

Abstract
Background
Corynebacterium glutamicum is a non-pathogenic bacterium widely used in industrial amino acid production and metabolic engineering research. Although the genome sequences of some C. glutamicum strains are available, comprehensive comparative genome analyses of these species have not been done. Six wild type C. glutamicum strains were sequenced using next-generation sequencing technology in our study. Together with 20 previously reported strains, we present a comprehensive comparative analysis of C. glutamicum genomes.
Results
By average nucleotide identity (ANI) analysis, we show that 10 strains, which were previously classified either in the genus Brevibacterium, or as some other species within the genus Corynebacterium, should be reclassified as members of the species C. glutamicum. C. glutamicum has an open pan-genome with 2359 core genes. An additional NAD+/NADP+ specific glutamate dehydrogenase (GDH) gene (gdh) was identified in the glutamate synthesis pathway of some C. glutamicum strains. For analyzing variations related to amino acid production, we have developed an efficient pipeline that includes three major steps: multi locus sequence typing (MLST), phylogenomic analysis based on single nucleotide polymorphisms (SNPs), and a thorough comparison of all genomic variation amongst ancestral or closely related wild type strains. This combined approach can provide new perspectives on the industrial use of C. glutamicum.
Conclusions
This is the first comprehensive comparative analysis of C. glutamicum genomes at the pan-genomic level. Whole genome comparison provides definitive evidence for classifying the members of this species. Identifying an aditional gdh gene in some C. glutamicum strains may accelerate further research on glutamate synthesis. Our proposed pipeline can provide a clear perspective, including the presumed ancestor, the strain breeding trajectory, and the genomic variations necessary to increase amino acid production in C. glutamicum.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3255-4) contains supplementary material, which is available to authorized users.
Keywords: Corynebacterium glutamicum, Pan-genome, Comparative genomics, Production of amino acids
Background
The non-spore-forming Gram-positive bacterium Corynebacterium glutamicum, a non-pathogenic species in the Corynebacterium genus, has been widely used for the industrial production of amino acids, because of its numerous and ideally suited attributes [1].
C. glutamicum was first discovered as a producer of glutamate. As early as the 1950s, strains accumulating glutamate in culture medium were isolated. One of them, M534, previously taxonomically named “Micrococcus glutamicus” and deposited as ATCC 13032 and NCIMB 10025, was designated as the C. glutamicum type strain [2]. In the 1960s and into the 1970s, several strains accumulating glutamate were isolated independently, including “Brevibacterium lactofermentum” ATCC 13869, “B. flavum” ATCC 14067, “C. acetoacidophilum” ATCC 13870, “C. crenatum” AS1.542, “C. pekinense” AS1.299, and “B. tianjinese” T6-13 [3–6]. According to previous reports and our recent research, these strains should all be classified as C. glutamicum species based on sharing roughly identical 16S rDNA sequences [5, 7].
Much research has been done on modifying C. glutamicum in various ways to make it more useful for humans. Classical strain breeding methods have been used to introduce mutations into the C. glutamicum genome since the 1950s. These breeding methods are based on random mutation and screening/selection techniques, and can be used to generate glutamate (as well as other amino acids, such as lysine) hyper-producing strains [8–12]. Metabolic engineering has been performed on C. glutamicum since the 1980s. These studies have focused on not only producing amino acids, but also on creating biosynthetic pathways for the production of many more chemicals, including succinate and 2,3-butanediol [13–16].
The genome sequences of 20 C. glutamicum strains were available previous to our study. The complete genome sequence of two type strain ATCC 13032 variants were initially published [17, 18]. The genome sequence of C. glutamicum R, a strain from a laboratory collection isolated in Japan, was subsequently reported [19]. The complete or draft genome sequences for many industrial producers, generated by conventional mutagenesis, have also been reported, including lysine producer B253 and glutamate producer S9114 [20, 21]. However, most of these strains have not been analyzed on a deep, genomic scale.
Recently, we have established a MLST scheme based on sequences of seven housekeeping genes of 17 strains for genotyping of C. glutamicum, which helps to understand the population structure of this bacterium [7]. MLST relies on allelic variants in conserved genes, so it can not give a comprehensive analysis of strains at the genomic level. Here, we report the genome sequences of six wild type C. glutamicum strains. Together with the 20 strains of previously available genome sequences, we have extended the genetic knowledge of this species, by performing a comparative analysis of 26 C. glutamicum strain genome sequences. These data allow for a pan-genomic description of C. glutamicum at the species level. We also analyzed the variations most likely related to amino acid production in several industrial strains.
Methods
Strains and next-generation genome sequencing
We sequenced the genome of six wild type strains for further research: ATCC 13869, ATCC 13870, B1, AS1.299, AS1.542 and T6-13. The strains were obtained from the CGMCC (China General Microbiological Culture Collection Center), CICC (China Center of Industrial Culture Collection), or SIIM (Shanghai Institute of Industrial microbiology) (Table 1 and Additional file 1: Table S1).
Table 1.
Detail Descriptions and allelic profile of the strains used in this study
