

Abstract
Two species of hairstreak butterflies from the genus Calycopis are known in the United States: C. cecrops and C. isobeon. Analysis of mitochondrial COI barcodes of Calycopis revealed cecrops-like specimens from the eastern US with atypical barcodes that were 2.6% different from either USA species, but similar to Central American Calycopis species. To address the possibility that the specimens with atypical barcodes represent an undescribed cryptic species, we sequenced complete genomes of 27 Calycopis specimens of four species: C. cecrops, C. isobeon, C. quintana and C. bactra. Some of these specimens were collected up to 60 years ago and preserved dry in museum collections, but nonetheless produced genomes as complete as fresh samples. Phylogenetic trees reconstructed using the whole mitochondrial and nuclear genomes were incongruent. While USA Calycopis with atypical barcodes grouped with Central American species C. quintana by mitochondria, nuclear genome trees placed them within typical USA C. cecrops in agreement with morphology, suggesting mitochondrial introgression. Nuclear genomes also show introgression, especially between C. cecrops and C. isobeon. About 2.3% of each C. cecrops genome has probably (p-value < 0.01, FDR < 0.1) introgressed from C. isobeon and about 3.4% of each C. isobeon genome may have come from C. cecrops. The introgressed regions are enriched in genes encoding transmembrane proteins, mitochondria-targeting proteins and components of the larval cuticle. This study provides the first example of mitochondrial introgression in Lepidoptera supported by complete genome sequencing. Our results caution about relying solely on COI barcodes and mitochondrial DNA for species identification or discovery.
Keywords: Lepidoptera, blues and hairstreaks, speciation, comparative genomics, phylogeny, taxonomy
1. Introduction
A 654 base-pair segment of mitochondrial DNA encoding the N-terminal half of cytochrome C oxidase subunit 1 has been proposed as a barcode to identify animal species [1]. This DNA barcode is indeed very effective at discriminating closely related and frequently cryptic species [2–4]. Barcode differences greater than 2% typically correspond to different biological species [1,5], although many exceptions have been reported [6]. A large library of over 5 million barcode sequences covering nearly 260 000 species has been assembled [7]. In addition to species identification, these barcodes have been successfully used for species discovery [8–10] and for association of males and females of animals with marked sexual dimorphism [11]. However, it has become clear that due to exchange of mitochondria between species [12], reliance on the COI barcode as a sole species identifier might be misleading [13–15]. In butterflies, one of the most striking examples of mitochondrial introgression is revealed in the genus Erynnis (family Hesperiidae), where barcodes of one eastern USA species are found in some specimens of an allopatric western USA species that is not even its closest relative [16]. While very insightful, the Erynnis study was based on DNA sequences of a single nuclear gene.
Here, using complete genomes, we demonstrate mitochondrial introgression in Calycopis species (family Lycaenidae). Being species-rich in the Neotropics, Calycopis in the United States is represented by two species: red-banded groundstreak (C. cecrops) and dusky-blue groundstreak (C. isobeon). Calycopis cecrops is a common species in the eastern half of the USA from Michigan and New York to Florida. In Texas and neighbouring states, C. cecrops overlaps in distribution with C. isobeon, which ranges southwards into Mexico and south to Panama [17]. Analysis of mitochondrial DNA COI barcodes of USA Calycopis revealed several specimens with atypical sequences that were 2.6% different from either of the two species. Their barcodes were more similar to Central American Calycopis species, but their wing patterns and male genitalia resembled C. cecrops. This raises the possibility that these specimens with atypical barcodes represent an undescribed cryptic Calycopis species in the USA. However, nuclear genomes suggest that the incongruence between morphology and barcode sequences probably resulted from mitochondria introgression.
2. Results and discussion
(a) Calycopis COI barcode conundrums
Comparison of COI barcodes of Calycopis cecrops, a common eastern USA hairstreak [18], revealed that several specimens from many states including Florida, Ohio, West Virginia, New Jersey, Maryland, Alabama and Louisiana did not group together with the rest of C. cecrops and C. isobeon (figure 1) due to a 2.6% sequence difference. These atypical barcodes were more similar (within 1%) to barcodes of other Calycopis species from Mexico and Costa Rica. To investigate whether the cecrops-like specimens with atypical barcodes represent a third species of Calycopis in the United States, we determined COI barcodes or barcode regions (ID tags) of 128 Calycopis specimens from across North and Central America and retrieved two additional sequences from GenBank. The majority of the sequenced barcodes were of the cecrops/isobeon type, but 16 specimens (approx. 17% of C. cecrops) from multiple locations in eastern USA had atypical barcodes (electronic supplementary material, table S1). In most of these localities, Calycopis with typical and atypical barcodes coexisted, and some were collected on the same day.
Figure 1.

Localities of specimens and their COI barcodes. Localities are shown for all barcoded specimens of C. cecrops (circles) and C. isobeon (squares), and specimens of C. quintana and C. bactra with complete genomes (star, the same locality for both). Localities where specimens with atypical barcodes (for cecrops/isobeon) were present are shown in red. A distance dendrogram built from the barcodes is shown in the middle, and alignment of barcodes with invariant positions removed is on the right. The most frequent nucleotides at each position are not shaded, next most frequent is shaded red, and the third is shaded green. Positions are numbered above the alignment. The last line indicates position in a codon. Protein sequence with invariant positions removed is shown to the right after ‘|’. Specimens with ‘NVG-’ numbers are sequenced in this study and GenBank accession is given for others. Specimens with complete genomes are marked with a dot to the right of the name and a number (1 to 27) that points to locality of the specimen on the map. (Online version in colour.)
