

Abstract
Genetic models for studying localized cell suicide that halt the spread of pathogen infection and immune response activation in plants include Arabidopsis accelerated-cell-death 11 mutant (acd11). In this mutant, sphingolipid homeostasis is disrupted via depletion of ACD11, a lipid transfer protein that is specific for ceramide 1-phosphate (C1P) and phyto-C1P. The C1P binding site in ACD11 and in human ceramide-1-phosphate transfer protein (CPTP) is surrounded by cationic residues. Here, we investigated the functional regulation of ACD11 and CPTP by anionic phosphoglycerides and found that 1-palmitoyl-2-oleoyl-phosphatidic acid or 1-palmitoyl-2-oleoyl-phosphatidylglycerol (≤15 mol %) in C1P source vesicles depressed C1P intermembrane transfer. By contrast, replacement with 1-palmitoyl-2-oleoyl-phosphatidylserine stimulated C1P transfer by ACD11 and CPTP. Notably, “soluble” phosphatidylserine (dihexanoyl-phosphatidylserine) failed to stimulate C1P transfer. Also, none of the anionic phosphoglycerides affected transfer action by human glycolipid lipid transfer protein (GLTP), which is glycolipid-specific and has few cationic residues near its glycolipid binding site. These findings provide the first evidence for a potential phosphoglyceride headgroup-specific regulatory interaction site(s) existing on the surface of any GLTP-fold and delineate new differences between GLTP superfamily members that are specific for C1P versus glycolipid.
Keywords: Arabidopsis thaliana, lipid trafficking, lipid-protein interaction, membrane biophysics, phosphatidic acid, phosphatidylglycerol, phosphatidylserine, protein-lipid interaction, sphingolipid
Introduction
Sphingolipids (SLs)4 regulate key physiological processes, including cell mitogenesis, growth, migration, and differentiation as well as stress-induced programmed cell death responses (autophagy and apoptosis) (1–4). Because SL synthesis occurs at distinct sites in cells, SL distribution and localization to various membrane organelles involves vesicle- and non-vesicule-mediated transport processes. When ceramide is initially glycosylated in the Golgi, nonvesicular transfer can occur via proteins characterized by a glycolipid transfer protein fold (GLTP-fold) (5–7). The GLTP-fold consists of multiple α-helices arranged in a two-layer “sandwich” topology to form a single sphingolipid binding site (8–12). A newly discovered family within the GLTP superfamily uses a modified GLTP-fold to selectively transfer ceramide 1-phosphate (C1P) rather than glycosphingolipids between membranes (12–14). Human C1P transfer protein (CPTP; 214 amino acids) is encoded by a three-exon transcript from the single-copy CPTP gene5 on chromosome 1 (locus 1p36.33). By contrast, human GLTP (209 amino acids) originates from a five-exon transcript from single-copy GLTP on chromosome 12 (locus 12q24.11) (15). The shared folding topology (but with different SL specificity generated from the limited sequence homologies of CPTP and GLTP) serves as a striking example of evolutionary convergence and emphasizes the structural premium placed on GLTP-fold conservation by eukaryotes.
CPTPs, like GLTPs, occur almost ubiquitously in eukaryotes but play key roles in controlling inflammation and programmed cell death processes (12–14). Arabidopsis thaliana contains a CPTP orthologue known as ACD11 that functions as a lipid transfer protein for C1P and phyto-C1P (13). The ACD11 name originates from the accelerated cell death (acd) phenotype observed upon disruption of the acd11 gene (16). In the Arabidopsis acd11 mutant, altered sphingolipid homeostasis manifests as moderately increased C1P and highly increased ceramide that help drive a programmed cell death response (13). Arabidopsis acd mutants provide genetic models for studying localized cell suicide that can halt the spread of pathogen infection and immune response activation in plants (2, 17, 18).
ACD11, CPTP, and GLTP are considered to be amphitropic proteins because their functionality involves translocation on/off membranes to bind and release the sphingolipid cargo (19–21). Amphitropic proteins often contain so-called lipid binding domains (LBDs) that bind specific phosphoglyceride headgroups within membranes to help target and tether to select cell membrane destinations (22–27). LBDs, such as the C1, C2, PH, PX, and FYVE domains, differ structurally from the GLTP-fold. In the case of ACD11, CPTP, and GLTP, the protein region surrounding the sphingolipid binding site contains differing numbers of tryptophans, tyrosines, lysines, and arginines, residues known to be concentrated in the membrane interaction regions of proteins (28–32). Clusters of positively charged Arg and Lys residues occur near the SL binding site in ACD11 and CPTP, but not in GLTP (12, 13). This raises the issue of whether the cationic residue clusters of ACD11 and CPTP might be topologically organized to engage specific phosphoglyceride headgroups during membrane interaction.
Herein, we report the discovery of stimulation of C1P transfer by ACD11 when phosphatidylcholine (PC) bilayer vesicles contain both phosphatidylserine (PS) and C1P. A PS stimulatory effect also was observed on human CPTP transfer of C1P. By contrast, slowdowns in the C1P transfer rate were observed in the presence of other anionic phosphoglycerides (phosphatidic acid, phosphatidylglycerol). Inclusion of PS had no effect on glycolipid transfer by human GLTP. Our findings suggest the presence of a potential PS-specific headgroup interaction site on the surface of C1P-specific ACD11 and CPTP.
Results
X-ray crystallography of ACD11 and CPTP previously revealed a sphingolipid headgroup recognition site containing a positively charged Arg/Lys triad that binds C1P in “transfer-viable” fashion (13, 14). Co-crystallization with other phosphate headgroup lipids showed altered non-transfer-viable interactions involving the same binding site. We initially determined whether fluorescence emission of the intrinsic Trp residues in ACD11 serves as an indicator of transfer-viable protein·lipid complex formation. The two Trp residues in ACD11 are located on the protein surface (Fig. 1a). Trp-145 in helix-6 is at a highly conserved location in the membrane interaction region of the GLTP-fold (12, 13) and is 16–19 Å from the bound C1P headgroup. Trp-206, which forms the C terminus, is closer to the bound C1P headgroup (7–9 Å) and is more favorably positioned for detecting environmentally induced fluorescence emission changes associated with C1P binding. Fig. 1b illustrates a typical Trp emission response for ACD11 upon titrating with C1P. The Trp emission λmax undergoes progressive blue-shifting (i.e. 347 to 342 nm), whereas the fluorescence intensity decreases in response to stepwise additions of N-octanoyl C1P dissolved in ethanol (titration increments <10 mol % total ACD11). A saturation response is eventually achieved, as λmax blue-shifts to ∼342 nm and the total Trp emission intensity declines by ∼20–30%. A similar but stronger response occurs during glycolipid uptake by GLTP and related orthologs that differ from ACD11 by having a Trp residue within the glycolipid binding site (33–35). Titration of ACD11 with sphingosine 1-phosphate or phosphatidic acid, which contain phosphate headgroups but are not transferred (13), induced no significant change in Trp emission λmax (Fig. 1, c and d). Other sphingolipids, such as ceramide, which is not transferred (13), elicited almost no change (Fig. 1e), whereas sphingomyelin, which is transferred slowly (36), elicited a weak response (Fig. 1f).
FIGURE 1.

C1P binding to ACD11 in solution. a, structure of ACD11 (13) showing the location of intrinsic Trp (W145 and W206) with respect to bound C1P (Protein Data Bank entry 4NTI). b, ACD11 Trp emission change induced by C1P uptake. N-Octanoyl-C1P was added stepwise (C1P step concentration = 0.08 μm in EtOH (1 μl)) to ACD11 (1 μm) stirring in buffer (sodium phosphate (pH 6.6) containing 150 mm NaCl) with 5-min incubation times between injections. The vertical arrow indicates response to increasing C1P concentration. Increments beyond the sixth addition (∼0.48 μm C1P) induce little change in Trp emission intensity or wavelength maximum (λmax blue shift), yielding a saturable binding response. c, same as b, but lipid titrant is sphingosine 1-phosphate; d, same as b, but lipid titrant is phosphatidic acid; e, same as b, but lipid titrant is ceramide; f, same as b, but lipid titrant is sphingomyelin.
