

Abstract
Plasmablasts are a highly differentiated, antibody secreting B cell subset whose prevalence correlates with disease activity in Multiple Sclerosis (MS). For most patients experiencing partial transverse myelitis (PTM), plasmablasts are elevated in the blood at the first clinical presentation of disease (known as clinically isolated syndrome or CIS). In this study we found that many of these peripheral plasmablasts are autoreactive and recognize primarily gray matter targets in brain tissue. These plasmablasts express antibodies that over-utilize immunoglobulin heavy chain V-region subgroup 4 (VH4) genes, and the highly mutated VH4+ plasmablast antibodies recognize intracellular antigens of neurons and astrocytes. Most of the autoreactive, highly mutated VH4+ plasmablast antibodies recognize only a portion of cortical neurons, indicating that the response may be specific to neuronal subgroups or layers. Furthermore, CIS-PTM patients with this plasmablast response also exhibit modest reactivity toward neuroantigens in the plasma IgG antibody pool. Taken together, these data indicate that expanded VH4+ peripheral plasmablasts in early MS patients recognize brain gray matter antigens. Peripheral plasmablasts may be participating in the autoimmune response associated with MS, and provide an interesting avenue for investigating the expansion of autoreactive B cells at the time of the first documented clinical event.
Keywords: Plasmablast, Multiple Sclerosis, autoantibody, B cell, antigen receptor genetics
Introduction
Multiple Sclerosis (MS) is an autoimmune disorder of the central nervous system (CNS) that results in a loss of neurological function [39, 60]. Throughout disease the adaptive immune system mediates brain inflammation and lesion formation in these patients [7, 43, 61]. Although MS was historically thought to be driven by T cells [44], evidence for the importance of B cells in MS produced a shift in this perspective [25, 26]. The efficacy of B cell depletion therapy (BCDT) [40, 50], the prevalence of B cells in type II MS lesions [61], and the inverse correlation between memory B cell repopulation and benefit from BCDT [1]all support the hypothesis that B cells play a central role in pathogenesis [25, 26].
One B cell subtype of particular interest is the plasmablast, which normally develops in the blood as a part of the antigen-stimulated memory B cell response to pathogens [30, 32, 55, 69]. Plasmablasts are identified by the upregulation of CD27 and simultaneous expression of CD38 and CD95, and secrete large amounts of antibody [3, 32, 34, 48, 71]. They also produce IL-10, which positively feeds back to promote the differentiation and expansion of IgG and IgM antibody secreting cells [41]. After 2-3 weeks, plasmablasts either undergo apoptosis or differentiate into long-lived plasma cells [30, 32, 55, 69], thus the presence of plasmablasts indicates an active B cell response and represents the fraction of B cells that are currently responding to stimulus. Plasmablasts also represent a large proportion (5-40%) of the B cell pool in the cerebrospinal fluid (CSF) of untreated early and established MS patients [58] and their frequency correlates with increased inflammation as assessed by MRI [18]. They are also elevated in the blood of patients experiencing their first clinical attack, and when left untreated, their frequency continues to rise [58].
Despite these observations, the role of plasmablasts and secreted antibodies in MS has been difficult to explicate [25, 54]. The function of secreted antibody was particularly questioned since BCDT begins to take effect before antibody titers are appreciably altered [40]. However the importance of the antibody goes beyond its secreted form, as membrane bound antibody is the primary factor in issuing B cell development and effector function [35, 64]. For example, B cells that lack cognate antigen recognition in germinal centers are removed from the B cell pool while those that engage cognate antigen are prompted towards activation, clonal expansion and affinity maturation [64, 101]. Thus, there is a critical relationship between the antibody expressed by B cells, the antigen recognized, and the B cell's potential to participate in an immune response.
Still, autoreactive antibodies secreted by plasmablasts or plasma cells may contribute to the disease by binding to self-antigens and mediating cell and tissue damage. Previous data from our laboratory demonstrated that B cells in the CSF of early and established MS patients express antibodies with a particular mutational pattern that bind gray matter targets in brain tissue, such as neurons and astrocytes [57]. Others have demonstrated that antibodies from clonally expanded MS CSF B cells that bind gray matter targets mediate complement deposition and damage to neurons in vitro, and pooled CSF antibodies from MS patients can mediate neurological dysfunction in vivo [12, 24].
Since we found that clinically isolated syndrome (CIS) patients display an expansion of plasmablasts during their first attack of partial transverse myelitis (PTM), we asked whether the peripheral plasmablasts from these patients harbor autoreactivity to CNS antigens. CIS-PTM patients are of particular interest as they display an expansion of plasmablasts [58], and focusing on this group increases homogeneity of the patients in the study. To this end, we isolated single peripheral plasmablasts from our CIS-PTM patients, cloned the expressed antibodies, and investigated the antibody's reactivity to brain antigens using a panel of methodologies. We found that antibodies expressed by plasmablasts from these early MS patients display high levels of reactivity for cellular and protein targets in the brain. Remarkably, only those antibodies that utilized variable heavy chain family 4 (VH4) genes bound strongly to brain antigens. Elevated levels of CNS reactive antibodies were detected in the plasma pool of many patients for whom CNS-reactive plasmablasts were detected. To our knowledge this is the first evidence for reactivity of peripheral plasmablasts from CIS-PTM patients to brain antigens, demonstrating their autoreactive nature.
Methods
Patient Sample Processing
Persons recruited for this study gave informed consent for the collection and utilization of blood according to the guidelines provided by the institutional review board at UTSWMC. Treatment naïve clinically isolated syndrome (CIS) patients with partial transverse myelitis symptoms (PTM) at high risk for developing MS, age and gender matched treatment naïve Neuromyelitis Optica (NMO) patients with established disease (used in the genetic analysis, cloning, and plasma antibody experiments), age and gender matched NMO patients on Cellcept therapy (used in the plasma antibody ELISA experiments), and age and gender matched healthy donors were included in this study (Table 1). CIS-PTM patients were defined as high risk for MS because the patients presented with at least one non-enhancing brain white matter lesion by MRI and the CSF was positive for oligoclonal banding or had a high IgG index. Average time to MS evolution was 12 months. NMO patients were diagnosed by the 2006 criteria and either ELISA or a cell-based assay was used to detect aquaporin-4 (AQP4) reactive antibodies in patient serum (Table 1). Only treatment naive NMO patients were used as comparators for immunoglobulin gene analysis and antibody cloning. Peripheral blood mononuclear cells (PBMCs) were isolated from the blood by Ficoll separation and stained with fluorescent antibodies as previously described [58]. B cells were gated from PBMCs as CD45+CD19+ cells, then memory B cells (CD19+CD27+) and plasmablasts (CD19+CD27hi, as defined by others [34, 48]) were sorted individually into 96-well plates using the BD FACSAria flow cytometer (BD Biosciences, San Jose, CA).
Table 1.
Patient information, Patients are grouped by diagnosis and whether they were further investigated by genetic analysis. Final columns list results of plasma ELISAs (Fig.6). Patients who were included in previous studies are denoted by a, b, or c. PB: plasmablast, CBA: cell based assay for aquaporin-4 reactivity, AZT: azathioprine
| Patient | Sex | Age | Diagnosis at Draw |
Current Diagnosis |
Relapses | MRI Changes |
Treatment at Draw |
Current Treatment |
Percent of PBs in Blood |
Percent of PBs in CSF |
PBs Sorted from Blood |
Number of Productive PB Sequences |
Plasma Brain ELISA |
Plasma Sy5y ELISA |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CIS Patients Included inGenetic Analysis | CIS924a | M | 69 | TM | TM | 0 | 0 | None | None | 7.1 | 33.3 | 144 | 47 | + | + |
| CIS799a | F | 28 | CIS | RRMS | 0 | 0 | None | Avonex | 5.74 | 13.3 | 190 | 68 | + | + | |
| CIS991a | F | 34 | CIS | TM | 0 | 0 | None | steroids | 3.3 | 15.4 | 73d | 25 | + | + | |
| CIS111abc | F | 62 | CIS | PPMS | 0 | 0 | None | Avonex | 3.04 | 5.77 | 190d | 58 | - | + | |
| CIS663a | F | 32 | TM | TM | 0 | 0 | None | None | 2.85 | 10.8 | 192 | 121 | - | + | |
| CIS431abc | F | 27 | CIS | RRMS | 1 | 1 | None | Gilenya | 2.07 | 35.9 | 190d | 81 | + | + | |
| ATM4 | M | 24 | CIS | RRMS | 0 | 0 | Prednisone | None | 1.54 | 16.1 | 176 | 28 | + | - | |
| CIS353abc | F | 58 | CIS | RRMS | 1 | 1 | None | Copaxone | 1.46 | 11.4 | 190d | 51 | + | + | |
| CIS683abc | F | 39 | CIS | RRMS | 0 | 0 | None | Tecfidera | 0.82 | 26.6 | 190d | 63 | - | - | |
|
| |||||||||||||||
| CIS Patients Not Included inGenetic Analysis | CIS287ac | F | 45 | TM | TM | 0 | 0 | None | Betaseron | 6.58 | 37.9 | - | - | ||
| CIS873ac | F | 19 | RRMS | RRMS | 0 | 0 | None | Avonex | 2.98 | 26 | + | + | |||
| CIS527ac | F | 43 | CIS | RRMS | 0 | 0 | None | Copaxone | 1.81 | 14.1 | - | - | |||
| CIS699a | F | 37 | CIS | RRMS | 0 | 0 | None | Avonex | 1.61 | 7.87 | - | + | |||
| CIS787ac | M | 33 | CIS | RRMS | 0 | 0 | None | Copaxone | 1.45 | 36.9 | - | + | |||
| CIS251ac | F | 53 | TM | Sarcoidosis | 0 | 0 | None | None | 1.43 | 2.57 | |||||
| CIS942a | F | 52 | CIS | CIS | 0 | 0 | None | Copaxone | 1.19 | 12.2 | - | - | |||
| CIS328ac | M | 32 | CIS | RRMS | 0 | 0 | None | Avonex | 0.64 | 2.41 | - | - | |||
| CIS371ac | F | 56 | CIS | CIS | 0 | 0 | None | Copaxone | 0.51 | 13.9 | |||||
|
| |||||||||||||||
| Patient | Sex | Age | Diagnosis at Draw | Current Diagnosis | AQP-4 Status | AQP-4 Test | Treatment at Draw | Current Treatment | Percent of PBs in Blood | Percent of PBs inCSF | PBs SortedFrom Blood | Number of Productive Sequences | Plasma Brain ELISA | Plasma Sy5y ELISA | |
|
| |||||||||||||||
| NMO Patientsin GeneticAnalysis | NM0.1 | F | 55 | NMO | NMO | + | ELISA | None | Cellcept | 3.47 | n/a | 95 | 31 | + | + |
| NM0.2 | F | 36 | NMO | NMO | + | ELISA | None | None | 0.22 | n/a | 83 | 15 | |||
| NM0.7 | F | 54 | NMO | NMO | + | CBA | None | Cellcept | 4.09 | n/a | 94 | 32 | |||
| NM0.8 | F | 64 | NMO | NMO | + | CBA | None | Rituxan | 2.43 | n/a | 94 | 25 | |||
|
| |||||||||||||||
| NMO Patients Not Included inGenetic Analysis | NM0.3 | F | 39 | NMO | NMO | - | CBA | Cellcept | Cellcept | 1.05 | n/a | - | - | ||
| NM0.4 | F | 61 | NMO | NMO | + | ELISA | Cellcept | Cellcept | 0.09 | n/a | - | - | |||
| NM0.5 | F | 41 | NMO | NMO | + | ELISA | Cellcept | Rituxan | 1.08 | n/a | - | - | |||
| NM0.6 | M | 47 | NMO | NMO | - | CBA | Cellcept | Rituxan | 1.01 | n/a | - | - | |||
| NM0.9 | F | 53 | NMO | NMO | - | CBA | Cellcept | Cellcept | 1.34 | n/a | - | - | |||
| NMO.10 | F | 61 | NMO | NMO | + | ELISA | Cellcept | Cellcept | 0.17 | n/a | - | - | |||
| NM0.31 | F | 46 | NMO | NMO | + | Unknown | AZT | Unknown | 0 | n/a | |||||
| NM0.33 | M | 38 | NMO | NMO | + | Unknown | Cellcept | Unknown | 5.97 | n/a | |||||
| NMO.70 | F | 45 | NMO | NMO | + | Unknown | AZT | Unknown | 4.52 | n/a | |||||
| NMO.260 | F | 47 | NMO | NMO | - | Unknown | Cellcept | Unknown | 3.02 | n/a | |||||
| NM0.626 | M | 36 | NMO | NMO | - | Unknown | AZT | Unknown | 2.15 | n/a | |||||
| NMO.740 | F | 29 | NMO | NMO | _ | Unknown | AZT | Unknown | 3.13 | n/a | |||||
| NM0.745 | F | 50 | NMO | NMO | Unknown | Unknown | Cellcept | Unknown | 1.66 | n/a | |||||
CSF and peripheral B cells previously studied by flow cytometry.
Peripheral B cells previously studied by genetic analysis.
CSF B cell previously studied by genetic analysis.
