

Abstract
Polymorphism of amyloid-β (Aβ) fibrils, implying different fibril structures, may play important pathological roles in Alzheimer's disease (AD). Morphologies of Aβ fibrils were found to be sensitive to fibrillation conditions. Herein, the Ser8-phosphorylated Aβ (pAβ), which is assumed to specially associate with symptomatic AD, is reported to modify the morphology, biophysical properties, cellular toxicity, and structures of Aβ fibrils. Under the same fibrillation conditions, pAβ favors the formation of fibrils (Fpβ), which are different from the wild-type Aβ fibrils (Fβ). Both Fβ and Fpβ fibrils show single predominant morphologies. Compared with Fβ, Fpβ exhibits higher propagation efficiency and higher neuronal cell toxicity. The residue-specific structural differences between the Fβ- and Fpβ-seeded Aβ fibrils were identified using magic angle spin NMR. Our results suggest a potential regulatory mechanism of phosphorylation on Aβ fibril formation in AD and imply that the post-translationally modified Aβ, especially the phosphorylated Aβ, may be an important target for the diagnosis or treatment of AD at specific stages.
Keywords: Alzheimer's disease, amyloid, fibril, phosphorylation, solid state NMR, morphology, structure
Introduction
Amyloid fibrils are β-sheet-enriched fibrillar aggregates with misfolded polypeptides and proteins. The formation and deposition of these fibrils are related to a variety of neurodegenerative diseases, including Alzheimer's disease (AD),3 Parkinson's disease, and Huntington disease among others (1–4). Amyloid fibrils derived from the same primary sequences of polypeptides or proteins usually show distinct morphologies in vitro (5, 6), and the morphologies of fibrils are sensitive to a variety of fibrillation conditions such as temperature (7), agitation (8), salt concentrations (9), surfactant (10), and seeding effects (11). Recent evidence revealed that amyloids can spread through a prion-like mechanism where fibrils seem to play important roles (12–14). Different fibrils with their specific morphologies, reminiscent of prion-like strains, may cause distinct pathological phenotypes and link different structures to the variations in disease transmission and pathology (15–17). Furthermore, fragmentation of fibrils, which always produces new ends for self- or cross-seeded fibrillation, is of critical importance for infectious amyloids (18, 19).
Senile plaques consisting of fibrillar Aβ are considered one of the important hallmarks in AD (20). The 40-residue and 42-residue Aβ peptides (i.e. Aβ40 and Aβ42, respectively) are the two main fibrillar species. Recently, Aβ has also been reported to exhibit prion-like propagation properties (21, 22). Distinct strains of Aβ were discerned in Alzheimer's patients (23–26). Different amyloid propagation properties and structural profiles of Aβ40 and Aβ42 mimic distinct amyloid strains (10, 27). The phenotypes induced by exogenous injection of Aβ-containing brain extracts from Alzheimer's patients were dependent on both the hosts and the sources of agents, suggesting that polymorphic Aβ strains might result in varying biological activities (25, 28, 29). Molecular structures of Aβ fibrils derived from Alzheimer's patients with distinct clinical histories were also different (24). This underlines that the structural variations of Aβ fibrils may correlate with the variations of pathological phenotypes (24). In addition, compared with the brain-derived Aβ fibrils, the synthetic fibrils showed lower prion activities and different molecular structures (23, 24, 30), implying that some crucial factors in vivo might account for different fibrillar polymorphisms and pathological phenotypes.