Formation of the ACD11·C1P complex indicated by the intrinsic Trp emission changes was verified by electrospray ionization mass spectrometry (ESI-MS). Fig. 2a shows the raw spectra obtained by direct infusion of ACD11·C1P complex solution under nondenaturing conditions. Deconvolution reveals that the positive ions (+8 and +9) correspond to monomeric complex (ACD11 + N-octanoyl C1P; 23,186 Da) plus small amounts of monomeric, C1P-free ACD11 (22,681 Da) (Fig. 2b). Compared with ESI-MS analyses of human GLTP and HET-C2 fungal GLTP binding of monohexosylceramide (33, 34), ACD11 showed stronger complexation based on the energy needed to disrupt the complex. This could reflect involvement of the positively charged Arg/Lys triad in binding the negatively charged phosphate of the C1P headgroup. No complexation could be detected with sphingomyelin, which is transferred slowly by ACD11 (36).
FIGURE 2.

Surface electrostatics surrounding the sphingolipid liganding site in ACD11, CPTP, and GLTP and sphingolipid intermembrane transfer measurement using a FRET-based assay. a, ESI-MS analysis of ACD11·C1P complex. Direct infusion under nondenaturing conditions results in two main positive charge states (+8 and +9) for the ACD11·N-octanoyl C1P complex. Traces of apo-ACD11 also are evident for the +8 and +9 charge states. The m/z label (x axis) represents mass divided by charge number. b, the transformed spectra indicate molecular masses of 22,681 Da for apo-ACD11 and 23,186 Da for the ACD11·N-octanoyl C1P complex, confirming a single binding site for C1P. c, schematic for in vitro measurement of SL intermembrane transfer via FRET loss. In the SL source vesicle, the AV-SL fluorescence signal (energy donor; red dots) is low due to FRET involving perylenoyl-PC (energy acceptor; green dots; nontransferable lipid). Loss of FRET occurs when AV-SL is removed by protein (catalytic amount) and transferred to the excess (10-fold) POPC receiver vesicles, resulting in a time-dependent increase in AV-SL emission signal. d, spectral emission changes for AV-SL/Per-PC energy transfer pair showing FRET loss induced by transfer protein activity. AV-C1P and Per-PC are at 1 mol % in SL source vesicles. Black, red, and green traces, successive scans of SL source vesicles (2.5-min intervals) showing signal stability; blue, cyan, and magenta traces, successive scans (2.5-min intervals) after the addition of excess POPC receiver vesicles; other spectral traces show the time-dependent emission changes in AV and Per (2-min intervals) after injection of transfer protein (2 μg).
The X-ray structures for ACD11 and human CPTP (13, 14) also show the sphingolipid binding sites surrounded by positively charged residue clusters that are absent in human GLTP (supplemental Fig. 1, a–c). This raised the issue of whether anionic membrane phospholipids influence the function of ACD11 and CPTP more strongly than GLTP because the membrane interaction region of the GLTP-fold encompasses the sphingolipid headgroup recognition site (12). To evaluate this possibility, we tested the effect of the negatively charged phospholipids PG, PS, and PA on sphingolipid departure rates from 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) vesicles by ACD11, CPTP, and GLTP. A well established approach (Fig. 2c) based on loss of fluorescence resonance energy transfer (FRET) was used to obtain real-time kinetic insights into lipid intermembrane transfer (13, 14, 37–40). In the assay, fluorescent anthrylvinyl (AV)-sphingolipid (energy donor) and 3-perylenoyl (Per)-PC (energy acceptor) are both incorporated into POPC vesicles either lacking or containing negatively charged phosphoglyceride. Fig. 2d illustrates the FRET response observed by excitation of AV-sphingolipid (AV-SL) at 370 nm, resulting in minimal AV emission (400–450 nm) and strong Per-PC emission (460–560 nm). The addition of protein plus excess POPC receiver vesicles (containing no lipid fluorophores) triggers a sudden, time-dependent loss of FRET as AV-SL departs while the nontransferable Per-PC remains in the SL source vesicles, consistent with AV-SL intermembrane transfer. With ACD11 or CPTP, the resulting increase in AV-C1P emission (Fig. 2d) enables monitoring of the AV-SL intermembrane transfer rate. If no transfer protein is added, almost no increase in AV emission is observed, confirming very slow spontaneous migration of AV-C1P to POPC receiver vesicles (41–43). If POPC receiver vesicles are omitted and protein amounts are increased by 20–30-fold, then only slight increases in AV-SL emission are observed, showing that AV-C1P binding to ACD11 does not account for FRET loss (e.g. see Ref. 14). With catalytic protein amounts, the “shuttle-like” protein action keeps AV-C1P transferring from the outer surfaces of the SL source vesicles to the receiver vesicles until dynamic equilibrium is reached (∼10 min).
The in vitro assay design is intended to reflect certain aspects of the physiological situation. In mammals, C1P is initially produced by ceramide kinase at select sites, such as the cytosolic face of the trans-Golgi, and is then transported to certain intracellular membranes. For this reason, C1P is localized initially only in the donor vesicles rather than in both the donor and acceptor vesicles. Anionic phosphoglycerides also often localize to intracellular membrane cytosolic faces (72), where they can encounter CPTP. Our experimental set-up takes these physiological factors into account.
C1P Transfer by ACD11 and CPTP Is Slowed by PA and PG but Accelerated by PS
Studies of the regulation of GLTP and its homologs by anionic phosphoglycerides previously focused on mammalian GLTP (38, 44, 45), the fungal HET-C2 GLTP (46), and the GLTPH domain of FAPP2 (35). With GLTP, including anionic phosphoglycerides, such as PA, PG, PS, and PI, in sphingolipid source vesicles impedes GalCer intermembrane transfer at low ionic strength due to enhanced membrane association by GLTP (i.e. Ka). However, adding salt to raise the ionic strength to physiologic or higher levels abrogates the enhanced membrane partitioning and restores GLTP transfer to rates similar to those observed when sphingolipid source vesicles lack negatively charged phosphoglycerides. The earlier studies involved GLTP purified from bovine brain (38). Here, ACD11 and CPTP functional regulation by anionic phosphoglycerides at physiologic ionic strength was studied using human GLTP expressed in Escherichia coli as a control (Fig. 3, a–i). AV-C1P kinetic transfer rates decreased nonlinearly in response to increasing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) concentrations ranging from 2 to 15 mol % in the sphingolipid source vesicles for both ACD11 (Fig. 3, a and d) and CPTP (Fig. 3, b and e). C1P transfer by both transfer proteins became maximally suppressed when POPA reached ∼4 mol %, whereas inclusion of only 2 mol % POPG resulted in C1P transfer reduction that remained low at the higher POPG molar fractions. Fig. 3, c and f, shows that increasing POPA and POPG concentrations (from 2 to 15 mol %) produced virtually no slowdown in the AV-GalCer transfer rate by human GLTP at physiologic ionic strength.
FIGURE 3.

Anionic phosphoglycerides exert differing effects on sphingolipid transfer by ACD11, CPTP, or GLTP. In each panel, traces represent AV-SL emission intensity measured at 415 nm as a function of time resulting from the loss of AV-SL/Per-PC FRET as AV-SL is transferred to POPC vesicles. Shown are POPA effects (0–15 mol %) (a–c) and POPG effects (0–15 mol %) (d–f) on ACD11, CPTP, and GLTP (2 μg), respectively. Per-PC (nontransferable) along with AV-C1P or AV-GalCer is present at 1 mol % in the SL source vesicles at time 0. g–i, POPS effects (0–15 mol %) on ACD11, CPTP, and GLTP (2 μg), respectively. Per-PC along with AV-C1P or AV-GalCer is present at 1 mol % in the SL source vesicles at time 0. j and k summarize the C1P transfer rate changes of ACD11 and CPTP induced by POPS, POPA, and POPG. The C1P transfer rates are expressed as pmol/min transferred from SL source to POPC vesicles as a function of different amounts of anionic phosphoglycerides (0–15 mol %) present in the SL source vesicles for ACD11 (a) and CPTP (b). Red, POPA; green, POPG; blue, POPS; gray, control. Error bars, S.D.
