

ABSTRACT
Corynebacterium glutamicum has been applied for the industrial production of various metabolites, such as amino acids. To understand the biosynthesis of the membrane protein in this bacterium, we investigated the process of signal recognition particle (SRP) assembly. SRP is found in all three domains of life and plays an important role in the membrane insertion of proteins. SRP RNA is initially transcribed as precursor molecules; however, relatively little is known about its maturation. In C. glutamicum, SRP consists of the Ffh protein and 4.5S RNA lacking an Alu domain. In this study, we found that 3′-to-5′ exoribonuclease, polynucleotide phosphorylase (PNPase), and two endo-type RNases, RNase E/G and YbeY, are involved in the 3′ maturation of 4.5S RNA in C. glutamicum. The mature form of 4.5S RNA was inefficiently formed in ΔrneG Δpnp mutant cells, suggesting the existence of an alternative pathway for the 3′ maturation of 4.5S RNA. Primer extension analysis also revealed that the 5′ mature end of 4.5S RNA corresponds to that of the transcriptional start site. Immunoprecipitated Ffh protein contained immature 4.5S RNA in Δpnp, ΔrneG, and ΔybeY mutants, suggesting that 4.5S RNA precursors can interact with Ffh. These results imply that the maturation of 4.5S RNA can be performed in the 4.5S RNA-Ffh complex.
IMPORTANCE Overproduction of a membrane protein, such as a transporter, is useful for engineering of strains of Corynebacterium glutamicum, which is a workhorse of amino acid production. To understand membrane protein biogenesis in this bacterium, we investigated the process of signal recognition particle (SRP) assembly. SRP contains the Ffh protein and SRP RNA and plays an important role in the membrane insertion of proteins. Although SRP RNA is highly conserved among the three domains of life, relatively little is known about its maturation. We show that PNPase, RNase E/G, and YbeY are involved in the 3′ maturation of the SRP RNA (4.5S RNA) in this bacterium. This indicates that 3′ end processing in this organism is different from that in other bacteria, such as Escherichia coli.
KEYWORDS: signal recognition particle (SRP), RNA processing, RNase E/G, PNPase, YbeY, Corynebacterium glutamicum
INTRODUCTION
The signal recognition particle (SRP) and its receptor are universally conserved, essential cellular machinery found in all three domains of life. SRP and its receptor catalyze the cotranslational targeting of membrane and secretory proteins (1). Eukaryotic SRP contains six proteins and a 300-nucleotide (nt) RNA (7SL RNA) (2). A 54-kDa protein subunit that binds to signal sequences and that is part of the 7SL RNA forms the core of the eukaryotic SRP (2). In contrast, bacterial SRP is much simpler than its eukaryotic counterpart, consisting of one protein named Ffh (fifty-four homolog) and SRP RNA (1). In bacteria, SRP binds to the hydrophobic amino-terminal signal sequence of the membrane protein and a few secreted proteins as it emerges from the ribosome. SRP then targets the nascent membrane protein-ribosome-mRNA complex to the SRP receptor (FtsY), located at the membrane. Subsequently, the membrane protein-ribosome-mRNA is transferred to the SecYEG channel, and then the membrane protein is inserted into the membrane (3). A GNRA-type tetraloop of the SRP RNA accelerates the initial SRP-FtsY assembly in the membrane-targeting process (4). The SRP RNA is also required for efficient GTPase activation, which controls the targeting of membrane proteins to the translocon (5, 6). Escherichia coli 4.5S RNA also interacts with elongation factor G (EF-G) and the 30S ribosomal subunit independently of Ffh (7).
The size and secondary structure of SRP RNA vary significantly. In eukaryotes and many Gram-positive bacteria, including Bacillus subtilis, the SRP RNA contains an Alu domain, which is composed of the 5′ and 3′ regions (8). In contrast, in many Gram-negative bacteria, including Escherichia coli, the SRP RNA does not contain an Alu domain (8). Some Gram-positive bacteria, including Mycobacterium tuberculosis and Corynebacterium glutamicum, also have a short SRP RNA lacking an Alu domain. The mammalian Alu domain is a protein-RNA complex, while bacterial Alu domains are protein free with significant extensions of the RNA (8). The Alu domains arrest translation elongation of membrane proteins by binding to the elongation factor binding site of ribosomes until targeting is complete (9). A recent study showed that internal Shine-Dalgarno-like elements compensate for the lack of SRP Alu domain-mediated arrest during membrane protein targeting in E. coli (9).
In E. coli, the SRP RNA, known as 4.5S RNA (114 nt), is processed by RNase P to generate a mature 5′ end (10). The mature 3′ end of the 4.5S RNA is generated by 3′-to-5′ exoribonuclease and RNases T, D, PH, and BN/Z (11). RNase T is relatively more important for maturation than the other RNases, and RNase II is the least effective (11). The B. subtilis SRP RNA (small cytoplasmic RNA [scRNA]), which contains an Alu domain, is more than twice the length (281 nt) of the E. coli 4.5S RNA (12). The 5′ mature end is directly generated by RNase III, and the mature 3′ end is generated by RNase Y and 3′ exonucleolytic trimming, mainly performed by RNase PH (12, 13). The mature 3′ end of scRNA can also be generated by inefficient RNase III cleavage (12, 13).
C. glutamicum is a Gram-positive, nonpathogenic soil bacterium which was first isolated in a screen for microorganisms that excrete l-glutamate (14). C. glutamicum is currently used for the industrial production of more than 2.5 million tons of the flavor enhancer l-glutamate and 1 million tons of the feed additive l-lysine per year (15). Far beyond traditional l-amino acid production, this bacterium has been widely applied in the industrial production of various other metabolites, including d-amino acids, organic acids, diamines, fuels, and proteins (16, 17). C. glutamicum belongs to the mycolic acid-containing actinomycetes, which also includes the genera Mycobacterium, Nocardia, and Rhodococcus. In addition to C. glutamicum, this group also contains many industrially important bacterial species. Streptomyces is best known for the production of secondary metabolites with antibiotic properties. Furthermore, many of these mycobacteria can be opportunistic pathogens of humans. However, in spite of these species' importance, the process of SRP assembly in this group is poorly understood. In this study, we identified the major process of 4.5S RNA maturation in C. glutamicum. The purification of Ffh proteins from RNase mutants suggests that precursors to 4.5S RNA can interact with Ffh.
RESULTS
PNPase, RNase E/G, and YbeY play a role in the maturation of C. glutamicum 4.5S RNA.