Recently, post-translational modifications of Aβ, such as phosphorylation and pyroglutamation, occurring in vivo have been found to promote the progression of AD (31, 32). Among different types of post-translational modifications, the phosphorylation of proteins plays crucial roles in protein folding (33). Phosphorylation can alter the structures of a protein and modulate its activities (33). Using Trp-cage as a model protein, Kardos et al. (34) reported that phosphorylation could serve as a conformational switch to trigger the transition from native to amyloid state. Significantly, we and other groups have demonstrated that phosphorylation is involved in the formation of low barrier hydrogen bond (35) and turn conformations (36) as well as the destabilization of β-hairpin structure (37). Furthermore, we have also reported that phosphorylation may modulate the fibrillation process of amyloid proteins, such as Tau and α-synuclein (38–40). Herein, we describe a novel regulatory function of phosphorylation at Ser8 on morphology, biophysical properties, cellular toxicity, and structures of the Aβ40 fibrils.
It has been shown that phosphorylation at Ser8 in Aβ has important roles in late onset sporadic AD (31, 41, 42). Phosphorylation at Ser8 was found in the brains of Alzheimer's patients in a hierarchical sequence and was specially suggested to be associated with symptomatic AD (43). Phosphorylation at Ser8 was modulated by protein kinase A (44). Additionally, this site-specific phosphorylation was known to accelerate the nucleation-dependent fibrillation of Aβ and to enhance the Aβ-mediated amyloid toxicity (44). Attenuation of Aβ clearance via insulin-degrading enzyme and angiotensin-converting enzyme induced by this phosphorylation was also reported (45). Furthermore, the phosphorylation at Ser8 could elevate numbers of strong hydrogen bonds in the N terminus of Aβ and increase the stability of the resulting pathogenic fibrils. The latter represents one of the crucial factors for disease progression in the brain (46). Despite all the functional importance, it is not clear whether this residue-specific phosphorylation can modify the morphologies and structures of Aβ fibrils, which are closely related to transmission and progression of AD.
Our present study demonstrates that Aβ40 with phosphorylation at Ser8 (pAβ) leads to a cross-β fibril (Fpβ) morphologically and structurally different from the wild-type Aβ fibril (Fβ). The Fpβ and Fβ show distinct single predominant morphologies and structures and have different biophysical properties and cellular toxicities. Residue-specific structural variations between the Fpβ- and Fβ-seeded 40-residue Aβ fibrils were determined using magic angle spinning (MAS) NMR spectroscopy. Our results show the effects of post-translational modifications on the polymorphism of Aβ fibrils and their potential correlations to the propagation properties and cellular toxicities of the fibrils. Phenomena of strains related to distinct amyloid polymorphism are the subject of extensive interest because of their correlation with different pathological phenotypes (10, 25, 26, 47, 48). The current study also strongly suggests the potential relationship between post-translational modifications and strain formation in vivo.
Results
The Fpβ Fibrils Have Different Morphologies Compared with the Fβ Fibrils
The Fβ and Fpβ fibrils were prepared using the same conditions with the same initial peptide concentration at 40 μm. Circular dichroism (CD) and Fourier transform infrared (FT-IR) spectroscopy were first used to study the structures of both monomers and fibrils. Fig. 1, A and B, shows that CD and FT-IR spectra of the unaggregated monomers of the wild-type Aβ40 and pAβ were very similar. The CD spectra (Fig. 1A) suggested that both monomeric Aβ40 and pAβ had mainly random coil structures. Furthermore, the FT-IR spectra (Fig. 1B) for both monomeric Aβ40 and pAβ were symmetric from 1580 to 1740 cm−1 with maximum peaks at about 1653 cm−1. Interestingly, we observed obvious differences in CD spectroscopy between the Fpβ and Fβ fibrils. The CD spectra of Fpβ and Fβ had minimum peaks at about 216 and 222 nm, respectively (Fig. 1A), indicating different β-sheet structures in their fibril cores (7, 49, 50). Compared to the Fpβ fibrils, the fibrillation of Fβ was accompanied by a reduction of CD spectral intensity presumably due to the increased scattering (50). Transmission electron microscopy (TEM) showed that the morphologies of both Fpβ and Fβ fibrils were homogenous. The Fpβ fibrils were mainly twisted cylindrical structures (Fig. 1D), which were distinct from the flat ribbon-like morphology for Fβ fibrils (Fig. 1C). Additionally, the FT-IR spectra (Fig. 1B) and X-ray diffraction (XRD) diffraction patterns (Fig. 1, E and F) revealed that both Fpβ and Fβ fibrils were rich in cross-β structures. The FT-IR spectra of both Fβ and Fpβ fibrils showed main peaks at ∼ 1628 nm−1 that were also consistent with previous studies on Aβ fibrils (51–53). However, two spectra were distinct in the loop or turn regions between 1640 and 1680 cm−1, indicating certain levels of structural variations between the two fibrils. The XRD pattern showed reflections at 4.7 and 10 Å for the inter- and intramolecular spacing, respectively, which characterized the cross-β structures for both Fβ and Fpβ fibrils (54). Taken together, we found that phosphorylation of Aβ40 at Ser8 could modify the morphologies of amyloid fibrils. Recent evidence has shown that different microenvironments modulate the free energy landscapes for Aβ folding into different morphological states (8, 25, 50). It is possible that the side-specific phosphorylation may alter the free energy barrier for the folding of Aβ40 and lead to a different morphology for the pAβ.
