

Abstract
Phosphoproteomic analysis of tumor samples has the potential to uncover significant insights into kinase signaling networks present in late stage prostate cancer that are complementary to genomic and transcriptomic approaches. Phosphoproteomics could potentially aid drug development in clinical trial design as well as provide utility for oncologists in the personalized therapeutic management of individual cancers through identifying novel biomarkers and druggable targets. Rapid advancement of targeted mass spectrometry platforms is underway to integrate phosphoproteomic technology with genomic assays to soon translate this information into the cancer clinic.
Keywords: Biomarkers, Clinical diagnostics, Clinical trial design, Kinase inhibitors, Pharmacotherapy, Phosphoproteomics, Precision medicine, Prostate cancer, Mass spectrometry
Current clinical state of prostate cancer
In 2016, the American Cancer Society estimates 181,890 new cases of prostate cancer in men in the United States, which ranks as the highest among cancers in men and second only to lung cancer in estimated deaths at 26,120 [1]. In advanced stages of the disease, androgen deprivation therapy (ADT) is the primary approach to reduce tumor burden. This hormonal therapy is effective initially, as assessed by the decline in prostate specific antigen (PSA) levels. However, this response is not durable and almost always results in relapse, termed castration resistant prostate cancer (CRPC). CRPC is often accompanied by the development of radiographically visible metastases. Metastatic castration-resistant prostate cancer (mCRPC) represents the lethal form of the disease where prognosis is poor with a median survival time of less than two years [2]. The best available treatment options for mCRPC patients, including second-generation antiandrogens and taxane-based chemotherapies, modestly improve survival by a few months [2–4]. This highlights the urgent need to identify more impactful agents and a patient-specific approach tailored to treat each tumor uniquely.
Unlike in early stage disease, where the development of the PSA test to screen patients for prostate cancer has both raised awareness and improved survival [1], finding useful biomarkers for patient stratification or therapy in mCRPC remains a clinical challenge [3, 4]. One issue is that prostate cancer has relatively low mutation rates when compared to other types of cancer [5, 6]. Nonetheless, several fascinating papers that analyzed the genomic and transcriptomic landscape of primary prostate cancer and mCRPC identified numerous point mutations, translocations, and amplifications [5–11]. While several of these mutations may be deemed actionable, no effective treatments have yet been developed that target a majority of these mutations, with the exception of DNA repair and androgen receptor (AR) mutations. Indeed, a recent clinical trial illustrated that patients with BRCA2 or ATM functional loss (deletion or mutation) responded favorably to olaparib whereas patients without these mutations did not [12]. Another breakthrough paper found that patients who exhibited resistance to abiraterone acetate or enzalutamide harbor the AR-V7 splice variant in their prostate circulating tumor cells (CTCs) [13]. These selected examples of novel biomarkers will no doubt improve the treatment landscape for patients with mCRPC. However, due to modest improvements in overall survival, it is important to consider other strategies such as proteomics and/or phosphoproteomics to illuminate our understanding of the pathways that drive this disease. These methods would complement nicely with existing genomic and transcriptomic information to prioritize the key drivers and corresponding treatments in mCRPC.
Integrating clinical omics
Since the turn of the century, genomic technologies have matured to a point where practicality is no longer in question. Advances in sequencing technologies have both reduced cost and increased effectiveness of obtaining genomic and transcriptomic data. In parallel, advances in the biomedical field has benefited from the increase in knowledge from sequencing. Projects such as The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) have provided a rich resource of genomic information for scientists to apply to their research. As a result, clinical practice has gradually transformed to incorporate genomics in diagnosis, patient stratification, and treatment selection [14]. Conversely, clinical proteomic and phosphoproteomic platforms have taken longer to develop, owing to the greater complexity and difficulty involved with analyzing proteins or phosphoproteins in very small tissue amounts (i.e., biopsy).