| No. | Group | ST | Strains | Synonym | Descriptions | Ancestorc | Chromosome/Draft contigsa | Genome size (bp) | C + G content (%) | Native Plasmid | atpA | dnaE | dnaK | fusA | leuA | odhA | rpoB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | ATCC13032 | C. glutamicum Type Strain (Kyowa Hakko) | – | NC_003450.1 | 3,309,401 | 53.81 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| 2 | 1 | 1 | ATCC13032 | C. glutamicum Type Strain (Bielefeld) | – | NC_006958.1 | 3,282,708 | 53.84 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| 3 | 1 | 1 | K51 | Substrain of ATCC13032 | ATCC13032 | NC_020519.1 | 3,309,400 | 53.8 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| 4 | 1 | 1 | MB001 | prophage-free variant of ATCC 13032 with a 6% reduced genome | ATCC13032 | NC_022040.1 | 3,079,253 | 54.21 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| 5 | 1 | 1 | ATCC21300 | Producing lysine, derived from ATCC13032 | ATCC13032 | DDBJ SRA: DRR001643 b | 3,243,227 | 53.84 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| 6 | 2 | 2 | ATCC13869 | B. lactofermentum | “wild-type B. lactofermentum” | – | LOQU01000000 | 3,311,939 | 54.25 | 4.5 kb/X03987.1 | 1 | 2 | 2 | 4 | 2 | 4 | 4 |
| 7 | 3 | 3 | ATCC13870 | C. acetoacidophilum | “wild-type C. acetoacidophilum” | – | LOQV01000000 | 3,360,227 | 54.02 | 4 | 6 | 5 | 5 | 6 | 1 | 1 | |
| 8 | 4 | 4 | ATCC14067 | B. flavum | “wild-type B. flavum” | – | AGQQ02000000 | 3,311,083 | 54.15 | 3 | 2 | 4 | 6 | 2 | 2 | 2 | |
| 9 | 4 | 5 | ATCC21493 | B. flavum | Producing arginine, derived from ATCC 14067 (SIIM B234) | ATCC14067 | LOQX01000000 | 3,275,235 | 54.10 | 3 | 2 | 4 | 6 | 2 | 5 | 2 | |
| 10 | 4 | 11 | SYPS-062 | L-serine overproduction | unknown | JXBH01000000 | 3,214,861 | 53.96 | 3 | 2 | 4 | 6 | 2 | 2 | 5 | ||
| 11 | 4 | 11 | SYPS-062-33a | L-serine overproduction, derived from SYPS-062 | unknown | JYEG01000000 | 3,211,995 | 53.95 | 3 | 2 | 4 | 6 | 2 | 2 | 5 | ||
| 12 | 4 | 4 | ATCC15168 | B. flavum | L-isoleucine production | unknown | CP011309 | 3,338,699 | 54.14 | 3 | 2 | 4 | 6 | 2 | 2 | 2 | |
| 13 | 5 | 6 | R | C. glutamicum isolated in Japan from a meadow soil sample | – | NC_009342.1 | 3,363,299 | 54.13 | 5 | 3 | 7 | 3 | 3 | 2 | 1 | ||
| 14 | 6 | 7 | AS1.299 | C. pekinense | “wild-type C. pekinense”, producing glutamate (=CICC 10119, SIIM B3) |
– | LOQS01000000 | 3,109,311 | 54.18 | 2 | 5 | 3 | 5 | 4 | 3 | 3 | |
| 15 | 7 | 8 | 617(B1) | A glutamate producing strain previously used in China(=CICC 10117, SIIM B1) | – | LOQY01000000 | 3,174,403 | 54.26 | 22 kb | 1 | 2 | 4 | 7 | 7 | 3 | 2 | |
| 16 | 7 | 13 | B253 | An important lysine-producing strain in China | unknown | CP010451 | 3,229,314 | 54.26 | 22 kb/CP010452 | 1 | 2 | 4 | 7 | 9 | 3 | 2 | |
| 17 | 8 | 9 | T6-13 | B. tianjinese | “wild-type B. tianjinese” (=CICC 20182, SIIM B226) | – | LOQW01000000 | 3,263,419 | 53.98 | 5 | 4 | 6 | 2 | 5 | 3 | 1 | |
| 18 | 8 | 9 | SCgG1 | Hyper-producing glutamate | unknown | NC_021351.1 | 3,350,620 | 53.93 | 5 | 4 | 6 | 2 | 5 | 3 | 1 | ||
| 19 | 8 | 9 | SCgG2 | Hyper-producing glutamate | unknown | NC_021352.1 | 3,350,619 | 53.93 | 5 | 4 | 6 | 2 | 5 | 3 | 1 | ||
| 20 | 8 | 9 | Z188 | Hyper-producing glutamate | unknown | AKXP01000000 | 3,283,833 | 53.93 | 5 | 4 | 6 | 2 | 5 | 3 | 1 | ||
| 21 | 8 | 9 | S9114 | A strain for industrial production of glutamate | T6-13 | AFYA01000000 | 3,262,889 | 53.90 | 5 | 4 | 6 | 2 | 5 | 3 | 1 | ||
| 22 | 8 | 9 | AS1.542 | C. crenatum | “wild-type C. crenatum” (=CICC10124, SIIM B6) | – | LOQT01000000 | 3,298,702 | 53.93 | 5 | 4 | 6 | 2 | 5 | 3 | 1 | |
| 23 | 8 | 10 | MT | C. crenatum | A mutant of AS1.542, producing arginine | AS1.542 | AQPS01000000 | 3,346,700 | 53.91 | 6 | 4 | 6 | 2 | 5 | 3 | 1 | |
| 24 | 8 | 10 | SYPA5-5 | C. crenatum | A mutant of AS1.542, producing arginine | AS1.542 | JPDH01000000 | 3,268,761 | 53.91 | 6 | 4 | 6 | 2 | 5 | 3 | 1 | |
| 25 | 9 | 12 | ATCC 21831(AR0) | Producing L-arginine | unknown | CP007722 | 3,192,886 | 54.14 | 17 kb/CP007723 | 7 | 7 | 2 | 8 | 8 | 1 | 1 | |
| 26 | 9 | 12 | AR1 | Producing L-arginine, derived from ATCC 21831 | unknown | CP007724 | 3,162,487 | 54.13 | 17 kb/CP007725 | 7 | 7 | 2 | 8 | 8 | 1 | 1 |
aDDBJ/EMBL/GenBank accession number
bSRA: Sequence Read Archive
cAccording to references, ATCC/CGMCC record or DDBJ/EMBL/GenBank record
Genomic DNA purifications were performed using an AxyPrep™ Bacterial Genomic DNA Miniprep Kit, according to the manufacturer’s manual. At least 2,000,000 read pairs were obtained from each sample, with paired-end libraries of an average insert size of 500 bp and an average read length of 100 bp, for a total length >400 Mb (130-fold coverage of the genome), using Illumina HiSeq2000 or Hiseq 2500 systems (performed by GBI, Shenzhen, China and/or Berry Genomics, Beijing, China). The raw sequence reads were sub-sampled to 2,000,000 read pairs, and trimmed to 1,822,466–1,962,257 read pairs (354,168,503–382,827,142 bases) by removing low quality bases using Trimmomatic 0.35 [22] with the parameters “LEADING:15 TRAILING:15 SLIDINGWINDOW:4:10 MINLEN:50” (Additional file 1: Table S1).
Genome assembly was performed with SPAdes 3.5.0 [23, 24], at an average coverage of 110–130 fold. The assembled contig sequences were evaluated using the QUAST Web interface [25]. Gene prediction and annotation were performed using Prokka 1.11 [26]. The C. glutamicum Type Strain ATCC 13032 (NC_003450.1) genome sequence was used to build a specific database for annotation. Unless otherwise specified, default parameters were used for these programs.
The genome sequences of other strains were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and other databases (see Table 1). As the previously published genome sequences were initially annotated with different tools, cut-offs, and over a time frame of 12 years, the sequences were all re-annotated using Prokka 1.11, as above.
16S rDNA, average nucleotide identity (ANI) and analysis
Primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) were used to identify 16S rDNA sequences before performing genome sequencing. Also, the 16S rDNA sequences were in silico extracted from the genome sequences.
Whole-genome ANI analysis was performed using the software Jspecies based on MUMmer with default parameters [27, 28]. Genome-to-genome distance and in-silico DDH (DNA-DNA hybridization) was calculated using GGDC 2.1 (http://ggdc.dsmz.de/) [29].
Pan-genome analysis
Pan-genome analysis, including a cluster analysis of functional genes, an estimation of the pan-genome profile, and a prediction of the number of dispensable genes when adding new genomes, was performed by the pan-genome analysis pipeline (PGAP) 1.12 [30]. The pan-genome profile image was drawn by PanGP 1.0.1 [31].
Phylogeny and MLST (Multi Locus Sequence Typing) study
Phylogenetic study was based on whole genome sequences, and was performed by the CVTree Web interface using a composition vector (CV) approach [32]. Alternatively, phylogenetic study was also performed using the genome-to-genome distance data with FastME 2.0 (http://atgc.lirmm.fr/fastme/) [33].