Memory B cells also sorted (productive/total sorted): CIS991: (49/95) CIS111: (14/94) CIS431: (71/188) CIS353: (54/190) CIS683: (61/188)
Single Cell Polymerase Chain Reaction and Immunoglobulin Gene Analysis
Individually sorted B cell subpopulations were flash frozen and lysed. Upon thawing, mRNA was reverse transcribed and immunoglobulin variable regions were amplified with multiple rounds of PCR as previously described [58]. Sanger sequencing was used at the UTSWMC sequencing core to generate the antibody variable domain reads. Sequence data was analyzed using the VDJserver online repertoire analysis tool (https://vdjserver.org/). Unproductive antibody rearrangements and truncated sequence reads (did not extend from the beginning of CDR1 to the first two codons of the J gene) were filtered out of the database. CIS-PTM and NMO sequence data was compared to healthy control CD19+ B cells provided by Peter Lipsky at UTSWMC [37, 67] and influenza responding plasmablasts from otherwise healthy donors as previously described [106]. GraphPad Prism software was used to determine the statistical significance of differences between groups and build graphs for figures. Frequencies were first subject to an arcsine transformation, as is appropriate for comparisons of frequencies, and non-parametric ANOVA was used with a post-hoc analysis to do pairwise comparison of patient groups with the healthy controls using the Dunnett multiple comparison method [108].
Antibody Cloning and Production
Plasmablasts from CIS-PTM and NMO patients expressing highly mutated VH4 or VH3 heavy chains were selected for production. The variable domains were synthesized (Integrated DNA Technologies, Corallville, IA) and bi-directionally cloned into an IgG1 backbone provided by Michel Nussenzweig at the Rockefeller University as previously described [93]. The 6 IgG1 antibodies cloned from one healthy donor used as controls were also previously described [51]. Betty Diamond at the Hofstra Northwell School of Medicine provided the DNA for two control IgG1 antibodies, B1 and G11, which serve as isotype negative and positive controls, respectively [110]. Protocol details are provided in the supplemental methods.
Tissue and Antigen ELISAs
Mouse tissues were used in this study to avoid epitope degradation that may occur when tissues are not immediately processed post mortem. Mouse brain and kidney lysates were made as described elsewhere [51] and the protocol was adapted from this reference. Protocol details are provided in the supplemental methods. In brief, plates were coated with 10 μg/mL of lysateor 1 μg/mL of purified antigen in bicarbonate buffer overnight, then blocked. Dilutions of rhAbs or patient plasma were added overnight, followed by incubation with biotinylated anti-human IgG (eBioscience, San Diego, CA) then streptavidin-HRP (BD Pharmigen, San Jose, CA) and detection by TMB substrate according to manufacturer recommendations (eBioscience, San Diego, CA). Background signal (typically 0.05 to 0.09, defined as wells without primary antibody) was subtracted from each measured well. Patient rhAbs were defined as positive by ELISA if the average absorbance of the rhAb at 20 and 10 μg/mL was more than two standard deviations higher than the average absorbance of the healthy donor rhAbs at 20 μg/mL.
Immunohistochemistry (IHC)
Healthy and diseased (experimental autoimmune encephalomyelitis [EAE] or stroke) mouse brains were preserved, cryosectioned, and used for immunohistochemistry as described previously [57, 86]. Protocol details are provided in the supplemental methods. Blinded experts in histology and pathology (co-authors RC, DR and AS) assessed staining of the rhAbs. Images for publication were prepared using Zeiss ZEN lite software (Zeiss, Oberkochen, Germany).
Immunocytochemistry
Hep2 ICC was performed with a Hep-2 Substrate Slide antinuclear antibody kit according to the manufacturer's instructions (MBL International, Woburn, MA). For SH-Sy5y staining, glass coverslips were coated with laminin, and SH-Sy5y were plated overnight. The following day, cells were fixed and blocked before adding primary rhAbs overnight. The next day, secondary antibodies were added followed by DAPI staining and imaging. Protocol details are provided in the Supplemental Methods.
Supplemental Methods
A separate document includes detailed methodology for the following: Antibody Cloning and Production, Tissue Antigen ELISAs, Immunohistochemistry (IHC), Stained Cell Enumeration of Mouse Cortical Sections, IHC Signal Affinity Verification, Immunocytochemistry (ICC), Cell Based Assay for AQP4 Binding, Cellular Fractionation and Western Blotting, and rhAb Binding by Flow Cytometry.
Results
Expansion of peripheral PBs is common to patients experiencing transverse myelitis symptoms
Previously, our laboratory demonstrated that peripheral plasmablasts are expanded in clinically isolated syndrome (CIS) patients experiencing their first partial transverse myelitis (PTM) attack [58]. To ascertain the extent of this increase, we determined the frequency of CD19+CD27high plasmablasts in PBMCs from CIS-PTM patients as compared to Neuromyelitis Optica (NMO) patients and healthy donors (Fig. 1a). Plasmablasts are typically expanded in NMO, a demyelinating neurological disease where patients similarly experience PTM, but do not classically exhibit the same pattern of brain inflammation as MS patients [78]. Therefore, these patients provided a relatively homogenous comparator for our CIS-PTM cohort, rather than patients with a variety of other neurological diseases. As expected, peripheral plasmablasts were expanded in NMO patients over steady state production found in healthy donors (2.12% vs 0.71%, p=0.01), and the frequency was similarly elevated in CIS-PTM patients(2.10% vs 0.71%, p=0.005) (Fig.1b).
Fig. 1. plasmablast expansion and genetics.
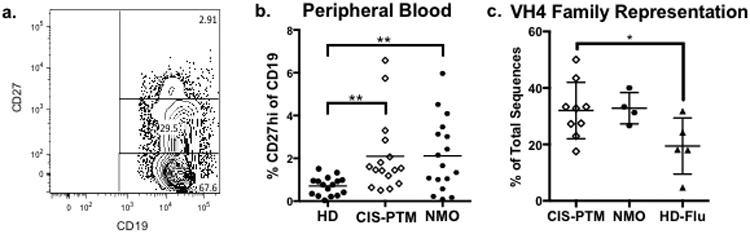
(a.) Representative flow cytometry plot for detecting CD19+CD27highplasmablasts from the peripheral blood of a CIS-PTM patient. (b.) Percentage of plasmablasts (CD19+ CD27high) of CD19+ B cells in blood samples from patient groups as measured by flow cytometry. Levels are elevated for CIS-PTM patients as compared to healthy donors, much like that of NMO patients. (c.) Representation of VH4 family genes in single cell PCR plasmablast repertoires of CIS-PTM and NMO patients as compared to plasmablasts responding to influenza infection in otherwise healthy donors. VH4 family gene usage is increased in CIS-PTM patients as compared to plasmablasts responding to influenza infection. NMO patients have a similar trend toward VH4 expansion, although it did not reach statistical significance. *p<0.05, **p<0.01
Expanded peripheral PBs in CIS-PTM patients over-utilize VH4 antibody genes
Considering the importance of the B cell receptor in the development and function of B cells, we then investigated the antibody gene repertoire of CIS-PTM plasmablasts for features of dysregulation. B cells generate their unique antigen receptor by the processes of immunoglobulin gene segment recombination, light and heavy chain pairing, and somatic hypermutation [56]. Previous data from our laboratory [16]demonstrated that CSF B cells of CIS-PTM patients at high risk to convert to MS tend to over-utilize one family of immunoglobulin gene segments, immunoglobulin heavy chain V-region subgroup 4 (VH4). To determine whether CIS-PTM peripheral plasmablasts also tend to utilize VH4 genes, we isolated and sequenced the variable domain of immunoglobulins from individual peripheral plasmablasts of CIS-PTM and NMO patients. Antibody repertoire data from these peripheral plasmablasts were compared to the ones deposited in published control databases of healthy total peripheral B cells [67] and flu-responding peripheral plasmablasts from otherwise healthy donors [106].
Approximately 19% of peripheral plasmablasts from healthy donors responding to influenza infection utilize VH4 genes (Fig.1c), which is a similar proportion to that of healthy donor total peripheral B cells (20%, Supplemental Fig. 1b) [37]. In contrast, 32% of peripheral plasmablasts from CIS-PTM patients utilize VH4 genes, which was statistically higher than flu-responding peripheral plasmablasts (CIS-PTM vs HD, 32% vs 19%, p=0.04) (Fig.1c). Plasmablasts from NMO patients demonstrated a similar trend toward expansion of VH4 usage, although this did not reach statistical significance (33%, p=0.13). When individual genes within the VH4 family are assessed, no single gene was expanded in CIS-PTM patients compared to controls, nor were there expanded genes in the other heavy chain variable domain families (Supplemental Fig. 1a). Similarly, no particular kappa light chain V gene or V gene family was overrepresented (Supplemental Fig. 2a, 2c). However, CIS-PTM plasmablasts more commonly expressed antibodies that utilized downstream light chain J gene segments in comparison to flu-responding plasmablasts (Supplemental Fig. 2d). JK5 was expanded in CIS-PTM plasmablasts in comparison to flu-responding plasmablasts (40% vs 11%, p=0.0065), while JK1 was under-utilized (4.5% vs 38%, p=0.0072). Common indicators of autoreactivity such as increased heavy chain CDR3 length and an over-representation of positively charged heavy chain CDR3 regions [102] were not present (Supplemental Fig. 1d, 1e). Additional antibody genetic analysis is provided in Supplemental Figures 1 and 2.
CIS-PTM peripheral plasmablasts bind strongly to brain antigens
We then sought to examine whether the antibodies expressed by the peripheral plasmablasts of CIS-PTM patients are autoreactive. To do this, recombinant human antibodies (rhAbs) from 38 peripheral plasmablasts were generated from seven treatment naive CIS-PTM patients (designated as CIS with a two-digit number). 10 rhAbs from four treatment naive NMO patients were also generated (designated as NMO with a two-digit number) and 6 rhAbs from one healthy donor were used as controls (designated as HD10 with a one- or two-digit number). All of the antibodies chosen for cloning were from the two most commonly represented heavy chain V-gene families (in both healthy donors and patients), VH3 and VH4, and had significant mutation accumulation that indicated they had undergone affinity maturation (Supplemental Table 1). Of the selected CIS-PTM plasmablasts from which the isotype could be determined, 85% belonged to the IgG isotype (Supplemental Table 1).
We first tested these rhAbs for binding to brain antigens using a mouse brain lysate ELISA. The six rhAbs from healthy donor peripheral B cells were not reactive in this assay (Fig. 2). In contrast, 29% of the CIS-PTM plasmablast rhAbs (11 out of 38) were highly reactive to the brain lysate, and 22% of the NMO rhAbs (2 out of 9) were similarly reactive. Of the seven CIS-PTM patients tested, six had one or two rhAbs that demonstrated strong reactivity to the lysate (Fig.2a), demonstrating that these antibodies can be found in many CIS-PTM patients.
Fig. 2. Brain lysate reactivity of plasmablast antibodies by ELISA.
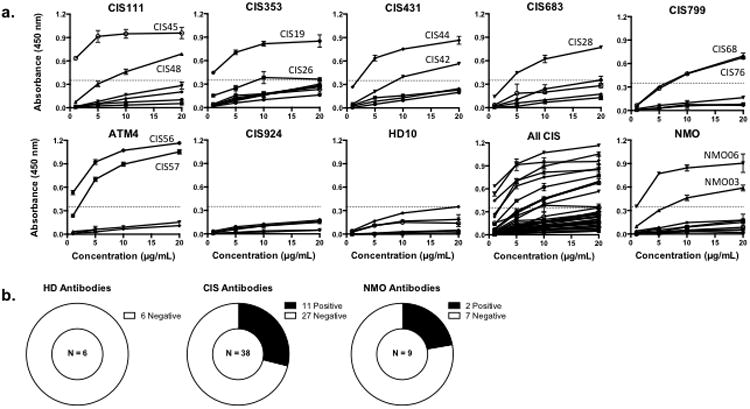
(a.) Brain ELISA results grouped by CIS-PTM patient, then by patient diagnosis (“All CIS”or“NMO”). Six of the seven CIS-PTM patients each contained two highly reactive rhAbs (33-50%), meaning the OD450 of the rhAb at 20 and 10 μg/mL was more than 2 standard deviations above the mean OD450 of healthy donor (HD) rhAbs at 20 μg/mL. The names of the positive rhAbs are designated for each titration curve. A dashed line in each graph represents the threshold for positive binding.(b.) Summary of ELISA results grouped by patient type. 11 of the 38 CIS-PTM rhAbs and 2 of the 9 NMO rhAbs bound strongly to the brain lysate.
The results of the mouse brain lysate ELISA were then confirmed using a commercially produced human brain lysate (Supplemental Fig. 3a, 3b). Of the 37 rhAbs from CIS-PTM patients tested in this ELISA, 19 of them (51 %) displayed positive reactivity to human brain lysate (Supplemental Fig. 3b). Nine of these bound both mouse and human brain lysate, while the remaining ten bound human brain lysate, but not mouse brain lysate. Of the nine rhAbs from the NMO patients, 3 of them (33 %) displayed positive reactivity to human brain lysate. Two of these bound both mouse and human brain lysate while one additional rhAb bound human brain lysate, but not mouse lysate (Supplemental Fig. 3b). This may be due to differences between human and mouse proteins, or it may be that the commercial human brain lysate contains a wider variety of antigens.
To investigate the specificity of the rhAbs, we tested 17 of the 19 rhAbs that were positive in the human brain lysate ELISA and 19 of the rhAbs that were negative in the human brain lysate ELISA (n = 36) for recognition of an in-house prepared mouse kidney lysate (Supplemental Fig. 3c, 3d). As expected, none of the healthy donor rhAbs bound to mouse kidney lysate. One of the NMO rhAbs displayed positive reactivity to mouse kidney lysate (Supplemental Fig. 3b). None of the brain-negative rhAbs from CIS-PTM patients (n = 19) displayed reactivity to mouse kidney lysate, but 5 of the 17 CIS-PTM rhAbs that displayed positive reactivity to brain lysate also displayed reactivity to mouse kidney lysate. Thus, 14 % of the CISPTM rhAbs (5 out of 36) and 11 % of the NMO rhAbs (1out of 9) are polyreactive.