The responses of ACD11 and CPTP to 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS) sharply contrasted to those elicited by POPA and POPG. Increasing the POPS in sphingolipid source vesicles over the same range (2–15 mol %) strongly stimulated C1P transfer rates (Fig. 3, g and h). With ACD11, the transfer rate more than doubled when POPS concentrations reached 10 mol % (Fig. 3j). With CPTP, the rate increases were slightly less at equivalent POPS molar fractions (Fig. 3k). By contrast, with human GLTP, no increase in the AV-GalCer transfer rate was observed over the same POPS concentration range (2–15 mol %) in the sphingolipid source vesicles (Fig. 3i). Neither calcium nor other divalent cations were needed to elicit the POPS stimulatory effect. Notably, however, when the POPC receiver (acceptor) vesicles contained the POPS instead of the C1P source (donor) vesicles, the increase in C1P was not observed for either ACD11 or CPTP (Fig. 4, a and b). To explain our findings, we hypothesized that the membrane interaction regions of ACD11 and CPTP possess a PS-specific headgroup interaction site but with no associated hydrophobic pocket for accommodating the PS acyl chains. Such an arrangement would enable the PS acyl chains to remain embedded in the membrane while the PS headgroup acts as a selective tethering/activation site for ACD11 and CPTP. To test this idea, we assessed ACD11 and CPTP for their ability to transfer AV-PS between vesicles, but none was observed (Fig. 4c). This lack of PS transfer is noteworthy because other lipid transfer proteins exist that can transfer more than one lipid type. Yeast Sec 14 phosphatidylinositol transfer protein contains overlapping binding/transfer sites for either phosphatidylinositol or phosphatidylcholine (47). Also, certain oxysterol-binding protein family members that originally were thought to bind/transfer only sterol derivatives have since been shown to also bind/transfer of PS and phosphatidylinositol 4-phosphate (48–53).
FIGURE 4.
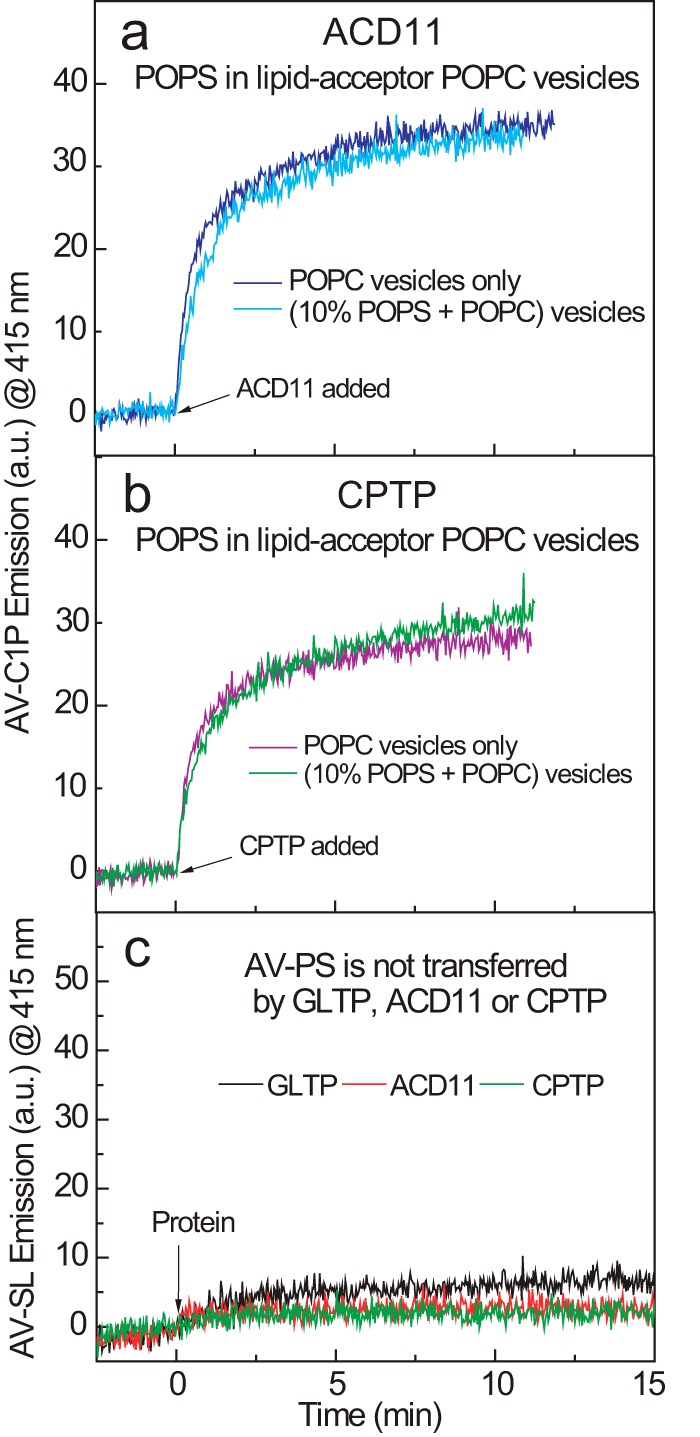
POPS in acceptor vesicles does not stimulate C1P transfer and POPS itself is not transferred. Traces for a and b represent AV-C1P emission intensity measured at 415 nm as a function of time resulting from the loss of AV-SL/Per-PC FRET as AV-SL is transferred to POPC vesicles. The presence of POPS (10 mol %) in POPC receiver (acceptor) vesicles (10-fold excess) does not stimulate AV-C1P transfer by ACD11 (a) or CPTP (b). c, traces represent AV-PS emission intensity measured at 415 nm as a function of time resulting from the loss of AV-PS/Per-PC FRET. The AV-PS transfer to POPC receiver (acceptor) vesicles (10-fold excess) by ACD11, CPTP, and GLTP is virtually nil.
Membrane Partitioning of ACD11
We initially evaluated using surface plasmon resonance at physiological ionic strength. After adsorbing vesicles of differing lipid composition to the lipophilic sensor chip, protein was introduced into the flow cell. Fig. 5a shows the enhanced partitioning to POPC/C1P (85:5) vesicles containing 10 mol % POPS or POPG by ACD11 compared with either POPC/C1P (95:5) or POPC vesicles. Using FRET, we further assessed ACD11 interaction with membranes of differing phosphoglyceride composition. Fig. 5 (b, c, and d) shows the spectral responses observed when ACD11 was titrated with increasing amounts of POPC/AV-PC (98:2) vesicles. Membrane interaction by ACD11 resulted in a nonlinear decline in the Trp emission intensity with increasing POPC/AV-PC vesicles in saturation-like fashion due to the spectral overlap of the intrinsic Trp emission of ACD11 (energy donor) and the AV excitation signal associated with PC (energy acceptor). Comparison of the interaction curves (Fig. 5d) shows that having POPS (10 mol %) in the PC membrane interface slightly enhanced FRET compared with the diminished FRET elicited by POPG. A similar outcome was observed when AV-C1P replaced the AC-PC in the POPC vesicles (data not shown).
FIGURE 5.

ACD11 partitioning to POPC vesicles containing POPS or POPG. a, surface plasmon resonance assessments of ACD11 adsorption and desorption membrane vesicles of differing lipid composition. POPC (red trace), POPC/C1P (95:5; green trace), POPC/C1P/POPS (80:5:15; blue trace), and POPC/C1P/POPG (80:5:15; orange trace) vesicles were prepared and adsorbed to a Sensor Chip L1 as described under “Experimental Procedures.” ACD11 injections of 46, 144, and 433 nmol into the 2 μl/min flow stream are indicated by red arrows; switches to buffer wash are shown by black arrows. b–d, FRET involving ACD11 intrinsic Trp (energy donor) and AV-PC (energy acceptor) in POPC vesicles enabled assessment of ACD11 partitioning to the membrane surface. ACD11 (0.5 μm) was titrated stepwise with POPC vesicles containing 2 mol % AV-PC to produce 0.13 μm incremental increases. b, control POPC vesicles lacking anionic phosphoglyceride; c, POPC vesicles containing 10 mol % POPS; d, POPC vesicles containing 10 mol % POPG; e, ACD11 partitioning curves to membrane vesicles consisting of POPC, POPC + POPS, or POPC + POPG.