FIGURE 1.
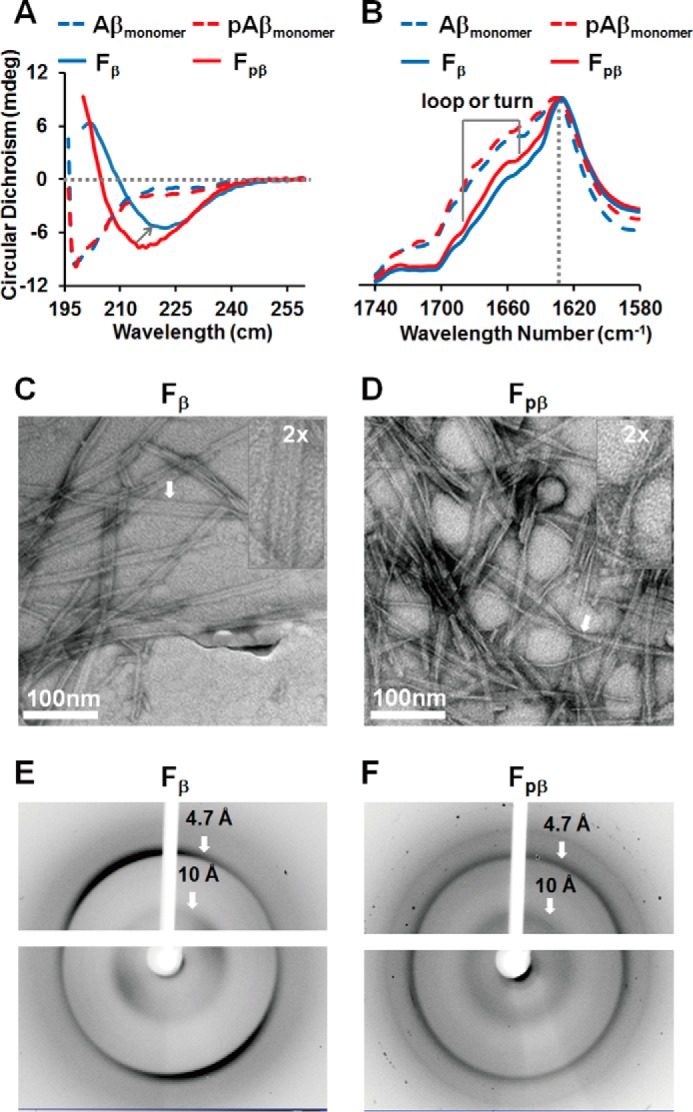
Phosphorylation induces pAβ misfolding into amyloid fibril morphology (Fpβ) distinct from wild-type Aβ (Fβ). A, CD spectra of unaggregated Aβ monomers (blue dashed line), pAβ monomers (red dashed line), Fβ fibrils (blue solid line), and Fpβ fibrils (red solid line). Both unaggregated Aβ and pAβ monomers mainly showed similar random coil structures (198 nm). Fβ and Fpβ fibrils showed β-sheet-enriched structures with a minimum peak at about 222 and 216 nm, respectively. B, FT-IR absorbance spectra of unaggregated Aβ monomers (blue dashed line), pAβ monomers (red dashed line), Fβ fibrils (blue solid line), and Fpβ fibrils (red solid line). Both unaggregated Aβ and pAβ monomers showed a symmetrical FT-IR spectrum with a maximum absorbance peak at 1653 cm−1.The Fβ and Fpβ fibrils showed the same maximum absorbance peak at 1628 cm−1 but small variations between 1640 and 1680 cm−1 that imply certain structural differences. C and D, TEM images of Fβ fibrils (C) and Fpβ fibrils (D). Fpβ exhibited a twisted cylindrical structure, whereas Fβ displayed a ribbon-like structure. E and F, XRD images of Fβ fibrils (E) and Fpβ fibrils (F). The reflections at 4.7 and 10 Å, respectively, indicated that both Fpβ and Fβ fibrils were rich in cross-β structures. mdeg, millidegrees.
Phosphorylation Plays Crucial Roles in Fpβ Formation