We also investigated 36 rhAbs from CIS-PTM patients for reactivity to whole native H1N1 influenza antigen (FLU), and ovalbumin (OVA) (Supplemental Fig. 4). Of the 12 CIS-PTM rhAbs we tested that demonstrated positive binding to human brain lysate but not kidney lysate in the ELISA, only one displayed positive reactivity to FLU and OVA (CIS59). Of the 5 CIS-PTM rhAbs we tested that demonstrated positive binding to both human brain lysate and kidney lysate in the ELISA, 3 displayed positive reactivity to FLU and OVA (CIS56, CIS57, CIS28). The remaining two polyreactive rhAbs (CIS48 and CIS42) were not reactive to either FLU or OVA. Thus, 92 % of the brain lysate specific rhAbs from CIS-PTM patients (11 out of 12) maintained their exclusivity for brain antigens while 60 % (3 out of 5) of the polyreactive rhAbs from CIS-PTM patients maintained their binding promiscuity. One of the NMO rhAbs that bound to kidney lysate also bound FLU, but none of the NMO antibodies bound to OVA.
Interestingly, when the CIS-PTM rhAbs are categorized by VH family, only VH4 utilizing rhAbs react strongly to brain lysate (Fig.3a, 3b). None of the rhAbs utilizing VH3 genes met our criteria for strong binding (Fig.3a, 3b) despite having similar mutation frequencies as the VH4+ rhAbs used in this study (Supplemental Table 1). Reactivity to brain lysate by ELISA was noted for rhAbs using six of the nine represented VH4 genes (Fig.3c), suggesting that the autoreactive plasmablast response occurs with multiple antibody genes, and possibly also many antibody specificities. Four of the nine VH4 genes were employed by rhAbs in both the positive and negative category (VH4-30, 4-39, 4-59, and 4-61), suggesting that other factors such as light chain pairing, CDR3 length and charge, or mutation accumulation govern whether antibodies with these antibodies will be autoreactive. Two of the nine genes (VH4-b and 4-38) were exclusively represented in the rhAbs that bound to brain lysate, and three of the nine genes (VH4-4, 4-31, and 4-34) were only utilized by rhAbs that were not reactive in this assay (Fig.3c). However, it should be noted that the relative representation of antigens in the lysate might encourage false negatives.
Fig. 3. Comparison of VH4 and VH3 rhAbs.
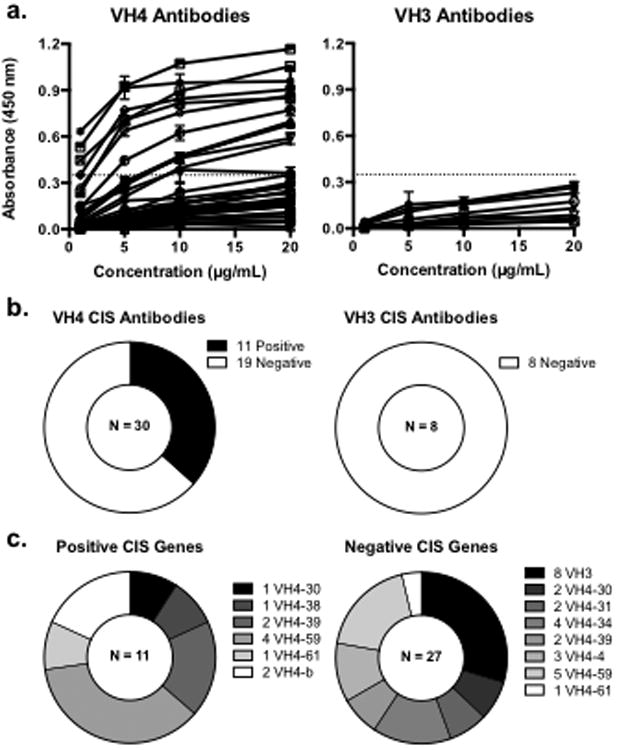
(a.) Brain ELISA results for CIS-PTM rhAbs grouped by heavy chain V gene family. A dashed line in each graph represents the threshold for positive binding. (b.) Summary of ELISA results for CIS-PTM rhAbs grouped by heavy chain V gene family. All VH3 rhAbs showed little reactivity of brain lysate, but many (37%) of the VH4 family rhAbs bound strongly to the lysate. (c.) V gene segments utilized by rhAbs that did and did not recognize brain lysate. Only VH4 family genes recognized whole brain lysate above the level of healthy donor antibodies, but only two of the VH4 genes were specifically found in the positive group. Four of the VH4 family genes represented were utilized by rhAbs in both the positive and negative group, and three others were used only by rhAbs that did not bind brain lysate.
CIS-PTM peripheral plasmablasts bind primarily to neurons and astrocytes
Next, we tested the binding of CIS-PTM rhAbs to brain tissues by fluorescent immunohistochemistry. We used brain tissues from healthy, experimental autoimmune encephalomyelitis (EAE) and stroke mice, which represent normal and inflamed brain tissues (Fig. 4, Supplemental Figures 4-11). In addition to theCIS-PTM rhAbs, NMO rhAbs (n=3) and two antibodies from lupus patients (B1 as a negative control and G11 as a positive control) were utilized as controls in this part of the study [110]. Though the specificities of B1 and G11 have not been fully characterized, B1 does not bind to multiple types of brain tissue [57], NMDA receptors [19], DNA, LPS, or recombinant human insulin [110]. In contrast, G11 is a polyreactive antibody that binds to DNA [110] as well as NMDA receptors [19]. As expected, B1 showed no reactivity to these brain tissues and G11 showed positive reactivity (Fig. 4 and data not shown). We found that the two structures most commonly recognized areastrocytes in the corpus callosum (marked by expression of GFAP) and neuronal bodies in the cortex (marked by NeuN). The rhAb CIS19 is presented in Fig. 4 as an example of binding to neuronal bodies, and rhAb CIS07 is presented in Fig. 4 as an example of binding to astrocytes. Overall, the majority of highly mutated VH4 expressing CIS-PTM plasmablast rhAbs recognized both neurons and astrocytes in multiple brain tissue types (Fig.4e, Supplemental Table 1, Supplemental Fig.6-12, 14a), with a smaller portion recognizing only astrocytes (Fig.4e, Supplemental Fig. 12, 14a).
Fig. 4. Plasmablast rhAbs react to neurons and glial cell in mouse brain.
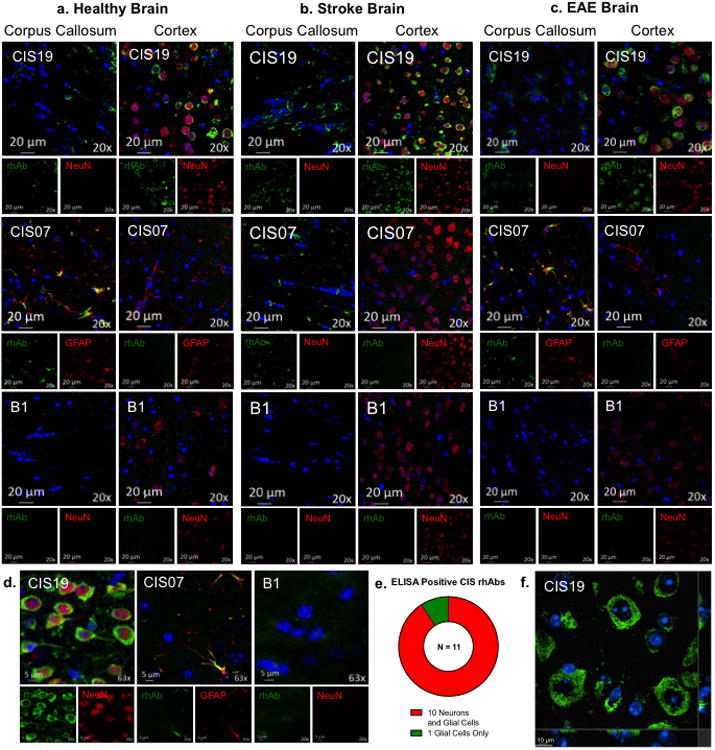
Example 20× images of example CIS-PTM rhAbs CIS19 and CIS07binding neurons and astrocytes in the white (corpus callosum, left) and gray (cortex, right) matter of healthy mouse brain (a.), stroke brain (b.) and EAE brain (c.) compared to the negative control antibody B1. Remaining images are in Supplemental Figures 6-13. RhAb staining is shown in green, DAPI is shown in blue, and NeuN is shown in red. Scale bar represents 20 μm. (d.) 63× images of example rhAbs CIS19 and CIS07 compared to the negative control antibody B1. RhAbs that recognized neurons did so in a ring-like pattern around the nucleus (as demonstrated by CIS19) and rhAbs that recognized astrocytes bound to cell body processes (as demonstrated by CIS07). Scale bar represents 5 μm. (e.) Of the 11 CIS-PTM rhAbs that were positive by mouse brain ELISA, one recognized exclusively glia and the remaining ten recognized both neurons and astrocytes. (f.) Orthogonal view of CIS19 staining in the cortex of healthy mouse brain, with only rhAb and DAPI overlay shown. Scale bar represents 10 μm.
Of those rhAbs that recognized neuronal cell bodies, the majority bound to the cytoplasm in a ring-like structure around the nucleus as illustrated by rhAb CIS19 (Fig.4a-d and 4f, top row and left-hand images). When viewed in the z-direction as in Fig. 4f and Supplemental Fig. 14c, it becomes clear that the green staining of these example rhAbs does not overlap with the blue staining of DAPI in the nucleus. Of those rhAbs that recognized astrocytes, the majority bound cell processes in the corpus callosum as illustrated by rhAb CIS07 (Fig.4a-d, middle images). Additionally the rhAbs were tested for binding to oligodendrocytes, but only CIS68 displayed co-localization with the oligodendrocyte marker PDGFR (Supplemental Fig. 13). In some of the cortical images there appeared to be recognition of neuropil, the areas of unmyelinated axons, dendrites and glial cells in parts of the tissue where cell bodies are less dense (Supplemental Fig. 7, 9, 11). However, lambda scan analysis indicated that the signal of the neuropil binding was no different than background autofluorescence, and is likely a staining artifact. Additional data from lambda scans verified that the staining observed is true positive signal (Supplemental Figure 14b).
Enumeration of Cortical Cells Stained by rhAbs
For the rhAbs that recognized neurons, rhAb binding was not observed in every NeuN stained neuron in the cortex. To quantify this effect we counted the number of cells stained by a particular rhAb as compared to the cells stained by NeuN (Supplemental Fig.15). By defining regions of interest (ROIs, Supplemental Fig.15a), measuring the signal intensity in that ROI, and setting a threshold for positive staining as compared to autofluorescence, the number of cells positive for rhAb or NeuN staining was determined. These values were used to create histogram plots of maximal signal intensity in each counted ROI (Supplemental Fig.15c) and obtain the percentage of cells that were stained by only the rhAb, only NeuN or both (Supplemental Fig.15b). Each rhAb shows a slightly different binding pattern, but the majority of all counted ROIs in the cortex were double positive for NeuN and rhAb staining (range: 56%—70%, Supplemental Fig.15d). For each rhAb there was also a substantial portion of neurons that do not stain with the rhAb (range: 19—37%, Supplemental Fig.15d), indicating that these rhAbs could be specific for neuronal groups or layers.
Peripheral plasmablasts from CIS-PTM patients recognize cytoplasmic and nuclear targets on human neurons
From the immunofluorescent staining of brain tissue, it appeared that the binding of CIS-PTM rhAbs was primarily located outside of the nucleus of these cells (Fig. 4, Supplemental Fig. 7, 9, 11). Indeed, orthogonal views demonstrated that rhAb binding did not overlap with the nuclear stain DAPI in cortical neurons stained by NeuN (Fig. 4f and Supplemental Fig. 14c). To further test this finding, we utilized immunocytochemistry (ICC) on a human neuroblastoma cell line (SH-Sy5y) as well as the human epithelial cell line (Hep-2) that is commonly used for detecting anti-nuclear antibodies in lupus patients (Fig.5, Supplemental Fig. 16-18) [88]. For both our CIS-PTM and NMO cohorts we found anti-nuclear rhAbs as well as rhAbs that bound outside of the nucleus of these cell lines (Fig.5a). As expected, negative control antibody B1 does not bind to the fixed and permeabilized SH-Sy5y cells (Fig. 5a) while G11, the positive control antibody, demonstrates strong binding across the entire body of the cell. Among 38 CIS-PTM rhAbs, 21 (55%) bound SH-Sy5y, including10 of the 11 CIS-PTM rhAbs that demonstrated strong binding by the brain lysate ELISA. Of these 21 CIS-PTM rhAbs that bound to targets outside the nuclei of these cells, only one bound to nuclear targets alone (Fig. 5b, Supplemental Fig. 16, 17). Of the 10 NMO rhAbs, 3 of them bound SH-Sy5y; one to nuclear targets alone and two to the cytoplasm. Only one of 6 healthy donor rhAbs showed cytoplasmic staining of SH-Sy5y (Supplemental Fig. 17), and none displayed positive binding of Hep-2 cells (Supplemental Fig. 18). One NMO plasmablast rhAb and 7 (20%) of the CIS-PTM plasmablast rhAbs bound the Hep-2 cells, but again most were not anti-nuclear in nature (Supplemental Fig. 18). Performing ICC on both SH-Sy5y and Hep-2 also permitted us to evaluate polyreactivity by this method. In so doing, we observed that 7 of the 21 CIS-PTM rhAbs bound Hep-2 cells (4 to cytoplasmic targets and 3 to nuclear targets) and were thus polyreactive (SH-Sy5y+Hep-2+). Of the 20 CIS-PTM rhAbs that displayed cytoplasmic binding to SH-Sy5y by ICC, 4 of them (20%) were polyreactive to cytoplasmic targets only (SH-Sy5ycytoplasmic+Hep-2cytoplasmic+). since they also displayed cytoplasmic binding to Hep-2 by ICC.