A high-throughput RNA sequencing (RNA-seq) approach suggested the genomic position of 4.5S RNA in C. glutamicum ATCC 13032 (18). M. tuberculosis 4.5S RNA (C8 small RNA) was mapped between tRNASer and Rv3722c, encoding aspartate aminotransferase (19). The orientation of the neighboring genes is conserved in both M. tuberculosis and C. glutamicum (Fig. 1A). To examine which RNases are involved in the maturation of C. glutamicum 4.5S RNA, we focused on all endo-type RNases except nonspecific RNases and RNase H, which is a DNA-RNA hybrid-specific RNase. We also selected two 3′-to-5′ exoribonuclease RNases, polynucleotide phosphorylase (PNPase) and RNase II/R, which are likely involved in RNA maturation. The RNases that exist in C. glutamicum are listed in Table S2 in the supplemental material. Our attempt to disrupt the rnpB gene, encoding the RNase P RNA component, failed. This suggests that RNase P is essential in C. glutamicum, as is the case in both E. coli and B. subtilis (20). We also constructed RNase J (CgR_1799), RNase Z (CgR_2416), RNase II/R (CgR_2111), and YbeY (CgR_2159) disruptants. Previously, we also constructed RNase E/G, RNase III, and PNPase disruptants (21, 22). The growth of the ybeY mutant was severely impaired. In an A-rich medium containing 1% (wt vol−1) glucose at 33°C, the growth rate of the wild-type strain was 0.83 ± 0.02 h−1, while the ΔybeY mutant strain grew at 0.47 ± 0.01 h−1. Normal growth was restored by complementation with pCRB52iP-ybeY, a plasmid carrying the wild-type ybeY gene (Fig. 1B). Next, we performed Northern blotting using the entire 4.5S RNA probe and total RNAs isolated from wild-type and ΔrneG, Δrnj, Δrnc, ΔybeY, ΔrnZ, Δpnp, and ΔrnR mutant cells and observed precursor RNAs in the Δpnp, ΔrneG, and ΔybeY mutants (Fig. 2A). In the case of M. tuberculosis, 4.5S RNA appeared as a large smear during the exponential phase, but in stationary phase, a distinct band corresponding to mature 4.5S RNA was seen (19). Therefore, we next performed Northern blotting using total RNAs isolated from C. glutamicum cultures in the exponential growth and stationary phases; however, here a difference was not observed (Fig. 2B). These data suggest that PNPase, RNase E/G, and YbeY are involved in the maturation of 4.5S RNA in C. glutamicum.
FIG 1.
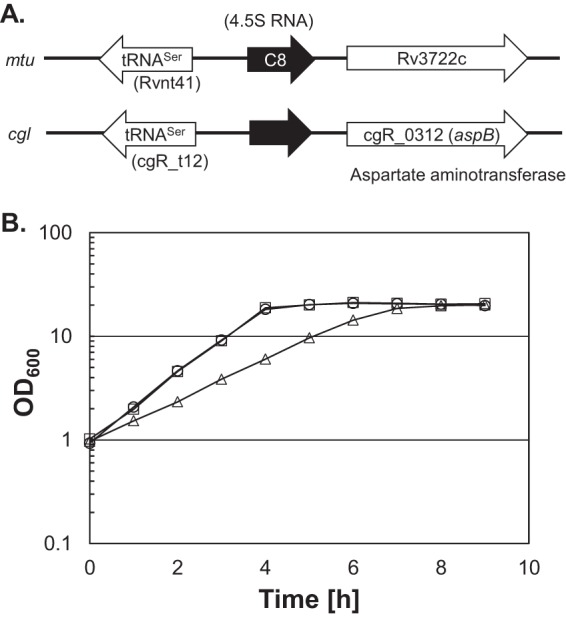
(A) Genomic positions of 4.5S RNA in M. tuberculosis (mtu) and C. glutamicum R (cgl). (B) Growth of the R strain (wild type) harboring pCRB52iP (vector plasmid), the ΔybeY strain harboring pCRB52iP, and the ΔybeY strain harboring pCRB52iP-ybeY. For the growth curve, overnight cultures of the wild type harboring pCRB52iP (circles), the ΔybeY strain harboring pCRB52iP (triangles), and the ΔybeY strain harboring pCRB52iP-ybeY (squares) grown on A-rich medium containing 1% glucose were washed and inoculated into fresh A-rich medium containing 1% glucose and 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Cell growth was monitored by measuring the OD600. Average values and standard deviations from at least three independent cultivations are shown.
FIG 2.
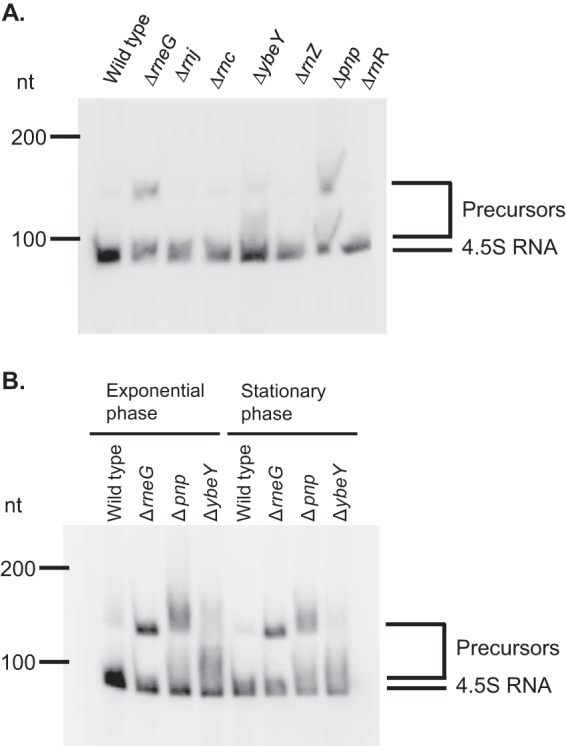
Accumulation of 4.5S RNA precursors in Δpnp, ΔrneG, and ΔybeY mutant cells. (A) Effect of the ΔrneG, Δrnj, Δrnc, ΔybeY, ΔrnZ, Δpnp, and ΔrnR mutations on the maturation of 4.5S RNA. Total RNAs from the exponentially growing R strain (wild type) and ΔrneG, Δrnj, Δrnc, ΔybeY, ΔrnZ, Δpnp, and ΔrnR mutant strains were analyzed by Northern hybridization using the entire 4.5S RNA probe. (B) Maturation of 4.5S RNA in exponential- and stationary-phase cultures. Total RNAs of C. glutamicum from exponential- and stationary-phase R strain (wild type) and ΔrneG, Δpnp, and ΔybeY mutant cells were analyzed by Northern hybridization using the entire 4.5S RNA probe. The DynaMarker RNA Low II molecular mass marker (BioDynamics Laboratory) was used to determine the lengths of the RNAs in some experiments. The experiments were carried out three times.
Mapping of the 5′ and 3′ ends of 4.5S RNA precursors.
To determine the 5′ end of the 4.5S RNA precursors, we performed primer extension analysis using total RNA isolated from the wild-type and Δpnp, ΔrneG, and ΔybeY mutant cells. As shown in Fig. 3A, only one 5′ end was detected in the wild type and the Δpnp, ΔrneG, and ΔybeY mutants. The detected 5′ end was located just downstream of the primary sigma factor SigA (σA) promoter (Fig. 3B). The consensus sequences of the SigA-dependent promoter were defined to be GNTANANTNG for the −10 region and TTGNCA for the −35 region (23), indicating that the 5′ mature end of 4.5S RNA is generated by transcription initiation at the first nucleotide in C. glutamicum.
FIG 3.