Even with these current limitations proteomics and phosphoproteomics have provided a wealth of information from clinically relevant cancer tissues. Work by us and the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC; previously the Clinical Proteomic Technology Assessment for Cancer) have published some groundbreaking papers in colorectal, ovarian, breast, and prostate cancer characterizing the genomic, proteomic, and/or phosphoproteomic landscapes in these diseases [15–19]. These studies have provided clues into the signaling networks related to each cancer type through incorporation of novel proteomic and phosphoproteomic data. Importantly, these studies have opened up new opportunities to develop and test computational strategies that will integrate proteomic or phosphoproteomic information with existing genomic data. Hence, a significant next step will be to extract clinically actionable information from these integrated approaches.
In prostate cancer, previous work by our group on the tyrosine phosphoproteome in mCRPC patients identified several activated tyrosine kinases and observed interpatient heterogeneity but similarity among metastatic sites within the same patient [19]. This may imply that evaluation of the phosphoproteome of a singly biopsy could reveal the crucial activated signaling pathways to inform treatment decisions or aid in the development of prognostic or predictive biomarkers during response or relapse to current therapies in mCRPC patients. The main issue, though, was devising a method to portray which kinases or pathways to prioritize from within the complex phosphoproteomic data. Our recent publication in Cell [16] attempted to resolve this issue through a defined, systematic approach that integrated several omic datasets from mCRPC patient tumor samples using a novel computational pipeline [20]. Our analysis revealed that inclusion of the phosphoproteomic data provided more functional pathway information after integration with genomics and transcriptomics versus the integration of only genomics and transcriptomics. Certain pathways (e.g., AKT/mTOR/MAPK, nuclear receptor, and cell cycle signaling) were found to be significantly enriched in mCRPC when the phosphoproteomic data was included but only marginally enriched when excluded [16]. To easily visualize specific patients’ signaling networks in the context of canonical cancer hallmark pathways, we created the phosphorylation-based cancer hallmarks using integrated personalized signatures (pCHIPS). It is important to note that every patient we evaluated had enrichment of at least four cancer hallmarks making prioritization of kinase pathways still very challenging.
To overcome this, we incorporated the pathway information to procure a set of targetable kinases predicted to have maximal effect on these cancer hallmarks. These patient-specific kinase hierarchies make it possible to stratify mCRPC patients according to these hierarchies using targeted kinase inhibitors, in combination with other agents, for maximum therapeutic potential [16]. Two major findings came from this approach: (1) Not every patient would be predicted to respond to the same kinase inhibitor (even though the same cancer hallmark pathway may be enriched) and (2) Involvement of the cell cycle pathway is very prominent in a majority of these patient samples suggesting that CDK4/6 kinase inhibitors may be highly efficacious clinically, especially in combination with other targeted agents. It should be mentioned that our phosphoproteomic data could also be influenced by activation (or repression) of phosphatases. Our focus was on activated kinases, but we cannot rule out the notion that hyperphosphorylation of these kinases (or their substrates) may be regulated by phosphatase activity. In addition to kinase inhibitors, our phosphoproteomic information could also help inform the selection of phosphatase inhibitors, which may provide a greater pleiotropic effect when compared to single agent kinase inhibitors, though none are currently FDA-approved [21]. While these results are exciting, further experimental validation in vitro and in vivo is needed to confirm the implicated pathways.
Clinical phosphoproteomics: opportunities and challenges
Kinase inhibitors have long been important players in the area of targeted therapies against cancer. However, clinical trials using kinase inhibitors where randomization is preferred over biomarker stratification have reported mixed results, with a majority of these studies not demonstrating any clinical benefit, especially in prostate cancer [22, 23]. An explanation for some of these clinical trial failures may result from primary or acquired drug resistance mechanisms. Many resistance pathways have been discovered during the administration of therapies targeting driver kinase mutations in several cancers and hence can reduce the durability of these targeted agents [24, 25]. For example, it has been observed that EGFR pathway activity is responsible for the continued growth and survival of BRAF (V600E) colon cancers resistant to vemurafenib, an observation not observed in BRAF (V600E) melanomas [26]. It is our view that future clinical trial successes will hinge on properly stratifying patients according to predicted drug response by utilizing biomarkers that accurately reflect the tumor’s biology. The incorporation of phosphoproteomics with genomics and transcriptomics could help characterize how a patient’s tumor responds to treatment as well as tease out any future mechanisms of resistance providing these necessary biomarkers. In that light, a soon to be initiated phase II clinical trial in prostate cancer (NCT03012321) will utilize genomics to pre-select patients with DNA repair mutations followed by randomization to evaluate the efficacy of olaparib alone or in combination with an androgen synthesis inhibitor, abiraterone acetate. As we move forward, it would be expected that biomarker-driven clinical trials will be the norm rather than the exception, providing clinicians with a method for stratifying late stage disease as well as selecting the appropriate therapy for each stratum (Fig. 1).
