

Abstract
Background
Promoting arousal by manipulating certain brain regions and/or neurotransmitters has been a recent research focus, with the goal of trying to improve recovery from general anesthesia. The present study tested the hypothesis that a single subanesthetic dose of ketamine during isoflurane anesthesia would increase cholinergic tone in the prefrontal cortex and accelerate recovery.
Methods
Adult male rats were implanted with electroencephalography electrodes (frontal, parietal, and occipital cortex) and a microdialysis guide cannula targeted for the prefrontal cortex. After establishing general anesthesia with isoflurane, animals were randomly assigned to receive a saline control or ketamine injection. When isoflurane was discontinued nearly 90 min after drug or saline administration, recovery from anesthesia was measured by experimenters and blinded observers. During the entire experiment, electrophysiological signals were recorded and acetylcholine was quantified by high performance liquid chromatography with electrochemical detection.
Results
A single dose of subanesthetic ketamine caused an initial 125% increase in burst suppression ratio (last isoflurane sample: 37.48 ± 24.11% versus isoflurane after ketamine injection: 84.36 ± 8.95%; p<0.0001), but also a significant 44% reduction in emergence time (saline: 877 ± 335 s versus ketamine: 494 ± 108 s; p=0.0005; n=10 per treatment). Furthermore, ketamine caused a significant 317% increase in cortical acetylcholine release (mean after ketamine injection: 0.18 ± 0.16 pmol versus ketamine recovery: 0.75 ± 0.41 pmol; p=0.0002) after isoflurane anesthesia was discontinued.
Conclusions
Administration of subanesthetic doses of ketamine during isoflurane anesthesia increases anesthetic depth but—paradoxically—accelerates the recovery of consciousness, possibly through cholinergic mechanisms.
INTRODUCTION
There has been a recent focus on the neurobiology of emergence from anesthesia, including attempts to reverse anesthetic effects or accelerate recovery through arousal-promoting interventions.1–8 In terms of reversing anesthetic effects, manipulations of the thalamus,1,2 the cholinergic system,9 and the dopaminergic system5 have predominated. Nicotine infused in central median thalamus has been shown to reverse the effects of sevoflurane,1 while infusing an antibody to a voltage-gated potassium channel in the same region reverses both sevoflurane and desflurane anesthesia.2 Increasing cholinergic tone through acetylcholinesterase inhibitors has been shown to induce electroencephalographic effects related to arousal and has reversed both sevoflurane- and propofol-induced unconsciousness in humans.10,11 Most recently, there has been a focus of dopaminergic modulation through the administration of methylphenidate,3,12,13 dopamine agonists,4 or direct electrical stimulation of the ventral tegmental area;5 Solt and colleagues have demonstrated that these pharmacological and nonpharmacological interventions induce reanimation in animals exposed to continuous propofol or isoflurane.8 In terms of accelerating recovery after anesthetic discontinuation, caffeine has been recently shown to reduce the time to emergence from both propofol and isoflurane.14
What has not been explored is the modulation of an anesthetic state to more closely resemble natural sleep. In humans, slow-wave sleep predominates earlier in the night, with increasing frequency and bout length of rapid eye movement sleep that builds to a crescendo as emergence from sleep approaches.15,16 Since rapid eye movement sleep is characterized by increased cortical activity and cholinergic tone,16,17 it has been proposed that it prepares the brain for wakefulness.15,18 This has also been suggested to be the case during ontogeny, as the proportion of time spent in rapid eye movement sleep is highest during the third trimester; the ontogenetic theory suggests that the activation patterns in utero prepare the brain for sensory input at birth.15,19–21
Ketamine anesthesia shares certain traits with rapid eye movement sleep, including high-frequency cortical activity, high cholinergic tone in the cortex, and dream states.22–24 We hypothesized that administration of subanesthetic ketamine during isoflurane anesthesia would increase cholinergic tone in prefrontal cortex, high-frequency cortical activity, and accelerate emergence once isoflurane was discontinued.
METHODS
Animals
All experiments were approved by the University of Michigan Committee on Use and Care of Animals and were conducted in accordance with The Public Health Service Policy on Humane Use and Care of Laboratory Animals (National Institutes of Health Publication 80-23). Adult (2-3 month old), male Sprague-Dawley rats (n=20, Harlan/Envigo, Indianapolis, IN) were housed in identical chambers under a 12-hour:12-hour light:dark cycle with continuous access to food and water. Animals were randomly divided into ketamine or saline groups.
Surgical Procedures
At least one week before the experiment, animals underwent head cap surgery. General anesthesia was induced with 4% isoflurane (Piramal Critical Care, Inc., Bethlehem, PA), which was measured continuously by spectrometry (Cardiocap™/5, Datex-Ohmeda, Louisville, CO). Once anesthetized, animals were placed in a Kopf Model 962 stereotaxic frame (David Kopf Instruments, Tujunga, CA) with a Kopf model 920 rat adaptor and rat anesthesia mask (Kopf Model 906). Isoflurane delivery was then reduced to 2.5%. During the entire surgery, vital signs were taken every 15 min and a heating pad (Far Infrared Warming Pad, Kent Scientific Corp., Torrington, CT) was used to keep the body temperature consistent (± 0.6 °C). Rats received a single dose of the non-steroidal anti-inflammatory drug carprofen (5 mg/kg, subcutaneous; Rimadyl®, Zoetis Inc., Kalamazoo, MI). Craniotomy was then performed and a CMA/11 guide cannula (CMA Microdialysis, Harvard Apparatus, Holliston, MA) was implanted above the prefrontal cortex at 3 mm anterior to bregma, 0.5 mm lateral to the midline, and 3.2 mm ventral to the skull surface.25 The guide cannula was placed either on the left or right side of the brain, to ensure that any observed effect was not due to brain laterality. In addition, six electrodes to record electroencephalogram (EEG) made out of stainless steel wire (A-M Systems, Inc., Carlsborg, WA) were placed at the following stereotaxic coordinates: one frontal, opposite side of the guide cannula, at 3.0 mm anterior to bregma and 2.5 lateral to the midline, two parietal at 4 mm posterior to bregma and 2.5 mm lateral to midline on both sides, two occipital at 8 mm posterior to bregma and 2.5 mm lateral to midline on both sides and one on the nasal commissure as the reference electrode. In addition, rats received two electromyography (EMG) electrodes placed into the neck muscle to evaluate muscle movement during emergence. All EEG electrodes were plugged into a plastic multichannel electrode pedestal (8K00022980IF and 8K00000363DC, Plastics One, Roanoke, VA). EMG electrodes were plugged into a second pedestal. Guide cannula and electrode arrays were fixed to the skull using dental acrylic (Stoelting Co, Woodlake, IL). Animals were then allowed to recover from surgery for 7 days and were thereafter conditioned to being housed in a Plexiglas recording chamber (Raturn, Bioanalytic Systems Inc., West Lafayette, IN).
Electroencephalographic Data Acquisition
EEG signals from 10 saline and 10 ketamine treated animals were amplified (× 5000) and filtered (0.1–300 Hz, Grass amplifier Model 15 LT, 15A54 Quad Amplifier, Warwick, RI). Data were digitized and recorded at a sampling rate of 1000 Hz using AcqKnowledge software (V4.1, MP150, Biopac Systems, Inc, Goleta, CA).