Recently, Rezaei-Ghaleh et al. (46, 55) have shown that phosphorylation at Ser8 could impel the undergoing changes in local conformational dynamics of Aβ40, induce the variations of N-terminal exposure of Aβ aggregates, and increase the fibril stability by promoting the formation of strong hydrogen bonds directly. We propose that different conformations of Fpβ fibrils may also be induced directly by phosphorylation. The thioflavin T (ThT) fluorescence kinetics measurement was first applied to determine the fibrillation rate of pAβ. ThT fluorescence kinetics measurements of 40 μm monomeric Aβ40 and pAβ were conducted at 37 °C with continuous shaking. Both Aβ40 and pAβ exhibited typical nucleation-dependent sigmoidal curves (Fig. 2). Similar to previous results from Kumar et al. (44), phosphorylation at Ser8 accelerated the fibrillation process (Fig. 2). The lag times of fibrillation for 40 μm pAβ and Aβ40 were 22 and 38 min, respectively. Furthermore, pAβ seemed to decrease the aggregation half-time (39.1 ± 9.0 min) compared with Aβ40 (57.6 ± 13.2 min). These results suggested that phosphorylation might either shorten or alter the fibrillation process of Aβ.
FIGURE 2.
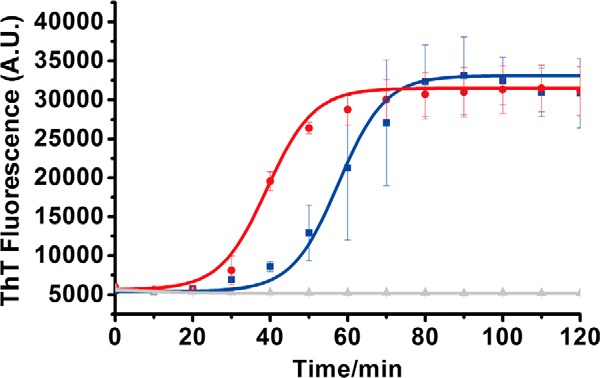
ThT fluorescence kinetic curves of pAβ and Aβ. For ThT fluorescence kinetics measurements, both 40 μm pAβ (red) and Aβ (blue) were used and replicated three times with 20 μm ThT in the presence of 20 mm Tris·HCl buffer, 150 mm NaCl, pH 7.4, with continuous shaking. The curves (solid curves) were fitted using a sigmoidal equation (50). Error bars (S.D.) were calculated from three independent experiments. A.U., arbitrary units.