Fig. 1.
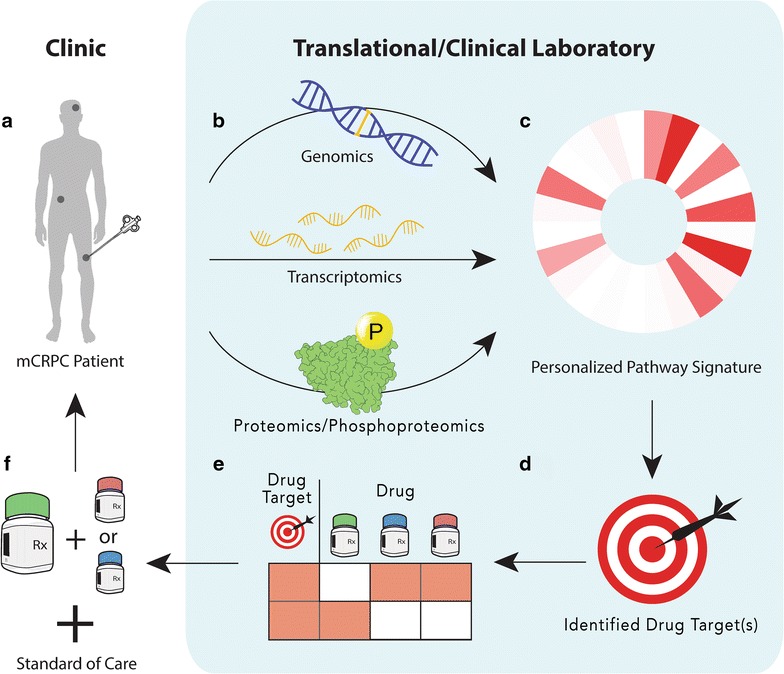
Conceptual diagram of the integration of phosphoproteomics into prostate cancer clinical management. a A biopsy of the tumor is taken from a metastatic castration-resistant prostate cancer (mCRPC) patient. b The biopsy specimen is processed to generate its omics data. c The omics data is integrated to produce a personalized pathway signature. d Drug targets are identified from this signature. e Available drugs are assessed for each drug target. f The drug or combinations of drugs that block the target are selected for treatment in addition to standard of care as an arm in a clinical trial
Phosphoproteomic technologies have made significant progress in the past decade with several platforms available: antibody-based assays (e.g., reverse phase protein arrays) and MS-based assays. MS-based assays are further delineated into discovery/global and targeted approaches [27]. Antibody-based approaches require low sample amounts, which makes them practical in the clinical setting where biopsies are typically performed. Furthermore, these assays do not require special instrumentation, easing their adoption into clinical laboratories. However, this approach is reliant on the availability of quality, specific antibodies, which can be more difficult when evaluating phosphorylation. MS-based approaches can bypass this limitation and perform multiplexed analysis of many proteins or phosphoproteins at once [27]. MS-based assays, specifically the targeted platform, carry great potential for future clinical utility especially in the space of diagnostic and predictive biomarker development. The global MS platform allows for relative quantitative comparisons of an unknown list of phosphopeptides between samples. The technique involves extracting proteins from the sample and digesting with a protease (usually trypsin plus Lys-C) into peptides and phosphopeptides [28]. Due to the relatively lower abundance of phosphoproteins, the phosphopeptides require enrichment from the complex mixture [29]. This can be done using immobilized metal affinity chromatography (IMAC) [30] such as TiO2 or antibodies (for phosphotyrosine enrichment) [31]. The enriched sample is then separated by liquid chromatography and detected via tandem MS. Detected peptides and phosphopeptides can be analyzed using software programs such as MaxQuant [32] or Skyline [33]. However, global MS is limited by its reproducibility due to the biased nature of the MS method [34]. Systemic bias in data collection arises from the complex digests and experimental design [34, 35]. Furthermore, phosphopeptides of higher abundance are sampled more frequently and precisely while phosphopeptides of lower abundance may be missed, creating a missing values problem [34, 35]. Efforts are ongoing in standardizing normalization strategies and data imputation methods to address these challenges both within experiments and across experiments [35, 36]. Targeted MS can bypass these limitations by measuring specifically annotated peptides that are constituent to a protein of interest. By using isotope-labeled standards and acquiring MS data specifically for the peptides of interest, targeted MS can precisely and reproducibly quantitate that protein in the sample by using previously mentioned programs such as Skyline [34]. Adoption of targeted MS to examine biomedical problems is growing and best practices are emerging to begin to standardize the field [37]. Crucial to this effort, the CPTAC network of laboratories demonstrated high reproducibility of this approach within and between laboratories as well as across instruments in measuring protein concentrations in plasma [38].