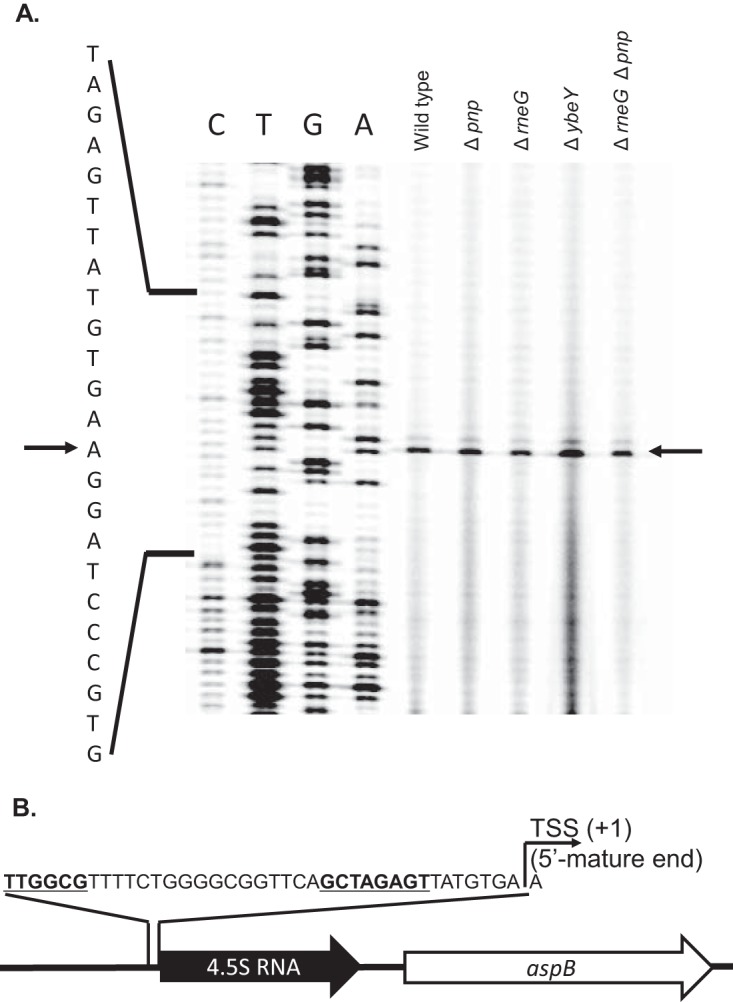
Mapping of the 5′ end of 4.5S RNA precursors. (A) Primer extension analysis of 4.5S RNA from wild-type and Δpnp, ΔrneG, and ΔybeY mutant cells. Primer extension products were derived from the total RNAs of wild-type and Δpnp, ΔrneG, ΔybeY, and ΔrneG Δpnp mutant cells grown on A-rich medium containing 1% glucose medium and separated on a denaturing polyacrylamide gel along with sequencing ladders (C, T, G, and A) obtained using the same primer. Parts of the sequence around the 4.5S RNA coding region are indicated on the left side. The transcriptional start site (TSS) is indicated by arrows. (B) Promoter sequence of 4.5S RNA. The genomic positions of 4.5S RNA and parts of the sequence around the promoter region of 4.5S RNA are shown. The transcriptional start site of the 4.5S RNA is defined as position +1. The experiment was carried out 4 times.
To identify the 3′ ends of 4.5S RNA molecules in wild-type and Δpnp, ΔrneG, and ΔybeY mutant cells, we performed 3′ rapid amplification of cDNA ends (RACE) analysis. For each strain, at least 100 clones were sequenced for each 3′ RACE-PCR product. The results of 3′ RACE analysis are summarized in Table 1. In the wild-type cells, the 3′ end of 66 clones was located 89 nucleotides (nt) downstream of the transcriptional start site (TSS) and is referred to as the +89 end. In addition to this 3′ end, other 3′ ends of 4.5S RNA were also detected in the wild type. In the wild type, the 3′ ends of 11 clones, 7 clones, and 14 clones were located at 100 nt, 124 nt, and 132 nt downstream of the TSS, respectively, and are referred to as the +100, +124, and +132 ends, respectively. This indicates that the most abundant and the shortest 3′ end (the +89 end) detected in the wild-type cells corresponds to the mature 3′ end of 4.5S RNA. In addition, a +124 end was not detected in Δpnp mutant cells. In the Δpnp mutant, the number of clones with a mature 3′ +89 end was reduced, while the number of clones with the longest 3′ end (the +132 end) was increased. In the Δpnp mutant, the 3′ ends of 35 clones were located at the +89 site, the 3′ ends of 17 clones were located at the +100 site, and the 3′ ends of 46 clones were located at the +132 site. The secondary structure of the 4.5S RNA precursor was predicted using RNA fold software (mfold [http://mfold.rna.albany.edu/?q=mfold/rna-folding-form]). As shown in Fig. 4, a GC-rich hairpin loop followed by a run of U residues was found at the 3′ end (position +132) of the transcript, which resembles typical bacterial rho-independent transcriptional terminators. These results indicate that the +132 end corresponds to the 3′ end of the primary transcript. In the ΔrneG mutant, the number of clones with the +89 end was reduced to 27, while the number of clones with the +124 end was increased to 38. Furthermore, in the ΔrneG mutant, only two clones with a +100 end were detected. This indicates that RNase E/G cleaves at the +100 site of the 4.5S RNA precursor. In the ΔybeY mutant, the number of clones with a +89 end was reduced, while the number of clones with a +100 end was increased. This indicates that the mature 3′ end is generated by the action of YbeY. In the ΔybeY mutant, the 3′ ends of 5 clones and 32 clones were located at the +89 and +100 sites, respectively.
TABLE 1.
Summary of 3′ RACE analysisa
| Position of 3′ end | No. of clones |
|||
|---|---|---|---|---|
| R | Δpnp mutant | ΔrneG mutant | ΔybeY mutant | |
| +89 | 66 | 35 | 27 | 5 |
| +90 | 0 | 0 | 0 | 8 |
| +91 | 0 | 0 | 0 | 3 |
| +92 | 0 | 0 | 0 | 7 |
| +93 | 0 | 1 | 0 | 0 |
| +94 | 0 | 0 | 1 | 0 |
| +95 | 0 | 0 | 1 | 0 |
| +96 | 1 | 0 | 0 | 0 |
| +97 | 1 | 1 | 2 | 4 |
| +98 | 0 | 1 | 1 | 2 |
| +99 | 0 | 0 | 1 | 5 |
| +100 | 11 | 17 | 2 | 32 |
| +101 | 0 | 1 | 0 | 4 |
| +102 | 2 | 2 | 1 | 8 |
| +103 | 0 | 0 | 3 | 0 |
| +123 | 0 | 0 | 6 | 0 |
| +124 | 7 | 0 | 38 | 10 |
| +125 | 0 | 0 | 7 | 0 |
| +132 | 14 | 46 | 10 | 15 |
| Total | 104 | 104 | 100 | 103 |
For each strain, at least 100 clones of each 3′ RACE-PCR product were sequenced. The numbers of sequenced clones are indicated in each column. The transcriptional start site of the 4.5S RNA transcript is defined as position +1.
FIG 4.