Segmentation and Quantification of Electroencephalographic Burst and Suppression
The raw EEG signals were exported into Matlab (version 2015b; MathWorks, Inc., Natick, MA) and downsampled to 250 Hz for further analysis. The spectral analysis was performed with fast Fourier transform to provide information on frequency content of bursts and artifacts in the EEG signals. The data were then transformed in three steps: (1) signals were bandpass filtered between 5 and 30 Hz through a 4-order Butterworth filter using a zero-phase forward and reverse algorithm; (2) the Hilbert transform of the bandpass signal was used to calculate the instantaneous amplitude to approximate the high frequency power,26 which was further smoothed with a moving average filter of 200 ms; (3) a threshold calculated from the manually labelled suppression periods (mean plus 3 or 4 standard deviations [SD] based on visual inspection) was applied to the transformed signal to yield a binary series of burst and suppression states for each rat. In this study, the minimum length of burst and suppression periods were set to 0.5 s, and burst suppression ratio was calculated as percentage of time spent in suppression of each 1 minute binary series. EEG epochs were analyzed from the beginning through the end of the 12.5 min duration of Acetylcholine (ACh) sample collection, to ensure parallel information for ACh sampling and EEG recording. To validate our results, we applied two alternative methods (variance-based method27 and nonlinear energy operator-based method28) to replace the amplitude approach in step (2). In the first alternative method, the standard deviation of the bandpassed signal was computed for each 200 ms interval with a 150 ms overlap; in the second method, the energy of the bandpassed signal was calculated by the nonlinear energy operator and smoothed using a 200 ms moving average. Since the three different types of analysis did not show any differences, we report only the first analysis for this study. Further, the detection methods were applied to different channels of EEG signals (monopolar: frontal, left/right–parietal, left/right–occipital; bipolar: left–right parietal; left–right occipital, left parietal–left occipital and right parietal–right occipital), and the channel was selected to be analyzed based on visual inspection. Frontal channel was selected in 18 of 20 rats, and left-parietal channel was used in the other two rats. No significant heterogeneity across the five recorded cortical sites was found. The frequency bandwidths in step (1) were varied: 5–30 Hz, 5–50 Hz, 3–125 Hz,26 and the difference of <8 Hz and >47 Hz,28 and found that 5–30 Hz and 5–50 Hz analyses yielded similar results, which were superior to the other two cases in almost all rats (in agreement with visual inspection). Furthermore, different values were tested for the threshold of mean+c*SD in step (3): c=3–7 and the one in best agreement with visual inspection was selected; c=3 for 8 and c=4 for 12 of 20 rats.
Power Spectral Analysis
The power spectrogram was calculated based on discrete Fourier transform with 2 s segment size and 1 s overlapping for each frequency bin (0.5–250 Hz with 0.5 Hz bin size; ‘spectrogram.m’ in Matlab signal processing toolbox; MathWorks Inc., Natick, MA). For each rat, the absolute power was calculated in each channel, and the spectrogram from the same channel used in the raw signal and expressed in a log scale.
To relate the EEG powers with changes of ACh level during the whole experiment, the spectrogram was estimated in non-overlapped 10 s bins using the Welch method, in order to achieve a better estimation of spectral density function. Furthermore, normalized power was calculated as the fraction of a specific frequency power in the total power over all frequency bands, and averaged across all EEG channels; the averaged power values are calculated for the following frequency bands: delta (0.5–4 Hz), theta (4–10 Hz), alpha (10–15 Hz), beta (15–25 Hz), low gamma (25–55 Hz), medium gamma (65–125 Hz), high gamma (125–175 Hz), and ultra-high gamma (185–250 Hz) at each studied sample, with the mean and SD values across all rats receiving either ketamine and saline.
Quantification of Acetylcholine Release in the Prefrontal Cortex
ACh release (pmol/12.5 min) in the prefrontal cortex was measured from nine saline- and eight ketamine-treated animals using high-performance liquid chromatography with electrochemical detection (Bioanalytical Systems Inc., West Lafayette, IN and Showa Denko America, Inc, New York, NY). Chromatograms were digitized and quantified using LC Real Time Analysis Program (Showa Denko America, Inc, New York, NY) in reference to a seven point standard curve ranging from 0.05 pmol to 1.0 pmol. CMA/11 microdialysis probes (CMA Microdialysis, Harvard Apparatus, Holliston, MA) were perfused continuously with Ringer’s solution (147 mM NaCl, 2.4 mM CaCl2, 4.0 mM KCl, 10 µM neostigmine; pH 6.0 ± 0.2). Salts for Ringer’s were purchased from Sigma-Aldrich (St. Louis, MO). Flow rate was held constant at 2.0 µl/min using a CMA/400 syringe pump (CMA Microdialysis, Harvard Apparatus, Holliston, MA). The microdialysis probe had a cuprophane membrane of 1 mm length, 0.24 mm diameter, and a molecular cutoff of 6,000 Dalton. Microdialysis samples were collected every 12.5 minutes (25 µl/sample) for subsequent quantification of ACh release from the prefrontal cortex. To ensure that changes seen in ACh release were not due to an artifact of intra-experimental variations in probe membrane function, in vitro probe recovery levels were compared between pre- and post-experimental measurements.
Experimental Design
Figure 1A illustrates the experimental design. On the day of the experiment, a microdialysis probe was inserted through the guide cannula into the prefrontal cortex, animals were connected to the EEG recording system, and placed into a modified Raturn (Figure 1B). The modification allows the Raturn to be sealed such that inhaled anesthetics can be administered while the animal behaves freely. During the entire experiment, ACh samples as well as vital signs were collected every 12.5 min with simultaneous EEG signal acquisition. During the time period spanning the first 3 ACh sample collection points (37.5 min), animals were kept awake with gentle handling. This ensures that all animals started the experiment in the same baseline state. Experiments started at approximately 12:00 pm (± 1 hour). After the baseline wake phase, the Raturn was sealed and filled with isoflurane in high-flow oxygen (10 L/min) until in- and outlet of the Raturn was reading 2.5% isoflurane for 2 min. After anesthetic induction, as defined by the loss of righting reflex, the animal was placed on its back and a temperature probe was inserted rectally through a door in the Raturn. Isoflurane levels were then kept at 1.5% throughout the rest of the experiment (note that 1.4% is the minimum alveolar concentration for isoflurane in rodents).29 After a dead space collection period of 6 min (the time it takes the sample to go through the probe and tubing) three prefrontal cortex ACh samples were collected during isoflurane anesthesia. After the third sample the Raturn door was opened and 25 mg/kg ketamine or an equivalent volume of saline was injected into the intraperitoneal (ip) space of the animal. This is significantly below anesthetic dosing levels, since 150mg/kg (ip) is required to induce loss of righting reflex in rats.24 After stable isoflurane levels and 6 min of dead space, seven samples were collected during isoflurane after ketamine or isoflurane after saline injection. After the 87.5 min of sample collection, isoflurane was discontinued and the time it took for the animal to right itself (the surrogate for emergence time) was measured while 10 ACh samples during this recovery period were collected. Emergence time was determined by two independent observers, at least one of which was blinded to the experimental condition.