Fig.5. Reactivity to Sy5y by ICC.
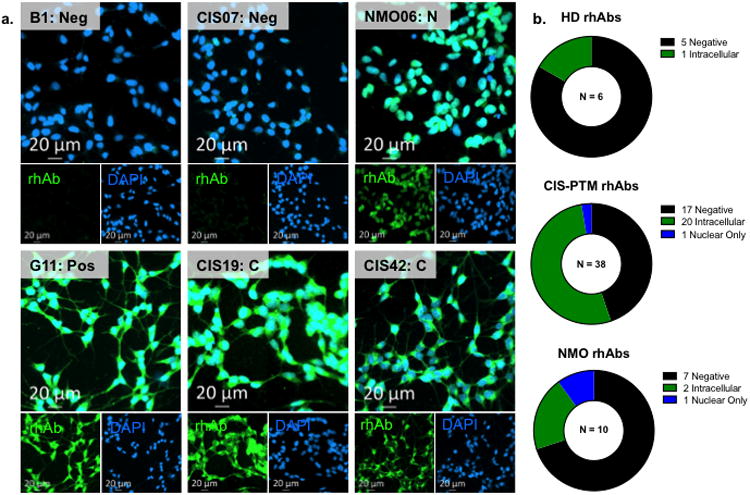
Immunocytochemistry (ICC) of rhAbs on the human neuroblastoma line, SH-Sy5y. (a.) Representative images ofCIS-PTM rhAbs, one NMO rhAb and the control rhAbs B1 and G11not binding or binding to SH-Sy5y neurons by ICC. B1 is an isotype negative control, and G11 is a positive control. Remaining images are in Supplemental Figs. 16 and 17. (b.) Summary of ICC staining of rhAbs on SH-Sy5y cells. Most healthy antibodies did not recognize SH-Sy5y, while over half of the CIS-PTM rhAbs and almost one third of NMO rhAbs bound to this cell line. Intracellular binding that included more than just the nucleus was most common, while a subset of rhAbs demonstrated nuclear binding.
To confirm the binding observed here, a western blot was run with select rhAbs with cellular fractionated lysate made from SH-Sy5y and Hep-2 cells (Supplemental Fig. 19). The positive control antibody G11 bound both the cytosol and nuclear/membrane fraction of both SH-Sy5y and Hep-2 cells, as was expected. The negative control rhAb R1 did not bind the lysates, as expected, as well as CIS05 which did not bind these cells in any other test. The only CIS-PTM rhAb that showed nuclear specificity by ICC, CIS10, also showed specificity for nuclear antigens by western blot. RhAbs CIS34, CIS42, and CIS46 bound both the cytoplasmic and the nuclear/membrane fractions of both SH-Sy5y and Hep-2 cells. These rhAbs may bind extracellular proteins, which would be included in the nuclear fraction cell lysate, or may bind some non-physiological linearized epitopes. NMO06, though reactive to SH-Sy5y by ICC, did not bind to the lysates, possibly due to the loss of conformational epitopes. Even so, the cytosolic CIS-PTM rhAb binding seen by ICC mirrors the previous result of cytosolic recognition of cortical neurons in mouse brain tissue (Fig. 4, Supplemental Fig. 14c), confirming the peripheral plasmablast response toward neuronal antigens in CIS-PTM patients.
Autoantibodies to AQP4 are detected in ∼75% of NMO patients [11, 109]. By standard clinical testing, all of the untreated NMO patients from whom rhAbs were cloned were positive for serum AQP4 antibodies (Table 1). However, none of the peripheral plasmablast rhAbs bound to AQP4 when tested on a transfected cell line (Supplemental Fig. 20). The expected frequency of AQP4 binding by peripheral B cells of NMO patients is unknown, but in the CSF, about half of clonally expanded B cells bind to AQP4 [10, 53]. This same frequency was observed in our two CSF rhAbs (R1 and R2) cloned from a CIS-PTM patient who later converted to NMO. AQP4 binding antibodies can utilize VH4, VH3, and VH2 family genes [11], so a larger study is needed to identify B cells that express AQP4 binding antibodies in the periphery of NMO patients.
CIS-PTM peripheral plasmablasts bind intracellular and extracellular antigens
The ICC methodology included the use of Triton-X, which could interfere with conformational epitopes of intracellular antigens. Thus, to evaluate whether the intracellular staining observed by ICC was towards conformational epitopes, we performed intracellular and cell surface staining of the human neuroblastoma cell line SH-Sy5y with select rhAbs and acquired data by flow cytometry. We chose to test one rhAb from each patient in the CIS-PTM cohort that displayed strong binding by ICC. As shown in Supplemental Fig. 21a, all 6 of the HD rhAbs, the CIS05 rhAb, and the negative control rhAb B1 showed no reactivity to intracellular and intact conformational antigens. In contrast, CIS19, CIS28, CIS42, CIS56 and the positive control rhAb G11 displayed reactivity to intracellular intact conformational antigens, while CIS49, CIS68 and CIS71 did not. Of these 7 CIS-PTM rhAbs that displayed strong binding by ICC, only one polyreactive rhAb (CIS56) showed reactivity to the cell surface of the SH-Sy5y neuroblastoma cell line (Supplemental Fig. 21b). A scatter plot of the geometric MFIs for both the intracellular and cell surface staining are presented in Supplemental Fig. 21c.
CIS-PTM plasmablast antibodies are well represented in the plasma pool
Even when expanded, plasmablasts are only a small portion of the peripheral B cell pool, and thus may have limited impact on the ongoing auto-reactivity associated with MS. To assess whether plasmablast expansion and detection of autoreactive antibodies in the blood occurred in the same patients, we tested plasma samples from 16 treatment naïve CIS-PTM patients, 7 NMO patients, and 8 healthy donors with plasmablasts responding to recent influenza vaccination for reactivity to brain lysate by ELISA (Fig.6). For comparison, the plasma antibodies were also tested for binding to native influenza antigen. Plasma from7 of the 16 treatment naïve CIS-PTM patients displayed antibody reactivity to the brain at least 2 standard deviations above the mean of healthy donors at 20 μg/mL (Fig.6b). Of the 7 CIS-PTM patients whose plasma displayed antibody reactivity to the brain lysate, 4 of them were CIS-PTM patients from which autoreactive rhAbs from single peripheral plasmablasts were identified (Table 1, Supplemental Table 1). Only one NMO patient's plasma displayed similar reactivity, and notably we were able to clone plasmablast rhAbs from this patient with high affinity for brain lysate (Supplemental Table 1, Fig. 2). Long-lived plasma cells in the bone marrow contribute substantially to the plasma antibody pools, and in the case of MS patients, modest affinity of the plasma antibody pools towards brain lysate may be due to a low frequency of autoreactive long-lived plasma cells in the bone marrow. Conceivably, plasmablasts in the CSF could also contribute to this reactivity as well. However, the fact that this affinity is modest may also be due to under-representation of antigens in the whole brain lysate pool. Thus, we also tested for binding to whole cell lysate made from the SH-Sy5y human neuroblastoma cells, since the majority of the autoreactive plasmablast rhAbs bind neurons. Here, 10 of the 16 CIS-PTM patients display plasma antibody reactivity at least 2 standard deviations above the mean of healthy donors at 20 μg/mL, with an apparent increase in affinity.
Fig.6. ELISAs with patient plasma.
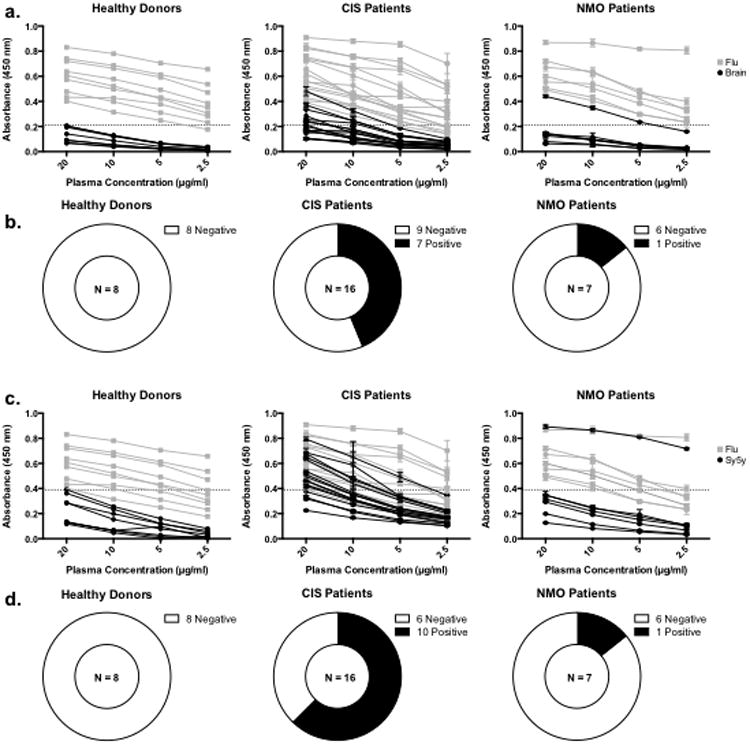
ELISA results with plasma taken from 8 healthy donors responding to influenza vaccination, 16 treatment naïve CIS-PTM patients, and 7 NMO patients. (a.) Absorbance data from plasma ELISAs on brain lysate and influenza antigen grouped by patient classification. Each black line represents ELISA data from an individual plasma antibody sample tested on brain lysate while each gray line represents ELISA data from an individual plasma antibody sample tested on influenza antigen. The dashed line on each graph represents the cutoff for positive binding (the average absorbance from healthy donor plasma on brain lysate at 20 μg/mL plus two standard deviations). (b.) Summary of brain lysate ELISAs with plasma antibodies. Plasma antibody samples from 1 NMO and 7 CIS-PTM patients were two standard deviations above the mean of healthy plasma antibody samples at 20 μg/mL. (c.) Absorbance data from plasma ELISAs on SH-Sy5y lysate and influenza antigen grouped by patient classification. Data are presented as in a, except that SH-Sy5y data is displayed in black. (d.) Summary of SH-Sy5y lysate ELISAs with plasma antibodies. Plasma antibody samples from 1 NMO and 10 CIS-PTM patients were two standard deviations above the mean of healthy plasma samples at 20 μg/mL.
Discussion
In this study, we discovered that expanded and highly mutated VH4+ peripheral plasmablasts from CIS-PTM patients experiencing their first documented clinical attack express antibodies that bind neurons and astrocytes. We and others previously demonstrated plasmablast expansion in the CSF is a common feature to many CIS-PTM, NMO and MS patients [18, 20, 58]. Here we extend that observation to demonstrate that the frequency of peripheral plasmablasts in our CIS-PTM cohort was similar to that of NMO patients, a CNS autoimmune disease in which patients also experience PTM, but classically without brain inflammation [78]. Peripheral plasmablast expansion is not a common feature reported by others, although our cohort displayed a high occurrence of plasmablast expansion in the periphery. This discrepancy may be partially explained by heterogeneity in the study group of the previous reports, as patients with both partial transverse myelitis (PTM) and optic neuritis (ON) symptoms are included, as well as patients at various stages of disease. Our study focuses only on patients experiencing their first clinical attack of PTM because we previously determined these patients are more likely to have a plasmablast expansion [58]. Interestingly, the four patients in our previous study who later displayed additional MRI activity had elevated frequencies of CD27high plasmablasts at the time of their initial clinical event, which recapitulated the finding that plasmablast frequency correlates with parenchymal inflammatory disease activity [18].
The antibody genetics of a B cell population can have a profound impact on our understanding of disease, as the development and function of a B cell is dependent on the antibody it expresses [14, 17, 64, 65, 70, 102]. In fact, antibody genetic studies have led to several key discoveries in MS that show expansion of particular genes, excessive receptor editing, dysregulation in B cell selection [6, 16, 22, 23, 37, 38, 54, 58, 67, 75-77, 79, 80, 83, 84, 96-98, 105], and even a mutational biomarker that identifies patients who will convert to MS with 86-92% accuracy [16, 84, 85]. Here, we demonstrate that VH4 genes are used more extensively by peripheral plasmablasts from CIS-PTM patients in comparison to a previously published healthy donor plasmablasts responding to influenza infection (Fig. 1b). However, the specific genes within the VH4 family were utilized at frequencies similar to these two control populations. This indicates that most VH4 family genes are slightly over-utilized in CIS-PTM patients, rather than particular VH4 genes driving the over-representation of the entire family. This data also suggests that VH4 expansion may be a generalized feature of patients with CNS diseases who experience PTM symptoms. Others have demonstrated VH4 family expansion in the CSF of MS patients [9, 76], which may suggest that VH4 expansion is an early and prolonged feature of particular CNS diseases. Interestingly, B cells from the CSF of NMO patients are dominated by an expansion of VH2 genes, and rarely VH4 genes [11].