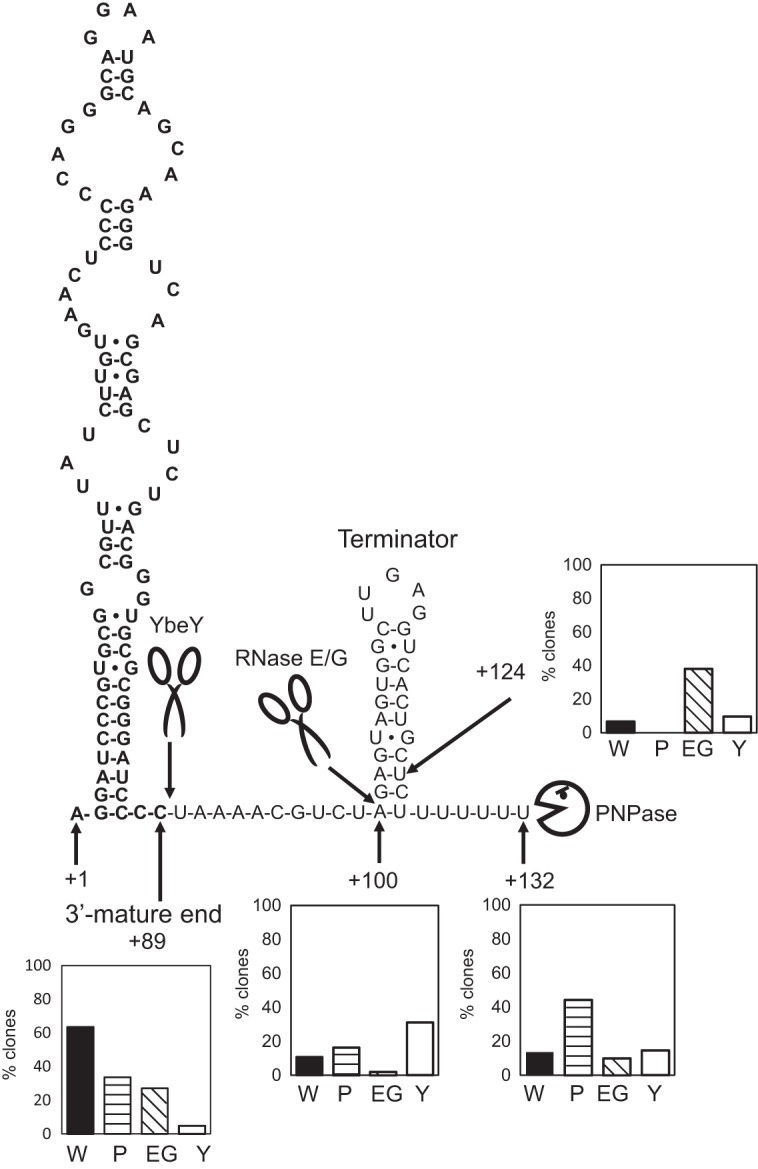
Model for the 3′ maturation of 4.5S RNA in C. glutamicum. The predicted secondary structure of the 4.5S RNA is shown. Bold letters indicate the mature 4.5S RNA sequence. Scissor symbols represent RNase E/G and YbeY. A Pac-Man symbol represents PNPase. Cleavage sites are indicated by arrows. RNA fold software (mfold [http://mfold.rna.albany.edu/?q=mfold/rna-folding-form]) was used to predict the secondary structure of the 4.5S RNA. The positions of the TSS (position +1) and the 3′ ends of 4.5S RNA precursors, determined by 3′ RACE analysis, are shown. The histograms represent the percentage of each sequenced clone of the different mutants listed in Table 1 having the main 3′ ends (the +89, +100, +124, and +132 ends), as determined by 3′ RACE analysis. The TSS of the 4.5S RNA is defined as position +1. W, wild type; P, Δpnp mutant; EG, ΔrneG mutant; Y, ΔybeY mutant. For details, see the text.
We also constructed ΔrneG Δpnp and ΔrneG ΔybeY double mutants. The growth of the ΔrneG Δpnp mutant was severely defective (Fig. 5). In an A-rich medium containing 1% (wt vol−1) glucose at 33°C, the growth rate of the ΔrneG mutant was 0.72 ± 0.02 h−1, the growth rate of the Δpnp mutant was 0.73 ± 0.01 h−1, and the growth rate of the ΔrneG Δpnp double mutant strain was 0.55 ± 0.01 h−1. We successfully obtained ΔrneG ΔybeY double mutants that grew significantly more slowly than the single ΔybeY mutant. The colonies of the ΔrneG ΔybeY double mutant required incubation on A-rich agar plates for 3 to 4 days before they became visible. However, after long-term cultivation (about 2 days) in liquid A-rich medium, the ΔrneG ΔybeY double mutant strain sometimes grew better, probably because of a spontaneous mutation. Therefore, we did not determine the growth rate of the ΔrneG ΔybeY double mutant strain. In addition, the Δpnp ΔybeY double mutant could not be generated, suggesting that the simultaneous disruption of ybeY and pnp is lethal in C. glutamicum. To verify that the precursor molecules have 3′ extensions, Northern blotting was performed using the 3′ spacer region-specific RNA probe. As shown in Fig. 6B, the precursor molecules were detected by the 3′ spacer region-specific RNA probe in the ΔrneG, Δpnp, ΔrneG Δpnp, and ΔrneG ΔybeY mutants. In addition, using the entire 4.5S RNA probe, an increased amount of the 4.5S RNA precursor was detected, while a decreased amount of mature 4.5S RNA was detected in the ΔrneG Δpnp mutant (Fig. 6B). These results indicate that PNPase, RNase E/G, and YbeY are required for the 3′ maturation of 4.5S RNA in C. glutamicum.
FIG 5.
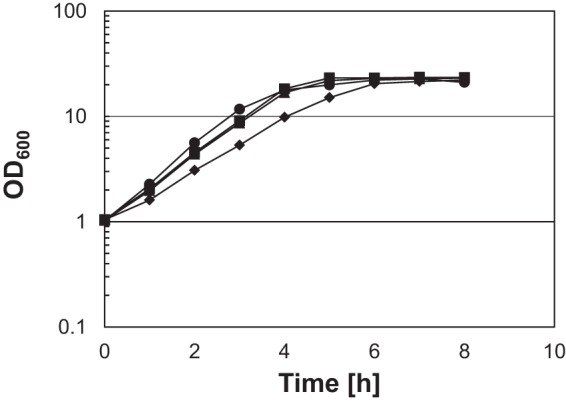
Growth of the wild type and ΔrneG, Δpnp, and ΔrneG Δpnp mutants. For the growth curve, overnight cultures of the wild type (circles), the ΔrneG mutant (triangles), the Δpnp mutant (squares), and the ΔrneG Δpnp mutant (diamonds) grown on A-rich medium containing 1% glucose were washed and inoculated into fresh A-rich medium containing 1% glucose. Cell growth was monitored by measuring the OD600. Average values and standard deviations from at least three independent cultivations are shown.
FIG 6.
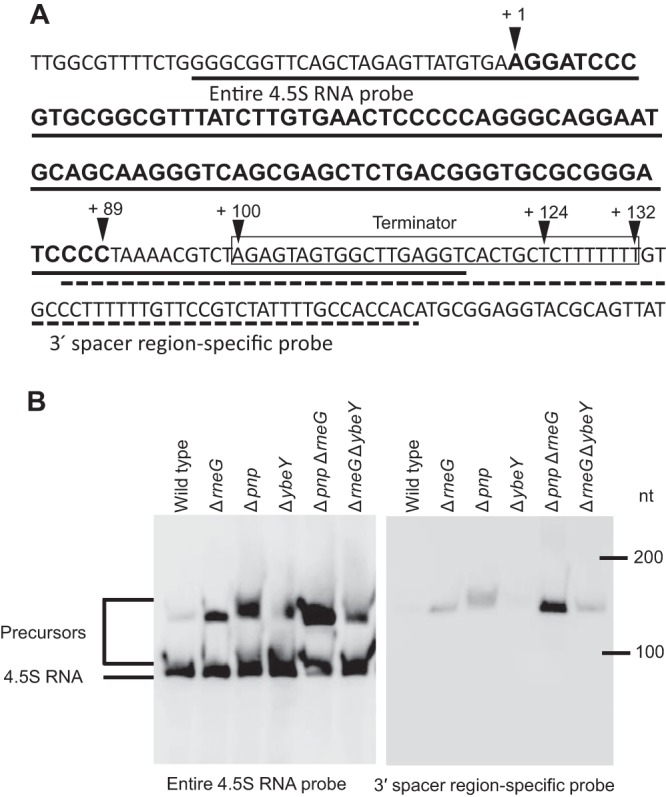
Northern blotting verifies that the precursor molecules have 3′ extensions. (A) The DNA sequence of the flanking region encoding 4.5S RNA is shown. The 4.5S RNA coding region is indicated by bold letters. The putative cleavage sites were determined by 3′ RACE analysis, and mature 5′ and 3′ ends are indicated by the arrowheads. The terminator sequence of 4.5S RNA is boxed. The region which is complementary to the entire 4.5S RNA probe is underlined with a solid line. The region which is complementary to the 3′ spacer region-specific probe is underlined with a dotted line. (B) Total RNAs from exponential-phase strain R (wild type) and ΔrneG, Δpnp, ΔybeY, ΔrneG Δpnp, and ΔrneG ΔybeY mutant cells were analyzed by Northern hybridization using the entire 4.5S RNA probe and the 3′ spacer region-specific RNA probe. The DynaMarker RNA Low II molecular mass marker (BioDynamics Laboratory) was used to determine the lengths of the RNAs in some experiments. The experiments were carried out three times.
