. In the presence of CO2 from ambient air LiOH·H2O then reacts further to form Li2CO3 (Supplementary Figs 4 and 5).
Figure 2. Comparison of crystal structures between LiOOH·H2O and LiOH·H2O.

Crystal structure of LiOOH·H2O as derived from (a) Rietveld refinement of synchrotron X-ray powder data and (b) from DFT refinement. (c) The crystal structure of LiOH·H2O given by Hermannson et al.44 Here the primitive cell is shown to emphasize the close relation between the structures of both phases. (O: red; H: grey, Li: magenta). Broken lines indicate the hydrogen bonds stabilizing the structures.
The Raman spectrum shown in Fig. 1b for the LiOOH·H2O sample reveals a distinct characteristic peak at around 860 cm−1, assigned to the stretching of O-O bond based on the density functional theory (DFT) calculations (Supplementary Figs 6 and 7). In comparison, the O–O bond stretching of Li2O2 and H2O2 molecules is observed at ∼790 and 877 cm−1 (Supplementary Fig. 8), respectively, just straddling that of LiOOH·H2O. Other fingerprint peaks for LiOOH·H2O were also observed at 80–150 cm−1, implying LiOOH·H2O is indeed a different species from LiOH and Li2O2. Characteristic IR responses of LiOOH·H2O were also detected in the FTIR measurement (Supplementary Fig. 9), where the peak at 1,643 cm−1 is identified as a H2O bending mode. Although other vibrations are not yet specified owing to a lack of reference data, the distinct spectra well evince LiOOH·H2O as a new species relevant to Li-O2 batteries.
Electrochemical properties of LiOOH
To investigate the catalytic effect of I3− on the oxidation of the above lithium compounds, rotating disk electrode (RDE) was employed to probe the reactions, in which the powders of LiOOH·H2O, Li2O2 and LiOH were dispersed in LiI electrolyte. As shown in Fig. 3a, in the presence of LiOOH·H2O suspension the limiting current for I− oxidation on RDE is enhanced by nearly 10 times as compared with those with LiOH and Li2O2. The direct oxidation of LiOOH·H2O on RDE could be excluded as it generated almost zero current in the absence of LiI (Fig. 3a). Thus, such a considerable enhancement is rationalized by the catalytic reaction between the formed I3− and LiOOH in the vicinity of RDE, which rapidly regenerates I−. In contrast, the presence of LiOH or Li2O2 suspension has little influence on the reaction of I3−, consistent with the titration experiment.
Figure 3. Electrochemical properties of LiOOH and the comparisons with LiOH and Li2O2.

(a) RDE measurements of 1 mM LiI in 0.1 M LiTFSI/DME with 30 mM LiOOH·H2O, Li2O2 or LiOH dispersed in the solution. For comparison, the same measurements were conducted in the absence of LiI or lithium compounds suspension in the electrolyte. The rotating rate was 1,200 r.p.m. and the scan rate was 0.01 V s−1. The inset illustrates the catalytic reaction between LiOOH and I3− upon RDE measurement. (b) The charging curves of Li-LiOOH, Li-Li2O2 and Li-LiOH cells. Lithium foil was used as anode. LiOOH·H2O precipitate, Li2O2 or LiOH powder in great excess to LiI in catholyte was loaded onto the cathode before the cells were assembled. The catholyte was 1 ml 0.5 M LiTFSI/DME containing 40 mM LiI. A LAGP membrane was used to segregate the two cell compartments. The cells were first charged at a constant current of 0.1 mA cm−2 for 21 h, and then followed by GITT measurement (2 h charging at the same current plus 10 min resting).
The reactions of the various lithium compounds with I3− were substantiated by battery charging test. The cell consists of a cathodic and an anodic compartment, which are separated by a piece of LAGP ceramic membrane (Supplementary Fig. 10a). The powders of LiOOH·H2O, Li2O2 and LiOH were loaded on the cathode (carbon felt) before it was fabricated into the cell. The use of Li+-conducting membrane is crucial as it prevents I3−, water and oxygen from crossing-over and parasitically reacting with the Li metal in the anodic side. As shown in Fig. 3b, the theoretical charging time of I− to I2 is ∼7 h, whereas all the three cells present significantly longer charging process (>30 h), indicating the lithium compounds are involved in the reactions contributing to the charging capacity. In the absence of the above lithium compounds, the reactions of I− in DME electrolyte exhibit two distinct voltage plateaus at ∼3.20 and 3.70 V, corresponding to the formation of I3− and higher order polyiodide to eventually I2, respectively (Supplementary Fig. 11)38. In comparison, the LiOOH cell reveals only one prolonged charging plateau at ∼3.20 V, which is rational in terms of the titration experiment that the formed I3− could instantaneously be reduced back to I− by LiOOH for extended charging, and the cell voltage is determined by the I−/I3− redox reaction on the electrode. So the overall reaction on the cathode only involves I−/I3−-mediated oxidation of LiOOH, and the capacity is limited by the quantity of material loaded. In comparison, the Li2O2 cell presents two charging plateaus resembling that of the pure LiI cell (Supplementary Fig. 11), but with the second plateau greatly extended. Such a phenomenon has previously been observed in redox flow lithium–oxygen battery (RFLOB)17 and is consistent with the titration that in aprotic electrolyte I3− is unable to rapidly oxidize Li2O2, which requires a stronger oxidizer such as I2 formed at the higher voltage plateau. The charging of the LiOH cell is fairly similar to that of Li2O2, except for slightly larger overpotentials at the higher voltage plateau, presumably a result of sluggish reaction between LiOH and I2, or more complex reactions22,23,24.