No C1P Transfer Acceleration by “Soluble” PS
To test whether POPS stimulates C1P transfer by ACD11 and CPTP by helping to optimally orient the C1P binding site of CPTP during membrane interaction, we investigated whether soluble PS with short acyl chains (dihexanoyl-PS) stimulates C1P transfer by ACD11 and CPTP. We reasoned that little or no activation by soluble PS should occur because its high aqueous solubility (critical micelle concentration ∼0.7 mm; see Ref. 54) is expected to lower its ability to remain embedded in the POPC bilayer vesicle compared with POPS. We found that replacing POPS with di-1,2-dihexanoyl-sn-glycero-3-phospho-l-serine (di-6:0 PS) in the sphingolipid source vesicles decreased C1P transfer rates by ACD11 (Fig. 6a) and CPTP (Fig. 6b), whereas relatively little effect was exerted on GalCer transfer by GLTP (data not shown). We also tested whether buffer containing di-6:0 PS affects fluorescent C1P transfer from sphingolipid donor POPC vesicles containing no POPS. Fig. 6 (c and d) shows that the C1P transfer rate by ACD11 and CPTP was neither decreased nor stimulated by stepwise increases in soluble PS that reached 100 μm, which represents a 100–120-fold increase over the 10 mol % POPS in POPC vesicles. Similarly, di-6:0 PS produced no enhancement of AV-GalCer transfer by GLTP (data not shown). Altogether, our findings are consistent with weak embedding of soluble PS in POPC membranes, thus rendering it ineffective for enhancing the oriented protein-membrane interaction needed to stimulate C1P uptake by ACD11 and CPTP.
FIGURE 6.

Soluble PS effects on AV-C1P transfer by ACD11 and CPTP. Traces in each panel represent AV-SL emission intensity measured at 415 nm as a function of time resulting from loss of AV-SL/Per-PC FRET as AV-SL is transferred to lipid acceptor POPC vesicles. a and b, effect of dihexanoyl-PS (0 to 10 mol %) on AV-C1P transfer by ACD11 and CPTP (2 μg; ∼0.1 nmol), respectively, when the dihexanoyl-PS is included as a lipid-donor vesicle component. c and d, effect of dihexanoyl-PS on AV-C1P transfer by ACD11 and CPTP (2 μg; ∼0.1 nmol), respectively, when the dihexanoyl-PS is included in the buffer (2.5-ml volume) at the indicated concentrations. The critical micelle concentration of dihexanoyl-PS is ∼700 μm.
Discussion
Our investigation provides the first evidence for the existence of a phosphoglyceride regulatory interaction site(s) on the GLTP-fold surface of a GLTP superfamily member. The GLTP-fold is a structural motif that uses an all α-helical, two-layer “sandwich” topology to form a single, distinct sphingolipid binding site (8, 11–14). Our data indicate that PS stimulates the sphingolipid transfer activity of C1P-specific GLTP-folds (i.e. plant ACD11 and human CPTP). This stimulation by PS is a special feature of the C1P-specific GLTP-fold and is not duplicated by the glycolipid-specific GLTP-fold (i.e. human GLTP). Other negatively charged phosphoglycerides (e.g. PA and PG) exert an opposing effect on ACD11 and CPTP function and slow the C1P transfer rate from sphingolipid source vesicles at physiological ionic strength. These same phosphoglycerides show no inhibitory effect on recombinant, glycolipid-specific human GLTP, consistent with earlier studies of GLTP purified from bovine brain (38). The findings indicate that potential post-translational processing differences are not responsible for the lack of response by GLTP to anionic phosphoglycerides. Previously, the essential role of FAPP2 in glycosphingolipid biosynthesis in the Golgi was shown to involve its C-terminal, glycolipid-specific GLTP homology domain (5–7, 35). However, the regulatory features controlling the membrane interaction of FAPP2 focused only on its N-terminal pleckstrin homology domain that interacts with phosphatidylinositol 4-phosphate without attention to the GLTP domain.
We expected that negatively charged phosphoglycerides embedded in PC membranes would down-regulate ACD11 and CPTP transfer activity by increasing protein affinity for the SL source vesicles because of the high density of positively charged residues in the membrane interaction regions surrounding the C1P recognition centers (13, 14). Indeed, surface plasmon resonance indicates enhanced partitioning of ACD11 to POPC vesicles containing either POPG or POPS compared with POPC or POPC/C1P (95:5) vesicles. However, ACD11 Trp-to-AV-PC FRET shows a diminished response when POPC vesicles contain POPG but a slightly enhanced response by POPC vesicles containing POPS compared with POPC vesicles. In this regard, it worth remembering that the FRET efficiency is affected by both distance and orientation of the fluorophores. The two Trp residues in ACD11 are located adjacent to the C1P binding site. We conclude that the enhanced partitioning driven by POPG leaves ACD11 in an orientation on the membrane that slows C1P uptake/transfer. By contrast, a more favorable orientation that helps enhance C1P uptake/transfer (and increases FRET efficiency) is elicited by PS.
Regarding POPA, a possibility also considered for the diminished C1P transfer induced is competition for the C1P binding site that impairs C1P uptake. Both PA and C1P have phosphate as a headgroup. Our earlier X-ray studies revealed distorted interaction of PA (compared with C1P) with the CPTP sphingolipid binding site, resulting in no PA transfer (14). We detected no AV-PA transfer by ACD11 or CPTP in control experiments (not shown), in agreement with our earlier findings (13, 14). Weak competition by PA for the C1P binding site cannot be absolutely ruled out, given the slowdown in AV-C1P transfer evident at higher POPA concentrations.
When evaluating lipid transfer processes, factors pertaining to the membrane also need consideration besides the protein structural features and simple charge-charge effects. The membrane serves as a matrix for both the “substrate” C1P and the “effector” anionic phosphoglycerides. In bilayer vesicles, the C1P located in the bilayer outer leaflet represents the sphingolipid pool that is accessible to the transfer protein. Spontaneous C1P transbilayer migration is expected to be highly restricted due to the unfavorable energetics of moving the phosphate polar headgroup through the nonpolar hydrocarbon matrix. The interactions of GLTP homologs with membranes occur in a nonperturbing and weakly penetrating manner that leaves the sphingolipid pool in the inner leaflet of the vesicle bilayer undisturbed and inaccessible to protein (14, 37, 44, 55). However, a condition known to influence the initial sphingolipid transbilayer distributions and expected to impact the C1P pool size in the bilayer outer leaflet is the lipid composition during formation of high curvature, small unilamellar vesicles (e.g. see Refs. 56 and 57). When the sphingolipid source vesicles are POPC, the observed 35% C1P transfer equilibrium value suggests that 35% of C1P localizes in the vesicle outer leaflet. This value is similar to that of GalCer (39, 56). When anionic phosphoglycerides are added to the mix, their influence also needs consideration. Because PA shares the same small phosphate headgroup as C1P (58), minimal alteration of the C1P transbilayer distribution and the outer leaflet pool size is expected compared with C1P source vesicles lacking PA. By contrast, the PG headgroup volume is significantly larger than that of C1P and only slightly smaller than that of PC (59). Thus, the PG transbilayer distribution, like that of PC, tends to remain mass-distributed with ∼65% in the outer leaflet of the donor vesicle bilayer (60). Compared with C1P source vesicles lacking POPG, relatively more of the small headgroup C1P is expected to be inaccessible to ACD11 and CPTP by virtue of C1P localization to the inner leaflet of the vesicles. The sudden and dramatic drop in the AV-C1P rate produced by low POPG concentrations in the donor vesicles seems consistent with this reasoning. These factors that influence C1P pool size and transbilayer distribution are noteworthy because the sphingolipid concentration in the membrane surface is known to influence the sphingolipid intermembrane transfer rate and transfer protein partitioning affinity for the membrane surface (38, 39, 44, 61).