4.5S RNA precursors were copurified with Ffh-FLAG proteins.
To understand how mature SRP is formed, we examined whether 4.5S RNA precursors are complexed with the Ffh protein. We first constructed strains harboring a modified ffh gene encoding a C-terminally FLAG-tagged Ffh protein. The chromosomally encoded ffh genes of the wild-type and ΔybeY, ΔrneG, Δpnp, and ΔrneG Δpnp mutant strains were replaced with the modified ffh gene. The newly constructed Ffh-FLAG strains were named FFHT1 (wild type), FFHT2 (Δpnp), FFHT3 (ΔrneG), FFHT4 (ΔybeY), and FFHT2 (ΔrneG Δpnp). Then, we purified the Ffh-FLAG proteins from the FFHT1, FFHT2, FFHT3, FFHT4, and FFHT5 strains. We also prepared the negative-control samples by the same procedures from the wild-type and ΔybeY, ΔrneG, Δpnp, and ΔrneG Δpnp mutant strains harboring a native ffh gene. Purification of the Ffh-FLAG protein was checked by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and a single band with a molecular mass of ∼60 kDa was observed (Fig. 7A). The size corresponded to that of Ffh, which has a molecular mass of 58 kDa. The purified Ffh-FLAG protein samples were subjected to Northern analysis to identify any 4.5S RNA-related molecules bound to it. As shown in Fig. 7B, both the 4.5S RNA precursors and mature forms were detected in the purified Ffh-FLAG proteins from the ΔybeY, ΔrneG, Δpnp, and ΔrneG Δpnp mutants. No band was detected in the negative-control strains harboring the wild-type ffh gene, using both the entire 4.5S RNA probe and the 3′ spacer region-specific RNA probe (data not shown). These results suggest that 4.5S RNA precursors can interact with Ffh.
FIG 7.
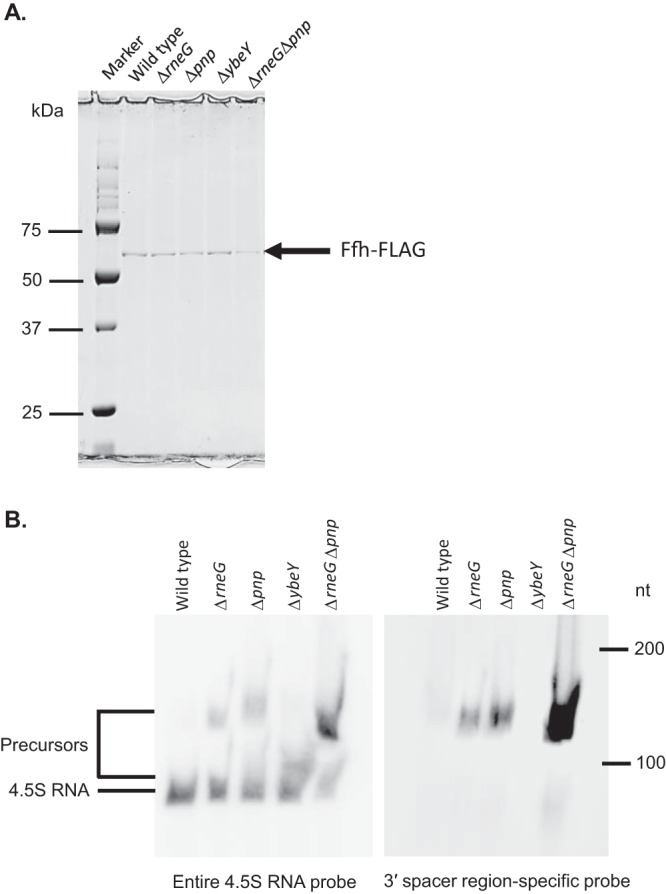
The Ffh protein interacts with 4.5S RNA precursors. (A) Purification of the Ffh-FLAG protein was examined by 10% SDS-PAGE. The position of the Ffh-FLAG protein is shown to the right of the gel. (B) Purified Ffh-FLAG proteins from exponentially growing FFHT1 (wild type), FFHT2 (Δpnp), FFHT3 (ΔrneG), FFHT4 (ΔybeY), and FFHT5 (ΔrneG Δpnp) cells were analyzed by Northern hybridization using the entire 4.5S RNA probe and the 3′ spacer region-specific RNA probe. The DynaMarker RNA Low II molecular mass marker (BioDynamics Laboratory) was used to determine the lengths of the RNAs in some experiments. The experiments were carried out three times.
DISCUSSION
In this study, we investigated the mechanism of 4.5S RNA maturation in C. glutamicum. We found that deletion of pnp, rneG, or ybeY resulted in the accumulation of 4.5S RNA precursors. We also showed that the 5′ mature end of 4.5S RNA is generated by the initiation of transcription at the first nucleotide in C. glutamicum. 3′ RACE analysis revealed that 3′ processing of 4.5S RNA is performed by PNPase, RNase E/G, and YbeY. Northern blotting using purified Ffh-FLAG protein showed that 4.5S RNA precursors can interact with Ffh.