In this study, we demonstrate that the expansion of VH4 utilization in peripheral plasmablasts translates to an increase in autoreactivity toward brain antigens. We used a brain lysate ELISA to initially screen rhAbs cloned from plasmablasts for binding to brain targets, but discovered that this assay could lead to false negatives. For example, several rhAbs that were not reactive in the brain lysate ELISA exhibited binding to neuronal cell bodies and astrocyte processes in the brain (Supplemental Table 1). However, it should be noted that the lysate preparation consists mainly of cytosolic and easily soluble proteins, such as the abundant myelin proteins [36]. Thus, the probability of identifying primary targets under-represented in the brain lysate such as non-myelin and hydrophobic targets is diminished. Indeed, more than half of all the CIS-PTM rhAbs displayed positive binding by the SH-Sy5y cell-based assays (Fig. 5, Supplemental Figs. 16, 17), while only a third displayed positive reactivity by SH-Sy5y lysate ELISA (Supplemental Fig. 5). Additionally many antibodies recognize conformational epitopes, and the degree to which a technique may alter these epitopes can profoundly affect the outcome. For example, tissue staining done with MS CSF rhAbs on formalin fixed paraffin embedded brain tissue yielded almost no positive staining [74], but positive staining is detected with similar rhAbs when tested on paraformaldehyde fixed and gently unmasked tissue [12, 57]. However this unmasking may denature some conformational epitopes. For these reasons, we agree that a multi-tiered pipeline for characterizing the autoreactive potential of antibodies is necessary, as suggested by others [109].
Autoantibodies to extracellular neuronal antigens are known to contribute to cognitive dysfunction in a variety of CNS disorders, including nerve hyper-excitability, limbic encephalitis, encephalopathy, and autoimmune disorders with neurological involvement such as systemic lupus erythematosus (SLE) and myasthenia gravis [42, 46, 47, 52, 72, 95]. Others have identified a wide variety of neuron, astrocyte and oligodendrocyte B cell autoantigens in MS that are expressed both extracellularly and intracellularly [4, 5, 15, 21, 27-29, 31, 33, 62, 68, 73, 87, 90-92, 94, 99, 103]. However, the majority of the autoreactive CIS-PTM plasmablast antibodies in our study recognized intracellular antigens expressed by neurons. Antibody recognition of intracellular antigens is only beginning to be understood as a means to drive autoimmunity [49, 59, 81, 89, 100, 107]. Most studies in SLE have focused on anti-nuclear antibodies since their presence correlates well with exacerbation of disease [81]. One leading thought is that anti-nuclear autoantibodies can enter the cell and exert cytotoxic effect there [49, 107]. Perhaps a similar effect could be expected from antibodies that bind cytoplasmic proteins in MS, although there are certainly other scenarios that should be considered [104]. As evidenced with orthogonal images, binding of our CIS-PTM plasmablast rhAbs within the cell was largely excluded from the nucleus.
Of note, many of our rhAbs were polyreactive and recognized targets independent of tissue origin. Recent work by others [13] demonstrated that antibodies contributing to the oligoclonal banding observed in MS patients are directed against ubiquitous intracellular proteins. However, eleven of the rhAbs we tested are brain-specific by ELISA (Brain+ Kidney- Flu- OVA-) and fourteen of the rhAbs we tested are brain-specific by ICC (Sy5y+ Hep-2-). Of the three NMO rhAbs we tested, only NMO03 was brain specific by ICC (Sy5y+Hep-2-). Future experiments on the ability of polyreactive and intracellular brain specific binding CIS-PTM plasmablast rhAbs to participate in a pathogenic response against primary human cells are needed to determine the role of these antibodies in MS.
When rhAb reactivity is considered in aggregate (Supplemental Table 1), it is interesting to note that while the CSF-derived rhAbs from these patients were largely reactive to neurons and astrocytes in the gray matter of the brain [57], many of the rhAbs generated from the peripheral plasmablasts were directed towards both gray and white matter targets. These data suggest that peripheral plasmablasts have a wider array of autoreactive specificities compared to CSF-derived B cells, although their specificity and significance to MS pathology remains unknown. Nevertheless, this scenario is consistent with an underlying dysregulation in tolerance of peripheral B cells in these CIS-PTM patients, and indeed others have demonstrated that there is a break in the peripheral tolerance checkpoint in MS patients [51]. The exact mechanism of CNS-reactive effector B cell development in the blood is still unknown, particularly as it relates to the importance of specific antigens driving the autoreactive plasmablast expansion. Indeed, this break in tolerance could involve both B cell intrinsic and extrinsic mechanisms. Furthermore the rapid return of memory B cells in the periphery following B cell depletion is a strong indicator of poor response to therapy [1], and further emphasizes the importance of studying the development of autoreactive B cells in the blood.
In MS patients, the blood brain barrier is often altered [63], allowing increased exchange of antigen-stimulated cells between the CNS and periphery [45, 66]. Although in our studies we did not detect clonal relatedness between peripheral blood and CSF B cells, others have found clonal overlap between these compartments in MS [8, 98]. Thus, one might predict that, as we observed, peripheral plasmablasts display autoreactivity towards CNS antigens. In MS and CIS patients, there may be an underlying open access to gray matter targets throughout the disease course, and access to white matter targets primarily duringdistinct points in time. It is also possible that there is less reactivity to white matter targets at later stages of disease due to immune response exhaustion to those targets [2, 82]. Alternatively, gray matter targets may be more immunogenic, considering the more extensive clonal expansion of CSF B cells that bind to these targets as compared to the peripheral plasmablasts [58, 75, 98]. Interestingly, others have demonstrated that antibodies targeting neurons from clonally expanded CSF B cells from MS patients mediate demyelination of axons in vitro, highlighting their potential to participate in the pathogenesis of disease [12]. Delineating the pathway by which autoreactive plasmablasts develop, persist and mediate pathogenesis in MS patients will greatly improve our understanding of the disease, and is particularly important given that the frequency of plasmablasts increases the longer that CIS-PTM patients are left untreated [58]. Of note, clonal overlap between the CSF and periphery has also been observed in NMO patients [20, 53], suggesting CNS autoreactivity should be evident among peripheral antigen experienced B cells from NMO patients. However, we observed that CNS autoreactivity, including AQP4, among the peripheral plasmablast rhAbs is less extensive in NMO patients. Although other studies demonstrated that approximately50 % of all clonally expanded CSF B cells from NMO patients bind AQP4 (range 3–97 %) [11, 53], the frequency of AQP4 binding by peripheral B cells from NMO patients is lower (19 % on average, range 0–40 %) [53]. Our data would support the concept that AQP4-reactive B cells may be enriched in the CNS of NMO patients rather than the periphery. Indeed, others have demonstrated that NMO plasmablasts express higher levels of the chemokine receptor CXCR3 during relapse, which may aid in retention of affinity-maturated clones in the CNS [20] and subsequent lack of detection in the periphery. Further studies are warranted to investigate this discordance in CNS autoreactivity by peripheral plasmablasts of PTM and NMO patients.
In summary, plasmablasts may be either direct perpetrators of autoimmunity or simply biomarkers of disease severity in MS [18], but they are certainly the footprints of previously activated B cells. The increased representation of VH4-utilizing autoreactive plasmablasts in the periphery of CIS-PTM patients demonstrates a B cell autoimmune response directed toward neurons and astrocytes early in disease. Their presence in the periphery proposes intriguing questions about the origin and function of autoreactive plasmablasts in CIS-PTM patients, and provides a unique avenue to explore the autoreactive B cell response in MS.
Supplementary Material
Supplemental Fig. 1. (a.) Usage of specific heavy chain V gene segments in patient repertoires for each cell population. CD19+ means total B cells, CD19+CD27+means memory B cells, and CD19+CD27hi means plasmablasts. Black bars designate healthy donor samples, dark gray bars designate CIS-PTM memory B cells, light gray bars designate CIS-PTM plasmablasts, white bars designate NMO plasmablasts, and striped bars designate flu-responding plasmablasts. (b.) V gene family representation in the patient repertoires for each cell population. (c.) J gene segment representation in patient repertoires for each cell population. (d.) The average CDR3 length for all heavy chain sequences per patient in each patient group. (e.) The average charge of heavy chain CDR3s as calculated by the net number of positive and negative amino acids per sequence. (f.) The abundance of antibody isotypes found in either plasmablasts or memory B cells for CIS-PTM and NMO patients.
Supplemental Fig. 2. (a.) Usage of specific kappa chain V gene segments in patient repertoires. CD19+ means total B cells, CD19+CD27+means memory B cells, and CD19+CD27hi means plasmablasts. Black bars designate healthy donor samples, dark gray bars designate CIS-PTM memory B cells, light gray bars designate CIS-PTM plasmablasts, white bars designate NMO plasmablasts, and striped bars designate flu-responding plasmablasts. (b.) Usage of specific lambda chain V gene segments in patient repertoires in NMO and CIS-PTM patient cell populations. (c.) Kappa family representation in patient repertoires for each cell population. (d.) Light chain J gene segment distribution in patient repertoires. (e.) The average CDR3 length for the light chain sequences in patient repertoires. (f.) The average CDR3 charge of the light chain as calculated by the net number of positive and negative amino acids.
Supplemental Fig. 3. (a.) Human brain lysate ELISA results grouped by patient. Dashed line represents the same cutoff used for positive binding. (b.) Summary of human brain ELISA results grouped by patient type. Half of the CIS-PTM plasmablasts rhAbs (51%) and one third of the NMO plasmablast rhAbs (33%) were reactive to human brain lysate, demonstrating even more brain reactive rhAbs in this assay. (c.) Kidney lysate ELISA results grouped by patient. Dashed line represents the same cutoff used for positive binding in the mouse brain lysate ELISA. (d.) Summary of kidney lysate ELISA results grouped by patient type. A similar proportion of CIS-PTM plasmablasts rhAbs (13%) and NMO plasmablast rhAbs (11%) were reactive to kidney lysate. None of the healthy donor rhAbs demonstrated binding to kidney lysate.
Supplemental Fig. 4. (a.) H1N1 influenza ELISA results grouped by patient. Dashed line represents the cutoff used to define positive binding. (b.) Summary of H1N1 influenza ELISA results grouped by patient type. A similar proportion of CIS-PTM plasmablasts rhAbs (17%) and NMO plasmablast rhAbs (13%) were reactive to H1N1 native whole viral antigen. None of the healthy donor rhAbs bound strongly to the virus. (c.) Ovalbumin ELISA results grouped by patient. Dashed line represents the cutoff used to define positive binding. (d.) Summary of ovalbumin ELISA results grouped by patient type. Some of the CIS-PTM plasmablasts rhAbs (14%) but none of the NMO plasmablast rhAbs bound to this irrelevant antigen.
Supplemental Fig. 5. (a.) SH-Sy5y (human neuroblastoma) lysate ELISA results grouped by patient. Dashed line represents the cutoff used to define positive binding. (b.) Summary of SH-Sy5y ELISA results grouped by patient type. A similar proportion of CIS-PTM plasmablasts rhAbs (32%) and NMO plasmablast rhAbs (33%) were reactive to SH-Sy5y lysate, demonstrating binding of many of the rhAbs to neuronal antigens. None of the healthy donor rhAbs demonstrated binding to SH-Sy5y lysate.
Supplemental Fig. 6. 20× images displaying rhAb reactivity to post-stroke mouse corpus callosum. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to unidentified glial cells in this white matter tissue.
Supplemental Fig. 7. 20× images displaying rhAb reactivity to post-stroke mouse cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to neurons, as the rhAb signal overlapped with NeuN signal in this gray matter tissue.
Supplemental Fig. 8. 20× images displaying rhAb reactivity to EAE mouse corpus callosum. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to unidentified glial cells in this white matter tissue.
Supplemental Fig. 9. 20× images displaying rhAb reactivity to EAE mouse cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to neurons, as the rhAb signal overlapped with NeuN signal in this gray matter tissue.
Supplemental Fig. 10. 20× images displaying rhAb reactivity to healthy mouse corpus callosum. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to unidentified glial cells in this white matter tissue.
Supplemental Fig. 11. 20× images displaying rhAb reactivity to healthy mouse cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to neurons, as the rhAb signal overlapped with NeuN signal in this gray matter tissue.
Supplemental Fig. 12. 20× images displaying rhAb reactivity to healthy and EAE mouse corpus callosum (CC) and cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and GFAP is shown in red to mark astrocytes. All of these rhAbs demonstrated binding to astrocytes, as the rhAb signal overlapped with GFAP signal in both gray and white matter tissue.
Supplemental Fig. 13. 20× images displaying rhAb reactivity to healthy and EAE mouse corpus callosum (CC) and cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and PDGFR is shown in red to mark oligodendrocytes. Most of the rhAbs did not bind to oligodendrocytes, as the rhAb signal did not overlap with PDGFR signal. However CIS68 did occasionally overlap with PDGFR in either the gray or white matter tissue (designated by yellow arrows).
Supplemental Fig. 14. (a.) Summary of all the CIS and NMO rhAbs tested for binding to mouse brain tissue. (b.) Lambda scans were performed to measure the signal intensity of staining observed and is displayed here as a ratio over background signal. Ratio values higher than 1.86 were considered to be true positive signal (ratio value for negative control rhAb B1). N designates areas where signal was not measured. (c.) Orthogonal views of example rhAbs on healthy mouse brain tissue. Scale bar indicates 10 μm, the leftmost column is an image of each rhAb with overlaid rhAb (green), NeuN (red) and DAPI (blue) staining with crosshairs to show the plane viewed beneath and to the right of the overlay. The column to the right of this is the same image without the crosshairs. The column to the right of this shows the same image with only the rhAb staining. The rightmost column shows the same image with only DAPI staining.
Supplemental Fig. 15. Enumeration of positively stained cortical cells from healthy mouse brain. (a.) Representative overlay image of NeuN and rhAb for enumeration with ROIs defined by faint yellow line. (b.) Percent of NeuN single positive (red), rhAb single positive (green), and double positive cells in the cortex. (c.) Histograms of maximal intensity in the red (NeuN) or green (rhAb) channel for ROIs with thresholds set for positive binding shown by the dashed lines in each graph. (d.) Table with the percent of ROIs in each cortical image stained with both NeuN and rhAb (red+green), only NeuN (red), or only the rhAb (green). Final column is the average percentage of ROIs in each category for all images measured for a particular rhAb.