Comparison of wing patterns of C. cecrops with typical and atypical barcodes did not reveal obvious differences (figure 2a; electronic supplementary material, figure S1): both possessed wide red discal bands on their ventral wing surface; and males had reduced blue areas on their dorsal hindwings. Moreover, male genitalia of specimens with atypical barcodes showed no meaningful differences from C. cecrops with typical barcodes (figure 2b; electronic supplementary material, figure S2). Thus, we did not find morphological support for distinctness of Calycopis with atypical barcodes, in contrast with our Hermeuptychia work [9]. However, it remained possible that these Calycopis are a cryptic species, which may differ from C. cecrops in aspects other than wing patterns and genitalia, such as caterpillar morphology and food source, similarly to Astraptes [8]. Alternatively, barcodes of Calycopis may have experienced introgression.
Figure 2.

Wings and male genitalia of C. cecrops specimens with typical and atypical barcodes. (a) Sex and collection abbreviation are shown by the dorsal and ventral images respectively, locality and sample ID below and between them. (b) Left lateral view is shown above and ventral view is shown below, species name is between the views, and locality with voucher code is below. Additional information for these specimens is in electronic supplementary material, table S1. (Online version in colour.)
In addition, neither C. cecrops nor C. isobeon clustered phenetically in the barcode distance dendrogram (figure 1b), and their barcodes revealed several haplotypes, all very similar to each other (figure 1c). In all barcoded C. isobeon specimens, position 84 was C, while the majority of C. cecrops had 84T. At position 561, most C. cecrops specimens have A and most C. isobeon specimens show C. However, there were several C. cecrops-like specimens, mostly from Florida and Texas, with 84C and 561C, similar to C. isobeon. It is challenging to distinguish C. cecrops and C. isobeon by wing patterns and genitalia [17] (electronic supplementary material, figures S2 and S3). Therefore, it is possible that some of the C. cecrops-like specimens might have been C. isobeon, which would significantly expand its known distribution range.
(b). Complete genomes of Calycopis
Comparison of nuclear genes could explain observed irregularities in mitochondria. Previously, only a couple of nuclear genes were utilized in such comparisons [16,19]. However, most of the nuclear genes do not have the phylogenetic signal to separate close species [20,21] and they can exchange between species just like mitochondria [22]. A small number of nuclear genes may be insufficient to differentiate between introgression and genuine emergence of a new species. Therefore, we sequenced complete genomes of 27 Calycopis specimens (marked with dots and numbered in figure 1b) of four species (electronic supplementary material, figure S3). Two species are from the United States: C. cecrops and C. isobeon. For C. isobeon, in addition to seven specimens from the USA, we also sequenced a specimen from Mexico and two specimens from Costa Rica (type locality). Among 15 sequenced C. cecrops specimens, two (from Ohio and West Virginia) possessed atypical barcodes. Two other species examined were C. quintana and C. bactra, one specimen of each, both from Belize. The barcodes of C. quintana are only 0.5–1% different from the atypical barcode of C. cecrops, but about 3% different from C. cecrops typical barcode. Calycopis bactra is a common Central American species with barcodes differing by more than 2.5% from the other three sequenced species of Calycopis.
The statistics for these genomes are in electronic supplementary material, table S2. The reads from each specimen cover the genome approximately 12.6 ± 2.1 times with two exceptions: specimen NVG-3306 was used to assemble the reference genome of Calycopis and was sequenced at a much higher coverage (80.2-fold); specimen NVG-1872, despite being rather recently collected (1980), contained only short DNA fragments and failed to produce sufficient data (the final coverage of 0.33-fold). The genomes for the rest of the C. cecrops specimens obtained by mapping are 88.7 ± 1.8% complete, and the genomes for C. isobeon specimens are 83.1 ± 2.7% complete. The genomes of C. quintana and C. bactra, which are more distantly related to the reference species C. cecrops, obtained by mapping and SNP calling, are less complete (81% and 79%, respectively). The portion of genomic regions that cannot be obtained in this way is related to the divergence between specimens and between species, leading to failure in aligning the reads properly to the reference genome. By contrast, the coding regions are more conserved between individuals and between species, and thus the coding regions (total of 20 533 236 bp consisting of 15 456 genes) obtained with this strategy are rather complete (96.7 ± 1.6%) for all species.
Seven specimens were collected in 2015 and preserved in RNAlater within several minutes after capture, ideal for genomics work. Others ranged from stored dry in a −20°C freezer (three specimens from 2014) and at room temperature in glassine envelopes (two specimens from 2013) to collected from 1959 to 2004 and pinned in museum collections. While genome quality generally deteriorates with specimen age, even the oldest specimen collected in November of 1959 (NVG-3034 from Costa Rica) yielded a genome of good quality (75.6% complete over the entire genome and 91.3% complete for the coding regions).
Complete mitochondrial genomes of these specimens revealed a picture similar to that from COI barcodes. Calycopis isobeon and C. cecrops with typical barcodes did not differ strongly from each other, although all typical C. cecrops mitochondria clustered together in the tree (figure 3b). As with COI barcodes, C. isobeon sequences were not monophyletic. Mitochondria of the two specimens with atypical barcodes (red in figure 3b) clustered with the C. quintana.
Figure 3.