The results of primer extension analysis and 3′ RACE analysis suggested a possible mechanism for 4.5S RNA maturation in C. glutamicum wild-type cells (Fig. 4). The 4.5S RNA precursor is first transcribed from a primary sigma factor (σA) promoter. The 5′ mature end of 4.5S RNA corresponded to the TSS. The predicted secondary structure of the 4.5S RNA primary transcript has a rho-independent transcriptional terminator at the 3′ end (position +132). The longest precursor accumulated in Δpnp mutant cells, as determined by Northern blotting (Fig. 2B and 6B). This suggests that PNPase processes the primary transcript. 3′ RACE analysis also supports this hypothesis because the clone with the longest 3′ end (the +132 end) was the most abundant 4.5S RNA precursor in Δpnp mutant cells (Table 1). It was reported that a strong stem-loop structure found at the 3′ end of RNA, which functions as the transcription terminator structure, is resistant to 3′-to-5′ exonucleolytic degradation in C. glutamicum (24). Thus, trimming of the 3′ spacer region of 4.5S RNA could be blocked by the stem-loop structure. PNPase is thought to remove the run of U residues, and the GC-rich hairpin loop is resistant to PNPase activity. In E. coli, PNPase requires a minimal 3′ overhang of 7 unpaired ribonucleotides for binding to RNA molecules without the help of poly(A) polymerase I (PAPI) (25). C. glutamicum has a PAPI orthologue encoded by the CgR_2974 gene. This PAPI orthologue may facilitate the 3′ processing of 4.5S RNA performed by PNPase in C. glutamicum. It has also been reported that PNPase adds a heteropolymeric tail to the 3′ ends of the RNA transcript (26) and plays a primary role in the posttranscriptional modification of RNA in a variety of bacteria (27). Therefore, it is also possible that PNPase can first attack the rho-independent terminator of 4.5S RNA by adding a heteropolymeric tail to the 3′ ends of the 4.5S RNA primary transcript. The +124 end was detected in wild-type cells, and the +124 end was the most abundant 3′ end in ΔrneG mutant cells (Table 1). The +124 end is located in close proximity to the stem-loop structure, suggesting that PNPase activity is blocked at this site. The +100 end was also detected in wild-type cells, and the +100 end was the most abundant 3′ end in ΔybeY mutant cells (Table 1). The +100 end was almost not detected in ΔrneG mutant cells (Table 1), suggesting that RNase E/G cleaves at the +100 site. Although RNase E/G cleaves single-stranded RNA, the RNase E/G cleavage site falls within the end of the double-stranded region. Since the precursor with a +124 end seems to be the main substrate for RNase E/G, the RNase E/G cleavage site (position +100) could turn out to be a single-stranded region after the removal of complementary residues by PNPase. 3′ RACE analysis showed that the mature 3′ end was significantly reduced in ΔybeY mutant cells, suggesting that YbeY generates the mature 3′ end of 4.5S RNA. In contrast, the precursor molecules were almost not detected in the ΔybeY mutant by Northern blotting using a 3′ spacer region-specific RNA probe (Fig. 6B). 3′ RACE analysis showed that 64% of the precursor molecules had less than 11 extra nucleotides in the ΔybeY mutant (Table 1). Since the 3′ spacer region-specific RNA probe covers only 14 nucleotides of these short precursor molecules, the 3′ probe might not detect the precursor molecules efficiently in the ΔybeY mutant (Fig. 6B). In many species, like E. coli and B. subtilis, the mature 3′ end of SRP RNA is generated by 3′-to-5′ exonucleolytic trimming (11, 12). However, our 3′ RACE analysis suggests that the mature 3′ end of the SRP RNA is generated by endoribonuclease YbeY. This is the first report showing that YbeY is involved in SRP RNA maturation. The 3′ RACE analysis suggested that an efficient processing pathway is performed by PNPase, RNase E/G, and YbeY, in that order. However, we could not conclude that YbeY cleaves most efficiently after RNase E/G cleavage since YbeY can cleave a certain portion of the 4.5S RNA precursor at any time. Therefore, it is also possible that these RNases process the 4.5S RNA precursor in alternative orders.
The immunoprecipitated Ffh protein contained immature 4.5S RNA in the Δpnp, ΔrneG, and ΔybeY mutants (Fig. 6B). This may suggest that the 3′ maturation of 4.5S RNA is performed in the 4.5S RNA-Ffh complex. This hypothesis is consistent with what has been seen for rRNA maturation. It was shown that mature 16S rRNA can be generated after the 16S rRNA precursor is assembled into 30S subunits and subsequently into 70S ribosomes in E. coli (28). A recent study showed that 17S rRNA, which is generated by RNase III action on the primary transcript of rRNAs, is a platform for ribosome biogenesis (29).
Even in the absence of the individual RNases or both PNPase and RNase E/G, the mature form of 4.5S RNA was detected (Fig. 2B and 6B). These results suggest that multiple pathways of entire SRP biogenesis exist. In ΔybeY mutant cells, several precursor bands or smear bands were detected (Fig. 2, 6B, and 7B). This may result from the action of another RNase which compensates for the lack of YbeY. It is also possible that there are other RNase E/G cleavage sites since YbeY is supposed to act after RNase E/G cleavage and RNase E/G can cleave the AU-rich single-stranded region. It is also possible that PNPase removes all extra nucleotides, including the rho-independent terminator at the 3′ spacer region. In the case of leuX tRNA maturation in E. coli, PNPase degrades through the entire rho-independent transcription terminator (30).
MATERIALS AND METHODS
Media and growth conditions.
C. glutamicum R (31) and its mutant strains used in this study are listed in Table 2. C. glutamicum was grown aerobically at 33°C with shaking at 180 rpm. C. glutamicum cells were precultured at 33°C overnight in nutrient-rich medium (A-rich medium) (32) supplemented with 1% (wt vol−1) glucose. The media were supplemented with 50 μg ml−1 kanamycin when necessary. Bacterial growth was monitored by determining the optical density at 600 nm (OD600). E. coli strain HST02 (TaKaRa) was used as a host for all genetic manipulations. The E. coli strains were cultivated at 37°C in LB medium supplemented with 50 μg ml−1 kanamycin when necessary.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| HST02 | F′ [traD36 proA+B+ lacIq lacZΔM15] Δ(lac-proAB) recA endA gyrA96 thi Δe14 [Δ(ymfJ-mcrA)] supE44 relA ΔdeoR Δ(mrr-hsdRMS-mcrBC) | TaKaRa |
| SCS110 | rpsL (Strr) thr leu endA thi-1 lacY galK galT ara tonA tsx dam dcm supE44D (lac-proAB) [F′ traD36 proAB lacIq lacZΔM15] | TaKaRa |
| C. glutamicum | ||
| R | Wild-type strain JCM18229 | 31 |
| ΔrneG mutant | R ΔrneG | 22 |
| Δrnc mutant | R Δrnc | 21 |
| Δpnp mutant | R Δpnp | 21 |
| Δrnj mutant | R Δrnj | Takemoto et al., unpublisheda |
| ΔrnR mutant | R ΔrnR | This study |
| ΔrnZ mutant | R ΔrnZ | This study |
| ΔybeY mutant | R ΔybeY | This study |
| ΔrneG Δpnp mutant | R ΔrneG Δpnp | This study |
| ΔrneG ΔybeY mutant | R ΔrneG ΔybeY | This study |
| FFHT1 | R with FLAG-tagged ffh | This study |
| FFHT2 | Δpnp mutant with FLAG-tagged ffh | This study |
| FFHT3 | ΔrneG mutant with FLAG-tagged ffh | This study |
| FFHT4 | ΔybeY mutant with FLAG-tagged ffh | This study |
| FFHT5 | ΔrneG Δpnp mutant with FLAG-tagged ffh | This study |
| Plasmids | ||
| pCRA725 | Kmr; suicide vector containing the Bacillus subtilis sacB gene | 33 |
| LKSΔrnR | pCRA725 with pnp fragment | This study |
| LKSΔrnZ | pCRA725 with rnZ fragment | This study |
| LKSΔybeY | pCRA725 with ybeY fragment | This study |
| LKSffh-FLAG | pCRA725 with a gene encoding Ffh-FLAG | This study |
| pCRB52iP | E. coli-C. glutamicum shuttle vector; Kmr ColE1 ori pBY503 ori tac promoter lacIq | 21 |
| pCRB52iP-ybeY | pCRB52iP with ybeY | This study |
| pSPT18 | Apr; pUC18-based vector containing T7 and SP6 promoters | Roche |
| pGEM-T Easy | Apr; vector | Promega |
N. Takemoto, Y. Tanaka, T. Maeda, N. Hamamoto, and M. Inui, unpublished data.
Bacterial strains and plasmids.