To eliminate the overpotentials imposed by the membrane and other cell components during the charging process, galvanostatic intermittent titration technique (GITT) measurement was performed with the cells after 20 h charging. The relaxed cell voltage is on average ∼2.95 and 3.55 V for the LiOOH and Li2O2 cells, and is slightly higher for the LiOH cell, broadly in agreement with the respective potential of I−/I3− and I3−/I2 as determined by voltammetry (Supplementary Fig. 12). The above charging tests corroborate the previous comparison of the three compounds with I3− and that LiOOH<Li2O2<LiOH in terms of oxidation capability.
Characterization of Li-O2 battery with water contamination
In order to verify the formation of LiOOH as an oxygen reduction product in moisture-contaminated Li-O2 battery, water was deliberately introduced into the aprotic catholyte of Li-O2 cells. As shown in Fig. 4a, the presence of water seems to be advantageous to the reduction of overpotential during the 10 h discharging process. With increasing water content in the electrolyte, the discharging plateau shifts upwards. The charging curves for all the cells predominantly present two voltage plateaus at ∼3.50 V and 3.85 V, respectively. The lower voltage plateau is assigned to the oxidation of I− to I3−, whereas owing to surface passivation of the electrode as generally observed in aprotic Li-oxygen batteries, the overpotential is considerably higher than that observed in Fig. 3b. The maximum charging time for I− to I3− is ∼4.5 h, so the extended charging would be a result of the catalytic reaction of I3− with the discharge product, in which LiOOH was identified by Raman spectroscopy when probing the electrode after 10 h discharge (Fig. 1b).
Figure 4. Electrochemical performance of Li-O2 cells with different H2O content in the catholyte.

(a) The charge–discharge curves of water-contaminated Li-O2 batteries. The catholyte consisted of 0.5 ml 50 mM LiI and 0.5 M LiTFSI in DME. Different amount of water was introduced in the catholyte. D:H denotes the volume ratio of DME and H2O. (b) The cyclic voltammograms of 2.5 mM LiI in 0.5 M LiTFSI/DME and 2.5 mM EVI2 in 0.5 M LiTFSI/ DEGDME-DMSO (1:1 v/v). Pt disc and plate were used as the working and counter electrode, respectively. The scan rate was 0.02 V s−1. (c) The charge–discharge curves of redox flow Li-O2 battery using EVI2 as the redox mediator. The catholyte consisted of 4 ml 15 mM EVI2 and 0.5 M LiTFSI in DEGDME-DMSO (1:1 v/v) with or without 9.1 vol.% H2O. The anolyte was 0.5 M LiTFSI in DEGDME. The current was set at 0.1 mA cm−2 for all the above galvanostatic measurements.
However, the prolonged additional 4–5 h charging time at ∼3.50 V could not account for the 10 h discharge, for which around half the discharge product seemingly remains intact with I3−. When the cells were further charged to a higher voltage, where the reaction of I3−/I2 prevails, a second voltage plateau appeared at ∼3.85 V with evidently extended charging for another 4–5 h. On the basis of the previous charging tests, either LiOH or Li2O2 may contribute to this process, whereas considering Li2O2 is instable in the presence of water, this extra capacity is deemed to be stemming from LiOH (or LiOH·H2O). This is reasonable in terms of reaction (1), and that the discharge product of aprotic Li-O2 battery, Li2O2, is converted into two distinct compounds co-existing in water-contaminated cells, of which LiOOH·H2O reacts with I3− at a lower voltage, whereas LiOH·H2O reacts with I2 at a higher voltage in a two-stage charging process. The presence of LiOOH and LiOH in the discharge product was confirmed by ATR-FTIR measurement (Supplementary Fig. 13), in which the characteristic peaks of LiOH and LiOOH·H2O are clearly identified upon redox-assisted ORR reaction in the presence of water.