The surprising and large stimulation in C1P transfer activity by anionic PS raises the question of what makes PS such a special regulator. This led us to look beyond simple positive charge density in the surface region near the C1P binding sites of ACD11 and CPTP and consider the existence of a PS headgroup-specific surface binding site. We reasoned that such an interaction site might function analogously to LBDs. Such domains (e.g. C1, C2, PH, PX, and FYVE) exist as modular structural elements within multidomain proteins. LBDs bind only the phosphoglyceride headgroup because there is no hydrophobic pocket for enveloping the lipid aliphatic chains. This arrangement keeps the lipid chains embedded in the membrane while the protein interacts with the phosphoglyceride headgroup. LBDs help target and tether various peripheral amphitropic proteins to select intracellular membrane sites (22–27). Our experimental data support the idea of POPS functioning as a membrane tethering site that helps orient ACD11 and CPTP in ways that optimize function. It is clear that neither ACD11 nor CPTP can transfer PS between membranes, indicating that the interaction mode is not “transfer-compatible” for PS. By contrast, C1P interaction with CPTP and ACD11 involves recognition of the phosphate-amide headgroup region via a mechanism that triggers uptake of the nonpolar aliphatic chains into a hydrophobic pocket, thus shielding much of the sphingolipid cargo from the aqueous milieu (13, 14). The lack of PS transfer by ACD11 or CPTP is consistent with the phosphoserine polar headgroup being the interaction focal point while the PS acyl chains remain embedded in the membrane matrix.
Notably, replacement of POPS with soluble PS (dihexanoyl-PS) results in no stimulation of C1P transfer by ACD11 and CPTP. Also, simply including soluble PS in the buffer of the transfer reaction at up to 1000-fold excess compared with protein, rather than as a component of the SL source vesicles, fails to stimulate C1P transfer activity. Thus, stimulation of C1P transfer activity requires that the phosphoserine headgroup remain firmly associated with the C1P source membrane. It also is noteworthy that inclusion of POPS in the receiver membrane vesicles does not enhance the C1P transfer rate by ACD11 or CPTP. Taken together, the findings suggest the presence of a PS-specific site on the ACD11 and CPTP surface that targets the phosphoserine headgroup and imply that the PS site could help optimize orientation of ACD11 or CPTP for C1P uptake during membrane interaction. Thus, we predict that the membrane interaction region surrounding the C1P binding site findings contains a PS headgroup binding site, as depicted in Fig. 7. Future studies will be needed to test this idea by structural approaches involving X-ray diffraction, NMR, or modeling coupled with functional analyses via point mutation. It will also be interesting to learn whether the interaction of ACD11 or CPTP with long-chain PS embedded in C1P-containing membranes facilitates protein conformational changes to enhance C1P uptake.
FIGURE 7.
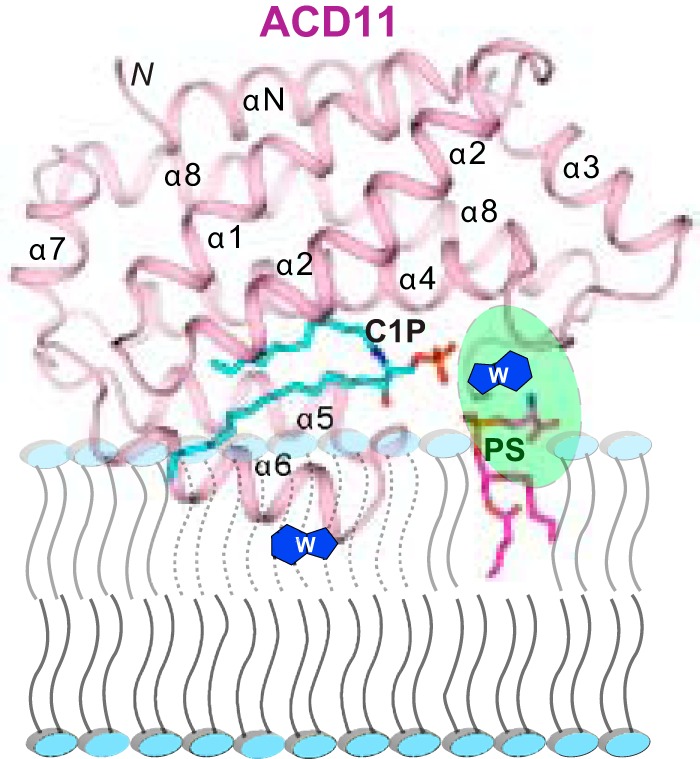
Model for putative PS-induced enhancement of ACD11. The model shows how the PS acyl chains remain embedded in the membrane while the PS headgroup (magenta) interacts with a putative surface site in ACD11 (ribbon representation) to help orient the C1P binding site to improve uptake of the C1P binding site and the membrane interaction region of ACD11.
From the physiological perspective, the discovery of PS-induced enhancement of C1P-specific GLTP homolog action provides insights into how ACD11 and CPTP might be targeted and site-specifically stimulated by certain membranes in plant and animal cells, respectively. PS plays important roles in cells. Exofacial PS exposure in activated blood platelets induces binding and activation of clotting factors, including factors V, VIII, and X and prothrombin (62). Intracellularly, PS occurs in the cytosol-facing surfaces of the plasma membrane, endosomes, and lysosomes, enabling docking and activation by important cytoplasmic signaling and fusogenic proteins with specific PS-binding domains (63, 64). Such proteins include the E3 ubiquitin-protein ligase NEDD4, various protein kinase C isoforms, several phospholipase C and D isoforms, phosphatidylinositol 3,4,5-trisphosphate phosphatase (PTEN), spectrin, and dysferlin (important in muscle repair) as well as certain synaptotagmin isoforms that participate in vesicular trafficking and fusion. ACD11, which regulates accelerated cell death in plants, and human CPTP, a regulator of pro-inflammatory eicosanoid production, can now be added to this growing list of proteins. It is noteworthy that CPTP intracellular enrichment occurs not only at the trans-Golgi but also on the cytoplasmic surfaces of endosomes and the plasma membrane, sites where PS also is enriched (14, 73).
Conclusions
C1P-specific lipid transfer proteins are known regulators of inflammation and programmed cell death, but mechanisms by which these proteins can be regulated have remained unknown until now. In the present study, significant stimulation of two C1P-specific lipid transfer proteins, at physiological ionic strength and in the absence of calcium, has been discovered when POPS, but not POPA or POPG, is embedded in POPC vesicles along with C1P. Notably, soluble PS that does not remain firmly embedded in the bilayer matrix produces no stimulatory effect on C1P transfer. By contrast, glycolipid-specific human GLTP activity is unaffected by all three anionic phosphoglycerides. The existence of a specific PS headgroup interaction site on the ACD11 surface is suggested where the long acyl chains of PS remain embedded in the bilayer during membrane interaction (Fig. 7). In this way, POPS could help orient and tether ACD11 and CPTP on the membrane surface to enhance interaction with C1P with PS-enriched membrane sites serving as ACD11 and CPTP activity “hot spots” in cells.
Experimental Procedures
Materials
POPC, POPS, 6:0 PS, POPA, and POPG were purchased from Avanti Polar Lipids and used without further purification. Lipids containing Per or AV fluorophore (e.g. Per-PC, AV-C1P, AV-GalCer, and AV-PS) were synthesized by lyso-lipid reacylation with ω-labeled 9-(3-perylenoyl)-nonanoyl or (11E)-12-(9-anthryl)-11-dodecenoyl chains and then purified (65–67).