Phylogenetic trees from nuclear and mitochondrial genomes of Calycopis. The trees are constructed on the concatenated alignments of the protein-coding genes in the (a) nuclear and (b) mitochondrial genomes. Both trees are unrooted. Each specimen is represented by two branches in the nuclear tree: father and mother copies of the genome (the copies are not phased and SNPs are assigned randomly to them; due to very poor coverage of specimen NVG-1872, many SNPs are impossible to call confidently and the terminal branches are very short). Name, voucher number and general locality are shown for each specimen. A number (1 to 27) points to locality of the specimen on the map shown in figure 1. Additional information about the specimens is in electronic supplementary material, table S1. Bootstrap support values above 70% are shown by the nodes, tree branches leading to C. cecrops specimens with atypical barcodes are shown in red and their names are highlighted in yellow. Coloured bars show how species are grouped in the trees. (Online version in colour.)
However, a tree constructed from all coding regions in nuclear genomes was different (figure 3a). First, both C. cecrops and C. isobeon were monophyletic with a large divergence between them (blue and green branches, respectively): (i) the length of internal branches connecting the clades of C. cecrops and C. isobeon in the phylogenetic tree is 0.00885, suggesting that about nine positions out of 1000 have changed between the ancestors of C. isobeon and C. cecrops, respectively (this distance is larger than the values we observed in other sister-species pairs [23,24]); (ii) the interspecific divergence between C. cecrops and C. isobeon (1.46 ± 0.13%) is much higher than the intraspecific divergence within C. cecrops and C. isobeon (0.87 ± 0.12% and 1.05 ± 0.19%, respectively). Second, the two C. cecrops specimens with atypical barcodes clustered deeply within C. cecrops clade (red in figure 3a) and grouped with the specimen from the closest locality (USA: Delaware). The C. quintana branch is well separated from the cecrops/isobeon clade. In summary, the nuclear genome tree matches expectations from specimen morphology, whereas the mitochondrial tree does not. These results strongly suggest that rather than being a separate species, Calycopis specimens with atypical COI barcodes belong to C. cecrops, which is in agreement with their genitalia morphology and wing patterns.
(c). Possible scenarios of mitochondria introgression
To test whether the differences in mitochondrial DNA can be explained by the differences in mutation rates rather than introgression, we calculated the likelihood of observing the current mitogenomes assuming only vertical descent (i.e. constraining all species to be monophyletic; RAxML, model: GTRGAMMA). The likelihood of this tree is significantly smaller (p-value < 0.0001) than the likelihood of the tree obtained without constraints on species monophyly. Retention of possible ancestral mitogenome dimorphism (typical and atypical mitogenomes) in C. cecrops is unlikely due to very strong (within 1%) similarity between atypical mitogenomes of C. cecrops and mitogenomes of C. quintana, a species not very closely related to C. cecrops.
The lack of expected mitochondrial DNA differences between well-differentiated species C. isobeon and C. cecrops and lower variability of C. cecrops mitochondria (figure 3b) suggest introgression of mitochondria from C. isobeon into C. cecrops followed by the replacement of the C. cecrops original mitochondria, possibly due to some selective advantage. Alternatively, the majority of C. cecrops specimens may carry the original mitochondria of this species, and the lack of divergence from C. isobeon mitogenomes may be explained by slow evolutionary rate or severe population bottlenecks that reduced the genetic diversity.
The atypical mitochondria may have been introgressed into C. cecrops populations from C. quintana or other C. quintana-like species. However, broader sampling of specimens to include all the Calycopis species and major populations will be needed to clarify the exact origin of these atypical mitochondria. Interestingly, current geographical ranges of C. cecrops and C. quintana do not overlap. Similarly, the two Erynnis species that experienced introgression are also allopatric [16]. It is conceivable that the atypical mitochondria of C. cecrops may represent remaining true ancestral mitochondria of C. cecrops, and they introgressed into C. quintana. However, this scenario is not likely because the evolutionary distances between mitogenomes of C. isobeon and atypical mitogenomes of C. cecrops are larger than the distances from either of them to mitogenomes of C. bactra, a species more distant from them both. Future genome-scale studies of additional Calycopis species and outgroups are in progress to further our understanding of introgression scenarios.
If the observed C. cecrops mitochondria represent introgression from C. isobeon (typical) and from C. quintana-like species (atypical), the original C. cecrops mitochondria were swept out of the population. Selective sweeps of mitochondria were studied in other species [25–27]. The best-documented cases are frequently associated with Wolbachia infection [25,26]. Wolbachia is a maternally transmitted symbiont that causes cytoplasmic incompatibility. Uninfected females do not produce viable offspring with Wolbachia-infected males, while infected females are compatible with all males [28,29]. Thus, the Wolbachia-infected females show selective advantage, spreading both Wolbachia and their mitochondria throughout the population.
Sequence reads obtained from Calycopis specimens did not reveal obvious Wolbachia-like sequences. However, many C. cecrops specimens contained copious sequences of bacteria in the Lactobacillaceae family. Several species in the Lactobacillaceae family are known gut symbionts [30,31]. The detritus-feeding Calycopis may need certain gut symbionts for digestion or detoxification. Therefore, beneficial gut symbionts introgressed from another species would confer selective advantage. Many symbionts, like the well-studied Wolbachia, are maternally transmitted. The introgressed beneficial gut symbionts will show strong linkage with the maternally inherited mitochondria, helping the introgressed mitochondria to spread throughout the population.
(d). Introgression in nuclear genomes of Calycopis
Complete genomes allow us to search for the signs of introgression between the sequenced species in their nuclear genomes. In each of the 24 specimens (specimen NVG-1872 was excluded from this analysis due to its poor completeness) of C. cecrops and C. isobeon, we detected genomic regions with significant probability (false discovery rate ≤0.1, p-value ≤0.01 and length ≥500 bp) of introgression from other Calycopis species. Overall, the fraction of introgressed regions is small, below 5% (table 1). Introgression is mostly between C. cecrops and C. isobeon, especially from the former to the latter. On average, 2.3% of each C. cecrops genome is from C. isobeon, while 3.4% of each C. isobeon genome comes from C. cecrops (table 1). These numbers probably represent lower bounds due to our conservative criteria for introgression detection and high similarity between genomes of the two species. Owing to this high similarity, it is impossible to find introgression in genomic regions that are almost identical (9.8%) between C. cecrops and C. isobeon, but could have exchanged between the two species.