When the charging curves in Fig. 4a are examined closely, one may notice multiple voltage steps at each voltage plateaus, which is ascribed to the direct oxidation of LiOOH·H2O and LiOH·H2O on the cathode alongside the reactions with redox mediators in the electrolyte. To avoid the complexity and ambiguity, a RFLOB cell was fabricated, which employed ethyl viologen diiodide (EVI2) as a bifunctional redox mediator for both the ORR and OER reactions (Fig. 4b). One significant advantage for RFLOB over the conventional Li-O2 battery is that upon discharging O2 is fed into a gas diffusion tank (GDT) in which it is reduced by EV+ when the catholyte circulates through (Supplementary Fig. 10c). As a result, the discharge product is chemically formed in the tank instead of being deposited on the electrode surface. Upon charging, the parallel reactions of these materials on the electrode are thus obviated. As the voltage profiles in Fig. 4c shows, the presence of water in the catholyte considerably reduces the cell overpotential, similar to that observed in static cells. The discharging process of the flow cells involves the reduction of EV2+ on the electrode and the associated ORR reaction in GDT. Upon charging, the dry cell exhibits two voltage plateaus with the first one relating to the oxidation of I− to I3−, and the second with extended capacity originating from the oxidation of I3− to I2 and the reaction between Li2O2 and I2, which is consistent with our previous study17. In comparison, although the water-contaminated flow cell also shows two clearly defined plateaus during the charging process, the capacities for both plateaus stretch nearly equally beyond that of the redox mediators, implying two distinct reactions with I3− and I2 take place at the two plateaus respectively.
The redox-targeting reactions of LiOOH with I3− and LiOH with I2 were further investigated with UV-Vis and mass spectrometry. After mixing with LiOOH·H2O suspension in DME, the characteristic absorption peak of I3− at 364 nm (extended to the visible region, Supplementary Fig. 14a) vanished. As a result, the solution became nearly colourless. Meanwhile, the mass spectrometric measurement in Supplementary Fig. 15a shows that O2 evolves instantaneously upon mixing I3− with LiOOH·H2O. In a separate test, after adding excessive LiOH into a solution of I2 in DME/H2O (10:1) and stirring for 1 h, the absorption of the I2 solution became fairly identical to that of I3− (Supplementary Fig. 14b). That is, the absorption at 400–500 nm was greatly attenuated with only the characteristic peak of I3− present, indicating the existence of I3− after reaction. We noticed IO3− was detected to be the main product of the reaction between I2 and LiOH in water-based electrolyte in the literature26. The involved reaction is, 3I2+6LiOH→5LiI+LiIO3+3H2O. Considering both I− and IO3− are colourless, the above reaction seems unlikely to be predominant in the DME/H2O (10:1)-based electrolyte. The mass spectrometric measurement of the reaction between LiOH and I2 was conducted in two different solutions. As shown in Supplementary Fig. 15b, oxygen evolution was observed instantaneously after I2 was injected into 2 M LiOH suspension in DME/H2O (10:1), further confirming the irrelevance of the above reaction in the DME/H2O (10:1)-based electrolyte. Therefore, O2 evolution is deemed part of the reactions of LiOOH with I3− and LiOH with I2 during the charging process of water-contaminated Li-O2 cells.
Two-stage charging reactions in water-contaminated Li-O2 cells
On the basis of the above analysis, the following reactions are thus proposed to expound the discharging and the two-stage charging processes in the water-contaminated cell:
Discharging process (ORR reaction):
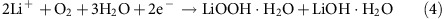 |
Charging process (OER reaction):
 |
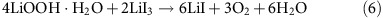 |
 |
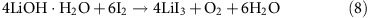 |
Reaction (4) indicates that equimolar amounts of LiOOH and LiOH are formed in the discharging process, which is evidenced by the identical capacity extension in the two-stage charging process in Fig. 4a,c. OOH− has been proposed in battery reactions in several studies as a result of H2O dissociation or electrolyte decomposition25,28,32,39, whereas none of them explicitly indicates LiOOH or LiOOH·H2O as a distinct discharge product, nor its structural and electrochemical properties. Besides, we have noticed that water has substantial influence on the morphology of the discharge product. When 9.1 vol.% water was added into the electrolyte, rod and cube-like crystals with clear edges were observed after discharge, whereas crystals formed in dry electrolyte tested in separate cells are agglomerated into round-shaped particulates (Supplementary Fig. 16). Reactions (5) and (7) describe the two-stage charging process, associated with the redox-targeting reactions with LiOOH·H2O (6) and LiOH·H2O (8), respectively. Reaction (6) represents a disproportionation reaction with ¾ of the peroxide being oxidized to O2, and the remaining ¼ reduced to H2O. Additional ¼ O2 is produced in reaction (8) via a 4-electron process, making up the total O2 consumption in the discharging reaction (4). A consequence of such two-step reactions is that both the voltage efficiency and energy efficiency are compromised.