Recombinant Protein Purification
Cloning, expression, and purification of ACD11, CPTP, and GLTP have been described previously (13, 14, 68–70). Briefly, Arabidopsis acd11 (NCBI NP_181016.1) and human CPTP (GenBankTM JN542538 and NP_077792.2) open reading frames were ligated into pET-SUMO vector (Invitrogen). Transformation of BL21 (DE3)-pLysS or -Star cells enabled expression of proteins N-terminally tagged with His6-SUMO (13, 14). Human GLTP ORF (GenBankTM AF209704) that was ligated into pET-30 Xa/LIC expression vector (Novagen) was used to transform BL21 cells for expression of GLTP N-terminally tagged with His6-S-peptide. Transformed cells were grown in Luria-Bertani medium at 37 °C for 6 h, induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside, and then incubated for 16–20 h at 15 °C. Affinity protein purification from soluble lysate was accomplished by nickel-nitrilotriacetic acid affinity chromatography. Cleavage of the N-terminal His6-SUMO tag was carried out with SUMO protease, Ulp1, overnight at 4 °C, whereas the His6-S-tag was removed from GLTP by factor Xa. Affinity repurification by nickel-nitrilotriacetic acid chromatography followed by FPLC gel filtration chromatography (HiLoad 16/60 Superdex-75 prep grade column; GE Healthcare), equilibrated in 25 mm Tris-HCl (pH 8.0) containing 100 mm NaCl and 1 mm DTT, yielded proteins with native sequences. Pooled peak fractions were concentrated by centrifugal concentrators (Vivaspin; 10 kDa cut-off). Protein purity was confirmed by SDS-PAGE (69, 71) before flash-freezing the pure proteins in buffer containing 15% glycerol and storing at −80 °C.
Sphingolipid Binding Specificity Assessment by Trp Emission Changes of ACD11
SL titrations were performed by adding aliquots (1-μl increments; C1P step concentration of 0.08 μm) dissolved in ethanol to ACD11 (1 μm, 2.5 ml) in sodium phosphate (pH 6.6) containing 150 mm NaCl with constant stirring as described previously (33). Measurements were performed using a SPEX FluoroLog-3 spectrofluorimeter (Horiba Scientific). Band passes for excitation and emission were 2 nm. The cuvette was temperature-controlled to ±0.1 °C (NesLab RTE-111, ThermoFisher). Emission spectra (305–500 nm) were corrected by subtraction of buffer and vesicle blanks. Inner filter effects were avoided by using low protein concentration (optical density at 295 nm <0.1). Excitation at 295 nm also eliminated fluorescence contributions from residues other than Trp.
Mass Spectrometry
The ACD11·C1P complex was prepared using the titration approach described above and concentrated by centrifugation (Vivaspin; 10 kDa cut-off). WT ACD11 and ACD11·C1P complex (10 μm) were analyzed using an Agilent 6210 LC/MS-TOF mass spectrometer in 5 mm NH4 acetate plus 5% methanol and infusing directly into the electrospray source, as described earlier for GLTP and HET-C2 (33, 34). Spectra were collected in positive mode over a 500–5000 m/z range using parameters optimized for complex stability (e.g. capillary, 3000 V; fragmentor, 300 V; skimmer, 60 V; octopole radio frequency, 300 V; octopole direct current, 32 V). Raw data were transformed into relative molecular masses using Agilent Time-of-Flight Protein BioConfirm software.
Protein-mediated Sphingolipid Intermembrane Transfer
To monitor sphingolipid intervesicular transfer, we used an established FRET approach (37). Sphingolipid source vesicles composed of POPC and containing 1 mol % AV-lipid (acyl chain ω-labeled with anthrylvinyl fluorophore, ((11E)-12-(9-anthryl)-11-dodecenoyl)) and 1 mol % 1-acyl-2-[9-(3-perylenoyl)-nonanoyl]-3-sn-glycero-3-phosphocholine (Per-PC) were prepared by rapid ethanol injection, as described previously (37, 38). When AV is excited at 370 nm, Per-PC (energy acceptor) emits because of energy transfer, whereas AV emission (energy donor) is minimal. Sonicated POPC vesicles that receive the transferred AV-sphingolipid are added to the stirred cuvette to establish the “no protein” baseline response in buffer consisting of 10 mm potassium phosphate (pH 6.6), 150 mm NaCl, and 0.2% EDTA. The addition of C1P transfer protein results in time-dependent FRET loss between AV and Per and results in an exponential increase in AV emission intensity as the protein transports AV-C1P away from the sphingolipid source vesicles and delivers to the POPC receiver vesicles present at 10-fold excess. The AV emission increase at ∼415 nm, relative to baseline fluorescence produced without protein, yields the AV-C1P transfer kinetics. The addition of Tween 20 detergent late in the kinetic time course provides the maximum AV intensity achievable upon “infinite” separation from 3-perylenoyl fluorophore. Maximum transfer, ΔF, represents the difference in emission intensity in the absence and presence of C1P late in the kinetic time course (>15 min). The initial lipid transfer rate, ν0, is obtained by nonlinear regression analyses (Origin version 7.0, OriginLab, Northampton, MA). The S.D. values are calculated at 95% confidence intervals, and R2 values are >0.96.
Protein Partitioning to Phosphoglyceride Membranes
Membrane partitioning by the sphingolipid transfer proteins was assessed by FRET and surface plasmon resonance. FRET from intrinsic Trp of ACD11 and CPTP (0.5 μm) to AV-PC (2 mol %) in POPC vesicles was measured before and after the addition of the AV-PC/POPC vesicles lacking or containing the indicated amounts of anionic phospholipid. Trp residues were selectively excited at 295 nm, and fluorescence spectral emission (304–525 nm) was monitored at 25 °C. Surface plasmon resonance was performed using a Biacore T200 system. Lipid vesicles of specified composition (0.5 mm), prepared by extrusion using 30 nm pore size membranes (33), were captured to a final surface density of 4000–7000 response units on a Sensor Chip L1 to establish the baseline prior to protein addition. Injections of increasing protein amounts or buffer were performed at 2 μl/min flow rates.
Author Contributions
X. Z. designed, performed, and analyzed all of the experiments and wrote the paper. Y.-G. G. and S. K. M. helped assess protein partitioning to membranes. D. K. S. designed and constructed ACD11 and CPTP expression vectors and performed the X-ray structure determinations essential for the experimental conception. I. A. B. and J. G. M. synthesized and purified the fluorescent lipids used in the experiments. L. M. B. and H. R. B. designed, performed, and analyzed the ESI-MS experiments. L. M. analyzed the structural data and contributed to the preparation of Figure 7. J. M., J. G. M., D. J. P., and R. E. B. analyzed and critically evaluated the results. R. E. B. conceived and designed the experiments, coordinated the study, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Helen Pike for expressing and purifying CPTP.
This work was supported in whole or part by NIGMS, National Institutes of Health (NIH), Grant GM45928 and NHLBI, NIH, Grants HL125353 and NCI CA121493; Russian Foundation for Basic Research Grant 015-04-07415; Danish Strategic Research Council Grant 09-067148; the Abby Rockefeller Mauze Trust; and the Maloris and Hormel Foundations. Portions of this work were presented at the 2015 American Chemical Society and 2015 American Society for Biochemistry and Molecular Biology Annual Meetings. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1.
GLTPD1 has been renamed CPTP by the HUGO Genome Nomenclature Committee.
- SL
- sphingolipid
- GLTP
- glycolipid lipid transfer protein
- C1P
- ceramide 1-phosphate
- LBD
- lipid binding domain
- PC
- phosphatidylcholine
- PS
- phosphatidylserine
- ESI
- electrospray ionization
- PG
- phosphatidylglycerol
- POPC
- 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine
- CPTP
- ceramide-1-phosphate transfer protein
- AV
- anthrylvinyl
- Per
- 3-perylenoyl
- GalCer
- galactosylceramide
- POPA
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol).