Table 1.
Percentages of genome that probably result from introgression in Calycopis genomes.
| measure | from C. bactraa (%) | from C. quintanab (%) | from C. isobeon (%) |
|---|---|---|---|
| to C. cecrops | |||
| average | 0.06 | 0.04 | 2.29 |
| standard deviation | 0.03 | 0.02 | 0.60 |
| minimum | 0.02 | 0.02 | 1.41 |
| maximum | 0.11 | 0.11 | 3.35 |
| quantity | from C. bactraa (%) | from C. quintanab (%) | from C. cecrops (%) |
|---|---|---|---|
| to C. isobeon | |||
| average | 0.10 | 0.07 | 3.40 |
| standard deviation | 0.05 | 0.03 | 0.80 |
| minimum | 0.03 | 0.03 | 2.58 |
| maximum | 0.21 | 0.14 | 4.76 |
aMore likely from C. bactra compared with C. quintana and C. isobeon. It is possible that these regions are introgressed from other C. bactra-like species not included in this study.
bMore likely from C. quintana compared with C. bactra and C. isobeon. It is possible that these regions are introgressed from other C. quintana-like species not included in this study.
The length of the introgressed regions between C. isobeon and C. cecrops varies from several hundreds to more than 100 000 base pairs. Introgression occurs with interspecific hybridization events, and the introgressed regions from another species will appear shorter in further generations due to recombination with genetic material of the same species. The known recombination rate in butterflies is in the range of 3 cM Mb−1 to 6 cM Mb−1 [32]. Given such range of recombination rate, in silico simulation shows that 100 000 bp introgressed regions probably result from hybridization events hundreds to thousands years ago. The longest introgressed region is about 173 300 bp and it is in specimen NVG-4166. Among C. isobeon specimens, NVG-4166 is morphologically unusual, with ventral hindwing pattern resembling C. cecrops in the absence of a red spot by the black spot at the tornus (see fig. 2 in [23]). Recent introgression with C. cecrops may explain this phenotypic similarity. The only other specimen with introgressed regions longer than 100 000 bp is C. isobeon specimen NVG-3978. This specimen is collected in a locality (Texas: Dallas, near White Rock Lake) where both C. cecrops and C. isobeon occur and recent introgression is expected.
Introgression from either C. quintana or C. bactra into C. cecrops and C. isobeon is more limited (below 0.1%). It is important to note that although these regions are more likely to originate from C. quintana or C. bactra than from C. isobeon or C. cecrops, they may be introgression from other C. quintana- or C. bactra-like species that are not sampled in this study. The length of introgressed regions from C. quintana or C. bactra is also generally shorter, with the longest region reaching 10 000 bp, indicating more ancient introgression. Calycopis isobeon, especially more southern specimens, show more introgression from C. bactra, a common species sympatric with C. isobeon in Mexico and Central America. Most notably, we do not detect more nuclear introgression from C. quintana in the two C. cecrops specimens with quintana-like mitochondria than in other C. cecrops or C. isobeon specimens. This is understandable because the sequenced C. quintana is male and lacks the maternal inherited W chromosome. In addition, although our reference genome is from a female specimen, the W chromosome is probably not assembled because the W chromosome of Lepidoptera is known to contain mostly repetitive regions and is difficult to assemble [33]. The W chromosome is not yet assembled for any model organisms of Lepidoptera, including Bombyx mori [34].
Analysis of introgressed coding regions from C. cecrops to C. isobeon reveals an abundance of transmembrane proteins (electronic supplementary material, table S3) such as sugar transporters, similar to what we observed in Phoebis sennae [23]. Transmembrane proteins, such as transporters, may function relatively independently from other proteins in the cell [35,36]. Therefore, they could be fully functional in a different genetic background and are not likely to cause Dobzhansky–Muller hybrid incompatibility. Many introgressed genes are components of larval cuticle, which may be related to the similar phenotypes of the caterpillars of the two species.
Interestingly, the genes introgressed from C. isobeon to C. cecrops individuals are the most enriched in mitochondria-targeting proteins (electronic supplementary material, table S4). The mitochondria-targeting proteins encoded by the nuclear genome need to function together with the proteins encoded by the mitochondrial genome. The lack of divergence between the ‘typical’ C. cecrops mitochondrial sequences and C. isobeon mitochondrial DNA lowered the fitness barrier of transferring mitochondria-targeting genes from C. isobeon to C. cecrops. In the case that the ‘typical’ C. cecrops mitochondria are introgressed from C. isobeon, the C. cecrops mitochondria-targeting proteins may not be fully compatible with the proteins encoded by the C. isobeon mitogenome. In this scenario, introgression of mitochondria-targeting genes from C. isobeon to C. cecrops may be selected for after the mitochondria introgression event.