Discussion
The above study unveils an intriguing picture of the ORR and OER reactions for water-contaminated Li-O2 batteries, for which the transfer of protons from water leads to the formation of a new compound—LiOOH·H2O, while the left-over OH− prompts the formation of an equimolar amount of LiOH (or LiOH·H2O). More precisely, such a situation might be called water contamination at ‘neutral' conditions. It would be interesting to consider the scenarios under varying proton concentrations. For instance, in weakly acidic condition, it is likely that 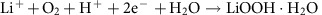 , so that LiOOH·H2O would again be the discharge product with pH-dependent equilibrium potential. However, when excessive protons present in the catholyte, it appears plausible that
, so that LiOOH·H2O would again be the discharge product with pH-dependent equilibrium potential. However, when excessive protons present in the catholyte, it appears plausible that 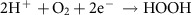 . That is, the remaining Li atom in LiOOH·H2O could be knocked out to form H2O2. At the other extreme, when extra OH− is introduced into the catholyte, the proton in LiOOH would be removed and as reported for water-based alkaline electrolytes40, the formation of Li2O2 would be favoured, for which the redox potential of the overall reaction becomes pH-independent. Figure 5 illustrates the plausible battery reactions of ‘proton-contaminated' Li-O2 cells which may predominate at different [H+]. We believe systematic studies on the impact of protons on the ORR and OER reactions would disclose deeper insights into the mechanistic understanding of the battery chemistry of Li-air batteries.
. That is, the remaining Li atom in LiOOH·H2O could be knocked out to form H2O2. At the other extreme, when extra OH− is introduced into the catholyte, the proton in LiOOH would be removed and as reported for water-based alkaline electrolytes40, the formation of Li2O2 would be favoured, for which the redox potential of the overall reaction becomes pH-independent. Figure 5 illustrates the plausible battery reactions of ‘proton-contaminated' Li-O2 cells which may predominate at different [H+]. We believe systematic studies on the impact of protons on the ORR and OER reactions would disclose deeper insights into the mechanistic understanding of the battery chemistry of Li-air batteries.
Figure 5. The proposed mechanism of proton-contaminated Li-O2 batteries and Pourbaix diagram with different proton concentration.

The proposed mechanisms of the charging and discharging processes in ‘proton-contaminated' aprotic Li-O2 battery at (a) acidic, (b) neutral and (c) basic conditions. Iodide is included to mediate the OER reaction. Elements in the ball-and-stick model: red-oxygen; purple-lithium; write-hydrogen; yellow-iodine. (d) A sketch of the Pourbaix diagram showing the predominant battery chemistries of Li-O2 cell at different [H+]. The 4-electron process shown in dotted line is just for reference and the potential relative to that of the two-electron process has no physical significance. HX is the acid introduced in the electrolyte.
Considering the LiOOH·H2O has greater reactivity towards the OER reaction, which substantially brings down the charging overpotential (Fig. 3b), it is of immediate importance to contemplate the implications of the new compound for the operation of the Li-O2 battery. However, this seems not intuitively straightforward. A formation of LiOOH·H2O during the discharging process requires protons which are not available from the anode. Although the moisture in air could be a natural source of protons, the accumulation of water in the catholyte during the charging process makes it unsustainable. In addition, the formation of LiOOH·H2O in water- or alcohol-containing electrolytes may provide an alternative approach of reactions for water-based Li-O2 battery, in which the 4-electron process is generally considered. As water could be precluded as a reactant from the 2-electron process of LiOOH (that is, in acidic and basic conditions), it may in theory boost the energy density and energy efficiency of water-based Li-O2 cells.
A distinct battery chemistry was discovered for water-contaminated Li-O2 battery, from which a new lithium compound—LiOOH·H2O, was identified as a predominant oxygen reduction product and structurally characterized. When iodide is used as the OER redox catalyst in the water-contaminated Li-O2 cell, the equimolar amounts of LiOOH·H2O and LiOH·H2O formed in the discharging process are oxidized stepwise by I3− and I2, leading to a two-stage charging process. This study discloses that the moisture fed into the cell does not pose immediate adverse impact to the battery operation, so long as the lithium anode is properly protected. On the basis of this new battery chemistry, a panoramic view of the ORR/OER reactions at different [H+] is conceived, which is anticipated to provide deeper insights into the mechanistic understanding of the chemistry of Li-air batteries. For that, we believe a more systematic study would be desired in future to understand the factors such as water content, [H+], type of redox mediators and so on, that influence the formation of LiOOH·H2O or other oxygen reduction products.