Figure 1. Study design.

A. Schematic time line illustrating the procedure for each animal in this study. Text above the boxes describes the manipulations performed during the experiment. The upper row of boxes describes the different states the animal is in, while the lower row of boxes illustrates the time line and number of ACh samples collected during the different time points. Throughout the entire experiment, electroencephalography (EEG) and electromyography (EMG) were recorded. ACh; Acetylcholine; ip: intraperitoneal; Iso: Isoflurane; LoRR: loss of righting response (surrogate for unconsciousness). B. Schematic shows a modified Raturn in which the animal can freely behave while EEG/EMG data are recorded and microdialysis sampling is performed during various levels of arousal.
Histologic Analysis
Three to seven days after the experiment, rats were deeply anesthetized and decapitated. Brains were removed, frozen, and sectioned coronally at a thickness of 40 µm. Serial prefrontal cortex sections were slide-mounted, dried, fixed with paraformaldehyde vapor at 80 °C, and stained with cresyl violet. Tissue sections were compared to a rat brain atlas25 to localize microdialysis sites.
Statistical Analysis
All data were evaluated with the input from the Center for Statistical Consultation and Research at the University of Michigan. Sample size was estimated as n=10 per treatment group based on previous experiments with similar design.24 In addition, after initial experiments, sample size was statistically calculated for 80% power with a two-tailed comparison. Burst suppression ratio was analyzed using a two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-hoc test. Changes in EEG power (for each frequency band), breathing rate, and core body temperature were analyzed using a two-way ANOVA with Sidak’s multiple comparisons test. Emergence time from anesthesia was analyzed by using a Mann-Whitney test. Drug effect on ACh release was analyzed using a two-way ANOVA with Tukey’s multiple comparisons post-hoc test for mean value comparisons and a two-way ANOVA with Bonferroni’s correction for timeline comparisons. Statistical analysis was performed using SAS v9.2 (SAS Institute Inc., Cary, NC) and Prism v7.0 programs (GraphPad Software, Inc., La Jolla, CA). A p-value less than 0.05 was considered statistically significant.
RESULTS
Histology
Serial coronal tissue sections of each brain were used to identify the stereotaxic coordinates of every microdialysis site relative to a rat brain atlas.25 Due to broken microdialysis probes, ACh release could not be measured in three animals, two from the ketamine and one from the saline group. The histological results described in this manuscript are from 17 rats in which microdialysis sites were localized within the prefrontal cortex (Figure 2).
Figure 2. Histologic confirmation of dialysis sites within the prefrontal cortex.

A. Vertical lines on a sagittal diagram of a rat brain25 depict the anterior-to-posterior range of the microdialysis sites within the prefrontal cortex. B. A cresyl violet-stained coronal brain section shows a typical microdialysis site within the prefrontal cortex. The arrow marks the most ventral position of the microdialysis membrane. C. The coronal plates were modified from a rat brain atlas25 to illustrate the location of the microdialysis sites within the prefrontal cortex. The size of the dialysis membrane is indicated by the cylinder drawn to scale, relative to the brain. Microdialysis probes of ketamine treated animals are in red, saline treated animals are in blue. Cg1: cingulate cortex area 1; Cl: caudal interstitial nucleus of the medial longitudinal fasciculus; DP: dorsal peducular cortex; fmi: forceps minor of the corpus callosum; IL: infralimbic cortex; M2: secondary motor cortex; MO: medial orbital cortex; PFC: prefrontal cortex; PrL: prelimbic cortex.
Effect of Ketamine on Electroencephalographic Burst Suppression
Burst suppression, a sign of deep anesthesia, was seen more frequently and with longer phases in ketamine treated animals. Figure 3A and B show representative EEG traces and power spectrograms for saline and ketamine, respectively, emphasizing the transition into burst suppression after ketamine was given. The traces show the timeline from the last isoflurane sample over the actual injection phase to the first two isoflurane samples after injection. Burst suppression ratio was significantly increased after injection of ketamine (Figure 3C). Two-way ANOVA revealed a significant difference between saline and ketamine (F(1,54)=47.78, p<0.0001), time points (last isoflurane only sample versus first and second isoflurane after injection [saline versus ketamine] sample); F(2,54)= 5.08, p=0.0095), and interaction (F(2,54)=12.63; p<0.0001). Tukey’s multiple comparisons post-hoc test demonstrated significant differences between the last isoflurane only sample and the first isoflurane after ketamine sample (p<0.0001) causing a significant 125% increase in burst suppression ratio. A significant 107% increase in burst suppression ratio was seen comparing the last isoflurane sample with the second isoflurane after ketamine sample (p=0.0005). Comparisons between the last isoflurane only sample and the first or second isoflurane after saline samples did not show any differences. The first and second isoflurane after ketamine samples showed a significant increase in burst suppression ratio when compared to isoflurane after saline samples 1 and 2 (p<0.0001 for both comparisons).
Figure 3. Effect of ketamine injection on electroencephalogram burst suppression ratio.

A and B show representative electroencephalographic (EEG) traces (upper) and power spectrogram (lower) during the transition phase from the last isoflurane-only sample up to the second isoflurane after injection sample for saline (A) and ketamine (B) treated animals. The EEG traces and spectrograms show four different phases divided by the vertical dotted lines. Color bar indicates normalized power in log scale in decibel (dB). C. Rats treated with ketamine showed a significant 125% increase (*) in burst suppression ratio during isoflurane after injection sample 1 and a 107% increase (*) during isoflurane after injection sample 2 when compared to the last isoflurane-only sample in the ketamine group (red data points). In addition, ketamine-treated animals showed a significant enhancement in burst suppression ratio compared to saline treated animals (red versus blue data points). Graph shows data as burst suppression ratio in percent ± SD. Iso: Isoflurane.
Effect of Ketamine on Emergence Time
Emergence time was defined as the time between the discontinuation of isoflurane anesthesia and the return of righting reflex. A second person who was blinded to the treatment reanalyzed and verified the wake up time independently through video and EEG/EMG recordings. Emergence time did not vary significantly (p=0.9861) between the two persons analyzing the data and reported time values differed14 ± 15 s, on average. Giving a subanesthetic dose of ketamine (25 mg/kg) during isoflurane anesthesia significantly (p=0.0005) decreased emergence time by 44% from 877 ± 335 s for saline to 494 ± 108 s for ketamine treated animals (Figure 4).
Figure 4.

Emergence time and electroencephalography during recovery. Animals receiving ketamine showed a significant 44% decrease in the time required to emerge from isoflurane anesthesia after it was discontinued. Blue: saline, red: ketamine; asterisk (*) define statistical significance with p <0.05.
Effect of Ketamine on Electroencephalographic Power during Recovery
To evaluate whether ketamine caused changes during the recovery phase, EEG power was analyzed using a two-way ANOVA followed by Sidak’s multiple comparison test for each frequency band (Figure 5A–H). Table 1 summarizes the statistical results. Ketamine treatment mainly affected EEG power of higher frequencies (low gamma, medium gamma, and high gamma) causing a significant increase (p<0.0001) for each of the three frequencies during the beginning of the recovery phase after isoflurane was discontinued (Figure 5 F, G, H).