(e). Taxonomic implications
On a series of 15 C. cecrops and 10 C. isobeon specimens across their distribution ranges, we confirm that the two taxa are well-differentiated species. Nuclear divergence between C. cecrops and C. isobeon is much higher (1.46% interspecific divergence in the coding region) than within each species (0.87% for C. cecrops and 1.05% for C. isobeon). Nuclear introgression level between species is low (2–3%), suggesting reproductive isolation. Sequenced specimens of C. isobeon included two from the type locality (Costa Rica, near the southern limit of C. isobeon distribution), one from Mexico and two from the western limits (USA: Big Bend National Park). The nuclear tree (figure 3a) does not reveal a profound hiatus between them, suggesting that it is currently sensible to treat all these populations as a single biological species, C. isobeon. Similarly, C. cecrops genomes from across its distribution range from north (USA: Ohio, West Virginia, Delaware) to south (USA: Florida and Texas) clustered together. Despite being from the same locality (Texas: Dallas, near White Rock Lake), C. cecrops NVG-5197 and C. isobeon NVG-3978 cluster deeply within their respective species (figure 3a) and the fraction of introgressed regions detected in these two specimens is not higher than in other specimens.
Calycopis quintana was listed as a subjective junior synonym of C. isobeon [37]. However, as the nuclear genomes demonstrate (figure 3a), C. isobeon and C. cecrops are sister species, while C. quintana diverges from both of them. Additionally, the male genitalia of C. cecrops and C. isobeon are featured by similar shape of the labides in lateral view [17]: distal margin of the labides lateral lobes is convex, and ventral margin is shorter than dorsal (electronic supplementary material, figure S3). In ventral view the distal margin of labides is concave. However, C. quintana labides are shaped differently: their distal margin is convex, and the ventral margin is relatively straight and is longer than the dorsal margin in lateral view (electronic supplementary material, figure S2, NVG-3029). In ventral view, the distal margin of labides is nearly straight. Calycopis quintana is smaller on average than C. isobeon and lacks areas of blue scales on wings above, characteristic of most C. isobeon specimens, especially females. Therefore, we reinstate C. quintana as a valid species.
3. Conclusion
We show that some Calycopis cannot be identified to the species level by their COI barcodes. Calycopis cecrops is associated with two distinct barcodes that differ by 2.6%. One of them is similar to the barcode of C. isobeon and another one is similar to the barcode of C. quintana. As evidenced by analysis of complete nuclear genomes that groups specimens in agreement with their morphology, the paraphyly of barcodes is the result of mitochondrial introgression. Our work cautions against the use of COI barcodes as a sole tool for species identification and discovery, and calls for more diligent analysis of either their morphology or their nuclear genomes. We give a definitive example of introgression, supported by complete nuclear genome sequencing that obscures the value of COI barcodes in particular and mitochondrial DNA in general for taxonomic work.
4. Material and methods
(a). Experimental procedures
All experimental procedures were the same as the ones we reported previously [9,23,38]. Briefly, bodies of specimens collected in the field were preserved in RNAlater solutions; wings and genitalia were placed in glassine envelopes. For dry specimens collected previously, abdomens were used to extract genomic DNA as described in [9,23]. Subsequently to DNA extraction, genitalia were dissected and photographed as described in [9]. Specimens were photographed with a D800 camera through a 105 mm f/2.8G AF-S VR Micro-Nikkor lens. Images were assembled and edited in Photoshop CS5.1. COI barcodes and their segments were obtained by Sanger sequencing after PCR amplification in 1 to 9 fragments depending on the specimen age as described in [9].
The information about specimens used in this study is in electronic supplementary material, table S1. The genomes of specimens NVG-3033, NVG-3306, NVG-3307, NVG-3515, NVG-3348, NVG-4166, NVG-3978 and NVG-3431 have been obtained and reported in our previous publication. We obtained the genomes of additional 19 Calycopis specimens in this study and thus a total of 27 specimens are with whole genome sequences (as shown in figure 3). All the genomic sequence reads for these specimens are deposited at NCBI SRA database under accession SRP071639.
(b). Genomes of Calycopis specimens and phylogenetic analysis
We mapped the sequencing reads of all Calycopis specimens to the reference genome (including the mitochondrial genome) using BWA [39] and detected SNPs using the Genome Analysis ToolKit (GATK) [40]. We deduced the sequences for each specimen based on the result of GATK. Based on the gene annotations of the reference genome and the SNP calls, we derived the alignments of protein-coding sequences. We used two sequences to represent the paternal and maternal DNA in each specimen. Heterozygous alleles were randomly assigned to either paternal or maternal DNA.
Alignments of all 16 456 nuclear protein-coding sequences and 13 mitochondrial protein-coding genes were concatenated to obtain an alignment of nuclear genes (20 533 236 positions) and an alignment of mitochondrial genes (11 181 positions), respectively. The two alignments were used to build both neighbour-joining trees with PHYLIP [41] based on the P-distances (percentage of different positions) between specimens and maximal-likelihood trees with RAxML (model: GTRGAMMA and MTZOA, respectively). Bootstrap resampling was performed to assign confidence levels for nodes in the maximal-likelihood tree. The COI barcode dendrogram was obtained with BioNJ using P-distances [42].
To estimate the likelihood of observing the mitochondrial DNA sequences under the assumption that mitochondria did not introgress between species, we constructed a phylogenetic tree of the mitogenomes using RAxML with the constraint on the tree topology that all species are monophyletic. To estimate statistical significance, the likelihood under this constraint is compared with the likelihood without the constraint on 1000 bootstrap samples of positions from mitogenome alignments.
(c). Estimating the fraction of cross-contaminating reads in specimens
We extracted the sequencing reads that were mapped to the COI barcode region in each specimen and compared their sequences with the barcode sequence of every Calycopis specimens in our dataset. We considered a read to be possible contamination if it showed higher similarity to the barcode sequences of other specimens. For each specimen, the fraction of such possibly contaminating reads in all the reads mapped to the COI barcode was determined (electronic supplementary material, table S2).