References
- 1. Hannun Y. A., and Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 2. Berkey R., Bendigeri D., and Xiao S. (2012) Sphingolipids and plant defense/disease: the “death” connection and beyond. Front. Plant Sci. 3, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hla T., and Dannenberg A. J. (2012) Sphingolipid signaling in metabolic disorders. Cell Metab. 16, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maceyka M., and Spiegel S. (2014) Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D'Angelo G., Polishchuk E., Di Tullio G., Santoro M., Di Campli A., Godi A., West G., Bielawski J., Chuang C.-C., van der Spoel A. C., Platt F. M., Hannun Y. A., Polishchuk R., Mattjus P., and De Matteis M. A. (2007) Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449, 62–67 [DOI] [PubMed] [Google Scholar]
- 6. D'Angelo G., Uemura T., Chuang C. C., Polishchuk E., Santoro M., Ohvo-Rekilä H., Sato T., Di Tullio G., Varriale A., D'Auria S., Daniele T., Capuani F., Johannes L., Mattjus P., Monti M., et al. (2013) Vesicular and non-vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 501, 116–120 [DOI] [PubMed] [Google Scholar]
- 7. Halter D., Neumann S., van Dijk S. M., Wolthoorn J., de Mazière A. M., Vieira O. V., Mattjus P., Klumperman J., van Meer G., and Sprong H. (2007) Pre- and post-Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J. Cell Biol. 179, 101–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malinina L., Malakhova M. L., Teplov A., Brown R. E., and Patel D. J. (2004) Structural basis for glycosphingolipid transfer specificity. Nature 430, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malinina L., Malakhova M. L., Kanack A. T., Lu M., Abagyan R., Brown R. E., and Patel D. J. (2006) The liganding mode of glycolipid transfer protein is controlled by glycosphingolipid structure. PLoS Biol. 4, e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Airenne T. T., Kidron H., Nymalm Y., Nylund M., West G., Mattjus P., and Salminen T. A. (2006) Structural evidence for adaptive ligand binding of glycolipid transfer protein. J. Mol. Biol. 355, 224–236 [DOI] [PubMed] [Google Scholar]
- 11. Brown R. E., and Mattjus P. (2007) Glycolipid transfer proteins. Biochim. Biophys. Acta 1771, 746–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Malinina L., Simanshu D. K., Zhai X., Samygina V. R., Kamlekar R., Kenoth R., Ochoa-Lizarralde B., Malakhova M. L., Molotkovsky J. G., Patel D. J., and Brown R. E. (2015) Sphingolipid transfer proteins defined by the GLTP-fold. Q. Rev. Biophys. 48, 281–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simanshu D. K., Zhai X., Munch D., Hofius D., Markham J. E., Bielawski J., Bielawska A., Malinina L., Molotkovsky J. G., Mundy J. W., Patel D. J., and Brown R. E. (2014) Arabidopsis accelerated-cell-death11 (ACD11) is a ceramide-1-phosphate transfer protein and intermediary regulator of phytoceramide levels. Cell Rep. 6, 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simanshu D. K., Kamlekar R. K., Wijesinghe D. S., Zou X., Zhai X., Mishra S. K., Molotkovsky J. G., Malinina L., Hinchcliffe E. H., Chalfant C. E., Brown R. E., and Patel D. J. (2013) Nonvesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoid production. Nature 500, 463–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou X., Chung T., Lin X., Malakhova M. L., Pike H. M., and Brown R. E. (2008) Human glycolipid transfer protein (GLTP) genes: organization, transcriptional status, and evolution. BMC Genomics 9, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brodersen P., Petersen M., Pike H. M., Olszak B., Skov S., Odum N., Jørgensen L. B., Brown R. E., and Mundy J. (2002) Knockout of Arabidopsis ACCELERATED-CELL-DEATH11 encoding a sphingosine transfer protein causes activation of programmed cell death and defense. Genes Dev. 16, 490–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang H., Yao N., Song J. T., Luo S., Lu H., and Greenberg J. T. (2003) Ceramides modulate programmed cell death in plants. Genes Dev. 17, 2636–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lam E. (2004) Controlled cell death, plant survival and development. Nat. Rev. Mol. Cell Biol. 5, 305–315 [DOI] [PubMed] [Google Scholar]
- 19. Burn P. (1988) Amphitropic proteins: a new class of membrane proteins. Trends Biochem. Sci. 13, 79–83 [DOI] [PubMed] [Google Scholar]
- 20. Johnson J. E., and Cornell R. B. (1999) Amphitropic proteins: regulation by reversible membrane interactions (review). Mol. Membr. Biol. 16, 217–235 [DOI] [PubMed] [Google Scholar]
- 21. Cornell R. B., and Taneva S. G. (2006) Amphipathic helices as mediators of the membrane interaction of amphitropic proteins, and as modulators of bilayer physical properties. Curr. Protein Pept. Sci. 7, 539–552 [DOI] [PubMed] [Google Scholar]
- 22. Cho W., and Stahelin R. V. (2005) Membrane protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34, 119–151 [DOI] [PubMed] [Google Scholar]
- 23. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell. Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 24. Stahelin R. V. (2009) Lipid binding domains: more than simple lipid effectors. J. Lipid Res. 50, S299–S304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moravcevic K., Oxley C. L., and Lemmon M. A. (2012) Conditional peripheral membrane proteins: facing up to limited specificity. Structure 20, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stahelin R. V., Scott J. L., and Frick C. T. (2014) Cellular and molecular interactions of phosphoinositides and peripheral proteins. Chem. Phys. Lipids 182, 3–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corbalan-Garcia S., and Gómez-Fernández J. C. (2014) Signaling through C2 domains: more than one lipid target. Biochim. Biophys. Acta 1838, 1536–1547 [DOI] [PubMed] [Google Scholar]
- 28. Wimley W. C., and White S. H. (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3, 842–848 [DOI] [PubMed] [Google Scholar]
- 29. Yau W.-M., Wimley W. C., Gawrisch K., and White S. H. (1998) The preference of tryptophan for membrane interfaces. Biochemistry 37, 14713–14718 [DOI] [PubMed] [Google Scholar]
- 30. Killian J. A., and von Heijne G. (2000) How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25, 429–434 [DOI] [PubMed] [Google Scholar]
- 31. MacCallum J. L., and Tieleman D. P. (2011) Hydrophobicity scales: a thermodynamic looking glass into lipid-protein interactions. Trends Biochem. Sci. 36, 653–662 [DOI] [PubMed] [Google Scholar]
- 32. Li L., Vorobyov I., and Allen T. W. (2013) The different interactions of lysine and arginine side chains with lipid membranes. J. Phys. Chem. B 117, 11906–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhai X., Malakhova M. L., Pike H. M., Benson L. M., Bergen H. R. 3rd, Sugár I. P., Malinina L., Patel D. J., and Brown R. E. (2009) Glycolipid acquisition by human glycolipid transfer protein dramatically alters intrinsic tryptophan fluorescence: insights into glycolipid binding affinity. J. Biol. Chem. 284, 13620–13628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kenoth R., Simanshu D. K., Kamlekar R.-K., Pike H. M., Molotkovsky J. G., Benson L. M., Bergen H. R. 3rd, Prendergast F. G., Malinina L., Venyaminov S. Y., Patel D. J., and Brown R. E. (2010) Structural determination and tryptophan fluorescence of heterokaryon incompatibility C2 protein (HET-C2), a fungal glycolipid transfer protein (GLTP), provide novel insights into glycolipid specificity and membrane interaction by the GLTP-fold. J. Biol. Chem. 285, 13066–13078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kamlekar R.-K., Simanshu D. K., Gao Y.-G., Kenoth R., Pike H. M., Prendergast F. G., Malinina L., Molotkovsky J. G., Venyaminov S. Y., Patel D. J., and Brown R. E. (2013) The glycolipid transfer protein (GLTP) domain of phosphoinositol 4-phosphate adaptor protein-2 (FAPP2): structure drives preference for simple neutral glycosphingolipids. Biochim. Biophys. Acta 1831, 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Petersen N. H. T., McKinney L. V., Pike H., Hofius D., Zakaria A., Brodersen P., Petersen M., Brown R. E., and Mundy J. (2008) Human GLTP and mutant forms of ACD11 suppress cell death in the Arabidopsis acd11 mutant. FEBS J. 275, 4378–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mattjus P., Molotkovsky J. G., Smaby J. M., and Brown R. E. (1999) A fluorescence resonance energy transfer approach for monitoring protein-mediated glycolipid transfer between vesicle membranes. Anal. Biochem. 268, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mattjus P., Pike H. M., Molotkovsky J. G., and Brown R. E. (2000) Charged membrane surfaces impede the protein-mediated transfer of glycosphingolipids between phospholipid bilayers. Biochemistry 39, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rao C. S., Lin X., Pike H. M., Molotkovsky J. G., and Brown R. E. (2004) Glycolipid transfer protein mediated transfer of glycosphingolipids between membranes: a model for action based on kinetic and thermodynamic analyses. Biochemistry 43, 13805–13815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Samygina V. R., Popov A. N., Cabo-Bilbao A., Ochoa-Lizarralde B., Goni-de-Cerio F., Zhai X., Molotkovsky J. G., Patel D. J., Brown R. E., and Malinina L. (2011) Enhance selectivity for sulfatide by engineered human glycolipid transfer protein. Structure 19, 1644–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown R. E. (1990) Spontaneous transfer of lipids between membranes. Subcell. Biochem. 16, 333–363 [DOI] [PubMed] [Google Scholar]