The MLST analysis was performed as in our previous report [7]. Seven housekeeping genes, including atpA, dnaE, dnaK, fusA, rpoB, leuA, and odhA, were selected for analysis according to our previous report[7] and referring to the genotyping scheme in C. diphtheriae, another species belonging to the same genus [34].
Comparative genome analysis
Comparative analysis was performed using BWA 0.7.10 [35–38] for mapping reads, Samtools 0.1.19 [36] for data interaction, and Tablet 1.14.4.10 [39] for assembly/mapping visualization. SnpEff 4.1e [40] was used for genetic variant annotation and effect prediction. Wombac 2.0 [41] was used to finds genome single nucleotide polymorphisms (SNPs) and build a phylogenomic tree for highly related strains. Whole-genome alignments were calculated using MUMmer 3.0 [28].
Nucleotide sequence accession numbers
This Whole Genome Shotgun sequences have been deposited at DDBJ/EMBL/GenBank under the accession numbers LOQS00000000, LOQT00000000, LOQU00000000, LOQV00000000, LOQW00000000, and LOQY00000000. The version described in this paper is version LOQS01000000, LOQT01000000, LOQU01000000, LOQV01000000, LOQW01000000 and LOQY01000000.
Results
16S rDNA sequence and average nucleotide identity (ANI) indicate that all 26 strains should be classified as C. glutamicum species
The 16S rRNA gene has become a common and trustworthy genetic marker for the study of bacterial taxonomy. All of the 26 strains listed in Table 1 harbor nearly identical 16S rDNA sequences, with a similarity >99%, which argues that all of the strains should be classified as C. glutamicum species [42].
Average nucleotide identity (ANI) based on entire genomes provides another appropriate gauge of bacterial species delineation. The strains listed in Table 1, including the type strain ATCC 13032, all show ANI values >97% (Additional file 2: Table S2) and estimated DDH >70% (Additional file 2: Table S3) to each other, providing additional and robust evidence that all of the strains should be classified as C. glutamicum. An ANI threshold range of 95–96% of and a DDH threshold of 70% for species demarcation has previously been suggested [27, 29, 42].
Overview of C. glutamicum genomes
The C. glutamicum genome ranges in size from 3.08 to 3.36 Mb. The GC content varies slightly, from 53.81 to 54.26%. Some of the strains harbor native plasmids, varying in size from 4.5 to 22 Kb (Table 1).
We found all finished C. glutamicum chromosome sequences to exhibit good synteny using MUMmer [28], although transposons and prophages are dispersed throughout the genomes (Additional file 3: Figure S1).
Phylogenetics shows the strains classified into nine groups
A phylogenetic tree constructed by CVTree [32] and the Genome Blast Distance Phylogeny approach (Additional file 2: Table S4) [29] shows the strains classified into nine separate groups (Fig. 1, Additional file 4: Figure S2). This classification is consistent with the dendrogram generated by the MLST method (13 sequence types, 9 groups, Table 1). In our previous report using the MLST method, eight groups were classified, based on 17 strains [7]. We have established a new group in the present study, which includes two additional strains, ATCC 21831 (AR0) and AR1, the genome sequences of which have been reported recently [43].
Fig. 1.
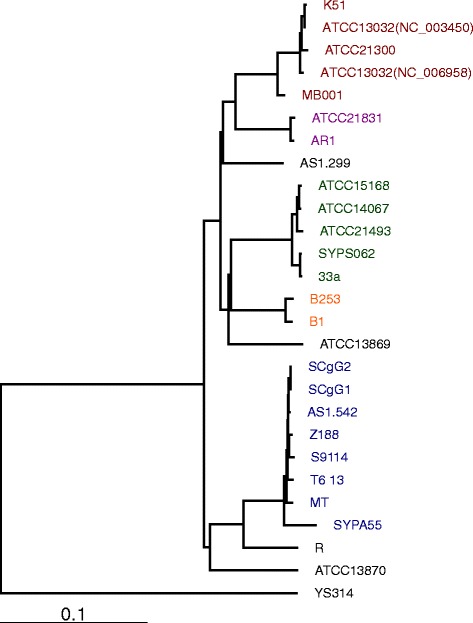
Phylogenetic trees based on the genome sequence of 26 C. glutamicum strains. YS314 was designated the out-group. The dendrogram was calculated by the CVTree Web interface using a composition vector (CV) approach. Figtree was used to draw the phylogenetic tree and produce the figure
Typically, each group contains one wild-type strain and several derived (or presumably derived) strains. For example, ATCC 14067 [44] and its derived strains ATCC 21493, ATCC 15168 are in the same group (Group 4, “B. flavum”). Two L-serine overproducers, SYPS-062 and SYPS-062-33a, also fall into this group, all potentially derived from the same ancestor, which would be closely related to ATCC 14067. Several groups contain only a single wild-type strain, as until now none of these derived strain genome sequences have been reported.
Group 8 and Group 9 are two exceptions. Group 8 contains two wild type strains (T6-13 and AS1.542) and their derived strains. Although T6-13 and AS1.542 have been considered as independent strains for a very long time, they have very similar genome sequences. Group 9 (ATCC 21831 and AR1) is another exception, containing two arginine-producing strains. We presume they derive from a corresponding wild type strain, the genome sequence of which has not yet been reported.
Pan/core -genome calculations
Based on the genome sequences of eight wild-type strains (ATCC 13032, ATCC 14067, ATCC 13869, ATCC 13870, R, AS1.299, AS1.542, and T6-13) C. glutamicum pan-genome parameters were calculated. A microbial pan-genome is defined as the full complement of genes in a bacterial species, and comprises the “core genome” containing genes present in all isolates of a species, and the “dispensable genome” containing genes present only in a subset of genomes. As shown in Fig. 2, the size of a species’ pan-genome can grow with the number of sequenced strains, indicating that the C. glutamicum has an “open” pan-genome. The pan-genome has a set of 2359 core genes. This gene number may be adjusted in the future, as draft genomes are finished and new genomes are added to the analyses.
Fig. 2.
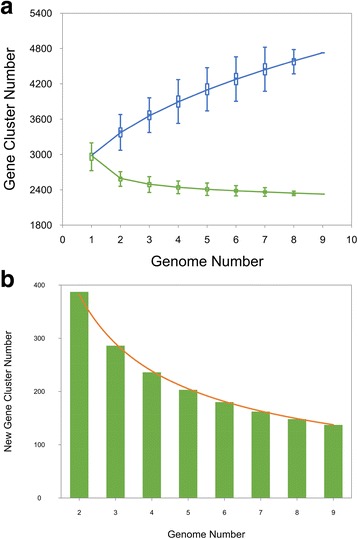
Pan-genome calculation of C. glutamicum using nine strains. a Core genes and pan genes calculation. The blue line shows the pan-genome development using, with the asymptotic value of y = 1161× x0.416 + 1821. The green line shows the core genes calculation, with the asymptotic value of y = 1364 × e(−0.802 × x) + 2359, where 2359 is the number of core genes regardless of how many genomes are added into the C. glutamicum pan-genome. b New (unique) genes of the pan-genome. The horizontal dashed line (orange) indicates the asymptotic value with the function of y = 612 × x-0.68. The figures were produced by PanGP
We exclusively considered the eight wild-type strains in our pan-genome calculations, and did not include other 18 strain genomes. We made this decision because some genes, especially genes related to by-products, as in some of the amino acid overproducing strains, might be artificially or naturally mutated, which may lead to miscalculated pan-genome results.