Methods
Materials
LiOH (98%, Sigma-Aldrich), Li2O2 (90%, Sigma-Aldrich), and H2O2 (35% (w/w) in H2O, Alfa Aesar) were used in the titration experiments. LiOH (98%, Sigma-Aldrich) and H2O2 (35% (w/w) in H2O, Alfa Aesar) were used for the preparation of LiOOH·H2O. DME(99%, Sigma-Aldrich) and dimethyl carbonate (anhydrous, >99%, Sigma-Aldrich) were used to get the precipitation of LiOOH·H2O. Diethylene glycol dimethyl ether (DEGDME, 99%, Sigma-Aldrich), DME (99%, Sigma-Aldrich), dimethyl sulfoxide (DMSO, 99.9%, Sigma-Aldrich) and lithium bis(trifluoromethane)sulfonimide (LiTFSI, 99.95%, Sigma-Aldrich) were used as solvent and lithium salt for electrolyte preparation. LiI (99%, Sigma-Aldrich) and EVI2 (99%, Sigma-Aldrich) were used as redox mediators in Li-O2 batteries. A Li+-conducting ceramic membrane (LAGP, area 2 cm × 2 cm, thickness 0.5 mm) was used as separator in Li-O2 batteries. Prior to use, all the above chemicals were stored in an argon-filled glove box without exposure to air.
Preparation of LiOOH·H2O
0.196 g LiOH (98%, Sigma-Aldrich) was added into 2.6 ml deionized water. Then the solution was stirred until LiOH powder was dissolved completely. With good stirring, 1.4 ml H2O2 solution (35% w/w, Alfa Aesar) were added dropwise into the above LiOH solution over 30 min, which resulted in 2 M LiOOH·H2O solution in water. In order to retrieve solid LiOOH·H2O from the solution, 2 ml DME were added dropwise into 1 ml of the above LiOOH·H2O solution with stirring for 10 min. During the process, a white LiOOH·H2O precipitate appeared and sedimented. In the RDE and battery measurements, 2 M LiOOH·H2O solution in water was employed directly to prepare suspension of LiOOH·H2O. For the Raman and XRD measurements, wet LiOOH·H2O particles were separated from the supernatant after centrifugation and gently dried in a vacuum oven at room temperature for 12 h.
Assembly of Li-O2 battery
Assembly of static Li-O2 battery: Lithium foil and carbon felt were used as the anode and cathode, respectively. The electrochemical cell was fabricated by sandwiching the lithium foil and carbon felt in a cell stack, in which the two electrodes were separated by a LAGP membrane mounted on a Teflon frame (Supplementary Fig. 6). The effective area of the membrane was 1 × 1 cm2. The anodic and cathodic end plates were made of stainless steel and titanium metal (with holes as O2 inlet and outlet), respectively, to prevent corrosion caused by the redox species. The anodic compartment was filled with electrolyte consisting of 0.5 M LiTFSI/DEGDME and the cathodic compartment was filled with 0.5–0.7 ml of 0.5 M LiTFSI/DME with around 40–50 mM LiI and varying quantity of water. For water-containing electrolyte, water was mixed uniformly with the electrolyte before injected into the cell. After the Li-O2 cell was assembled, the electrolyte was introduced into the cell, which was then tested in oxygen bag filled with pure oxygen.
Assembly of redox flow Li-O2 battery: Redox flow Li-O2 cell consists of a battery stack and a GDT. The procedure of fabricating the cell stack is the same to the static Li-O2 cell. The cell stack was connected to the GDT tank by Teflon tubing, through which the catholyte was circulated between the cell and GDT tank by a peristaltic pump (Supplementary Fig. 6). The anolyte was 0.5 M LiTFSI/DEGDME. The catholyte was 15 mM EVI2 in 0.5 M LiTFSI/DEGDME-DMSO (1:1 v/v) with or without 9.1 vol.% H2O. DMSO was used to reduce the volatility of the catholyte. The volume of catholyte was 4 ml. Constant O2 flow was provided to the GDT tank (O2 pressure ∼1 atm) during the discharging process.
Electrochemical measurements
The RDE measurements were conducted by using a PINE AFMSRCE rotator. The electrolyte was 1 mM LiI in 0.1 M LiTFSI/DME with 30 mM LiOOH·H2O, Li2O2 or LiOH dispersed in the solution. For comparison, the same measurements were conducted in the absence of LiI or lithium compounds suspension in the electrolyte solution. Pt disc (diameter 12 mm) and Pt plate were used as working and counter electrode, respectively. And Ag/AgNO3 electrode was used as reference electrode. The rotating rate was 1,200 r.p.m. and the scan rate was 0.01 V s−1.
The cyclic voltammetry measurements for 2.5 mM LiI in 0.5 M LiTFSI/DME and 2.5 mM EVI2 in 0.5 M LiTFSI/DEGDME-DMSO (1:1 v/v) were conducted with a scan rate of 0.02 V s−1. The working electrode was Pt disc electrode. Both counter and reference electrodes were Li metal. Differential pulse voltammetry measurements of LiI were conducted at a variety of water contents. The electrolyte was 5 mM LiI in 0.5 M LiTFSI/DME with different water contents (DME:H2O are 100:0, 100:1, 100:5, 100:10, 100:20, 100:30, 100:50, 100:100, v/v). The working and the counter electrode were Pt disc and Pt plate electrode, respectively. The reference electrode was Ag/AgNO3 electrode. The step potential is 0.005 V and the modulation amplitude is 0.025 V. All the above measurements were performed on an Autolab electrochemical workstation (Metrohm Autolab, PGSTAT302N). The charge and discharge tests were conducted on an Arbin battery tester. The battery was tested at a constant current of 0.1 mA cm−2.