Figure 5.

EEG spectral analysis shows the power for all frequency bands throughout the 10 recovery samples. The averaged power values were calculated for the following frequency bands: A) delta (0.5–4 Hz), B) theta (4–10 Hz), C) alpha (10–15 Hz), D) beta (15–25 Hz), E) low gamma ( 25–55 Hz), F) medium gamma (65–125 Hz), G) high gamma (125–175 Hz), and H) ultra-high gamma (185–250 Hz) with the mean and SD values across all the rats injected with either ketamine or saline. Blue line shows data from saline-treated animals; red line shows data from ketamine-treated animals. Asterisks (*) define statistical significance with p <0.05. Iso: Isoflurane.
Table 1.
Statistical analysis for EEG power frequencies during recovery
| Delta (0.5–4 Hz) | Theta (4–10 Hz) | Alpha (10–15 Hz) | Beta (15–25 Hz) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F(DFn, DFd) | p-value | F(DFn, DFd) | p-value | F(DFn, DFd) | p-value | F(DFn, DFd) | p-value | |||||
| Treatment | F(1,180) = 11.42 | * | 0.0009 | F(1,180) = 4.188 | * | 0.0422 | F (1, 180) = 20.95 | * | <0.0001 | F (1, 180) = 2.329 | ns | 0.1288 |
| Time Point | F(9,180) = 0.5377 | ns | 0.8457 | F(9,180) = 0.2997 | ns | 0.9741 | F (9, 180) = 1.504 | ns | 0.1495 | F (9, 180) = 1.566 | ns | 0.1285 |
| Interaction | F(9,180) = 2.961 | * | 0.0026 | F(9,180) = 2.481 | * | 0.0108 | F (9, 180) = 1.189 | ns | 0.3047 | F (9, 180) = 1.43 | ns | 0.1782 |
| Sidak's multiple comparisons test |
Saline vs Ketamine | p-value | Saline vs Ketamine | p-value | Saline vs Ketamine | p-value | Saline vs Ketamine | p-value | ||||
| Recovery 1 | * | <0.0001 | Recovery 1 | * | <0.0001 | Recovery 1 | ns | 0.8837 | Recovery 1 | * | 0.0033 | |
| Recovery 2 | ns | 0.1898 | Recovery 2 | ns | 0.7652 | Recovery 2 | ns | 0.9060 | Recovery 2 | ns | >0.9999 | |
| Recovery 3 | ns | 0.9459 | Recovery 3 | ns | >0.9999 | Recovery 3 | ns | 0.9060 | Recovery 3 | ns | >0.9999 | |
| Recovery 4 | ns | 0.9986 | Recovery 4 | ns | >0.9999 | Recovery 4 | ns | 0.7420 | Recovery 4 | ns | >0.9999 | |
| Recovery 5 | ns | 0.9874 | Recovery 5 | ns | >0.9999 | Recovery 5 | ns | 0.7061 | Recovery 5 | ns | 0.9859 | |
| Recovery 6 | ns | >0.9999 | Recovery 6 | ns | >0.9999 | Recovery 6 | ns | 0.4399 | Recovery 6 | ns | >0.9999 | |
| Recovery 7 | ns | 0.9932 | Recovery 7 | ns | 0.9761 | Recovery 7 | ns | 0.5824 | Recovery 7 | ns | >0.9999 | |
| Recovery 8 | ns | >0.9999 | Recovery 8 | ns | >0.9999 | Recovery 8 | ns | 0.3532 | Recovery 8 | ns | 0.9989 | |
| Recovery 9 | ns | >0.9999 | Recovery 9 | ns | >0.9999 | Recovery 9 | * | 0.0319 | Recovery 9 | ns | >0.9999 | |
| Recovery 10 | ns | >0.9999 | Recovery 10 | ns | >0.9999 | Recovery 10 | ns | 0.7925 | Recovery 10 | ns | >0.9999 | |
| Two-way ANOVA | Low Gamma (25–55 Hz) | Medium Gamma (65–125 Hz) | High Gamma (125–175 Hz) | Ultra-High Gamma (185–250 Hz) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F(DFn, DFd) | p-value | F(DFn, DFd) | p-value | F(DFn, DFd) | p-value | F(DFn, DFd) | p-value | |||||
| Treatment | F (1, 180) = 122.3 | * | <0.0001 | F (1, 180) = 60.65 | * | <0.0001 | F (1, 180) = 49.38 | * | <0.0001 | F (9, 180) = 1.938 | * | 0.0492 |
| Time Point | F (9, 180) = 10.61 | * | <0.0001 | F (9, 180) = 8.266 | * | <0.0001 | F (9, 180) = 4.846 | * | <0.0001 | F (9, 180) = 0.9208 | ns | 0.5083 |
| Interaction | F (9, 180) = 5.305 | * | <0.0001 | F (9, 180) = 2.787 | * | 0.0044 | F (9, 180) = 3.06 | * | 0.0020 | F (1, 180) = 3.58 | * | 0.0601 |
| Sidak's multiple comparisons test |
Saline vs Ketamine | p-value | Saline vs Ketamine | p-value | Saline vs Ketamine | p-value | Saline vs Ketamine | p-value | ||||
| Recovery 1 | * | <0.0001 | Recovery 1 | * | <0.0001 | Recovery 1 | ns | 0.7918 | Recovery 1 | ns | 0.2143 | |
| Recovery 2 | * | <0.0001 | Recovery 2 | * | <0.0001 | Recovery 2 | * | <0.0001 | Recovery 2 | ns | 0.8167 | |
| Recovery 3 | * | <0.0001 | Recovery 3 | * | 0.0167 | Recovery 3 | * | <0.0001 | Recovery 3 | ns | >0.9999 | |
| Recovery 4 | * | 0.0007 | Recovery 4 | ns | 0.4190 | Recovery 4 | ns | 0.0575 | Recovery 4 | ns | 0.9998 | |
| Recovery 5 | * | 0.0010 | Recovery 5 | ns | 0.0645 | Recovery 5 | ns | 0.0575 | Recovery 5 | ns | >0.9999 | |
| Recovery 6 | ns | 0.1676 | Recovery 6 | ns | 0.9671 | Recovery 6 | ns | 0.8743 | Recovery 6 | ns | >0.9999 | |
| Recovery 7 | ns | 0.3230 | Recovery 7 | ns | 0.4864 | Recovery 7 | ns | 0.6089 | Recovery 7 | ns | 0.9998 | |
| Recovery 8 | ns | 0.4702 | Recovery 8 | ns | 0.3560 | Recovery 8 | ns | 0.9896 | Recovery 8 | ns | 0.9998 | |
| Recovery 9 | ns | 0.9088 | Recovery 9 | ns | 0.1643 | Recovery 9 | ns | 0.9212 | Recovery 9 | * | 0.0200 | |
| Recovery 10 | ns | >0.9999 | Recovery 10 | ns | 0.9999 | Recovery 10 | ns | >0.9999 | Recovery 10 | ns | 0.6067 | |
Two-way ANOVA with Sidak’s multiple comparisons test.
P-value <0.05 considered significant (*) different;
ns: non-significant; vs: versus.