Dispensable genes: glutamate dehydrogenase (gdh) genes and the PS2 surface (S)-layer gene (cspB)
We will illustrate with two dispensable genes of notice that have been thoroughly analyzed in C. glutamicum, those encoding glutamate dehydrogenase (gdh) and the PS2 S-layer (cspB).
Glutamate dehydrogenase, which catalyzes the reversible NAD (P)+ −linked oxidative deamination of glutamate into alpha-ketoglutarate and ammonia, is an important branch-point enzyme for glutamate synthesis [45]. Several C. glutamicum strains only have an NADP+ specific glutamate dehydrogenase gene (EC 1.4.1.4). However, others not only have a NADP+ specific glutamate dehydrogenase gene, but also have a glutamate dehydrogenase gene compatible with both NAD+ and NADP+ (EC 1.4.1.3) (Table 2). The latter is not a pseudogene, at least in the glutamate-producing strain S9114, as two glutamate dehydrogenases have been physically isolated from it [46].
Table 2.
Glutamate dehydrogenase(GDH) and cspB genes detected in strains
| Group | Strain | Synonym | GDH-NADP+
(EC 1.4.1.4) |
GDH-NAD+
(EC 1.4.1.3) |
cspB |
|---|---|---|---|---|---|
| 1 | ATCC13032 | + | - | - | |
| 2 | ATCC13869 | B. lactofermentum | + | + | + |
| 3 | ATCC13870 | C. acetoacidophilum | + | + | + |
| 4 | ATCC14067 | B. flavum | + | + | + |
| 5 | R | + | + | + | |
| 6 | AS1.299 | C. pekinense | + | - | + |
| 7 | B1(617) | + | - | + | |
| 8 | T6-13 | B. tianjinese | + | + | + |
| 8 | AS1.542 | C. crenatum | + | + | + |
| 9 | ATCC21831 (AR0) | + | - | - |
The C. glutamicum PS2 S-layer cspB gene is located on a 6 Kb genomic island absent from the type strain ATCC 13032 [47, 48]. According to our comparative genomic analysis, the genomic island harboring cspB exists in most strains, and is only absent in ATCC 13032 and ATCC 21831 and their derived strains (Table 2). These two groups are quite close to each other in our phylogenetic tree (Fig. 1).
Variations likely related to amino acid production
That genomic variation most likely related to amino acid production may be the most interesting thing that a C. glutamicum pan-genomic analysis can offer. The ATCC 13032-derived lysine-producing strain ATCC 21300 has been analyzed in depth [12]. However, detailed analyses of many other strains have not been reported. The next section briefly describes some of these strains.
Lysine-producing strain B253
B253 is an important lysine-producing strain [21]. The genome consists of a circular chromosome and a plasmid. Compared with the genome of C. glutamicum ATCC 13032, about 46,000 mutations (insertions or deletions [InDels] and SNPs) are detected (Additional file 5: Dataset 1), with most of the key genes potentially relevant to lysine synthesis gaining one or more mutations [21]. According to our MLST analysis, B253 has a profile very similar to B1’s (profile of B253: 1-2-4-7-9-3-2, profile of B1: 1-2-4-7-9-3-3, with only a 1 bp difference in the leuA sequence), so B253 may be naturally or artificially derived from B1. By comparing the genome sequence of B253 with B1, only 432 mutations are detected (Additional file 5: Dataset 1). Three of these mutations, which are likely relevant to lysine production, were manually identified and confirmed by mapping reads to reference genome sequence (Table 3). (a) The aspartokinase gene lysC harbors an in-frame deletion (Leu329 to Gln330) and a missense mutation (Gly359Asp) that could be key mutations related to L-lysine production. (b) The stop gaining nonsense mutation in hom (homoserine dehydrogenase) could result in cutting off the metabolic flux toward threonine, methionine, or isoleucine, accompanied with a spontaneous increase in metabolic flux toward lysine. Phenotype annotation shows B253 to be a homoserine auxotroph.
Table 3.
SNP and InDel distribution in amino acid biosynthetic pathway
| Strains | Production | Ref. genome | SNP and InDel in genes | Gene description |
|---|---|---|---|---|
| ATCC21300 | lysine | ATCC13032 |
ppc: upstream -1 A deletion; …… |
ppc: phosphoenolpyruvate carboxylase |
| B253 | lysine | B1 |
lysC: p.Leu329_Gln330del (inframe deletion), p.Gly359Asp; hom: p.Gln399* stop gained |
lysC: Aspartokinase hom: Homoserine dehydrogenase |
| ATCC21493 | arginine | ATCC14067 | KIQ_011285: p.Gly159Asp; KIQ_013990: p.Arg390Cys; KIQ_009960: Ala701Thr p.Ala378Thr |
KIQ_011285: arginine repressor KIQ_013990: glutamate_dehydrogenase odhA(KIQ_009960): 2-oxoglutarate dehydrogenase E1/E2 component |
| SYPS-062 | serine | ATCC14067 | KIQ_000725: p.Leu103Phe; KIQ_012535: p.Glu251Lys, p.Arg422Gln; KIQ_009375: p.Asp394Asn; KIQ_009610: upstream-9 C->T |
KIQ_000725: serine acetyltransferase KIQ_012535: serine dehydratase KIQ_009375: serine_hydroxymethyltransferase KIQ_009610: phosphoglycerate mutase KIQ_014800: pyruvate dehydrogenase E1 |
| SYPS-062-33a | serine | ATCC14067 | KIQ_000725: p.Leu103Phe; KIQ_012535: p.Glu251Lys, p.Arg422Gln; KIQ_009375: p.Asp394Asn; KIQ_009610: upstream-9 C->T; KIQ_014800: p.His594Tyr |
|
| ATCC15168 | isoleucine | ATCC14067 | KIQ_005265: p.Ser248Phe; KIQ_012240: p.Gly186Arg |
KIQ_005265:2-isopropylmalate synthase; KIQ_012240: phosphoenolpyruvate carboxylase |
| MT | arginine | AS1.542 |
argR: p.Gln37*stop gained; odhA: p.Ala170Thr; argC; p.Gly134Glu |
argR Arginine repressor argC: N-acetyl-gamma-glutamyl-phosphate reductase argG: Argininosuccinate synthase argF: Ornithine carbamoyltransferase odhA: 2-oxoglutarate dehydrogenase E1/E2 component |
| SYPA5-5 | arginine | AS1.542 |
argR: p.Gln37* stop gained; odhA: p.Ala170Thr; argC: p.Gly134Glu, p.Asp123Asn; argG: p.Ile219Thr; argF: p.Ala191fs |
|
| SCgG1 | glutamate | T6-13 |
dapA: p.Glu293Lys; ppc: p.Ala433Thr |
dapA: 4-hydroxy-tetrahydrodipicolinate synthase ppc: phosphoenolpyruvate carboxylase ykuT(yggB): putative MscS family protein YkuT aceF: Dihydrolipoyllysine-residue acetyltransferasecomponent of pyruvate dehydrogenase complex |
| SCgG2 | glutamate | T6-13 |
dapA: p.Glu293Lys; ppc: p.Ala433Thr |
|
| Z188 | glutamate | T6-13 |
dapA: p.Glu293Lys; ppc: p.Ala433Thr; ykuT: p.Glu350Lys |
|
| S9114 | glutamate | T6-13 |
dapA: p.Glu293Lys; ppc: p.Ala433Thr; ykuT: p.Glu350Lys; aceF: p.Glu216Asp, p.Glu344Gln, p.Lys365 Pro369del |
According to previous report, introduction of hom Val59Ala and lysC Thr311Ile mutations into the wild-type strain leads to an accumulation of 75 g/L of L-lysine [49]. We presume that B253 may share the same mechanism of L-lysine production.