Supplemental Fig. 16. Most of the remaining images of CIS-PTM rhAbs binding to SH-Sy5y cells by ICC. Binding of the rhAbs is displayed in green while the nuclear stain DAPI is shown in blue. The scale bar in each indicates 20 μm.
Supplemental Fig. 17. Remaining images of CIS-PTM, NMO and HD rhAbs binding to SH-Sy5y cells by ICC, plus controls. Binding of the rhAbs is displayed in green while the nuclear stain DAPI is shown in blue. The scale bar in each indicates 20 μm. R1, R2, and CSF10 served as additional negative and positive controls, as published in our previous study [57].
Supplemental Fig. 18. (a.) All images of rhAb binding to Hep-2 cells by ICC. Counterstain EvoBlue is shown in red while rhAb staining is shown in green (overlay is yellow). (b.) Summary of rhAb binding to Hep-2 cells. None of the healthy donor rhAbs bound to Hep-2 cells, but a small proportion of the CIS-PTM and NMO rhAbs did.
Supplemental Fig. 19. Western blots with cellular fractions from SH-Sy5y and Hep-2 cells and human brain lysate blotted with select rhAbs. Lanes from left to right are: (1) protein ladder, (2) nuclear and extracellular membrane (M+N) fraction of SH-Sy5y, (3) cytosolic (C) fraction of SH-Sy5y, (4) nuclear and extracellular membrane (M+N) fraction of Hep-2, (5) cytosolic (C) fraction of Hep-2, and (6) total human brain lysate. To demonstrate clear separation of cytosol from the nuclear/membrane cellular fraction, commercial antibodies to Histone H3 (H3, 15 kDa) and Heat Shock Protein 90 (Hsp90, 90 kDa) were used. Histone H3 was only detected in the nuclear fractions and Hsp90 was only detected in the cytosolic fractions, demonstrating clean separation of these cellular compartments. A commercial antibody to Tubulin was used to demonstrate that protein is loaded into each lane.
Supplemental Fig. 20. Binding of antibodies to AQP4-transfected HEK293 cells. Antibody binding is displayed in red while EGFP labeled AQP4 is displayed in green. Control serum from a strongly positive patient, a weakly positive patient and a negative patient are shown to compare to the two rhAbs cloned from the CSF of a CIS-PTM patient that later converted to NMO (R1 and R2). None of the NMO peripheral plasmablast rhAbs bound AQP4 in this assay (data not shown). Images are captured at 100× and show one to three representative stained cells.
Supplemental Fig. 21. Flow cytometry testing of rhAb binding to SH-Sy5y. Histograms depict rhAb staining in black outline without fill overlaid with a no primary antibody control in gray. (a.) Histograms of rhAbs binding to intracellular antigens of SH-Sy5y cells by flow cytometry. (b.) Histograms of rhAbs binding to intracellular and extracellular antigens of SH-Sy5y cells by flow cytometry. (c.) Geometric mean fluorescence intensity (gMFI) of rhAbs binding to intracellular and extracellular antigens of SH-Sy5y by flow cytometry.
Supplemental Table 1. All CIS-PTM and NMO rhAbs cloned for this study are listed, grouped by patient. Displayed is the heavy chain V gene segment (VH), heavy chain J gene segment (JH), light chain V gene segment (VL), light chain J gene segment (JL), number of mutations in the heavy chain (HC Mut), and antibody isotype for each sequence. The result of each test performed with the rhAb follows and is listed as positive (+), negative (-), with the cell type recognized (N for neuron, G for glial cell), or pattern of recognition (N for nuclear, C for cytoplasmic, I for intracellular, E for extracellular). VH4+ rhAbs are listed in bold font, VH3+ rhAbs are listed in regular font, rhAbs utilizing kappa light chains are not highlighted, and rhAbs utilizing lambda light chains are highlighted in gray.
Acknowledgments
This project was funded by a grant from the National MS Society and National Institute of Health, national research service award 5 T32 AI005284-38 from the National Institutes of Allergy and Infectious Disease, and the authors would like to thank the patients and healthy donors who gave samples for this study. Angela Mobley assisted with flow cytometry and sorting and Genevieve Konopka provided the SH-Sy5y cells. Betty Diamond at the Hofstra Northwell School of Medicine Department of Molecular Medicine provided the lupus control antibodies. We also thank E. Sally Ward at Texas A&M University for helpful discussion of this manuscript.
Footnotes
Conflicts of Interest: Nancy Monson reports patent US 8,394,583 B2 on MSPrecise™, a diagnostic tool for predicting conversion to MS.
References
- 1.Adlowitz DG, Barnard J, Biear JN, Cistrone C, Owen T, Wang W, Palanichamy A, Ezealah E, Campbell D, Wei C, et al. Expansion of Activated Peripheral Blood Memory B Cells in Rheumatoid Arthritis, Impact of B Cell Depletion Therapy, and Biomarkers of Response. PLoS One. 2015;10(6):e0128269. doi: 10.1371/journal.pone.0128269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11(4):289–95. doi: 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- 3.Avery DT, Ellyard JI, Mackay F, Corcoran LM, Hodgkin PD, Tangye SG. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J Immunol. 2005;174(7):4034–42. doi: 10.4049/jimmunol.174.7.4034. [DOI] [PubMed] [Google Scholar]
- 4.Ayoglu B, Mitsios N, Kockum I, Khademi M, Zandian A, Sjoberg R, Forsstrom B, Bredenberg J, Lima Bomfim I, Holmgren E, et al. Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Proc Natl Acad Sci U S A. 2016;113(8):2188–93. doi: 10.1073/pnas.1518553113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banki K, Colombo E, Sia F, Halladay D, Mattson DH, Tatum AH, Massa PT, Phillips PE, Perl A. Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J Exp Med. 1994;180(5):1649–63. doi: 10.1084/jem.180.5.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, von Budingen HC. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol. 2014;75(2):266–76. doi: 10.1002/ana.24088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar-Or A, Antel JP. Central nervous system inflammation across the age span. Curr Opin Neurol. 2016 doi: 10.1097/WCO.0000000000000331. [DOI] [PubMed] [Google Scholar]
- 8.Beltran E, Obermeier B, Moser M, Coret F, Simo-Castello M, Bosca I, Perez-Miralles F, Villar LM, Senel M, Tumani H, et al. Intrathecal somatic hypermutation of IgM in multiple sclerosis and neuroinflammation. Brain. 2014;137(Pt 10):2703–14. doi: 10.1093/brain/awu205. [DOI] [PubMed] [Google Scholar]
- 9.Bennett JL, Haubold K, Ritchie AM, Edwards SJ, Burgoon M, Shearer AJ, Gilden DH, Owens GP. CSF IgG heavy-chain bias in patients at the time of a clinically isolated syndrome. J Neuroimmunol. 2008;199(1-2):126–32. doi: 10.1016/j.jneuroim.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617–29. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett JL, O'Connor KC, Bar-Or A, Zamvil SS, Hemmer B, Tedder TF, von Budingen HC, Stuve O, Yeaman MR, Smith TJ, et al. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e104. doi: 10.1212/NXI.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blauth K, Soltys J, Matschulat A, Reiter CR, Ritchie A, Baird NL, Bennett JL, Owens GP. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid cause demyelination of spinal cord explants. Acta Neuropathol. 2015;130(6):765–81. doi: 10.1007/s00401-015-1500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandle SM, Obermeier B, Senel M, Bruder J, Mentele R, Khademi M, Olsson T, Tumani H, Kristoferitsch W, Lottspeich F, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc Natl Acad Sci U S A. 2016;113(28):7864–9. doi: 10.1073/pnas.1522730113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brezinschek HP, Foster SJ, Brezinschek RI, Dorner T, Domiati-Saad R, Lipsky PE. Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on human peripheral CD5(+)/IgM+ and CD5(-)/IgM+ B cells. J Clin Invest. 1997;99(10):2488–501. doi: 10.1172/JCI119433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bronstein JM, Lallone RL, Seitz RS, Ellison GW, Myers LW. A humoral response to oligodendrocyte-specific protein in MS: a potential molecular mimic. Neurology. 1999;53(1):154–61. doi: 10.1212/wnl.53.1.154. [DOI] [PubMed] [Google Scholar]
- 16.Cameron EM, Spencer S, Lazarini J, Harp CT, Ward ES, Burgoon M, Owens GP, Racke MK, Bennett JL, Frohman EM, et al. Potential of a unique antibody gene signature to predict conversion to clinically definite multiple sclerosis. J Neuroimmunol. 2009;213(1-2):123–30. doi: 10.1016/j.jneuroim.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casellas R, Shih TA, Kleinewietfeld M, Rakonjac J, Nemazee D, Rajewsky K, Nussenzweig MC. Contribution of receptor editing to the antibody repertoire. Science. 2001;291(5508):1541–4. doi: 10.1126/science.1056600. [DOI] [PubMed] [Google Scholar]
- 18.Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, Sommer N, Hartung HP, Hemmer B. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain. 2005;128(Pt 7):1667–76. doi: 10.1093/brain/awh486. [DOI] [PubMed] [Google Scholar]
- 19.Chang EH, Volpe BT, Mackay M, Aranow C, Watson P, Kowal C, Storbeck J, Mattis P, Berlin R, Chen H, et al. Selective Impairment of Spatial Cognition Caused by Autoantibodies to the N-Methyl-d-Aspartate Receptor. EBioMedicine. 2015;2(7):755–64. doi: 10.1016/j.ebiom.2015.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chihara N, Aranami T, Oki S, Matsuoka T, Nakamura M, Kishida H, Yokoyama K, Kuroiwa Y, Hattori N, Okamoto T, et al. Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS One. 2013;8(12):e83036. doi: 10.1371/journal.pone.0083036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colombo E, Banki K, Tatum AH, Daucher J, Ferrante P, Murray RS, Phillips PE, Perl A. Comparative analysis of antibody and cell-mediated autoimmunity to transaldolase and myelin basic protein in patients with multiple sclerosis. J Clin Invest. 1997;99(6):1238–50. doi: 10.1172/JCI119281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colombo M, Dono M, Gazzola P, Chiorazzi N, Mancardi G, Ferrarini M. Maintenance of B lymphocyte-related clones in the cerebrospinal fluid of multiple sclerosis patients. Eur J Immunol. 2003;33(12):3433–8. doi: 10.1002/eji.200324144. [DOI] [PubMed] [Google Scholar]
- 23.Colombo M, Dono M, Gazzola P, Roncella S, Valetto A, Chiorazzi N, Mancardi GL, Ferrarini M. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J Immunol. 2000;164(5):2782–9. doi: 10.4049/jimmunol.164.5.2782. [DOI] [PubMed] [Google Scholar]
- 24.Cristofanilli M, Rosenthal H, Cymring B, Gratch D, Pagano B, Xie B, Sadiq SA. Progressive multiple sclerosis cerebrospinal fluid induces inflammatory demyelination, axonal loss, and astrogliosis in mice. Exp Neurol. 2014;261:620–32. doi: 10.1016/j.expneurol.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 25.Cross AH, Waubant E. MS and the B cell controversy. Biochim Biophys Acta. 2011;1812(2):231–8. doi: 10.1016/j.bbadis.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 26.Disanto G, Morahan JM, Barnett MH, Giovannoni G, Ramagopalan SV. The evidence for a role of B cells in multiple sclerosis. Neurology. 2012;78(11):823–32. doi: 10.1212/WNL.0b013e318249f6f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eikelenboom MJ, Petzold A, Lazeron RH, Silber E, Sharief M, Thompson EJ, Barkhof F, Giovannoni G, Polman CH, Uitdehaag BM. Multiple sclerosis: Neurofilament light chain antibodies are correlated to cerebral atrophy. Neurology. 2003;60(2):219–23. doi: 10.1212/01.wnl.0000041496.58127.e3. [DOI] [PubMed] [Google Scholar]
- 28.Elliott C, Lindner M, Arthur A, Brennan K, Jarius S, Hussey J, Chan A, Stroet A, Olsson T, Willison H, et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain. 2012;135(Pt 6):1819–33. doi: 10.1093/brain/aws105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endo T, Scott DD, Stewart SS, Kundu SK, Marcus DM. Antibodies to glycosphingolipids in patients with multiple sclerosis and SLE. J Immunol. 1984;132(4):1793–7. [PubMed] [Google Scholar]
- 30.Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cell subsets to long-term survival niches. Semin Immunol. 2008;20(1):49–58. doi: 10.1016/j.smim.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Fialova L, Bartos A, Svarcova J, Zimova D, Kotoucova J, Malbohan I. Serum and cerebrospinal fluid light neurofilaments and antibodies against them in clinically isolated syndrome and multiple sclerosis. J Neuroimmunol. 2013;262(1-2):113–20. doi: 10.1016/j.jneuroim.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Fink K. Origin and Function of Circulating Plasmablasts during Acute Viral Infections. Front Immunol. 2012;3:78. doi: 10.3389/fimmu.2012.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraussen J, Claes N, de Bock L, Somers V. Targets of the humoral autoimmune response in multiple sclerosis. Autoimmun Rev. 2014;13(11):1126–37. doi: 10.1016/j.autrev.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Frolich D, Giesecke C, Mei HE, Reiter K, Daridon C, Lipsky PE, Dorner T. Secondary immunization generates clonally related antigen-specific plasma cells and memory B cells. J Immunol. 2010;185(5):3103–10. doi: 10.4049/jimmunol.1000911. [DOI] [PubMed] [Google Scholar]
- 35.Gauld SB, Dal Porto JM, Cambier JC. B cell antigen receptor signaling: roles in cell development and disease. Science. 2002;296(5573):1641–2. doi: 10.1126/science.1071546. [DOI] [PubMed] [Google Scholar]
- 36.Siegel George J, A BW, Albers R Wayne, Fisher Stephen K, Uhler Michael D. Basic Neurochemistry, Molecular, Cellular and Medical Aspects. Philadelphia: Lippincott-Raven; 1999. [Google Scholar]
- 37.Harp C, Lee J, Lambracht-Washington D, Cameron E, Olsen G, Frohman E, Racke M, Monson N. Cerebrospinal fluid B cells from multiple sclerosis patients are subject to normal germinal center selection. J Neuroimmunol. 2007;183(1-2):189–99. doi: 10.1016/j.jneuroim.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haubold K, Owens GP, Kaur P, Ritchie AM, Gilden DH, Bennett JL. B-lymphocyte and plasma cell clonal expansion in monosymptomatic optic neuritis cerebrospinal fluid. Ann Neurol. 2004;56(1):97–107. doi: 10.1002/ana.20152. [DOI] [PubMed] [Google Scholar]
- 39.Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: Prospects and promise. Ann Neurol. 2013;74(3):317–27. doi: 10.1002/ana.24009. [DOI] [PubMed] [Google Scholar]
- 40.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 41.Heine G, Drozdenko G, Grun JR, Chang HD, Radbruch A, Worm M. Autocrine IL-10 promotes human B-cell differentiation into IgM- or IgG-secreting plasmablasts. Eur J Immunol. 2014;44(6):1615–21. doi: 10.1002/eji.201343822. [DOI] [PubMed] [Google Scholar]
- 42.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7(3):365–8. doi: 10.1038/85520. [DOI] [PubMed] [Google Scholar]
- 43.Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016;15(3):317–31. doi: 10.1016/S1474-4422(15)00313-0. [DOI] [PubMed] [Google Scholar]
- 44.Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 2015 doi: 10.1016/S1474-4422(15)00334-8. [DOI] [PubMed] [Google Scholar]
- 45.Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. 2011;1812(2):220–30. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci U S A. 2006;103(3):678–83. doi: 10.1073/pnas.0510055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, Peles E, Buckley C, Lang B, Vincent A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain. 2010;133(9):2734–48. doi: 10.1093/brain/awq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, Burmester GR, Hiepe F, Dorner T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis. 2010;69(1):305–8. doi: 10.1136/ard.2008.096495. [DOI] [PubMed] [Google Scholar]