Effect of Ketamine on Breathing Rate and Body Temperature
To verify that the decrease in emergence time was not caused by an increase in respiration we analyzed the breathing rate, which was measured every 12.5 min throughout the entire experiment. Using a two-way ANOVA no significant difference in breathing rate between saline and ketamine treated animals was found (p=0.1672; Figure 6A).
Figure 6. Breathing rate and core body temperature are not affected by ketamine treatment.
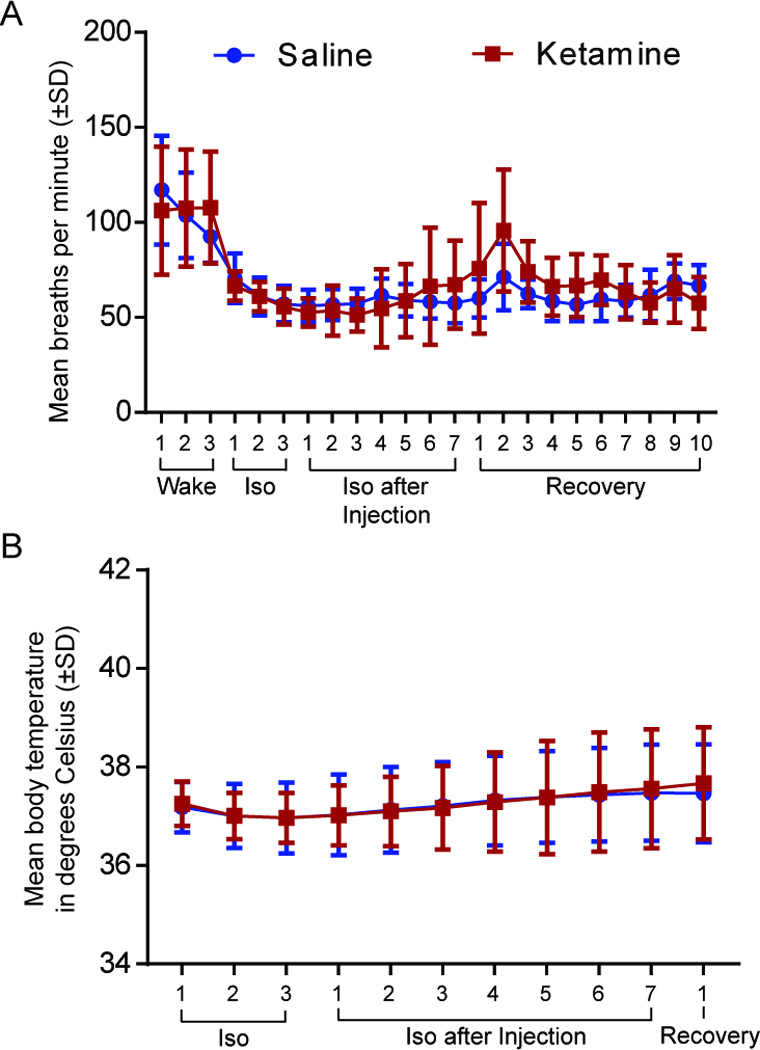
A. There is no difference in breathing rate in minutes (± SD) between saline- (blue) and ketamine- (red) treated animals. B. Body temperature in degrees Celsius (± SD) does not vary between saline and ketamine injected animals. Iso: Isoflurane.
In addition, body temperature was measured during general anesthesia to evaluate the effects of ketamine. After the animal lost consciousness, a rectal temperature probe was inserted and measurements of body core temperature were taken every 12.5 min (at each ACh sampling) until the animal emerged from anesthesia. A two-way ANOVA showed no significant difference between saline and ketamine treated animals in core body temperature (p=0.8292, Figure 6B).
Effect of Ketamine on Acetylcholine Release
A two-way ANOVA revealed a significant difference in ACh release from the prefrontal cortex across the various time points (wake, isoflurane, isoflurane after injection, and recovery) (F(3,60)=16.85, p<0.0001) and in the time points and treatment (saline versus ketamine) interaction (F(3,60)=2.884, p=0.0430) (Figure 7A). Tukey’s multiple comparisons test demonstrates that ACh release from the prefrontal cortex is significantly decreased (around 70%) when animals are under isoflurane anesthesia (p(saline)=0.0095, p(ketamine)=0.0139). Neither saline nor ketamine injection changed the suppressed ACh values during isoflurane anesthesia, which remained significantly lower in comparison to the wake phase (p[saline]=0.0095), p[ketamine]=0.0365). After isoflurane was discontinued, ketamine caused a significant (p<0.0002) 317% increase in ACh release during the recovery phase compared with exposure to isoflurane. During this recovery phase, ketamine induced a significant (p<0.0283) 103% increase in ACh release when compared with animals treated with saline. Overall, ketamine increased ACh release by 34% during the recovery phase when compared with the wake phase, while animals treated with saline showed a 35% decrease in ACh release during recovery compared with the wake phase.
Figure 7. Mean acetylcholine (ACh) release in the prefrontal cortex during saline and ketamine injection.

A. No difference was seen between the two treatment groups during the wake phase. ACh release in the prefrontal cortex was significantly decreased by isoflurane administration (*). Injection of ketamine during isoflurane anesthesia did not result in any change of ACh levels compared to saline. However, after isoflurane was discontinued, ACh significantly increased (+) in ketamine treated animals (n=8) during recovery phase while animals in the saline group showed no significant difference. Compared to the saline group (n=9), animals injected with ketamine showed a significant increase in ACh release during the recovery phase (#). Data are shown as mean ACh release in pmol from prefrontal cortex (+SD). B. ACh release timeline in the prefrontal cortex. No difference was seen between saline and ketamine injection during the wake, isoflurane, and isoflurane after injection phase. However, ketamine treated animals showed a significant increase in ACh release within the prefrontal cortex for the first 62.5 min during the recovery phase after discontinuation of isoflurane. Data are shown as mean ACh release from the prefrontal cortex (± SD). Iso: Isoflurane; PFC: prefrontal cortex. Blue line: saline treatment, red line: ketamine treatment; statistical significance (*, +, #) defined as p <0.05.
A timeline analysis (Figure 7B) for ACh release using a two-way ANOVA revealed significant differences in treatment (saline versus ketamine) (F (1, 345) = 43.08, p<0.0001), in the time points (F (22, 345) = 12.42, p<0.0001) and the interaction between drug treatment and time points (F (22, 345) = 3.192, p<0.0001). Bonferroni’s multiple comparisons post-hoc test reveals significant increases in ACh release in the ketamine-treated group during recovery samples 1 (p=0.0222), 2 (p<0.0001), 3 (p=0.0009), 4 (p=0.0333), and 5 (p=0.01), which represents the first 62.5 min after discontinuing isoflurane anesthesia.
DISCUSSION
The data presented in this study demonstrate that a single ip injection of subanesthetic ketamine during isoflurane anesthesia causes 1) a significant increase in burst suppression ratio, 2) a significant reduction in emergence time once isoflurane was discontinued, 3) significant increases in high-frequency cortical activation during recovery from anesthesia, and 4) a significant increase in ACh release from the prefrontal cortex during the recovery period. These findings are unique because we would expect an adjunct drug that modulates levels of consciousness to either deepen a general anesthetic state or facilitate recovery. By contrast, subanesthetic ketamine both deepens the anesthetic state and facilitates recovery. This “paradoxical emergence” also challenges the intuition that increased time spent in burst suppression necessarily results in prolonged recovery.