Other characterizations
Raman spectra were measured by a confocal Raman system with 532 nm laser excitation (WITec Instruments Corp, Germany). Samples for Raman spectroscopic measurement include gently dried LiOOH·H2O powder, powders of the as-purchased LiOH and Li2O2, as well as 35 % H2O2 solution. The Raman spectrum of the cathode (carbon felt) in the Li-O2 batteries was also measured immediately after disassembling the fully discharged static Li-O2 cells. For the Li-O2 cells with water-free electrolyte, the cathode was washed and dried in a vacuum chamber prior to Raman measurement. All the above samples were loaded on a piece of sapphire for Raman measurement. FTIR spectra were collected via PerkinElmer Frontier MIR/FIR system by 16 scans with a nominal resolution of 1 cm−1 through an ATR mode. In the in situ measurement of the redox-targeting reaction product between EV+ and O2, a flow cell with two inlets and one outlet (two for liquid and one for oxygen) was used. UV-Vis spectroscopic measurements were conducted on a UV-Vis spectrophotometer (Shimadzu 1800). Mass spectrometric measurements were conducted on a Hiden analytical QGA (HAS-301-1376A). There are one outlet and two inlets of the reactor for carrying gas and injecting reactant.
The high-energy XRD measurements were performed on the beam line 11-ID-C at the Advanced Photon Source, Argonne National Laboratory. A monochromator with a Si (113) single crystal was used to provide an X-ray beam with the energy of 115 keV. High-energy X-ray with a beam size of 0.2 mm × 0.2 mm and wavelength of 0.10725 Å was used to obtain two-dimensional diffraction patterns in the transmission geometry. X-rays were collected with a Perkin-Elmer large-area detector placed at 1,800 mm from the sample. The obtained two-dimensional diffraction patterns were calibrated using a standard CeO2 sample and converted to one-dimensional patterns using Fit2D software. In order to figure out the structure of the obtained LiOOH·H2O sample, the high-resolution X-ray powder diffraction pattern of LiOOH·H2O was taken at 11-BM, Advanced Photon Source, Argonne National Lab, whereas the wavelength of the X-ray is 0.41423 Å. The samples were measured in air without protection. For the stability test, XRD measurements were carried out on a Bruker D8 with Cu Kα1 radiation (λ=0.154059, nm). The samples were measured in air without protection.
Theoretical calculations
DFT calculations were carried out with B3LYP hybrid exchange-correlation functional in combination with the quadruple-zeta polarized valence basis set augmented with diffuse functions, aug-cc-pVQZ, using the Gaussian 09 programme suite41,42. The scaled quantum mechanics force field procedure was used to analyse vibrational bands of all fundamentals. The calculated frequencies were scaled by a factor of 0.9852 for frequencies below 2,000 cm−1 (ref. 43). Scaling harmonic vibrational frequencies is an effective way to facilitate comparison with experimentally observed frequencies. A scaling factor of 0.9852 was recommended for the B3LYP/aug-cc-pVQZ level of theory for which the corresponding root mean square error relative to the experimental frequencies was report to be 8 cm−1 (ref. 43). The calculated scaled frequencies for the O–O stretch are: 838.0 (LiOOH·H2O), 827.6 (LiOOLi), 934.9 (HOOH) cm−1. The structure and vibration frequencies of LiOOH·H2O are also verified against partial phonon density of states generated using Phonopy package with Vienna ab initio simulation package and GGA-PBE exchange and correlation potential.
Data availability
The authors declare that data supporting the findings of this study are available within the paper and its supplementary information file or from the corresponding author on reasonable request.
Additional information
How to cite this article: Zhu, Y. G. et al. Proton enhanced dynamic battery chemistry for aprotic lithium–oxygen batteries. Nat. Commun. 8, 14308 doi: 10.1038/ncomms14308 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Supplementary Figures 1-16, Supplementary Tables 1-2 and Supplementary References
Acknowledgments
This research was supported by the National Research Foundation, Prime Minister's Office, Singapore, under its Competitive Research Program (CRP Awards No NRF-CRP8-2011-04 and NRF-CRP10-2012-06). We thank Dr Du Yuan for his assistance on the FTIR measurement. We thank Professor Hui Ying Yang and Dr Linfeng Sun from Singapore University of Technology and Design for their assistance on Raman measurement.
Footnotes
The authors declare no competing financial interests.