- 42. Brown R. E. (1992) Spontaneous lipid transfer between organized lipid assemblies. Biochim. Biophys. Acta 1113, 375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lev S. (2010) Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat. Rev. Mol. Cell Biol. 11, 739–750 [DOI] [PubMed] [Google Scholar]
- 44. Rao C. S., Chung T., Pike H. M., and Brown R. E. (2005) Glycolipid transfer protein interaction with bilayer vesicles: modulation by changing lipid composition. Biophys. J. 89, 4017–4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nylund M., Fortelius C., Palonen E. K., Molotkovsky J. G., and Mattjus P. (2007) Membrane curvature effects on glycolipid transfer activity. Langmuir 23, 11726–11733 [DOI] [PubMed] [Google Scholar]
- 46. Mattjus P., Turcq B., Pike H. M., Molotkovsky J. G., and Brown R. E. (2003) Glycolipid intermembrane transfer is accelerate by HET-C2, a filamentous fungus gene product involved in the cell-cell incompatibility response. Biochemistry 42, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schaaf G., Ortlund E. A., Tyeryar K. R., Mousley C. J., Ile K. E., Garrett T. A., Ren J., Woolls M. J., Raetz C. R. H., Redinbo M. R., and Bankaitis V. A. (2008) Functional anatomy of phospholipid binding and regulation of phosphoinositide homeostasis by proteins of the Sec14 superfamily. Mol. Cell 29, 191–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Saint-Jean M., Delfosse V., Douguet D., Chicanne G., Payrastre B., Bourguet W., Antonny B., and Drin G. (2011) Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J. Cell Biol. 195, 965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maeda K., Anand K., Chiapparino A., Kumar A., Poletto M., Kaksonen M., and Gavin A.-C. (2013) Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature 501, 257–261 [DOI] [PubMed] [Google Scholar]
- 50. Olkkonen V. M. (2013) OSBP-related proteins: liganding by glycerophospholipids opens new insight into their function. Molecules 18, 13666–13679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Olkkonen V. M., and Li S. (2013) Oxysterol-binding proteins: sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog. Lipid Res. 52, 529–538 [DOI] [PubMed] [Google Scholar]
- 52. Tong J., Yang H., Yang H., Eom S. H., and Im Y. J. (2013) Structure of Osh3 reveals a conserved mode of phosphoinositide binding in oxysterol-binding proteins. Structure 21, 1203–1213 [DOI] [PubMed] [Google Scholar]
- 53. Drin G. (2014) Topological regulation of lipid balance in cells. Annu. Rev. Biochem. 83, 51–77 [DOI] [PubMed] [Google Scholar]
- 54. Koppaka V., Wang J., Banerjee M., and Lentz B. R. (1996) Soluble phospholipids enhance factor Xa-catalyzed prothrombin activation in solution. Biochemistry 35, 7482–7491 [DOI] [PubMed] [Google Scholar]
- 55. Kamlekar R.-K., Gao Y., Kenoth R., Molotkovsky J. G., Prendergast F. G., Malinina L., Patel D. J., Wessels W. S., Venyaminov S. Y., and Brown R. E. (2010) Human GLTP: three distinct functions for the three tryptophans in a novel peripheral amphitropic fold. Biophys. J. 99, 2626–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mattjus P., Malewicz B., Valiyaveettil J. T., Baumann W. J., Bittman R., and Brown R. E. (2002) Sphingomyelin modulates the transbilayer distribution of galactosylceramide in phospholipid membranes. J. Biol. Chem. 277, 19476–19481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Malewicz B., Valiyaveettil J. T., Jacob K., Byun H.-S., Mattjus P., Baumann W. J., Bittman R., and Brown R. E. (2005) The 3-hydroxy group and 4,5-trans double bond of sphingomyelin are essential for modulation of galactosylceramide transmembrane asymmetry. Biophys. J. 88, 2670–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kooijman E. E., and Burger K. N. J. (2009) Biophysics and function of phosphatidic acid: a molecular perspective. Biochim. Biophys. Acta 1791, 881–888 [DOI] [PubMed] [Google Scholar]
- 59. Boscia A. L., Treece B. W., Mohammadyani D., Klein-Seetharaman J., Braun A. R., Wassenaar T. A., Klösgen B., and Tristram-Nagle S. (2014) X-ray structure, thermodynamics, elastic properties and MD simulations of cardiolipin/dimyristoylphosphatidylcholine mixed membranes. Chem. Phys. Lipids 178, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nordlund J. R., Schmidt C. F., and Thompson T. E. (1981) Transbilayer distribution in small unilamellar phosphatidylglycerol-phosphatidylcholine vesicles. Biochemistry 20, 6415–6420 [DOI] [PubMed] [Google Scholar]
- 61. Ohvo-Rekilä H., and Mattjus P. (2011) Monitoring glycolipid transfer protein activity and membrane interaction with the surface plasmon resonance technique. Biochim. Biophys. Acta 1808, 47–54 [DOI] [PubMed] [Google Scholar]
- 62. Leventis P. A., and Grinstein S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427 [DOI] [PubMed] [Google Scholar]
- 63. Yeung T., Gilbert G. E., Shi J., Silvius J., Kapus A., and Grinstein S. (2008) Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 [DOI] [PubMed] [Google Scholar]
- 64. Yeung T., Heit B., Dubuisson J.-F., Fairn G. D., Chiu B., Inman R., Kapus A., Swanson M., and Grinstein S. (2009) Contribution of phosphatidylserine to membrane surface charge and protein targeting during phagosome maturation. J. Cell Biol. 185, 917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bergelson L. D., Molotkovsky J. G., and Manevich Y. M. (1985) Lipid-specific fluorescent probes in studies of biological membranes. Chem. Phys. Lipids 37, 165–195 [DOI] [PubMed] [Google Scholar]
- 66. Molotkovsky J. G., Mikhalyov I. I., Imbs A. B., and Bergelson L. D. (1991) Synthesis and characterization of new fluorescent glycolipid probes: molecular organization of glycosphingolipids in mixed composition lipid bilayer. Chem. Phys. Lipids 58, 199–212 [Google Scholar]
- 67. Boldyrev I. A., Brown R. E., and Molotkovsky J. G. (2013) An expedient synthesis of fluorescent labeled ceramide-1-phosphate analogues. Russ. J. Bioorganic Chem. 39, 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lin X., Mattjus P., Pike H. M., Windebank A. J., and Brown R. E. (2000) Cloning and expression of glycolipid transfer protein from bovine and porcine brain. J. Biol. Chem. 275, 5104–5110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li X.-M., Malakhova M. L., Lin X., Pike H. M., Chung T., Molotkovsky J. G., and Brown R. E. (2004) Human glycolipid transfer protein: probing conformation using fluorescence spectroscopy, Biochemistry 43, 10285–10294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Malakhova M. L., Malinina L., Pike H. M., Kanack A. T., Patel D. J., and Brown R. E. (2005) Point mutational analysis of the liganding site in human glycolipid transfer protein. J. Biol. Chem. 280, 26312–26320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brown R. E., Jarvis K. L., and Hyland K. J. (1990) Purification and characterization of glycolipid transfer protein from bovine brain. Biochim. Biophys. Acta 1044, 77–83 [DOI] [PubMed] [Google Scholar]
- 72. Holthuis J. C. M., and Menon A. K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57 [DOI] [PubMed] [Google Scholar]
- 73. Hankins H. M., Baldridge R. D., Xu P., and Graham T. R. (2015) Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic 16, 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.