Based on known molecular mechanisms of ketamine and the observed microdialysis results, we suggest that effects on glutamatergic and cholinergic systems might, in part, explain the findings. Ketamine and isoflurane had a combined effect (either additive or synergistic) on the observed depth of anesthesia, as evidenced by the increased burst suppression ratio during the isoflurane after ketamine phase. Similar effects were described previously with the addition of ketamine to propofol administration.30,31 This phenomenon is potentially attributable to ketamine’s molecular actions as an antagonist of the glutamatergic N-methyl-D-aspartate (NMDA) receptor.32–35 In support of this, a similar effect was described in a study by Scheller et al.,36 in which the non-competitive NMDA receptor antagonist MK-801 was given to isoflurane-anesthetized rabbits, resulting in EEG burst suppression.
Furthermore, isoflurane was shown to have a dominant and suppressive effect on cholinergic neurons, as evidenced by the continuous suppression of acetylcholine levels in the prefrontal cortex despite the administration of ketamine, which is well known to activate subcortical arousal centers37 and increase cortical cholinergic tone when given alone.38,24 The discontinuation of isoflurane allowed (to adopt a metaphor from genetics) the derepression of ketamine’s effects, resulting in increased cholinergic tone during recovery and potentially accelerating emergence.
More than 300 million patients worldwide undergo major surgery each year, the majority with general anesthesia.39 Although general anesthetics are powerful and effective tools to reversibly modulate levels of consciousness, prolonged or abnormal recovery from general anesthesia can have significant consequences.40–42 Although anesthesiologists have pharmacological tools to antagonize specific drugs acting at specific receptors (e.g., benzodiazepines),43 only recently has research focused on reversing anesthetic effects by promoting arousal systems.1–3,12,4–6,8 This has been typically accomplished by either pharmacological manipulation using catecholamine reuptake inhibitors,3,12 dopamine agonists,4 central nervous system stimulants,13 and nicotine,1,3,12,4,6 or by electrical stimulation of specific brain regions such as the ventral tegmental area5 or parabrachial nucleus.8 In many of these studies, resumption of righting response (as a surrogate for emergence) was restored while the animal was still exposed to general anesthetics and a direct arousal promoting effect was observed. The current study is unique and advances the field because it revealed that the addition of a second anesthetic drug could accelerate the recovery from the first after enhancing its effect (i.e., deepening the anesthetic state). We specifically chose ketamine because it shares some traits with rapid eye movement sleep, including cortical activation, increased cholinergic tone, and dream states.38,16,23,17,24
Several studies indicate that ACh is critical for wakefulness and cognitive function44–46,17 and that the cholinergic arousal system is involved in the emergence from general anesthesia. Alkire et al. showed that injection of nicotine into the central medial thalamus induced resumption of righting during sevoflurane anesthesia.1 In humans, physostigmine, a cholinesterase inhibitor, can reverse loss of consciousness from sevoflurane11 and propofol10 anesthesia. Hudetz and colleagues9 evaluated intraventricular administration of the acetylcholinesterase inhibitor neostigmine and found an increase in cross-approximate entropy of the EEG and signs of arousal. This supports ketamine’s observed effect on cortical cholinergic tone during the recovery phase as a plausible mediator of accelerated recovery.
Ketamine appears to affect “neural inertia,” defined by Kelz and colleagues as a barrier to behavioral state transitions.47 Unlike past studies of neural inertia, ketamine appears to have dynamic effects depending on the neurochemical milieu of the brain. When ketamine is added to isoflurane anesthesia, it increases neural inertia, as evidenced by an increase in burst suppression ratio and a reduced variability in electroencephalographic state. However, ketamine reduces neural inertia after discontinuation of isoflurane, lowering resistance to the transition into wakefulness, and narrowing the variability in emergence time. As such, ketamine might be a unique tool to probe new dimensions of the concept of neural inertia.
Previous data have shown that subanesthetic injections of ketamine in rats48,49 and humans50 increase EEG power in the gamma bandwidth. In this study, the EEG power increase in various gamma frequencies did not occur directly after injection of ketamine, but nearly 90 min later when isoflurane was discontinued. This effect suggests that the excitatory properties of ketamine when given alone24 are blunted by the effect of isoflurane.31 Interestingly, higher-frequency gamma activation appears to be increased in ketamine treated animals slightly before the animal emerges from anesthesia, while in saline treated animals increase in gamma activity is not only less pronounced, but appears directly at the time of emergence (Figure 5 as well as Supplemental Digital Content 1 and 2, showing normalized power spectrogram of all saline- and ketamine-treated animals). These data suggest that an increase in higher-frequency cortical activity contributes to arousal, which is consistent with our recent findings.51 The fact that breathing rate and core body temperature did not differ between saline- and ketamine-treated animals indicates that the reduction in emergence time is not due to pharmacokinetics (i.e., increased isoflurane clearance with increased respiration) or the known sympathomimetic effects of ketamine.52,53
Limitations
Isoflurane and ketamine have complex pharmacological and neural effects; the nature of their interactions at the circuit and behavioral level remain to be clarified. Analyzing only the effect of the cholinergic system on emergence from general anesthesia is a limitation of this study, since multiple neurotransmitter systems are involved in the recovery process.9,4 Furthermore, the prefrontal cortex is not the only brain region included in the cholinergic arousal system. Therefore, further assessment of whether ACh in the prefrontal cortex plays a causal role in emergence time is needed as well as a multimodal analysis evaluating the interactions between different neurotransmitters and brain regions. It will also be necessary to verify that ketamine actually improves post-anesthetic cognition and not merely reanimation. The time point at which ketamine was injected was arbitrary. Further studies are needed to investigate how the results of this study are affected by changes in the time at which ketamine is administered. Finally, the clinical relevance of this phenomenon is unknown. Additional studies with surgical patients in the routine perioperative period (which often includes multiple psychoactive drugs and pain) are necessary.
Conclusion
The administration of subanesthetic ketamine enhances the effects of isoflurane as an anesthetic but accelerates emergence, possibly through cholinergic mechanisms and high-frequency activity in the cortex. This “paradoxical emergence” has compelling implications at both the clinical and neuroscientific level. Further work is required to assess the effects of these drug interactions on neural circuits and the relevance for improved perioperative care.
Supplementary Material
Summary Statement.
Administration of subanesthetic ketamine during isoflurane anesthesia increases anesthetic depth, but—paradoxically—accelerates emergence, possibly through cholinergic mechanisms.
Acknowledgments
Funding: Supported by grant R01GM111293 from the National Institutes of Health, Bethesda, Maryland, USA, and the Department of Anesthesiology at the University of Michigan, Ann Arbor, Michigan, USA.