Author contributions Q.W. conceived the study and supervised the work. Y.G.Z. designed and performed most of the experiments. Q.L., Y. Rong and Y. Ren conducted synchrotron XRD measurements and data analysis. S.A. solved the crystal structure of LiOOH·H2O. C.J. provided some scientific suggestion. X.X. prepared the LAGP membrane. J.Y., L.-J.Y., A.K. and H.C. did the DFT calculations. Y.G.Z., S.A. and Q.W. wrote the paper.
References
- Girishkumar G., McCloskey B., Luntz A., Swanson S. & Wilcke W. Lithium-air battery: promise and challenges. J. Phys. Chem. Lett. 1, 2193–2203 (2010). [Google Scholar]
- Abraham K. M. & Jiang Z. A polymer electrolyte-based rechargeable lithium/oxygen battery. J. Electrochem. Soc. 143, 1–5 (1996). [Google Scholar]
- Read J. Ether-based electrolytes for the lithium/oxygen organic electrolyte battery. J. Electrochem. Soc. 153, A96–A100 (2006). [Google Scholar]
- Ogasawara T., Débart A., Holzapfel M., Novák P. & Bruce P. G. Rechargeable Li2O2 electrode for lithium batteries. J. Am. Chem. Soc. 128, 1390–1393 (2006). [DOI] [PubMed] [Google Scholar]
- Ottakam Thotiyl M. M., Freunberger S. A., Peng Z. & Bruce P. G. The carbon electrode in nonaqueous Li-O2 cells. J. Am. Chem. Soc. 135, 494–500 (2012). [DOI] [PubMed] [Google Scholar]
- Lu Y.-C et al. Platinum-gold nanoparticles: a highly active bifunctional electrocatalyst for rechargeable lithium-air batteries. J. Am. Chem. Soc. 132, 12170–12171 (2010). [DOI] [PubMed] [Google Scholar]
- Liu S. et al. Direct growth of flower-like δ-MnO2 on three-dimensional graphene for high-performance rechargeable Li-O2 batteries. Adv. Energy Mater. 4, 1301960 (2014). [Google Scholar]
- Thotiyl M. M. O. et al. A stable cathode for the aprotic Li-O2 battery. Nat. Mater. 12, 1050–1056 (2013). [DOI] [PubMed] [Google Scholar]
- Li F. et al. Superior performance of a Li P2 battery with metallic RuO2 hollow spheres as the carbon-free cathode. Adv. Energy Mater. 5, 1500294 (2015). [Google Scholar]
- Chen Y., Freunberger S. A., Peng Z., Fontaine O. & Bruce P. G. Charging a Li-O2 battery using a redox mediator. Nat. Chem. 5, 489–494 (2013). [DOI] [PubMed] [Google Scholar]
- Bergner B. J., Schürmann A., Peppler K., Garsuch A. & Janek J. TEMPO: a mobile catalyst for rechargeable Li-O2 batteries. J. Am. Chem. Soc. 136, 15054–15064 (2014). [DOI] [PubMed] [Google Scholar]
- Lim H. D. et al. Superior rechargeability and efficiency of lithium-oxygen batteries: hierarchical air electrode architecture combined with a soluble catalyst. Angew. Chem. Int. Ed. 53, 3926–3931 (2014). [DOI] [PubMed] [Google Scholar]
- Sun D. et al. A solution-phase bifunctional catalyst for lithium-oxygen batteries. J. Am. Chem. Soc. 136, 8941–8946 (2014). [DOI] [PubMed] [Google Scholar]
- Feng N., He P. & Zhou H. Enabling catalytic oxidation of Li2O2 at the liquid–solid interface: the evolution of an aprotic Li-O2 battery. ChemSusChem 8, 600–602 (2015). [DOI] [PubMed] [Google Scholar]
- Kwak W.-J et al. Understanding the behavior of Li-oxygen cells containing LiI. J. Mater. Chem. A 3, 8855–8864 (2015). [Google Scholar]
- Liu T. et al. Cycling Li-O2 batteries via LiOH formation and decomposition. Science 350, 530–533 (2015). [DOI] [PubMed] [Google Scholar]
- Zhu Y. G. et al. Dual redox catalysts for oxygen reduction and evolution reactions: towards a redox flow Li–O2 battery. Chem. Commun. 51, 9451–9454 (2015). [DOI] [PubMed] [Google Scholar]
- Gao X., Chen Y., Johnson L. & Bruce P. G. Promoting solution phase discharge in Li-O2 batteries containing weakly solvating electrolyte solutions. Nat. Mater. 15, 882–888 (2016). [DOI] [PubMed] [Google Scholar]
- Zhu Y. G., Wang X., Jia C., Yang J. & Wang Q. Redox-mediated ORR and OER reactions: redox flow lithium oxygen batteries enabled with a pair of soluble redox catalysts. ACS Catal 6, 6191–6197 (2016). [Google Scholar]
- Aetukuri N. B. et al. Solvating additives drive solution-mediated electrochemistry and enhance toroid growth in non-aqueous Li–O2 batteries. Nat. Chem. 7, 50–56 (2015). [DOI] [PubMed] [Google Scholar]
- Schwenke K. U., Metzger M., Restle T., Piana M. & Gasteiger H. A. The influence of water and protons on Li2O2 crystal growth in aprotic Li-O2 cells. J. Electrochem. Soc. 162, A573–A584 (2015). [Google Scholar]