ATCC 14067 and related strains
ATCC 21493 is an arginine-producing strain derived from the wild-type strain “B. flavum” ATCC 14067. A Gly159Asp mutation in argR (KIQ_011285, arginine repressor, ArgR) may be a key mutation in the production of arginine, as we presume this mutation leads to the inactivation or reduction in the activity of ArgR, with a resulting increase in L-arginine biosynthetic enzyme activities and L-arginine production. Two mutations (Ala701Thr and Ala378Thr) in odhA (KIQ_009960, E1o subunit of the 2-oxoglutarate dehydrogenase complex) may be other key mutations, possibly altering metabolic flux, increasing it toward glutamate and arginine (Table 3) [50].
ATCC 15168 is an isoleucine-producing strain derived from ATCC 14067. We presume two mutations relate to isoleucine production: (a) Ser248Phe mutation in the 2-isopropylmalate synthase leuA gene (KIQ_005265) is likely relevant to branch amino acid synthesis. (b) Gly186Arg mutation in the phosphoenolpyruvate carboxylase gene ppc (KIQ_012240) may increase metabolic flux toward the TCA cycle (Table 3).
SYPS-062 is a serine-producing strain obtained from a mud culture collection [51, 52]. According to our MLST analysis, SYPS-062 may be naturally derived from an ancestor closely related to ATCC 14067. D-3-phosphoglycerate dehydrogenase (serA) is a key enzyme in serine biosynthesis. The SYPS-062 serA sequence in GenBank (HQ329183) shows two mutations compared with ATCC 14067’s genome sequence. However, the SYPS-062 and SYPS-062-33a genome sequences show no divergence from ATCC 14047 in this gene. It is interesting. Furthermore, several other mutations have been detected in three genes related to serine metabolism [(a) KIQ_000725: serine acetyltransferase, (b) KIQ_012535: serine dehydratase, (c) KIQ_009375: serine_hydroxymethyltransferase]. (d) We have also detected a C → T mutation 9 bp upstream of the phosphoglycerate mutase gene (KIQ_009610), which may reduce metabolic flux to pyruvate, subsequently accumulating 3-phosphoglycerate, which is a direct precursor in serine biosynthesis (Table 3).
SYPS-062-33a was derived from SYPS-062 by random mutation [53]. We presume a key mutation for its increased serine production is a His594Tyr mutation in the pyruvate dehydrogenase E1 component aceE gene, which may reduce pyruvate to acetyl coenzyme A activity, and increase the accumulation of pyruvate and other glycolysis metabolites, including 3-phosphoglycerate. Reported by-products, alanine and valine, which are derived from pyruvate, increased in the analysis [53]. This may be the result of pyruvate accumulation (Table 3).
AS1.542, T6-13, and related strains
AS1.542 and T6-13 are the “wild type” strains of “C. crenatum” and “B. tianjinese”.
Although T6-13 and AS1.542 have been considered as independent strains since sometime in the 1960–1970s, they have very similar genome sequences. Comparative genomic analysis showed that much less SNPs and InDels were detected between T6-13 and AS1.542 than comparing them with derivative strains, such as S9114 and MT (Fig. 3).
Fig. 3.
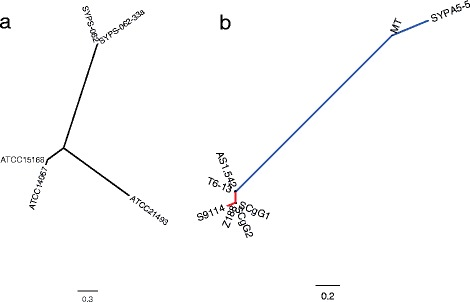
Phylogenomic trees of ATCC 14067, AS1.542, T6-13, and related strains. a ATCC 14067 and related strains. b AS1.542, T6-13, and related strains. The blue lines show the branch from AS1.542 to the arginine-producing strains MT and SYPA5-5; the red lines show the branch from T6-13 to the glutamate-producing strains SCgG1, SCgG2, Z188,and S9114. Wombac was used to finds genome SNPs and build phylogenomic trees for these strains. Figtree was used to draw the phylogenetic trees and produce the figures
MT and SYPA5-5 are arginine-producing strains [54]. AS1.542 is the probable ancestral strain. These two strains share several mutations when comparing with AS1.542, including: (a) a stop gaining nonsense mutation (Gln37stop) in argR, which could be a key mutation for L-arginine production; (b) a missense mutation (Ala170Thr) in odhA, which may play key roles in altering metabolic flux, increasing the flux toward glutamate and arginine; (c) a missense mutation (Gly134Glu) in argC, which may result in increased L-arginine production (Table 3). SYPA5-5 has gained several particular mutations in the arginine synthesis genes, including (a) Asp123Asn in argC; (b) Ile219Thr in argG; (c) Ala191framshift in argF (Table 3).
SCgG1, SCgG2, Z188, and S9114 are glutamate-producing strains. S9114 was derived from T6-13 [11, 20]. SCgG1, SCgG2, and Z188 are all soil isolates from China (the NCBI BioSample database: http://www.ncbi.nlm.nih.gov/biosample). According to our phylogenic study, SCgG1, SCgG2, and Z188 all cluster together, very close to S9114 (Fig. 3). It is an interesting result. We hypothesize that these isolates’ oil samples may have been contaminated by fermentation broth. Several mutations could be benefit glutamate production (Table 3), including: (a) Ala433Thr in ppc, by increasing the metabolic flux from PEP toward the TCA; (b) Glu216Asp, Glu344Gln, and Lys365 to Pro369 deletion in aceF, by decreasing metabolic flux from pyruvate toward acetyl coenzyme A; (c) Glu350Lys in ykuT, by increasing glutamate export; (d) Glu293Lys in dapA, by reducing lysine production.
Discussion
C. glutamicum strains are widely used for the industrial production of amino acids. Analyses of these strains have two major objectives: to provide (1) an overview genomic analysis and pan-genomic study of the species; and (2) a direct comparison between the amino acid producing strains to their ancestors, for the study of variations likely related to amino acid production. Analyses at this level have not been yet reported.
Similarity on 16S rDNA sequences indicated that several strains previously regarded as Brevibacterium, and as different Corynebacterium species, should be classified as C. glutamicum [5, 7]. ANI and DDH results support that conclusion. All of the strains listed in Table 1 should be classified as C. glutamicum species. The strains were primarily isolated independently toward the same goal of selecting for glutamate production. However, it is quite interesting that these strains all fall into the same species, as they differ significantly in several phenotypic characteristics, and were previously given distinct taxonomic species and/or genera names.