- 49.Jang JY, Jeong JG, Jun HR, Lee SC, Kim JS, Kim YS, Kwon MH. A nucleic acid-hydrolyzing antibody penetrates into cells via caveolae-mediated endocytosis, localizes in the cytosol and exhibits cytotoxicity. Cell Mol Life Sci. 2009;66(11-12):1985–97. doi: 10.1007/s00018-009-9179-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kappos L, Li D, Calabresi PA, O'Connor P, Bar-Or A, Barkhof F, Yin M, Leppert D, Glanzman R, Tinbergen J, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779–87. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 51.Kinnunen T, C N, Morbach H, Cantaert T, Lynch M, Preston-Hurlburt P, Herold KC, Hafler DA, C OCK, Meffre E. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest. doi: 10.1172/JCI68775. 20131558-8238 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, Volpe BT. Cognition and immunity; antibody impairs memory. Immunity. 2004;21(2):179–88. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 53.Kowarik MC, Dzieciatkowska M, Wemlinger S, Ritchie AM, Hemmer B, Owens GP, Bennett JL. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J Neuroinflammation. 2015;12:19. doi: 10.1186/s12974-015-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. 2012;8(11):613–23. doi: 10.1038/nrneurol.2012.203. [DOI] [PubMed] [Google Scholar]
- 55.Lee FE, Halliley JL, Walsh EE, Moscatiello AP, Kmush BL, Falsey AR, Randall TD, Kaminiski DA, Miller RK, Sanz I. Circulating human antibody-secreting cells during vaccinations and respiratory viral infections are characterized by high specificity and lack of bystander effect. J Immunol. 2011;186(9):5514–21. doi: 10.4049/jimmunol.1002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004;18(1):1–11. doi: 10.1101/gad.1161904. [DOI] [PubMed] [Google Scholar]
- 57.Ligocki AJ, Rivas JR, Rounds WH, Guzman AA, Li M, Spadaro M, Lahey L, Chen D, Henson PM, Graves D, et al. A Distinct Class of Antibodies May Be an Indicator of Gray Matter Autoimmunity in Early and Established Relapsing Remitting Multiple Sclerosis Patients. ASN Neuro. doi: 10.1177/1759091415609613. LID - 10.1177/1759091415609613 [doi] LID - 1759091415609613 [pii] 20151759-0914 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ligocki AJ, Rounds WH, Cameron EM, Harp CT, Frohman EM, Courtney AM, Vernino S, Cowell LG, Greenberg B, Monson NL. Expansion of CD27high plasmablasts in transverse myelitis patients that utilize VH4 and JH6 genes and undergo extensive somatic hypermutation. Genes Immun. doi: 10.1038/gene.2013.18. 20131476-5470 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lim PL, Zouali M. Pathogenic autoantibodies: emerging insights into tissue injury. Immunol Lett. 2006;103(1):17–26. doi: 10.1016/j.imlet.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 60.Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86. doi: 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 62.Mathiesen T, von Holst H, Fredrikson S, Wirsen G, Hederstedt B, Norrby E, Sundqvist VA, Wahren B. Total, anti-viral, and anti-myelin IgG subclass reactivity in inflammatory diseases of the central nervous system. J Neurol. 1989;236(4):238–42. doi: 10.1007/BF00314506. [DOI] [PubMed] [Google Scholar]
- 63.McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, Klein RS. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172(3):799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meffre E, Casellas R, Nussenzweig MC. Antibody regulation of B cell development. Nat Immunol. 2000;1(5):379–85. doi: 10.1038/80816. [DOI] [PubMed] [Google Scholar]
- 65.Meffre E, Davis E, Schiff C, Cunningham-Rundles C, Ivashkiv LB, Staudt LM, Young JW, Nussenzweig MC. Circulating human B cells that express surrogate light chains and edited receptors. Nat Immunol. 2000;1(3):207–13. doi: 10.1038/79739. [DOI] [PubMed] [Google Scholar]
- 66.Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. 2003;9(6):540–9. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- 67.Monson NL, Brezinschek HP, Brezinschek RI, Mobley A, Vaughan GK, Frohman EM, Racke MK, Lipsky PE. Receptor revision and atypical mutational characteristics in clonally expanded B cells from the cerebrospinal fluid of recently diagnosed multiple sclerosis patients. J Neuroimmunol. 2005;158(1-2):170–81. doi: 10.1016/j.jneuroim.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 68.Morris-Downes MM, McCormack K, Baker D, Sivaprasad D, Natkunarajah J, Amor S. Encephalitogenic and immunogenic potential of myelin-associated glycoprotein (MAG), oligodendrocyte-specific glycoprotein (OSP) and 2',3'-cyclic nucleotide 3'-phosphodiesterase (CNPase) in ABH and SJL mice. J Neuroimmunol. 2002;122(1-2):20–33. doi: 10.1016/s0165-5728(01)00460-x. [DOI] [PubMed] [Google Scholar]
- 69.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15(3):160–71. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- 70.Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, Lipsky PE, Radbruch A, Dorner T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165(10):5970–9. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 71.Odendahl M, jacobi A, Hansen A, Fesit E, Hiepe F, Burmester G, Lipsky P, Radbruch A, Dorner T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165:5970–9. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 72.Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren SI. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol. 2005;12(5):392–8. doi: 10.1111/j.1468-1331.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 73.Onoue H, Satoh JI, Ogawa M, Tabunoki H, Yamamura T. Detection of anti-Nogo receptor autoantibody in the serum of multiple sclerosis and controls. Acta Neurol Scand. 2007;115(3):153–60. doi: 10.1111/j.1600-0404.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 74.Owens GP, Bennett JL, Lassmann H, O'Connor KC, Ritchie AM, Shearer A, Lam C, Yu X, Birlea M, DuPree C, et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann Neurol. 2009;65(6):639–49. doi: 10.1002/ana.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol. 2003;171(5):2725–33. doi: 10.4049/jimmunol.171.5.2725. [DOI] [PubMed] [Google Scholar]
- 76.Owens GP, Winges KM, Ritchie AM, Edwards S, Burgoon MP, Lehnhoff L, Nielsen K, Corboy J, Gilden DH, Bennett JL. VH4 gene segments dominate the intrathecal humoral immune response in multiple sclerosis. J Immunol. 2007;179(9):6343–51. doi: 10.4049/jimmunol.179.9.6343. [DOI] [PubMed] [Google Scholar]
- 77.Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, Sundar PD, Telman D, Zhao LZ, Derstine M, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med. 2014;6(248):248ra106. doi: 10.1126/scitranslmed.3008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parratt JD, Prineas JW. Neuromyelitis optica: a demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult Scler. 2010;16(10):1156–72. doi: 10.1177/1352458510382324. [DOI] [PubMed] [Google Scholar]
- 79.Qin Y, Duquette P, Zhang Y, Olek M, Da RR, Richardson J, Antel JP, Talbot P, Cashman NR, Tourtellotte WW, et al. Intrathecal B-cell clonal expansion, an early sign of humoral immunity, in the cerebrospinal fluid of patients with clinically isolated syndrome suggestive of multiple sclerosis. Lab Invest. 2003;83(7):1081–8. doi: 10.1097/01.lab.0000077008.24259.0d. [DOI] [PubMed] [Google Scholar]
- 80.Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest. 1998;102(5):1045–50. doi: 10.1172/JCI3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Racanelli V, Prete M, Musaraj G, Dammacco F, Perosa F. Autoantibodies to intracellular antigens: generation and pathogenetic role. Autoimmun Rev. 2011;10(8):503–8. doi: 10.1016/j.autrev.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 82.Ratts RB, Karandikar NJ, Hussain RZ, Choy J, Northrop SC, Lovett-Racke AE, Racke MK. Phenotypic characterization of autoreactive T cells in multiple sclerosis. J Neuroimmunol. 2006;178(1-2):100–10. doi: 10.1016/j.jneuroim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 83.Ritchie AM, Gilden DH, Williamson RA, Burgoon MP, Yu X, Helm K, Corboy JR, Owens GP. Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol. 2004;173(1):649–56. doi: 10.4049/jimmunol.173.1.649. [DOI] [PubMed] [Google Scholar]
- 84.Rounds WH, Ligocki AJ, Levin MK, Greenberg BM, Bigwood DW, Eastman EM, Cowell LG, Monson NL. The antibody genetics of multiple sclerosis: comparing next-generation sequencing to sanger sequencing. Front Neurol. 2014;5:166. doi: 10.3389/fneur.2014.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rounds WH, Salinas EA, Wilks TB, 2nd, Levin MK, Ligocki AJ, Ionete C, Pardo CA, Vernino S, Greenberg BM, Bigwood DW, et al. MSPrecise: A molecular diagnostic test for multiple sclerosis using next generation sequencing. Gene. 2015;572(2):191–7. doi: 10.1016/j.gene.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Selvaraj UM, Ortega SB, Hu R, Gilchrist R, Kong X, Partin A, Plautz EJ, Klein RS, Gidday JM, Stowe AM. Preconditioning-induced CXCL12 upregulation minimizes leukocyte infiltration after stroke in ischemia-tolerant mice. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16639327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Silber E, Semra YK, Gregson NA, Sharief MK. Patients with progressive multiple sclerosis have elevated antibodies to neurofilament subunit. Neurology. 2002;58(9):1372–81. doi: 10.1212/wnl.58.9.1372. [DOI] [PubMed] [Google Scholar]
- 88.Solomon DH, Kavanaugh AJ, Schur PH American College of Rheumatology Ad Hoc Committee on Immunologic Testing G. Evidence-based guidelines for the use of immunologic tests: antinuclear antibody testing. Arthritis Rheum. 2002;47(4):434–44. doi: 10.1002/art.10561. [DOI] [PubMed] [Google Scholar]
- 89.Song YC, Sun GH, Lee TP, Huang JC, Yu CL, Chen CH, Tang SJ, Sun KH. Arginines in the CDR of anti-dsDNA autoantibodies facilitate cell internalization via electrostatic interactions. Eur J Immunol. 2008;38(11):3178–90. doi: 10.1002/eji.200838678. [DOI] [PubMed] [Google Scholar]
- 90.Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, Rothhammer V, Chan A, Gold R, Berthele A, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367(2):115–23. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stevens A, Weller M, Wietholter H. CSF and serum ganglioside antibody patterns in MS. Acta Neurol Scand. 1992;86(5):485–9. doi: 10.1111/j.1600-0404.1992.tb05129.x. [DOI] [PubMed] [Google Scholar]
- 92.Szmyrka-Kaczmarek M, Pokryszko-Dragan A, Pawlik B, Gruszka E, Korman L, Podemski R, Wiland P, Szechinski J. Antinuclear and antiphospholipid antibodies in patients with multiple sclerosis. Lupus. 2012;21(4):412–20. doi: 10.1177/0961203311427550. [DOI] [PubMed] [Google Scholar]
- 93.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329(1-2):112–24. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trotter J. NG2-positive cells in CNS function and the pathological role of antibodies against NG2 in demyelinating diseases. J Neurol Sci. 2005;233(1-2):37–42. doi: 10.1016/j.jns.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 95.Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, Clover L, Parkinson A, Bien CG, Omer S, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127(Pt 3):701–12. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- 96.von Budingen HC, Gulati M, Kuenzle S, Fischer K, Rupprecht TA, Goebels N. Clonally expanded plasma cells in the cerebrospinal fluid of patients with central nervous system autoimmune demyelination produce “oligoclonal bands”. J Neuroimmunol. 2010;218(1-2):134–9. doi: 10.1016/j.jneuroim.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 97.von Budingen HC, Harrer MD, Kuenzle S, Meier M, Goebels N. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin-specific antibodies. Eur J Immunol. 2008;38(7):2014–23. doi: 10.1002/eji.200737784. [DOI] [PubMed] [Google Scholar]
- 98.von Budingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, Cree BA, Gourraud PA, Schwartzburg A, Huerta G, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest. 2012;122(12):4533–43. doi: 10.1172/JCI63842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vu T, Myers LW, Ellison GW, Mendoza F, Bronstein JM. T-cell responses to oligodendrocyte-specific protein in multiple sclerosis. J Neurosci Res. 2001;66(3):506–9. doi: 10.1002/jnr.1241. [DOI] [PubMed] [Google Scholar]
- 100.Waldman M, Madaio MP. Pathogenic autoantibodies in lupus nephritis. Lupus. 2005;14(1):19–24. doi: 10.1191/0961203305lu2054oa. [DOI] [PubMed] [Google Scholar]
- 101.Wang LD, Clark MR. B-cell antigen-receptor signalling in lymphocyte development. Immunology. 2003;110(4):411–20. doi: 10.1111/j.1365-2567.2003.01756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 103.Willis SN, Stathopoulos P, Chastre A, Compton SD, Hafler DA, O'Connor KC. Investigating the Antigen Specificity of Multiple Sclerosis Central Nervous System-Derived Immunoglobulins. Front Immunol. 2015;6:600. doi: 10.3389/fimmu.2015.00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Winger RC, Zamvil SS. Antibodies in multiple sclerosis oligoclonal bands target debris. Proc Natl Acad Sci U S A. 2016;113(28):7696–8. doi: 10.1073/pnas.1609246113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Winges KM, Gilden DH, Bennett JL, Yu X, Ritchie AM, Owens GP. Analysis of multiple sclerosis cerebrospinal fluid reveals a continuum of clonally related antibody-secreting cells that are predominantly plasma blasts. J Neuroimmunol. 2007;192(1-2):226–34. doi: 10.1016/j.jneuroim.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 106.Wrammert J, K D, Li GM, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med. doi: 10.1084/jem.20101352. 20111540-9538 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yanase K, Smith RM, Puccetti A, Jarett L, Madaio MP. Receptor-mediated cellular entry of nuclear localizing anti-DNA antibodies via myosin 1. J Clin Invest. 1997;100(1):25–31. doi: 10.1172/JCI119517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zar JH. Biostatistical Analysis. Upper Saddle River, NJ: Prentice Hall Inc; 2010. [Google Scholar]
- 109.Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e110. doi: 10.1212/NXI.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang J, Jacobi AM, Wang T, Berlin R, Volpe BT, Diamond B. Polyreactive autoantibodies in systemic lupus erythematosus have pathogenic potential. J Autoimmun. 2009;33(3-4):270–4. doi: 10.1016/j.jaut.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. (a.) Usage of specific heavy chain V gene segments in patient repertoires for each cell population. CD19+ means total B cells, CD19+CD27+means memory B cells, and CD19+CD27hi means plasmablasts. Black bars designate healthy donor samples, dark gray bars designate CIS-PTM memory B cells, light gray bars designate CIS-PTM plasmablasts, white bars designate NMO plasmablasts, and striped bars designate flu-responding plasmablasts. (b.) V gene family representation in the patient repertoires for each cell population. (c.) J gene segment representation in patient repertoires for each cell population. (d.) The average CDR3 length for all heavy chain sequences per patient in each patient group. (e.) The average charge of heavy chain CDR3s as calculated by the net number of positive and negative amino acids per sequence. (f.) The abundance of antibody isotypes found in either plasmablasts or memory B cells for CIS-PTM and NMO patients.