The fraction of contaminating reads in each specimen is below 2% with an average of 0.8%, which allowed us to estimate the possibility of false detection of introgressed region due to cross-contamination. We artificially introduced no more than 2% cross-contaminating reads from different species to each specimen and performed the same procedure to detect inrogressed regions in the genome (discussed below). This low level of cross-contamination did not result in any false detection of introgression in mitochondrial genome. Although it did occasionally result in false detection of nuclear genome introgression, the rate of this error is below 1 × 10−7. This error rate is far below the level of introgression we detected in each specimen (1.5–5.1%), therefore the little amount of cross-sample contamination would not affect our conclusions.
(d). Detection of nuclear introgression
As observed in the human population, introgression from other species (such as from Neanderthals to modern humans) frequently appears as rare alleles showing linkage disequilibrium [43], and mitochondria of C. cecrops fall in this category. Our approach uses three properties of introgressed regions. First, an introgressed region in a specimen should be more similar in sequence to another species than to most other specimens of its own species. Second, introgressed alleles are usually less common. Third, a different evolutionary scenario that can cause a certain allele in one species to be more similar to the alleles in another species is incomplete lineage sorting (ILS). ILS happens during speciation, but introgression is a more recent event that happens after speciation. Therefore, introgressed alleles will show higher linkage disequilibrium compared with those originated from ILS.
The speciation time for several pairs of butterfly sister species [20,23] dated to about one million generations ago. Sequence divergence between the Calycopis species is larger than that between those species. Therefore we estimated their speciation time to be one to several million generations ago. Recombination rate in Lepidoptera is 3–6 cM Mb−1 per generation [32], and thus there should have been three to six recombination events per 100 bp after 1 million generations. Therefore, the SNPs inherited from the ancestral population are expected to show linkage over a very short range: no more than 300 bp based on our simulation of recombination. Therefore, if alleles that are more similar to other species are linked over longer ranges, it suggests that they arise from introgression rather than ILS.
We divided each scaffold of at least 1000 bp into 100 bp windows. For each specimen and in each 100 bp window, we calculated the difference in log-likelihood for its sequence to be sampled from another species j, and the log-likelihood for it to be from its own species i. The log-likelihood difference over this 100 bp window is the sum of log-likelihood differences for each position. At each position, the log-likelihood difference for a certain nucleotide k (A, T, G or C) to be sampled from species j but not species i is defined as
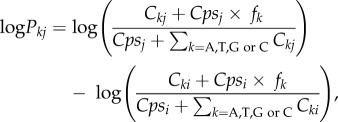 |
where Ckj is the count of nucleotide k in species j at this position; Cki is the count of nucleotide k in species i at this position; fk is the average fraction of nucleotide k among all species; and Cpsi and Cpsj are the pseudo-counts to avoid zero likelihood when a certain nucleotide is lacking in a species. The pseudo-count for species i is defined as
where n is the number of possible nucleotide types observed in all species at this position. Each specimen may have two possible nucleotide types at each position (heterozygous). In that case, the nucleotide type showing a higher log-likelihood difference is taken because a specimen does not have to be (and is even less likely to be) homozygous in the region with introgression.
For convenience, we term this log-likelihood difference as an introgression score. In genomic regions that have not significantly diverged between species, the introgression scores will be affected by errors or mutations in individual specimens. To avoid overestimation of introgression, we assign average introgression scores over the whole genome to windows in weakly diverged (average divergence less than 0.5%) genomic regions. The introgression scores are mostly negative, because each specimen is more similar to other specimens of the same species in most of its genomic regions. Windows with positive scores are candidates for introgression. In each scaffold, adjacent candidate introgressed windows are joined into segments that maximize the sum of introgression scores over the window to be joined.
To assign a p-value to a potentially introgressed segment, we divide it into blocks of 100 bp and shuffle these blocks between specimens in the alignment to produce a random distribution of introgression scores. The p-value is calculated as the fraction of shuffled samples with higher introgression score than the one computed from the non-shuffled alignment. Only segments with p-value lower than 0.05 were considered for further analysis.
Some segments show introgression from more than one species. They are assigned to the most likely species indicated by a higher introgression score. To reduce false positives due to the large number of genomic segments being tested, we performed false discovery rate tests. For each specimen, we sorted the segments possibly introgressed from a certain species by their p-values, and from the top of the sorted list, the false discovery rates were calculated as
where Nt is the total number of 100 bp windows for which the introgression scores can be calculated and Nc is number of 100 bp windows in the segments with lower p-values. Once the Q-value reaches 0.1, segments with higher p-values are considered failing the false discovery rate test. Segments with p-values lower than 0.01, Q-values lower than 0.1 and length of at least 500 bp were considered as introgressed.
Protein-coding genes that overlap with introgressed segments in over 25% of their base pairs in the exons were considered to be introgressed genes. Their functional properties were studied using GO terms. The enriched GO terms associated with these introgressed genes are identified using binomial tests: m = the number of introgressed genes that were associated with this GO term in all specimens, N = number of introgressed genes in all specimens, p = the probability for this GO term to be associated with any gene family.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgement
We acknowledge Texas Parks and Wildlife Department (Natural Resources Program Director David H. Riskind) for permit #08-02Rev that allows collection of materials in Texas State Parks. We are indebted to the United States National Park Service and the personnel of Big Bend National Park (in particular, to wildlife biologist Raymond Skiles) for assistance and continuing support with research permits (BIBE-2004-SCI-0011) that enabled discovery of C. isobeon in the park. We thank R. Dustin Schaeffer for proofreading of the manuscript; John M. Burns and Brian Harris (Smithsonian Institution), Edward G. Riley (Texas A & M University), Rebekah Shuman Baquiran and Crystal Maier (The Field Museum of Natural History) for facilitating access to the collections and stimulating discussions; and John A. Shuey, Jeff R. Slotten, Bill R. Dempwolf and Bryan E. Reynolds for collecting additional specimens used in the project. Q.C. was a Howard Hughes Medical Institute International Student Research fellow.