Pan-genomic analysis of the wild-type C. glutamicum strains indicate that this species has an “open” pan-genome with a set of 2359 core genes, which is larger than the other members of this genus with available data, C. diphtheriae (1632) and C. pseudotuberculosis (1504) [55, 56]. Dispensable and strain-specific genes often relate to strain specific phenotypes, such as sensitivity to specific phages [57].
Pan-genomic analysis can provide useful insights on genome reduction. A top-down reduction of a bacterial genome to construct a minimal chassis is an important concept in synthetic biology [58]. This approach has been accomplished with many strains including Escherichia coli and C. glutamicum. A prophage-free variant of C. glutamicum ATCC 13032 with a 6% reduced genome has been constructed [59]. Recently, 41 C. glutamicum gene clusters ranging from 3.7 to 49.7 Kb in length were determined as target sites for deletion and 36 of them were successfully deleted. A combinatory deletion of all irrelevant gene clusters further decreased the size of the native genome by about 722 Kb (22%) down to 2561 Kb [60]. Subsequent C. glutamicum top-down reduction research can be guided by pan-genomic analyses.
In particular, we looked at dispensable genes: the NAD+/NADP+ dependent glutamate dehydrogenase gdh genes and PS2 S-layer cspB gene, which are absent in the type strain ATCC 13032. We first noticed that many C. glutamicum strains possess a functional NAD+/NADP+ dependent glutamate dehydrogenase gene. More attention should be paid to whether metabolic models based on ATCC 13032 are fully accurate or not, when researching the metabolic flux of these strains. Our hypothesis is that more C. glutamicum strains useful for the industrial production of glutamate, arginine, or proline will fall into those groups with two functional gdh genes. These results may provide hints regarding the importance of choosing the most appropriate beginning strain in glutamate production selection breeding experiments.
PS2 is a structural protein of the surface (S)-layer, encoded by the cspB gene, which forms a solid two-dimensional para-crystalline array surrounding the entire cell. A reconstituted double mutant (ΔcspBΔpbp1a) showed improved recombinant antibody-binding fragments (Fab) secretion [48]. The cspB gene is only absent in ATCC 13032, ATCC 21831 and derivatives of them, suggesting that these strains may have different protein secretion machinery.
We have built an efficient pipeline for analysis amino-acid-producing C. glutamicum strains (Fig. 4). Perhaps the most interesting thing to come out of C. glutamicum genome analysis may be the identification of those variations that likely relate to amino acid production. This pipeline is designed for toward this purpose. First, MLST is used to determine the presumed ancestor. Both MLST and whole genome phylogenetics would work for this purpose. We recommend MLST, as it is simple, and can be performed using either genome sequences or PCR fragments. Second, phylogenomic analysis of the strains using SNPs can give a direct view of the relationship to other strains and provide trajectories in strain breeding. Using the corresponding wild-type strain as a reference genome sequence, the results can provide a clear view of the relationship between the strains of interest and other related strains. Finally, all genetic variation, including SNPs, InDels, and SVs (structural variations), can be determined and annotated. This approach should provide a clearer molecular view of possible amino acid production mechanisms. We also presume that this pipeline should be useful for other industrial strains, such as Corynebacterium ammoniagenes, Bacillus subtilis, and Xanthomonas campestris.
Fig. 4.
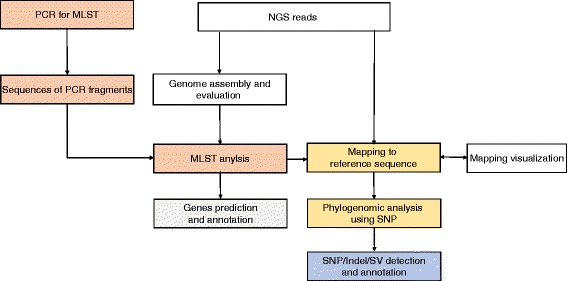
Pipeline for genome sequence analysis of amino acid producing C. glutamicum strains. The major steps are marked in red (MLST), yellow (phylogenomic analysis using SNPs) and blue (SNPs/Indels/SVs detection and annotation)
Clear information regarding industrial strains’ ancestry and breeding processes is occasionally missing after long-term utilization and preservation. This may hinder the discovery of amino acid hyper-production mechanisms in these strains. Therefore, the first and the most important step in the analysis of such strains should be MLST to determine which group the strain belongs to. The most closely related wild-type strain is ascertained to be the presumed ancestor, and performs as a suitable reference genome sequence for further research.
A deeper, more mechanistic view regarding amino acid producing strains is available using our pipeline. B253, for example, is a lysine-producing strain, and its genome, therefore, contains various mutations relevant to lysine production [21]. When compared with the type strain ATCC 13032, most genes for lysine biosynthesis are seen to have one or more mutations. This conclusion provides little help in understanding lysine production mechanisms, however, as it is almost impossible to recognize which mutations are actually relevant. Nonetheless, using our pipeline, B253 falls into the B1 group, indicating that B253 was most likely derived from B1 or an ancestor close to B1. When comparing B253 with B1, two key mutations are identified in lysC and hom. In fact, most other variation between B253 and ATCC 13032 is just general variation between different groups, probably unrelated to lysine production. We have reported and submitted to GenBank the genome sequence of six wild type strains, providing basic data for subsequent comparative analyses. Phylogenomic analysis using the SNPs of whole or core genomes from related strains will provide clear information about the strain breeding process. SCgG1, SCgG2, and Z188 are glutamate-producing strains with available genome sequences, but without clear genetic information. According to our results, the three should be related to an intermediate strain in the breeding of S9114 [20].
Conclusions
This is the first comprehensive comparative analysis of C. glutamicum genomes at the pan-genomic level. Whole genome comparison provides definitive evidence for classifying the members of this species. Identifying an alternative gdh gene in some C. glutamicum strains may accelerate further research on glutamate synthesis. Our proposed pipeline can provide a clear perspective, including the presumed ancestor, the strain breeding trajectory, and the genomic variations necessary to increase amino acid production in C. glutamicum.
Acknowledgements
We thank Prof. Ji-Bin Sun (Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences) for providing the draft genome sequences of several strains. We also thank Prof. Xuan Li (Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) for support on high-throughput genome sequencing.
Declaration
This article has been published as part of BMC Genomics Volume 18 Supplement 1, 2016: Proceedings of the 27th International Conference on Genome Informatics: genomics. The full contents of the supplement are available online at http://bmcgenomics.biomedcentral.com/articles/supplements/volume-18-supplement-1.
Funding
This work was partially supported by the National Basic Research Program (973 Program) of China (2014CB745100) (SY), the National Natural Science Foundation of China (31500068) (JY) and the National Key Technologies R&D Program of China (2012AA022101).
Availability of data and materials
This Whole Genome Shotgun sequences have been deposited at DDBJ/EMBL/GenBank under the accession numbers LOQS00000000, LOQT00000000, LOQU00000000, LOQV00000000, LOQW00000000, and LOQY00000000. The version described in this paper is version LOQS01000000, LOQT01000000, LOQU01000000, LOQV01000000, LOQW01000000 and LOQY01000000.