As phosphoproteomic technologies mature we foresee a paradigm shift. Clinicians will order tests that evaluate pathway activity as well as mutational status and will apply these results in real-time in the clinic. Until that day arrives, more work is still needed in the field of phosphoproteomics that improve upon some of the same sets of challenges that early work in the genomics field faced nearly 15 years ago. Phosphoproteomic MS platforms require greater amounts of sample as well as greater upfront investment on infrastructure in terms of equipment and operator expertise [27]. Furthermore, the assay workflow is lengthy, limiting reproducibility as well as practicality. We anticipate that many of these issues will be solved with continual technological advancement. Reducing the sample requirement amount will make the platform more practical for biopsy-based tests in the clinic. Indeed, a recent landmark paper demonstrated reproducible detection and quantification of over 2000 proteins from 18 clinical biopsy samples by utilizing novel pressure cycling technology and sequential window acquisition of all theoretical fragment ion mass spectra (SWATH-MS) [39]. While exciting, further investigation is necessary to determine if phosphopeptides can be detected in the same manner due to the relatively lower abundance of phosphopeptides when compared to total peptides. Efforts to simplify the workflow [40] will reduce the turn-around time between sample collection and results, enabling clinicians to more quickly evaluate and determine the next step in disease management.
Conclusions
While MS-based targeted phosphoproteomics is in preclinical stages of research and development, we believe that the eventual translation of this technology will open new doors in the clinical setting. Phosphoproteomics, as an integrative approach with genomics and other omics data, may have a future hand in addressing the challenges of prostate cancer (and other cancer) diagnoses and drug development by identifying actionable pathways. The technology would also pave the way for a more comprehensive field of pharmaco-omics to rationally select and modify a patient’s drug therapy for different diseases that have low mutation burden.
Authors’ contributions
LCC, VMT, SG, and JMD wrote the commentary. All authors read and approved of the final manuscript.
Acknowledgements
We thank all the co-authors from the Drake, Paull et al. paper (Cell 2016), especially members of the Dr. Owen N. Witte and Dr. Joshua M. Stuart laboratories and the University of Michigan rapid autopsy program. The computational algorithm described in this manuscript was developed by Dr. Evan O. Paull in the laboratory of Dr. Stuart (University of California, Santa Cruz). We also thank Sharon Feng for her digital design expertise in producing the figure.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
As the senior author, I, Justin M. Drake, hereby give consent to publish this commentary.
Funding
LCC is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32 GM008339. SG is supported by the NCI R01 CA169182 and P30 CA072720, Hugs for Brady Foundation, Val Skinner Foundation, and Breast Cancer Research Foundation. JMD is supported by the Department of Defense Prostate Cancer Research Program W81XWH-15-1-0236, Prostate Cancer Foundation Young Investigator Award, and by a grant from the New Jersey Health Foundation.