Data accessibility
The sequencing data are deposited to the NCBI SRA database under accession SRP071639.
Authors' contributions
Q.C. designed the experiments and performed the computational analyses; J.S. carried out the experiments; R.K.R. curated specimens, conceived the project and supervised the analysis; P.A.O. initiated the COI barcoding of Calycopis that resulted in discovery of atypical barcodes; D.B. and Z.O. supervised the experimental studies; N.V.G. directed the project. All authors wrote the manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported in part by the National Institutes of Health (GM094575 to N.V.G.) and the Welch Foundation (I-1505 to N.V.G.).
References
- 1.Hebert PD, Cywinska A, Ball SL, deWaard JR. 2003. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 270, 313–321. ( 10.1098/rspb.2002.2218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinca V, Zakharov EV, Hebert PD, Vila R. 2011. Complete DNA barcode reference library for a country's butterfly fauna reveals high performance for temperate Europe. Proc. R. Soc. B 278, 347–355. ( 10.1098/rspb.2010.1089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rougerie R, Naumann S, Nassig WA. 2012. Morphology and molecules reveal unexpected cryptic diversity in the enigmatic genus Sinobirma Bryk, 1944 (Lepidoptera: Saturniidae). PLoS ONE 7, e43920 ( 10.1371/journal.pone.0043920) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lara A, Ponce de Leon JL, Rodriguez R, Casane D, Cote G, Bernatchez L, Garcia-Machado E. 2010. DNA barcoding of Cuban freshwater fishes: evidence for cryptic species and taxonomic conflicts. Mol. Ecol. Resour. 10, 421–430. ( 10.1111/j.1755-0998.2009.02785.x) [DOI] [PubMed] [Google Scholar]
- 5.Wilson JJ, Sing KW, Sofian-Azirun M. 2013. Building a DNA barcode reference library for the true butterflies (Lepidoptera) of Peninsula Malaysia: what about the subspecies? PLoS ONE 8, e79969 ( 10.1371/journal.pone.0079969) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns JM, Janzen DH, Hajibabaei M, Hallwachs WH, Paul DN. 2007. DNA barcodes of closely related (but morphologically and ecologically distinct) species of skipper butterflies (Hesperiidae) can differ by only one to three nucleotides. J. Lepidopterists’ Soc. 61, 138–153. [Google Scholar]
- 7.Ratnasingham S, Hebert PD. 2007. bold: The Barcode of Life Data System (http://www.barcodinglife.org/). Mol. Ecol. Notes 7, 355–364. ( 10.1111/j.1471-8286.2007.01678.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebert PD, Penton EH, Burns JM, Janzen DH, Hallwachs W. 2004. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl Acad. Sci. USA 101, 14 812–14 817. ( 10.1073/pnas.0406166101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cong Q, Grishin NV. 2014. A new Hermeuptychia (Lepidoptera, Nymphalidae, Satyrinae) is sympatric and synchronic with H. sosybius in southeast US coastal plains, while another new Hermeuptychia species—not hermes—inhabits south Texas and northeast Mexico. Zookeys 379, 43–91. ( 10.3897/zookeys.379.6394) [DOI] [Google Scholar]
- 10.Bertrand C, Janzen DH, Hallwachs W, Burns JM, Gibson JF, Shokralla S, Hajibabaei M. 2014. Mitochondrial and nuclear phylogenetic analysis with Sanger and next-generation sequencing shows that, in Area de Conservacion Guanacaste, northwestern Costa Rica, the skipper butterfly named Urbanus belli (family Hesperiidae) comprises three morphologically cryptic species. BMC Evol. Biol. 14, 153–170. ( 10.1186/1471-2148-14-153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheffield CS, Hebert PD, Kevan PG, Packer L. 2009. DNA barcoding a regional bee (Hymenoptera: Apoidea) fauna and its potential for ecological studies. Mol. Ecol. Resour. 9(Suppl s1), 196–207. ( 10.1111/j.1755-0998.2009.02645.x) [DOI] [PubMed] [Google Scholar]
- 12.Harrison RG, Larson EL. 2014. Hybridization, introgression, and the nature of species boundaries. J. Hered. 105, 795–809. ( 10.1093/jhered/esu033) [DOI] [PubMed] [Google Scholar]
- 13.Meier R, Shiyang K, Vaidya G, Ng PK. 2006. DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst. Biol. 55, 715–728. ( 10.1080/10635150600969864) [DOI] [PubMed] [Google Scholar]
- 14.Vences M, Thomas M, Bonett RM, Vieites DR. 2005. Deciphering amphibian diversity through DNA barcoding: chances and challenges. Phil. Trans. R. Soc. B 360, 1859–1868. ( 10.1098/rstb.2005.1717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein PZ, DeSalle R. 2011. Integrating DNA barcode data and taxonomic practice: determination, discovery, and description. Bioessays 33, 135–147. ( 10.1002/bies.201000036) [DOI] [PubMed] [Google Scholar]
- 16.Zakharov EV, Lobo NF, Nowak C, Hellmann JJ. 2009. Introgression as a likely cause of mtDNA paraphyly in two allopatric skippers (Lepidoptera: Hesperiidae). Heredity (Edinb) 102, 590–599. ( 10.1038/hdy.2009.26) [DOI] [PubMed] [Google Scholar]
- 17.Field WD, Smithsonian I. 1967. Preliminary revision of butterflies of the genus Calycopis Scudder (Lycaenidae: Theclinae). Washington, DC: Smithsonian Press. [Google Scholar]