The strains and plasmids used in this study are listed in Table 2. The oligonucleotide primers used in this study are listed in Table S1 in the supplemental material. To construct ybeY (CgR_2159) expression plasmid pCRB52iP-ybeY, the DNA fragment containing the coding region of ybeY was amplified by PCR using genomic DNA as the template and cloned into E. coli-C. glutamicum shuttle vector plasmid pCRB52iP (21). All gene-modified strains were constructed as described previously (21). A suicide vector, pCRA725, carrying the sacB gene was used for markerless gene deletion and gene modification (33). Using plasmids LKSΔrnZ, LKSΔybeY, and LKSΔrnR and C. glutamicum strain R as a wild type, ΔrnZ, ΔybeY, and ΔrnR strains were constructed by two-step homologous recombination, as described previously (34). The DNA fragment encoding the ffh gene fused with a FLAG tag (DYKDDDK) was cloned into plasmid pCRA725 (33), yielding LKSffh-FLAG. Using the LKSffh-FLAG plasmid, the ffh (CgR_1945)-FLAG strains were also constructed by two-step homologous recombination. The FLAG tag (DYKDDDK) was added at the C terminus of ffh.
Total RNA purification.
Overnight cultures grown on A-rich medium containing 1% glucose at 33°C were washed and then inoculated into fresh A-rich medium containing 1% glucose. Two volumes of RNA Protect bacterial reagent (Qiagen) were added directly to 1 volume of exponentially growing cultures at an OD600 of ∼4 (in logarithmic phase) to stabilize cellular RNAs. The cells were harvested by centrifugation at 5,000 × g for 10 min at 25°C, and total cellular RNAs were isolated using an miRNeasy minikit (Qiagen) according to the manufacturer's instructions. Total RNA was treated with DNase I at 37°C.
Northern hybridization.
Northern hybridization was performed as described previously (34). A digoxigenin (DIG)-labeled antisense RNA probe targeting the 4.5S RNA coding region (entire 4.5S RNA probe) and the 3′ spacer region (3′ spacer region-specific RNA probe) was produced according to the manufacturer's instructions (Roche). To generate a hybridization probe, specific DNA fragments were amplified by PCR using the primers listed in Table S1. The amplified fragments were cloned into plasmid pSPT18 (Roche). The correctness of the cloned DNA fragments was confirmed by DNA sequencing. The newly constructed plasmids were used as a template for synthesis of the DIG-labeled antisense RNA probes. Northern hybridization was performed according to the procedures described in the DIG Northern starter kit (Roche Diagnostics). Briefly, 2 μg of total RNA or RNA that was copurified with the Ffh-FLAG protein was fractionated on denaturing polyacrylamide gels (8.3 M urea, 6% gel) in 1× TBE buffer (90 mM Tris-borate, 2 mM EDTA, pH 8.0). The hybridization temperature was 68°C. Positive hybridization bands were detected using the CDP-Star reagent (Roche Diagnostics) with exposure times of between 10 s and 5 min.
Primer extension analysis.
Primer extension was carried out using appropriate gene-specific primers and 5 μg total RNA. Briefly, 1.5 pmol IRD700-labeled fluorescein primer 5′-CTCGCTGACCCTTGCTGCATTCC-3′, which was complementary to a region within the 4.5S RNA coding region, was hybridized to RNA at 65°C for 5 min, and cDNA was synthesized at 50°C for 60 min using a SuperScript III first-strand synthesis system for reverse transcription-PCR (Invitrogen) according to the manufacturer's instructions. The template for the sequencing ladders was PCR amplified using the primers 5′-CTAGAACGGCTGCAACACAT-3′ and 5′-CCTGAATTCGTGGTGGCAAAATAGA-3′. A 1.0-μl aliquot of the sample was heat denatured and loaded onto a denaturing 5.5% (wt vol−1) polyacrylamide sequencing gel in parallel with the sequencing ladder and run in a LI-COR DNA sequencer (Aloka model 4000).
3′ RACE-PCR.
To map the 3′ ends of 4.5S RNA precursors, 3′ rapid amplification of cDNA ends (RACE) analysis was carried out using a SMARTer RACE cDNA amplification kit (Clontech). The cDNA was amplified with universal primer A (supplied with the kit) and gene-specific primer 5′-GATCCCGTGCGGCGTTTATCTTGTG-3′. The resulting PCR product was cloned into a pGEM-T Easy vector (Promega). For each strain, at least 100 clones of each 3′ RACE-PCR product were sequenced.
Purification of Ffh.
Expression and purification of the Ffh-FLAG protein were performed as described previously (35). Briefly, exponentially growing C. glutamicum cultures (100 ml) in A-rich medium with 1% glucose were collected by centrifugation, washed twice with Tris-buffered saline (20 mM Tris-HCl [pH 7.5], 150 mM NaCl), and stored at −80°C. The pellets were resuspended in 0.5 ml FLAG buffer (20 mM Tris-HCl [pH 8.0], 200 mM KCl, 5 mM MgCl2, 10% glycerol, 0.1% SDS) containing protease inhibitor cocktail (Roche Antiprotease Mini). The cell suspension was transferred to a 15-ml tube containing 300 mg of 0.1-mm zirconia-silica beads (BioSpec). Cell disruption was conducted using a Bioruptor UCD-250 sonicator (Cosmo Bio) under the following conditions: 10 cycles of 5 s on and 5 s off at high power at 4°C. The cell lysate was collected and centrifuged (20,000 × g for 30 min at 4°C) to prepare the cell extract. The amount of total protein was measured by a Bio-Rad protein assay (Bio-Rad) using bovine serum albumin (BSA) as a standard. The cell extract was incubated with DNase I (TaKaRa) for 60 min at 4°C. The incubated extract was subjected to immunoprecipitation with 80 μl of anti-FLAG M2-agarose (Sigma). The mixture was incubated for 90 min on a rotating platform at 4°C. The beads were washed five times with FLAG buffer. Immunoprecipitated complexes were eluted from beads by treatment with 240 μl of immunoprecipitation buffer with 100 ng μl−1 of 3× FLAG-peptide (Sigma). Purification of the Ffh-FLAG protein was checked using sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Coomassie brilliant blue staining. The purified Ffh-FLAG protein samples were further used for Northern analysis to detect copurified RNA with the Ffh-FLAG protein.
Supplementary Material
ACKNOWLEDGMENTS
We thank Norihiko Takemoto for generously sharing the Δrnj strain.
This work was partially supported by a grant from the Ministry of Economy, Trade and Industry of Japan, entrusted by the New Energy and Industrial Technology Development Organization (NEDO); a grant-in-aid for Scientific Research (B) (20380047 to M.W.) from the Japan Society for the Promotion of Science; and a grant from the Global COE Program of the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00798-16.