Abbreviations
- ADT
androgen deprivation therapy
- AR
androgen receptor
- CPTAC
Clinical Proteomic Tumor Analysis Consortium
- CTC
circulating tumor cell
- ICGC
International Cancer Genome Consortium
- IMAC
immobilized metal affinity chromatography
- mCRPC
metastatic castration-resistant prostate cancer
- MS
mass spectrometry
- pCHIPS
Phosphorylation-based Cancer Hallmarks using Integrated Personalized Signatures
- PSA
prostate-specific antigen
- SWATH-MS
sequential window acquisition of all theoretical fragment ion mass spectra
- TCGA
The Cancer Genome Atlas
Contributor Information
Larry C. Cheng, Email: larry.c.cheng@rutgers.edu
Victor M. Tan, Email: vt187@scarletmail.rutgers.edu
Shridar Ganesan, Email: ganesash@cinj.rutgers.edu.
Justin M. Drake, Email: justin.drake@cinj.rutgers.edu
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Lowrance WT, Roth BJ, Kirkby E, Murad MH, Cookson MS. Castration-resistant prostate cancer: AUA guideline amendment 2015. J Urol. 2016;195(5):1444–1452. doi: 10.1016/j.juro.2015.10.086. [DOI] [PubMed] [Google Scholar]
- 3.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32(49):5501–5511. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silberstein JL, Pal SK, Lewis B, Sartor O. Current clinical challenges in prostate cancer. Transl Androl Urol. 2013;2(3):122–136. doi: 10.3978/j.issn.2223-4683.2013.09.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 8.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153(3):666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network Electronic address scmo, cancer genome atlas research N. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and Olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doroshow JH, Kummar S. Translational research in oncology–10 years of progress and future prospects. Nat Rev Clin Oncol. 2014;11(11):649–662. doi: 10.1038/nrclinonc.2014.158. [DOI] [PubMed] [Google Scholar]
- 15.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, et al. phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell. 2016;166(4):1041–1054. doi: 10.1016/j.cell.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166(3):755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci USA. 2013;110(49):E4762–E4769. doi: 10.1073/pnas.1319948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paull EO, Carlin DE, Niepel M, Sorger PK, Haussler D, Stuart JM. Discovering causal pathways linking genomic events to transcriptional states using tied diffusion through interacting events (TieDIE) Bioinformatics. 2013;29(21):2757–2764. doi: 10.1093/bioinformatics/btt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lazo JS, Sharlow ER. Drugging undruggable molecular cancer targets. Annu Rev Pharmacol Toxicol. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- 22.Araujo JC, Trudel GC, Saad F, Armstrong AJ, Yu EY, Bellmunt J, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 2013;14(13):1307–1316. doi: 10.1016/S1470-2045(13)70479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34(25):3005–3013. doi: 10.1200/JCO.2015.65.5597. [DOI] [PubMed] [Google Scholar]
- 24.Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27(3):397–408. doi: 10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 26.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 27.Noujaim J, Payne LS, Judson I, Jones RL, Huang PH. Phosphoproteomics in translational research: a sarcoma perspective. Ann Oncol. 2016;27(5):787–794. doi: 10.1093/annonc/mdw030. [DOI] [PubMed] [Google Scholar]
- 28.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 29.Riley NM, Coon JJ. Phosphoproteomics in the age of rapid and deep proteome profiling. Anal Chem. 2016;88(1):74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20(3):301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 31.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23(1):94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 32.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 33.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;52(22):3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karpievitch YV, Dabney AR, Smith RD. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 2012;13(Suppl 16):S5. doi: 10.1186/1471-2105-13-S16-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kauko O, Laajala TD, Jumppanen M, Hintsanen P, Suni V, Haapaniemi P, et al. Label-free quantitative phosphoproteomics with novel pairwise abundance normalization reveals synergistic RAS and CIP2A signaling. Sci Rep. 2015;5:13099. doi: 10.1038/srep13099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13(3):907–917. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27(7):633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo T, Kouvonen P, Koh CC, Gillet LC, Wolski WE, Rost HL, et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21(4):407–413. doi: 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jersie-Christensen RR, Sultan A, Olsen JV. Simple and reproducible sample preparation for single-shot phosphoproteomics with high sensitivity. Methods Mol Biol. 2016;1355:251–260. doi: 10.1007/978-1-4939-3049-4_17. [DOI] [PubMed] [Google Scholar]