Supplemental Fig. 2. (a.) Usage of specific kappa chain V gene segments in patient repertoires. CD19+ means total B cells, CD19+CD27+means memory B cells, and CD19+CD27hi means plasmablasts. Black bars designate healthy donor samples, dark gray bars designate CIS-PTM memory B cells, light gray bars designate CIS-PTM plasmablasts, white bars designate NMO plasmablasts, and striped bars designate flu-responding plasmablasts. (b.) Usage of specific lambda chain V gene segments in patient repertoires in NMO and CIS-PTM patient cell populations. (c.) Kappa family representation in patient repertoires for each cell population. (d.) Light chain J gene segment distribution in patient repertoires. (e.) The average CDR3 length for the light chain sequences in patient repertoires. (f.) The average CDR3 charge of the light chain as calculated by the net number of positive and negative amino acids.
Supplemental Fig. 3. (a.) Human brain lysate ELISA results grouped by patient. Dashed line represents the same cutoff used for positive binding. (b.) Summary of human brain ELISA results grouped by patient type. Half of the CIS-PTM plasmablasts rhAbs (51%) and one third of the NMO plasmablast rhAbs (33%) were reactive to human brain lysate, demonstrating even more brain reactive rhAbs in this assay. (c.) Kidney lysate ELISA results grouped by patient. Dashed line represents the same cutoff used for positive binding in the mouse brain lysate ELISA. (d.) Summary of kidney lysate ELISA results grouped by patient type. A similar proportion of CIS-PTM plasmablasts rhAbs (13%) and NMO plasmablast rhAbs (11%) were reactive to kidney lysate. None of the healthy donor rhAbs demonstrated binding to kidney lysate.
Supplemental Fig. 4. (a.) H1N1 influenza ELISA results grouped by patient. Dashed line represents the cutoff used to define positive binding. (b.) Summary of H1N1 influenza ELISA results grouped by patient type. A similar proportion of CIS-PTM plasmablasts rhAbs (17%) and NMO plasmablast rhAbs (13%) were reactive to H1N1 native whole viral antigen. None of the healthy donor rhAbs bound strongly to the virus. (c.) Ovalbumin ELISA results grouped by patient. Dashed line represents the cutoff used to define positive binding. (d.) Summary of ovalbumin ELISA results grouped by patient type. Some of the CIS-PTM plasmablasts rhAbs (14%) but none of the NMO plasmablast rhAbs bound to this irrelevant antigen.
Supplemental Fig. 5. (a.) SH-Sy5y (human neuroblastoma) lysate ELISA results grouped by patient. Dashed line represents the cutoff used to define positive binding. (b.) Summary of SH-Sy5y ELISA results grouped by patient type. A similar proportion of CIS-PTM plasmablasts rhAbs (32%) and NMO plasmablast rhAbs (33%) were reactive to SH-Sy5y lysate, demonstrating binding of many of the rhAbs to neuronal antigens. None of the healthy donor rhAbs demonstrated binding to SH-Sy5y lysate.
Supplemental Fig. 6. 20× images displaying rhAb reactivity to post-stroke mouse corpus callosum. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to unidentified glial cells in this white matter tissue.
Supplemental Fig. 7. 20× images displaying rhAb reactivity to post-stroke mouse cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to neurons, as the rhAb signal overlapped with NeuN signal in this gray matter tissue.
Supplemental Fig. 8. 20× images displaying rhAb reactivity to EAE mouse corpus callosum. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to unidentified glial cells in this white matter tissue.
Supplemental Fig. 9. 20× images displaying rhAb reactivity to EAE mouse cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to neurons, as the rhAb signal overlapped with NeuN signal in this gray matter tissue.
Supplemental Fig. 10. 20× images displaying rhAb reactivity to healthy mouse corpus callosum. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to unidentified glial cells in this white matter tissue.
Supplemental Fig. 11. 20× images displaying rhAb reactivity to healthy mouse cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and NeuN is shown in red to mark neurons. Most of the rhAbs demonstrated binding to neurons, as the rhAb signal overlapped with NeuN signal in this gray matter tissue.
Supplemental Fig. 12. 20× images displaying rhAb reactivity to healthy and EAE mouse corpus callosum (CC) and cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and GFAP is shown in red to mark astrocytes. All of these rhAbs demonstrated binding to astrocytes, as the rhAb signal overlapped with GFAP signal in both gray and white matter tissue.
Supplemental Fig. 13. 20× images displaying rhAb reactivity to healthy and EAE mouse corpus callosum (CC) and cortex. In each image the scale bar indicates 20 μm, DAPI nuclear stain is shown in blue, rhAb is shown in green, and PDGFR is shown in red to mark oligodendrocytes. Most of the rhAbs did not bind to oligodendrocytes, as the rhAb signal did not overlap with PDGFR signal. However CIS68 did occasionally overlap with PDGFR in either the gray or white matter tissue (designated by yellow arrows).
Supplemental Fig. 14. (a.) Summary of all the CIS and NMO rhAbs tested for binding to mouse brain tissue. (b.) Lambda scans were performed to measure the signal intensity of staining observed and is displayed here as a ratio over background signal. Ratio values higher than 1.86 were considered to be true positive signal (ratio value for negative control rhAb B1). N designates areas where signal was not measured. (c.) Orthogonal views of example rhAbs on healthy mouse brain tissue. Scale bar indicates 10 μm, the leftmost column is an image of each rhAb with overlaid rhAb (green), NeuN (red) and DAPI (blue) staining with crosshairs to show the plane viewed beneath and to the right of the overlay. The column to the right of this is the same image without the crosshairs. The column to the right of this shows the same image with only the rhAb staining. The rightmost column shows the same image with only DAPI staining.
Supplemental Fig. 15. Enumeration of positively stained cortical cells from healthy mouse brain. (a.) Representative overlay image of NeuN and rhAb for enumeration with ROIs defined by faint yellow line. (b.) Percent of NeuN single positive (red), rhAb single positive (green), and double positive cells in the cortex. (c.) Histograms of maximal intensity in the red (NeuN) or green (rhAb) channel for ROIs with thresholds set for positive binding shown by the dashed lines in each graph. (d.) Table with the percent of ROIs in each cortical image stained with both NeuN and rhAb (red+green), only NeuN (red), or only the rhAb (green). Final column is the average percentage of ROIs in each category for all images measured for a particular rhAb.
Supplemental Fig. 16. Most of the remaining images of CIS-PTM rhAbs binding to SH-Sy5y cells by ICC. Binding of the rhAbs is displayed in green while the nuclear stain DAPI is shown in blue. The scale bar in each indicates 20 μm.
Supplemental Fig. 17. Remaining images of CIS-PTM, NMO and HD rhAbs binding to SH-Sy5y cells by ICC, plus controls. Binding of the rhAbs is displayed in green while the nuclear stain DAPI is shown in blue. The scale bar in each indicates 20 μm. R1, R2, and CSF10 served as additional negative and positive controls, as published in our previous study [57].
Supplemental Fig. 18. (a.) All images of rhAb binding to Hep-2 cells by ICC. Counterstain EvoBlue is shown in red while rhAb staining is shown in green (overlay is yellow). (b.) Summary of rhAb binding to Hep-2 cells. None of the healthy donor rhAbs bound to Hep-2 cells, but a small proportion of the CIS-PTM and NMO rhAbs did.
Supplemental Fig. 19. Western blots with cellular fractions from SH-Sy5y and Hep-2 cells and human brain lysate blotted with select rhAbs. Lanes from left to right are: (1) protein ladder, (2) nuclear and extracellular membrane (M+N) fraction of SH-Sy5y, (3) cytosolic (C) fraction of SH-Sy5y, (4) nuclear and extracellular membrane (M+N) fraction of Hep-2, (5) cytosolic (C) fraction of Hep-2, and (6) total human brain lysate. To demonstrate clear separation of cytosol from the nuclear/membrane cellular fraction, commercial antibodies to Histone H3 (H3, 15 kDa) and Heat Shock Protein 90 (Hsp90, 90 kDa) were used. Histone H3 was only detected in the nuclear fractions and Hsp90 was only detected in the cytosolic fractions, demonstrating clean separation of these cellular compartments. A commercial antibody to Tubulin was used to demonstrate that protein is loaded into each lane.
Supplemental Fig. 20. Binding of antibodies to AQP4-transfected HEK293 cells. Antibody binding is displayed in red while EGFP labeled AQP4 is displayed in green. Control serum from a strongly positive patient, a weakly positive patient and a negative patient are shown to compare to the two rhAbs cloned from the CSF of a CIS-PTM patient that later converted to NMO (R1 and R2). None of the NMO peripheral plasmablast rhAbs bound AQP4 in this assay (data not shown). Images are captured at 100× and show one to three representative stained cells.
Supplemental Fig. 21. Flow cytometry testing of rhAb binding to SH-Sy5y. Histograms depict rhAb staining in black outline without fill overlaid with a no primary antibody control in gray. (a.) Histograms of rhAbs binding to intracellular antigens of SH-Sy5y cells by flow cytometry. (b.) Histograms of rhAbs binding to intracellular and extracellular antigens of SH-Sy5y cells by flow cytometry. (c.) Geometric mean fluorescence intensity (gMFI) of rhAbs binding to intracellular and extracellular antigens of SH-Sy5y by flow cytometry.
Supplemental Table 1. All CIS-PTM and NMO rhAbs cloned for this study are listed, grouped by patient. Displayed is the heavy chain V gene segment (VH), heavy chain J gene segment (JH), light chain V gene segment (VL), light chain J gene segment (JL), number of mutations in the heavy chain (HC Mut), and antibody isotype for each sequence. The result of each test performed with the rhAb follows and is listed as positive (+), negative (-), with the cell type recognized (N for neuron, G for glial cell), or pattern of recognition (N for nuclear, C for cytoplasmic, I for intracellular, E for extracellular). VH4+ rhAbs are listed in bold font, VH3+ rhAbs are listed in regular font, rhAbs utilizing kappa light chains are not highlighted, and rhAbs utilizing lambda light chains are highlighted in gray.