REFERENCES
- 1.Akopian D, Shen K, Zhang X, Shan SO. 2013. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem 82:693–721. doi: 10.1146/annurev-biochem-072711-164732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wild K, Sinning I. 2014. RNA gymnastics in mammalian signal recognition particle assembly. RNA Biol 11:1330–1334. doi: 10.1080/15476286.2014.996457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalbey RE, Chen M. 2004. Sec-translocase mediated membrane protein biogenesis. Biochim Biophys Acta 1694:37–53. doi: 10.1016/j.bbamcr.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Shen K, Shan SO. 2010. Transient tether between the SRP RNA and SRP receptor ensures efficient cargo delivery during cotranslational protein targeting. Proc Natl Acad Sci U S A 107:7698–7703. doi: 10.1073/pnas.1002968107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen K, Arslan S, Akopian D, Ha T, Shan SO. 2012. Activated GTPase movement on an RNA scaffold drives co-translational protein targeting. Nature 492:271–275. doi: 10.1038/nature11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voigts-Hoffmann F, Schmitz N, Shen K, Shan SO, Ataide SF, Ban N. 2013. The structural basis of FtsY recruitment and GTPase activation by SRP RNA. Mol Cell 52:643–654. doi: 10.1016/j.molcel.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu SQ, Jockel J, Beinker P, Warnecke J, Semenkov YP, Rodnina MV, Wintermeyer W. 2005. Conformation of 4.5S RNA in the signal recognition particle and on the 30S ribosomal subunit. RNA 11:1374–1384. doi: 10.1261/rna.7219805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kempf G, Wild K, Sinning I. 2014. Structure of the complete bacterial SRP Alu domain. Nucleic Acids Res 42:12284–12294. doi: 10.1093/nar/gku883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fluman N, Navon S, Bibi E, Pilpel Y. 2014. mRNA-programmed translation pauses in the targeting of E. coli membrane proteins. eLife 3:e03440. doi: 10.7554/eLife.03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peck-Miller KA, Altman S. 1991. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol 221:1–5. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Pandit S, Deutscher MP. 1998. 3′ Exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc Natl Acad Sci U S A 95:2856–2861. doi: 10.1073/pnas.95.6.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilet L, DiChiara JM, Figaro S, Bechhofer DH, Condon C. 2015. Small stable RNA maturation and turnover in Bacillus subtilis. Mol Microbiol 95:270–282. doi: 10.1111/mmi.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oguro A, Kakeshita H, Nakamura K, Yamane K, Wang W, Bechhofer DH. 1998. Bacillus subtilis RNase III cleaves both 5′- and 3′-sites of the small cytoplasmic RNA precursor. J Biol Chem 273:19542–19547. doi: 10.1074/jbc.273.31.19542. [DOI] [PubMed] [Google Scholar]
- 14.Kinoshita S, Udaka S, Shimono M. 1957. Studies on the amino acid fermentation. I. Production of l-glutamic acid by various microorganisms. J Gen Appl Microbiol 3:193–205. [PubMed] [Google Scholar]
- 15.Hirasawa T, Shimizu H. 2016. Recent advances in amino acid production by microbial cells. Curr Opin Biotechnol 42:133–146. doi: 10.1016/j.copbio.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Becker J, Wittmann C. 2012. Bio-based production of chemicals, materials and fuels—Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. doi: 10.1016/j.copbio.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 17.Matsuda Y, Itaya H, Kitahara Y, Theresia NM, Kutukova EA, Yomantas YA, Date M, Kikuchi Y, Wachi M. 2014. Double mutation of cell wall proteins CspB and PBP1a increases secretion of the antibody Fab fragment from Corynebacterium glutamicum. Microb Cell Fact 13:56. doi: 10.1186/1475-2859-13-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mentz A, Neshat A, Pfeifer-Sancar K, Pühler A, Rückert C, Kalinowski J. 2013. Comprehensive discovery and characterization of small RNAs in Corynebacterium glutamicum ATCC 13032. BMC Genomics 14:714. doi: 10.1186/1471-2164-14-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnvig KB, Young DB. 2009. Identification of small RNAs in Mycobacterium tuberculosis. Mol Microbiol 73:397–408. doi: 10.1111/j.1365-2958.2009.06777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Condon C, Putzer H. 2002. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res 30:5339–5346. doi: 10.1093/nar/gkf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeda T, Tanaka Y, Takemoto N, Hamamoto N, Inui M. 2016. RNase III mediated cleavage of the coding region of mraZ mRNA is required for efficient cell division in Corynebacterium glutamicum. Mol Microbiol 99:1149–1166. doi: 10.1111/mmi.13295. [DOI] [PubMed] [Google Scholar]
- 22.Takemoto N, Tanaka Y, Inui M. 2015. Rho and RNase play a central role in FMN riboswitch regulation in Corynebacterium glutamicum. Nucleic Acids Res 43:520–529. doi: 10.1093/nar/gku1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pátek M, Nešvera J. 2011. Sigma factors and promoters in Corynebacterium glutamicum. J Biotechnol 154:101–113. doi: 10.1016/j.jbiotec.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Maeda T, Wachi M. 2012. 3′ untranslated region-dependent degradation of the aceA mRNA, encoding the glyoxylate cycle enzyme isocitrate lyase, by RNase E/G in Corynebacterium glutamicum. Appl Environ Microbiol 78:8753–8761. doi: 10.1128/AEM.02304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, Moreira RN, Pobre V, Reis FP, Saramago M, Silva IJ, Viegas SC. 2010. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev 34:883–923. doi: 10.1111/j.1574-6976.2010.00242.x. [DOI] [PubMed] [Google Scholar]
- 26.Mohanty BK, Kushner SR. 2006. The majority of Escherichia coli mRNAs undergo post-transcriptional modification in exponentially growing cells. Nucleic Acids Res 34:5695–5704. doi: 10.1093/nar/gkl684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohanty BK, Kushner SR. 2000. Polynucleotide phosphorylase functions both as a 3′ → 5′ exonuclease and a poly(A) polymerase in Escherichia coli. Proc Natl Acad Sci U S A 97:11966–11971. doi: 10.1073/pnas.220295997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Pandit S, Deutscher MP. 1999. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J 18:2878–2885. doi: 10.1093/emboj/18.10.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta N, Culver GM. 2014. Multiple in vivo pathways for Escherichia coli small ribosomal subunit assembly occur on one pre-rRNA. Nat Struct Mol Biol 21:937–943. doi: 10.1038/nsmb.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohanty BK, Kushner SR. 2010. Processing of the Escherichia coli leuX tRNA transcript, encoding tRNA(Leu5), requires either the 3′ → 5′ exoribonuclease polynucleotide phosphorylase or RNase P to remove the Rho-independent transcription terminator. Nucleic Acids Res 38:597–607. doi: 10.1093/nar/gkp997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, Vertès AA, Inui M. 2007. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058. doi: 10.1099/mic.0.2006/003657-0. [DOI] [PubMed] [Google Scholar]
- 32.Inui M, Murakami S, Okino S, Kawaguchi H, Vertès AA, Yukawa H. 2004. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J Mol Microbiol Biotechnol 7:182–196. doi: 10.1159/000079827. [DOI] [PubMed] [Google Scholar]
- 33.Inui M, Kawaguchi H, Murakami S, Vertès AA, Yukawa H. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J Mol Microbiol Biotechnol 8:243–254. [DOI] [PubMed] [Google Scholar]
- 34.Maeda T, Wachi M. 2012. Corynebacterium glutamicum RNase E/G-type endoribonuclease encoded by NCgl2281 is involved in the 5′ maturation of 5S rRNA. Arch Microbiol 194:65–73. doi: 10.1007/s00203-011-0728-3. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka Y, Takemoto N, Ito T, Teramoto H, Yukawa H, Inui M. 2014. Genome-wide analysis of the role of global transcriptional regulator GntR1 in Corynebacterium glutamicum. J Bacteriol 196:3249–3258. doi: 10.1128/JB.01860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.