- Viswanathan V. et al. Comment on ‘Cycling Li-O2 batteries via LiOH formation and decomposition'. Science 352, 667–667 (2016). [DOI] [PubMed] [Google Scholar]
- Shen Y., Zhang W., Chou S.-L & Dou S.-X. Comment on ‘Cycling Li-O2 batteries via LiOH formation and decomposition'. Science 352, 667–667 (2016). [DOI] [PubMed] [Google Scholar]
- Liu T. et al. Response to comment on ‘Cycling Li-O2 batteries via LiOH formation and decomposition'. Science 352, 667–667 (2016). [DOI] [PubMed] [Google Scholar]
- Kwak W.-J et al. Li-O2 cells with LiBr as an electrolyte and a redox mediator. Energy Environ. Sci. 9, 2334–2345 (2016). [Google Scholar]
- Burke C. M. et al. Implications of 4 e–oxygen reduction via iodide redox mediation in Li-O2 batteries. ACS Energy Lett. 1, 747–756 (2016). [Google Scholar]
- Zhang W. et al. Promoting Li2O2 oxidation via solvent-assisted redox shuttle process for low overpotential Li-O2 battery. Nano Energy 30, 43–51 (2016). [Google Scholar]
- Li F. et al. The water catalysis at oxygen cathodes of lithium-oxygen cells. Nat. Commun. 6, 7843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. et al. A synergistic system for lithium-oxygen batteries in humid atmosphere integrating a composite cathode and a hydrophobic ionic liquid-based electrolyte. Adv. Funct. Mater. 26, 3291–3298 (2016). [Google Scholar]
- Aurbach D., McCloskey B. D., Nazar L. F. & Bruce P. G. Advances in understanding mechanisms underpinning lithium–air batteries. Nat. Energy 1, 16128 (2016). [Google Scholar]
- Choban A., Yurchuk I. & Lyavinets A. Effect of base nature on the oxidation of dimethyl sulfoxide with hydrogen peroxide in superbasic media. Russ. J. Gen. Chem. 82, 247–250 (2012). [Google Scholar]
- Black R. et al. Screening for superoxide reactivity in Li-O2 batteries: effect on Li2O2/LiOH crystallization. J. Am. Chem. Soc. 134, 2902–2905 (2012). [DOI] [PubMed] [Google Scholar]
- Cohen A. J. Observations on several compounds of lithium and oxygen. I1a,1b. J. Am. Chem. Soc. 74, 3762–3764 (1952). [Google Scholar]
- Vol'nov I. & Petrocelli A. Peroxides, superoxides, and ozonides of alkali and alkaline earth metals Springer (1966). [Google Scholar]
- Adams S. Practical considerations in determining bond valence parameters Springer (2013). [Google Scholar]
- Adams S. & Rao R. P. Simulated defect and interface engineering for high power Li electrode materials. Solid State Ion. 184, 57–61 (2011). [Google Scholar]
- Adams S. & Rao R. P. High power lithium ion battery materials by computational design. Phys. Status Solidi (a) 208, 1746–1753 (2011). [Google Scholar]
- Behl W. K. & Chin D. T. Electrochemical overcharge protection of rechargeable lithium batteries i. kinetics of iodide/tri-iodide/iodine redox reactions on platinum in solutions. J. Electrochem. Soc. 135, 16–21 (1988). [Google Scholar]
- Staszak-Jirkovský J. et al. Water as a promoter and catalyst for dioxygen electrochemistry in aqueous and organic media. ACS Catal. 5, 6600–6607 (2015). [Google Scholar]
- Matsui M. et al. A novel aqueous lithium-oxygen cell based on the oxygen-peroxide redox couple. Chem. Commun. 51, 3189–3192 (2015). [DOI] [PubMed] [Google Scholar]
- Frisch M. et al. Gaussian 09, revision A. 02 Gaussian, Inc. (2009). [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- Sinha P., Boesch S. E., Gu C., Wheeler R. A. & Wilson A. K. Harmonic vibrational frequencies: scaling factors for HF, B3LYP, and MP2 methods in combination with correlation consistent basis sets. J. Phys. Chem. A 108, 9213–9217 (2004). [Google Scholar]
- Hermansson K. & Thomas J. O. The experimental electron density in lithium hydroxide monohydrate. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 38, 2555–2563 (1982). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-16, Supplementary Tables 1-2 and Supplementary References
Data Availability Statement
The authors declare that data supporting the findings of this study are available within the paper and its supplementary information file or from the corresponding author on reasonable request.