We thank Max Kelz, M.D., Ph.D. (David E. Longnecker Professor, University of Pennsylvania) for suggesting the term “paradoxical emergence” (which parallels “paradoxical sleep”) to describe this phenomenon. For technical and expert assistance the authors thank Romi Ajluni (Undergraduate), Mary A. Norat (Senior Research Associate), and Giancarlo Vanini, M.D. (Assistant Professor) from the Department of Anesthesiology, and Chris Andrews, Ph.D., from the Center for Statistical Consultation and Research, University of Michigan.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest
REFERENCES
- 1.Alkire MT, McReynolds JR, Hahn EL, Trivedi AN. Thalamic microinjection of nicotine reverses sevoflurane-induced loss of righting reflex in the rat. Anesthesiology. 2007;107:264–272. doi: 10.1097/01.anes.0000270741.33766.24. [DOI] [PubMed] [Google Scholar]
- 2.Alkire MT, Asher CD, Franciscus AM, Hahn EL. Thalamic microinfusion of antibody to a voltage-gated potassium channel restores consciousness during anesthesia. Anesthesiology. 2009;110:766–773. doi: 10.1097/aln.0b013e31819c461c. [DOI] [PubMed] [Google Scholar]
- 3.Solt K, Cotten JF, Cimenser A, Wong KF, Chemali JJ, Brown EN. Methylphenidate actively induces emergence from general anesthesia. Anesthesiology. 2011;115:791–803. doi: 10.1097/ALN.0b013e31822e92e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor NE, Chemali JJ, Brown EN, Solt K. Activation of D1 dopamine receptors induces emergence from isoflurane general anesthesia. Anesthesiology. 2013;118:30–39. doi: 10.1097/ALN.0b013e318278c896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solt K, Van Dort CJ, Chemali JJ, Taylor NE, Kenny JD, Brown EN. Electrical stimulation of the ventral tegmental area induces reanimation from general anesthesia. Anesthesiology. 2014;121:311–319. doi: 10.1097/ALN.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kenny JD, Taylor NE, Brown EN, Solt K. Dextroamphetamine (but not atomoxetine) induces reanimation from general anesthesia: Implications for the roles of dopamine and norepinephrine in active emergence. PLoS One. 2015;10:e0131914. doi: 10.1371/journal.pone.0131914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarnal V, Vlisides PE, Mashour GA. The neurobiology of anesthetic emergence. J Neurosurg Anesthesiol. 2015;122:307–316. doi: 10.1097/ANA.0000000000000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muindi F, Kenny JD, Taylor NE, Solt K, Wilson MA, Brown EN, Van Dort CJ. Electrical stimulation of the parabrachial nucleus induces reanimation from isoflurane general anesthesia. Behav Brain Res. 2016;306:20–25. doi: 10.1016/j.bbr.2016.03.021. [DOI] [PubMed] [Google Scholar]
- 9.Hudetz AG, Wood JD, Kampine JP. Cholinergic reversal of isoflurane anesthesia in rats as measured by cross-approximate entropy of the electroencephalogram. Anesthesiology. 2003;99:1125–1131. doi: 10.1097/00000542-200311000-00019. [DOI] [PubMed] [Google Scholar]
- 10.Meuret P, Backman SB, Bonhomme V, Plourde G, Fiset P. Physostigmine reverses propofol-induced unconsciousness and attenuation of the auditory steady state response and bispectral index in human volunteers. Anesthesiology. 2000;93:708–717. doi: 10.1097/00000542-200009000-00020. [DOI] [PubMed] [Google Scholar]
- 11.Plourde G, Chartrand D, Fiset P, Font S, Backman SB. Antagonism of sevoflurane anaesthesia by physostigmine: effects on the auditory steady-state response and bispectral index. Br J Anaesth. 2003;91:583–586. doi: 10.1093/bja/aeg209. [DOI] [PubMed] [Google Scholar]
- 12.Chemali JJ, Van Dort CJ, Brown EN, Solt K. Active emergence from propofol general anesthesia is induced by methylphenidate. Anesthesiology. 2012;116:998–1005. doi: 10.1097/ALN.0b013e3182518bfc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kenny JD, Chemali JJ, Cotten JF, Van Dort CJ, Kim SE, Ba D, Taylor NE, Brown EN, Solt K. Physostigmine and methylphenidate induce distinct arousal states during isoflurane general anesthesia in rats. Anesth Analg. 2016 doi: 10.1213/ANE.0000000000001234. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Q, Fong R, Mason P, Fox AP, Xie Z. Caffeine accelerates recovery from general anesthesia. J Neurophysiol. 2014;111:1331–1340. doi: 10.1152/jn.00792.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roffwarg HP, Muzio JN, Dement WC. Ontogenetic development of the human sleep-dream cycle. Science. 1966;152:604–619. doi: 10.1126/science.152.3722.604. [DOI] [PubMed] [Google Scholar]
- 16.McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med. 2007;8:302–330. doi: 10.1016/j.sleep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–1187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klemm WR. Why does REM sleep occur? A wake-up hypothesis. Front Syst Neurosci. 2011;5:73. doi: 10.3389/fnsys.2011.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirmiran M, Maas YG, Ariagno RL. Development of fetal and neonatal sleep and circadian rhythms. Sleep Med Rev. 2003;7:321–334. doi: 10.1053/smrv.2002.0243. [DOI] [PubMed] [Google Scholar]
- 20.Hobson JA. REM sleep and dreaming: towards a theory of protoconsciousness. Nat Rev Neurosci. 2009;10:803–813. doi: 10.1038/nrn2716. [DOI] [PubMed] [Google Scholar]
- 21.Van den Bergh BR, Mulder EJ. Fetal sleep organization: a biological precursor of self-regulation in childhood and adolescence? Biol Psychol. 2012;89:584–590. doi: 10.1016/j.biopsycho.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Grace RF. The effect of variable-dose diazepam on dreaming and emergence phenomena in 400 cases of ketamine-fentanyl anaesthesia. Anaesthesia. 2003;58:904–910. doi: 10.1046/j.1365-2044.2003.03341.x. [DOI] [PubMed] [Google Scholar]
- 23.Blagrove M, Morgan CJ, Curran HV, Bromley L, Brandner B. The incidence of unpleasant dreams after sub-anaesthetic ketamine. Psychopharmacology (Berl) 2009;203:109–120. doi: 10.1007/s00213-008-1377-3. [DOI] [PubMed] [Google Scholar]
- 24.Pal D, Hambrecht-Wiedbusch VS, Silverstein BH, Mashour GA. Electroencephalographic coherence and cortical acetylcholine during ketamine-induced unconsciousness. Br J Anaesth. 2015;114:979–989. doi: 10.1093/bja/aev095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th. London: Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- 26.Lewis LD, Ching S, Weiner VS, Peterfreund RA, Eskandar EN, Cash SS, Brown EN, Purdon PL. Local cortical dynamics of burst suppression in the anaesthetized brain. Brain. 2013;136:2727–2737. doi: 10.1093/brain/awt174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman WE, Edelman G. Comparison of isoflurane and desflurane anesthetic depth using burst suppression of the electroencephalogram in neurosurgical patients. Anesth Analg. 1995;81:811–816. doi: 10.1097/00000539-199510000-00026. [DOI] [PubMed] [Google Scholar]