Authors’ contributions
JY and SY designed the study. JY performed the data analysis. JY and SY wrote the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
Strains sequenced in this study. (PDF 37 kb)
ANI analysis results; Table S3: in-silico DDH (DNA-DNA hybridization) analysis results; Table S4: Genome-to-genome distance analysis results. (PDF 60 kb)
Genome-wide alignment of selected C. glutamicum strains in an all-versus-all manner to ATCC 13032: MB001 (A), ATCC 15168 (B), R (C), B253 (D), SCgG1 (E), and ATCC 21831 (F). Matches in the forward strand are in red and those in the reverse strand are in blue. (PDF 394 kb)
Phylogenetic trees based on the genome sequence of 26 C. glutamicum strains using the Genome Blast Distance Phylogeny approach. YS314 was designated the out-group. (PDF 2 kb)
Mutations (InDels and SNPs) detected in B253 and annotations, by using ATCC 13032 or B1 as a reference genome sequence. (XLS 8150 kb)
References
- 1.Vertes AA, Inui M, Yukawa H. Postgenomic approaches to using corynebacteria as biocatalysts. Annu Rev Microbiol. 2012;66:521–550. doi: 10.1146/annurev-micro-010312-105506. [DOI] [PubMed] [Google Scholar]
- 2.Kinoshita S, Nakayama K, Akita S. Taxonomical study of glutamic acid accumulating bacteria, Micrococcus glutamicus nov. sp. Bull Agric Chem Soc Jpn. 1958;22:176–185. [Google Scholar]
- 3.Chen Q, Zhang Z-Y, Li L-G. A new L-glutamic acid-producing species of Corynebacterium (In Chinese with English abstract) Acta Microbiologica Sinica (Wei Sheng Wu Xue Bao) 1973;13(1):1–6. [Google Scholar]
- 4.Chen Q, Li L-G. Studies on L-glutamic acid producing bacteria AS 1.542; I. Identification of strain AS 1.542 (In Chinese with English abstract) Acta Microbiologica Sinica (Wei Sheng Wu Xue Bao) 1975;15(2):119–124. [Google Scholar]
- 5.Liebl W, Ehrmann M, Ludwig W, Schleifer KH. Transfer of Brevibacterium divaricatum DSM 20297T, “Brevibacterium flavum” DSM 20411, “Brevibacterium lactofermentum” DSM 20412 and DSM 1412, and Corynebacterium glutamicum and their distinction by rRNA gene restriction patterns. Int J Syst Bacteriol. 1991;41(2):255–260. doi: 10.1099/00207713-41-2-255. [DOI] [PubMed] [Google Scholar]
- 6.Hu X-z, Shen T-y. Researching history of glutamic acid produced by 617 brevis (In Chinese with English abstract) Industrial Microbiology(Gong Ye Wei Sheng Wu) 2006;36(2):4–6. [Google Scholar]
- 7.Yang J, Kong Y, Yang S. Genotyping of amino acid-producing Corynebacterium glutamicum strains based on multi-locus sequence typing (MLST) scheme. Bioresources and Bioprocessing. 2015;2(1). http://bioresourcesbioprocessing.springeropen.com/articles/10.1186/s40643-014-0030-8.
- 8.Institute of Microbiology Chinese Academy of Sciences. Hangzhou Glutamate Factory Study on the production of lysine by auxotrophic mutant of Corynebacterium pekinense AS1.299 (In Chinese) Microbiology China (Wei Sheng Wu Xue Tong Bao) 1974;1(1):7–11. [Google Scholar]
- 9.Zhang K, Liu Y. Studies on Glutamate Dehydrogenase from Brevibacterium Tianjinese T6-13 (In Chinese with English abstract) Acta Microbiologica Sinica (Wei Sheng Wu Xue Bao) 1991;31(4):281–286. [Google Scholar]
- 10.Vallino JJ, Stephanopoulos G. Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol Bioeng. 1993;41(6):633–646. doi: 10.1002/bit.260410606. [DOI] [PubMed] [Google Scholar]
- 11.Yun F, Zhou W. Breeding and Application of a Strain of High Glutamic Acid Yielding Bacterium S9114 (In Chinese with English abstract). Journal Of South China University of Technology (Natural Science). 1994;22(1):56–62.
- 12.Lee CS, Nam JY, Son ES, Kwon OC, Han W, Cho JY, Park YJ. Next-generation sequencing-based genome-wide mutation analysis of L-lysine-producing Corynebacterium glutamicum ATCC 21300 strain. J Microbiol. 2012;50(5):860–863. doi: 10.1007/s12275-012-2109-2. [DOI] [PubMed] [Google Scholar]
- 13.Otten A, Brocker M, Bott M. Metabolic engineering of Corynebacterium glutamicum for the production of itaconate. Metab Eng. 2015;30:156–165. doi: 10.1016/j.ymben.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Rados D, Carvalho AL, Wieschalka S, Neves AR, Blombach B, Eikmanns BJ, Santos H. Engineering Corynebacterium glutamicum for the production of 2,3-butanediol. Microb Cell Fact. 2015;14(1):171. doi: 10.1186/s12934-015-0362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Litsanov B, Kabus A, Brocker M, Bott M. Efficient aerobic succinate production from glucose in minimal medium with Corynebacterium glutamicum. Microb Biotechnol. 2012;5(1):116–128. doi: 10.1111/j.1751-7915.2011.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J, Sim SJ, Bott M, Um Y, Oh MK, Woo HM. Succinate production from CO(2)-grown microalgal biomass as carbon source using engineered Corynebacterium glutamicum through consolidated bioprocessing. Sci Rep. 2014;4:5819. doi: 10.1038/srep05819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda M, Nakagawa S. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol. 2003;62(2–3):99–109. doi: 10.1007/s00253-003-1328-1. [DOI] [PubMed] [Google Scholar]
- 18.Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, et al. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003;104(1–3):5–25. doi: 10.1016/S0168-1656(03)00154-8. [DOI] [PubMed] [Google Scholar]
- 19.Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, et al. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology. 2007;153(Pt 4):1042–1058. doi: 10.1099/mic.0.2006/003657-0. [DOI] [PubMed] [Google Scholar]
- 20.Lv Y, Wu Z, Han S, Lin Y, Zheng S. Genome sequence of Corynebacterium glutamicum S9114, a strain for industrial production of glutamate. J Bacteriol. 2011;193(21):6096–6097. doi: 10.1128/JB.06074-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Li P, Zheng P, Zhou W, Chen N, Sun J. Complete genome sequence of Corynebacterium glutamicum B253, a Chinese lysine-producing strain. J Biotechnol. 2015;207:10–11. doi: 10.1016/j.jbiotec.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 22.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 2013;20(10):714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 27.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106(45):19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14(1):60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. PGAP: pan-genomes analysis pipeline. Bioinformatics. 2012;28(3):416–418. doi: 10.1093/bioinformatics/btr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Jia X, Yang J, Ling Y, Zhang Z, Yu J, Wu J, Xiao J. PanGP: a tool for quickly analyzing bacterial pan-genome profile. Bioinformatics. 