- 18.Scott JA. 1986. The butterflies of North America: a natural history and field guide. Stanford, CA: Stanford University Press. [Google Scholar]
- 19.Kodandaramaiah U, Simonsen TJ, Bromilow S, Wahlberg N, Sperling F. 2013. Deceptive single-locus taxonomy and phylogeography: Wolbachia-associated divergence in mitochondrial DNA is not reflected in morphology and nuclear markers in a butterfly species. Ecol. Evol. 3, 5167–5176. ( 10.1002/ece3.886) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cong Q, Borek D, Otwinowski Z, Grishin NV. 2015. Tiger swallowtail genome reveals mechanisms for speciation and caterpillar chemical defense. Cell Rep. 10, 910–919. ( 10.1016/j.celrep.2015.01.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontaine MC, et al. 2015. Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347, 42–48. ( 10.1126/science.1258524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heliconius Genome Consortium. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cong Q, Shen J, Warren AD, Borek D, Otwinowski Z, Grishin NV. 2016. Speciation in cloudless sulphurs gleaned from complete genomes. Genome Biol. Evol. 8, 915–931. ( 10.1093/gbe/evw045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cong Q, Shen J, Borek D, Robbins RK, Otwinowski Z, Grishin NV. 2016. Complete genomes of Hairstreak butterflies, their speciation, and nucleo-mitochondrial incongruence. Sci. Rep. 6, 24863 ( 10.1038/srep24863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiggins FM. 2003. Male-killing Wolbachia and mitochondrial DNA: selective sweeps, hybrid introgression and parasite population dynamics. Genetics 164, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raychoudhury R, Grillenberger BK, Gadau J, Bijlsma R, van de Zande L, Werren JH, Beukeboom LW. 2010. Phylogeography of Nasonia vitripennis (Hymenoptera) indicates a mitochondrial-Wolbachia sweep in North America. Heredity (Edinb) 104, 318–326. ( 10.1038/hdy.2009.160) [DOI] [PubMed] [Google Scholar]
- 27.Irwin DE, Rubtsov AS, Panov EN. 2009. Mitochondrial introgression and replacement between yellowhammers (Emberiza citrinella) and pine buntings (Emberiza leucocephalos) (Aves: Passeriformes). Biol. J. Linn. Soc. 98, 422–438. ( 10.1111/j.1095-8312.2009.01282.x) [DOI] [Google Scholar]
- 28.Yen JH, Barr AR. 1971. New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens L. Nature 232, 657–658. ( 10.1038/232657a0) [DOI] [PubMed] [Google Scholar]
- 29.Werren JH. 1997. Biology of Wolbachia. Annu. Rev. Entomol. 42, 587–609. ( 10.1146/annurev.ento.42.1.587) [DOI] [PubMed] [Google Scholar]
- 30.Vasquez A, Forsgren E, Fries I, Paxton RJ, Flaberg E, Szekely L, Olofsson TC. 2012. Symbionts as major modulators of insect health: lactic acid bacteria and honeybees. PLoS ONE 7, e33188 ( 10.1371/journal.pone.0033188) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frese SA, et al. 2011. The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genet. 7, e1001314 ( 10.1371/journal.pgen.1001314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilfert L, Gadau J, Schmid-Hempel P. 2007. Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity (Edinb) 98, 189–197. ( 10.1038/sj.hdy.6800950) [DOI] [PubMed] [Google Scholar]
- 33.Sahara K, Yoshido A, Traut W. 2012. Sex chromosome evolution in moths and butterflies. Chromosome Res. 20, 83–94. ( 10.1007/s10577-011-9262-z) [DOI] [PubMed] [Google Scholar]
- 34.Kiuchi T, et al. 2014. A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature 509, 633–636. ( 10.1038/nature13315) [DOI] [PubMed] [Google Scholar]
- 35.Kihara D, Kanehisa M. 2000. Tandem clusters of membrane proteins in complete genome sequences. Genome Res. 10, 731–743. ( 10.1101/gr.10.6.731) [DOI] [PubMed] [Google Scholar]
- 36.Boll M, Foltz M, Rubio-Aliaga I, Daniel H. 2003. A cluster of proton/amino acid transporter genes in the human and mouse genomes. Genomics 82, 47–56. ( 10.1016/S0888-7543(03)00099-5) [DOI] [PubMed] [Google Scholar]
- 37.Robbins RK. 2004. Lycaenidae. Theclinae. Tribe Eumaeini. In Checklist: Part 4A Hesperioidea—Papilionoidea Volume 5A. (ed. Lamas G.), pp. 118–137. Gainesville, FL: Association for Tropical Lepidoptera/Scientific Publishers. [Google Scholar]
- 38.Shiraiwa K, Cong Q, Grishin NV. 2014. A new Heraclides swallowtail (Lepidoptera, Papilionidae) from North America is recognized by the pattern on its neck. Zookeys 468, 85–135. ( 10.3897/zookeys.468.8565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. ( 10.1093/bioinformatics/btp698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. ( 10.1038/ng.806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Felsenstein J. 1989. PHYLIP—Phylogeny Inference Package (Version 3.2). 5, 164–166.
- 42.Gascuel O. 1997. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695. ( 10.1093/oxfordjournals.molbev.a025808) [DOI] [PubMed] [Google Scholar]
- 43.Racimo F, Sankararaman S, Nielsen R, Huerta-Sanchez E. 2015. Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 16, 359–371. ( 10.1038/nrg3936) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data are deposited to the NCBI SRA database under accession SRP071639.