- 28.Sarkela M, Mustola S, Seppanen T, Koskinen M, Lepola P, Suominen K, Juvonen T, Tolvanen-Laakso H, Jantti V. Automatic analysis and monitoring of burst suppression in anesthesia. J Clin Monit Comput. 2002;17:125–134. doi: 10.1023/a:1016393904439. [DOI] [PubMed] [Google Scholar]
- 29.Pal D, Walton ME, Lipinski WJ, Koch LG, Lydic R, Britton SL, Mashour GA. Determination of minimum alveolar concentration for isoflurane and sevoflurane in a rodent model of human metabolic syndrome. Anesth Analg. 2012;114:297–302. doi: 10.1213/ANE.0b013e31823ede22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hui TW, Short TG, Hong W, Suen T, Gin T, Plummer J. Additive interactions between propofol and ketamine when used for anesthesia induction in female patients. Anesthesiology. 1995;82:641–648. doi: 10.1097/00000542-199503000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Albanese J, Arnaud S, Rey M, Thomachot L, Alliez B, Martin C. Ketamine decreases intracranial pressure and electroencephalographic activity in traumatic brain injury patients during propofol sedation. Anesthesiology. 1997;87:1328–1334. doi: 10.1097/00000542-199712000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Anis NA, Berry SC, Burton NR, Lodge D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol. 1983;79:565–575. doi: 10.1111/j.1476-5381.1983.tb11031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomson AM, West DC, Lodge D. An N-methylaspartate receptor-mediated synapse in rat cerebral cortex: a site of action of ketamine? Nature. 1985;313:479–481. doi: 10.1038/313479a0. [DOI] [PubMed] [Google Scholar]
- 34.Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology. 1997;86:903–917. doi: 10.1097/00000542-199704000-00021. [DOI] [PubMed] [Google Scholar]
- 35.Ogata J, Shiraishi M, Namba T, Smothers CT, Woodward JJ, Harris RA. Effects of anesthetics on mutant N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2006;318:434–443. doi: 10.1124/jpet.106.101691. [DOI] [PubMed] [Google Scholar]
- 36.Scheller MS, Zornow MH, Fleischer JE, Shearman GT, Greber TF. The noncompetitive N-methyl-D-aspartate receptor antagonist, MK-801 profoundly reduces volatile anesthetic requirements in rabbits. Neuropharmacology. 1989;28:677–681. doi: 10.1016/0028-3908(89)90150-0. [DOI] [PubMed] [Google Scholar]
- 37.Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, Saper CB. Role of endogenous sleep-wake and analgesic systems in anesthesia. J Comp Neurol. 2008;508:648–662. doi: 10.1002/cne.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikuchi T, Wang Y, Shinbori H, Sato K, Okumura F. Effects of ketamine and pentobarbitone on acetylcholine release from the rat frontal cortex in vivo. Br J Anaesth. 1997;79:128–130. doi: 10.1093/bja/79.1.128. [DOI] [PubMed] [Google Scholar]
- 39.Weiser TG, Haynes AB, Molina G, Lipsitz SR, Esquivel MM, Uribe-Leitz T, Fu R, Azad T, Chao TE, Berry WR, Gawande AA. Estimate of the global volume of surgery in 2012: an assessment supporting improved health outcomes. Lancet. 2015;385(Suppl 2):S11. doi: 10.1016/S0140-6736(15)60806-6. [DOI] [PubMed] [Google Scholar]
- 40.Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, Rabbitt P, Jolles J, Larsen K, Hanning CD, Langeron O, Johnson T, Lauven PM, Kristensen PA, Biedler A, van Beem H, Fraidakis O, Silverstein JH, Beneken JE, Gravenstein JS. Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet. 1998;351:857–861. doi: 10.1016/s0140-6736(97)07382-0. [DOI] [PubMed] [Google Scholar]
- 41.Pratico C, Quattrone D, Lucanto T, Amato A, Penna O, Roscitano C, Fodale V. Drugs of anesthesia acting on central cholinergic system may cause post-operative cognitive dysfunction and delirium. Med Hypotheses. 2005;65:972–982. doi: 10.1016/j.mehy.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 42.Saczynski JS, Marcantonio ER, Quach L, Fong TG, Gross A, Inouye SK, Jones RN. Cognitive trajectories after postoperative delirium. N Engl J Med. 2012;367:30–39. doi: 10.1056/NEJMoa1112923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safavynia SA, Keating G, Speigel I, Fidler JA, Kreuzer M, Rye DB, Jenkins A, Garcia PS. Effects of gamma-Aminobutyric Acid Type A Receptor Modulation by Flumazenil on Emergence from General Anesthesia. Anesthesiology. 2016;125:147–158. doi: 10.1097/ALN.0000000000001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marrosu F, Portas C, Mascia MS, Casu MA, Fa M, Giagheddu M, Imperato A, Gessa GL. Microdialysis measurement of cortical and hippocampal acetylcholine release during sleep-wake cycle in freely moving cats. Brain Res. 1995;671:329–332. doi: 10.1016/0006-8993(94)01399-3. [DOI] [PubMed] [Google Scholar]
- 45.Lydic R, Baghdoyan HA. Sleep, anesthesiology, and the neurobiology of arousal state control. Anesthesiology. 2005;103:1268–1295. doi: 10.1097/00000542-200512000-00024. [DOI] [PubMed] [Google Scholar]
- 46.Platt B, Riedel G. The cholinergic system, EEG and sleep. Behav Brain Res. 2011;221:499–504. doi: 10.1016/j.bbr.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 47.Friedman EB, Sun Y, Moore JT, Hung HT, Meng QC, Perera P, Joiner WJ, Thomas SA, Eckenhoff RG, Sehgal A, Kelz MB. A conserved behavioral state barrier impedes transitions between anesthetic-induced unconsciousness and wakefulness: evidence for neural inertia. PLoS One. 2010;5:e11903. doi: 10.1371/journal.pone.0011903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hakami T, Jones NC, Tolmacheva EA, Gaudias J, Chaumont J, Salzberg M, O'Brien TJ, Pinault D. NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS One. 2009;4:e6755. doi: 10.1371/journal.pone.0006755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicolas MJ, Lopez-Azcarate J, Valencia M, Alegre M, Perez-Alcazar M, Iriarte J, Artieda J. Ketamine-induced oscillations in the motor circuit of the rat basal ganglia. PLoS One. 2011;6:e21814. doi: 10.1371/journal.pone.0021814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong LE, Summerfelt A, Buchanan RW, O'Donnell P, Thaker GK, Weiler MA, Lahti AC. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology. 2010;35:632–640. doi: 10.1038/npp.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pal D, Silverstein BH, Lee H, Mashour GA. Neural correlates of wakefulness, sleep, and general anesthesia: an experimental study in rat. Anesthesiology. 2016;125:929–942. doi: 10.1097/ALN.0000000000001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White PF, Way WL, Trevor AJ. Ketamine--its pharmacology and therapeutic uses. Anesthesiology. 1982;56:119–136. doi: 10.1097/00000542-198202000-00007. [DOI] [PubMed] [Google Scholar]
- 53.Svenson JE, Abernathy MK. Ketamine for prehospital use: new look at an old drug. Am J Emerg Med. 2007;25:977–980. doi: 10.1016/j.ajem.2007.02.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.