2014;30(9):1297–1299. doi: 10.1093/bioinformatics/btu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Z, Hao B. CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Res. 2009;37(Web Server issue):W174–W178. doi: 10.1093/nar/gkp278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefort V, Desper R, Gascuel O. FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol. 2015;32(10):2798–2800. doi: 10.1093/molbev/msv150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolt F, Cassiday P, Tondella ML, Dezoysa A, Efstratiou A, Sing A, Zasada A, Bernard K, Guiso N, Badell E, et al. Multilocus sequence typing identifies evidence for recombination and two distinct lineages of Corynebacterium diphtheriae. J Clin Microbiol. 2010;48(11):4177–4185. doi: 10.1128/JCM.00274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics. 2012;28(14):1838–1844. doi: 10.1093/bioinformatics/bts280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. Tablet--next generation sequence assembly visualization. Bioinformatics. 2010;26(3):401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.wombac: Rapid core genome SNP alignments from multiple bacterial genomes [https://github.com/tseemann/wombac; http://www.vicbioinformatics.com/software.wombac.shtml]
- 42.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64(Pt 2):346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 43.Park SH, Kim HU, Kim TY, Park JS, Kim SS, Lee SY. Metabolic engineering of Corynebacterium glutamicum for L-arginine production. Nat Commun. 2014;5:4618. doi: 10.1038/ncomms5618. [DOI] [PubMed] [Google Scholar]
- 44.Lv Y, Liao J, Wu Z, Han S, Lin Y, Zheng S. Genome sequence of Corynebacterium glutamicum ATCC 14067, which provides insight into amino acid biosynthesis in coryneform bacteria. J Bacteriol. 2012;194(3):742–743. doi: 10.1128/JB.06514-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Son HF, Kim IK, Kim KJ. Structural insights into domain movement and cofactor specificity of glutamate dehydrogenase from Corynebacterium glutamicum. Biochem Biophys Res Commun. 2015;459(3):387–392. doi: 10.1016/j.bbrc.2015.02.109. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Song X, Yang PP, Duan ZY, Mao ZG. Purification and characterization of glutamate dehydrogenase. from Corynebacterium glutamicum S9114. Sheng wu gong cheng xue bao = Chinese Journal of Biotechnology. 2003;19(6):725–729. [PubMed] [Google Scholar]
- 47.Hansmeier N, Albersmeier A, Tauch A, Damberg T, Ros R, Anselmetti D, Puhler A, Kalinowski J. The surface (S)-layer gene cspB of Corynebacterium glutamicum is transcriptionally activated by a LuxR-type regulator and located on a 6 kb genomic island absent from the type strain ATCC 13032. Microbiology. 2006;152(Pt 4):923–935. doi: 10.1099/mic.0.28673-0. [DOI] [PubMed] [Google Scholar]
- 48.Matsuda Y, Itaya H, Kitahara Y, Theresia NM, Kutukova EA, Yomantas YA, Date M, Kikuchi Y, Wachi M. Double mutation of cell wall proteins CspB and PBP1a increases secretion of the antibody Fab fragment from Corynebacterium glutamicum. Microb Cell Fact. 2014;13(1):56. doi: 10.1186/1475-2859-13-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ikeda M, Ohnishi J, Mitsuhashi S. Genome breeding of an amino acid-producing Corynebacterium glutamicum mutant. 2005. pp. 179–190. [Google Scholar]
- 50.Kim J, Hirasawa T, Sato Y, Nagahisa K, Furusawa C, Shimizu H. Effect of odhA overexpression and odhA antisense RNA expression on Tween-40-triggered glutamate production by Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2009;81(6):1097–1106. doi: 10.1007/s00253-008-1743-4. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X, Xu G, Li H, Dou W, Xu Z. Effect of cofactor folate on the growth of Corynebacterium glutamicum SYPS-062 and L-serine accumulation. Appl Biochem Biotechnol. 2014;173(7):1607–1617. doi: 10.1007/s12010-014-0945-8. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Q, Zhang X, Luo Y, Guo W, Xu G, Shi J, Xu Z. l-Serine overproduction with minimization of by-product synthesis by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2014;99(4):1665–1673. doi: 10.1007/s00253-014-6243-0. [DOI] [PubMed] [Google Scholar]
- 53.Xu G, Zhu Q, Luo Y, Zhang X, Guo W, Dou W, Li H, Xu H, Zhang X, Xu Z. Enhanced production of l-serine by deleting sdaA combined with modifying and overexpressing serA in a mutant of Corynebacterium glutamicum SYPS-062 from sucrose. Biochem Eng J. 2015;103:60–67. doi: 10.1016/j.bej.2015.06.009. [DOI] [Google Scholar]
- 54.Dou W, Xu M, Cai D, Zhang X, Rao Z, Xu Z. Improvement of L-arginine production by overexpression of a bifunctional ornithine acetyltransferase in Corynebacterium crenatum. Appl Biochem Biotechnol. 2011;165(3–4):845–855. doi: 10.1007/s12010-011-9302-3. [DOI] [PubMed] [Google Scholar]
- 55.Mokrousov I, Soares SC, Silva A, Trost E, Blom J, Ramos R, Carneiro A, Ali A, Santos AR, Pinto AC, et al. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the Biovar ovis and equi Strains. PLoS One. 2013;8(1):e53818. doi: 10.1371/journal.pone.0053818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trost E, Blom J, de Castro Soares S, Huang IH, Al-Dilaimi A, Schroder J, Jaenicke S, Dorella FA, Rocha FS, Miyoshi A, et al. Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia. J Bacteriol. 2012;194(12):3199–3215. doi: 10.1128/JB.00183-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ge B-Z, Wang J-X, Zhu S-J, Si X-D. Identification of glutamic acid producing strains by phages (In Chinese with English abstract) Virol Sin. 1991;6(3):256–259. [Google Scholar]
- 58.Xue X, Wang T, Jiang P, Shao Y, Zhou M, Zhong L, Wu R, Zhou J, Xia H, Zhao G, et al. MEGA (Multiple Essential Genes Assembling) deletion and replacement method for genome reduction in Escherichia coli. ACS Synth Biol. 2015;4(6):700–706. doi: 10.1021/sb500324p. [DOI] [PubMed] [Google Scholar]
- 59.Baumgart M, Unthan S, Ruckert C, Sivalingam J, Grunberger A, Kalinowski J, Bott M, Noack S, Frunzke J. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl Environ Microbiol. 2013;79(19):6006–6015. doi: 10.1128/AEM.01634-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Bruhl N, Bartsch A, Bott M, Wiechert W, Marin K, et al. Chassis organism from Corynebacterium glutamicum--a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J. 2015;10(2):290–301. doi: 10.1002/biot.201400041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This Whole Genome Shotgun sequences have been deposited at DDBJ/EMBL/GenBank under the accession numbers LOQS00000000, LOQT00000000, LOQU00000000, LOQV00000000, LOQW00000000, and LOQY00000000. The version described in this paper is version LOQS01000000, LOQT01000000, LOQU01000000, LOQV01000000, LOQW01000000 and LOQY01000000.