Solid-state 31P NMR spectra for the Fpβ fibrils and the monomeric pAβ peptides are shown in Fig. 3. Both spectra were recorded with similar quantities of samples (∼8 mg) and the same hydration level (∼1 μl/mg). The direct polarization 31P spectroscopy allowed quantitative comparison of the peak intensities between the two samples. The fact that the 31P intensity was greatly enhanced in the Fpβ sample suggested that the phosphate group at Ser8 was located in an ordered environment upon fibrillation compared with the monomeric peptides. Furthermore, different from the monomeric pAβ sample, which might form amorphous aggregates because of the rehydration, the fibrillar Fpβ 31P spectrum showed only one predominant peak, suggesting a single local conformation for the side chain of Ser8.
FIGURE 3.
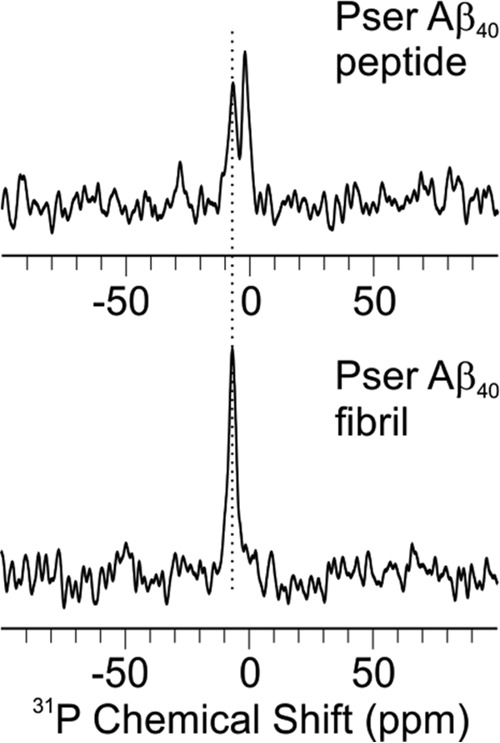
31P MAS NMR spectra for pAβ peptides (top) and fibrils (bottom). Both spectra were acquired with 1024-scan signal averaging and processed with 10-Hz Gaussian line broadening.
To further uncover the roles of phosphorylation in Fpβ fibrils, the alkaline phosphatase dephosphorylation assay was applied (56). We utilized calf intestinal alkaline phosphatase (CIP) purified from calf intestinal mucosa that can remove phosphate groups from phosphorylated species (57). Previous work has suggested the involvement of phosphorylated Ser8 in specific hydrogen bonding to the hydrophobic core of fibrils (46, 55). In these cases, the phosphate group might be restricted, and efficiency of dephosphorylation might decrease. In our experimental design, the monomeric pAβ (Fig. 4, A and C) and Fpβ fibrils (Fig. 4B) were treated with a series of CIP solutions with different enzymatic concentrations for 30 min, and then the dephosphorylated pAβ bands were traced using Tricine-SDS-PAGE (Fig. 4, A and B) and native PAGE (Fig. 4C). Only Tricine-SDS-PAGE was used to trace the bands after dephosphorylation of fibrils (Fig. 4B) because fibrils could not be disassociated and enter into the gel in native PAGE. The results showed that dephosphorylation of pAβ generated Aβ40 in a concentration-dependent manner. Furthermore, the phosphate groups on Ser8 in monomeric pAβ could be cleaved by CIP much more easily than those in Fpβ fibrils (Fig. 4, compare the corresponding bands in A with those in B). Even at a lower concentration of CIP (0.1 unit of CIP enzymatic activity), monomeric pAβ was almost completely dephosphorylated. For Fpβ fibrils, however, the phosphate groups were only partially removed even at high CIP concentrations, such as 1 and 10 units of CIP enzymatic activity (i.e. Fig. 4, two bands for both pAβ and Aβ in B, second and third columns, compared with only a single band for Aβ in the corresponding columns in A). Therefore, we identified that Fpβ fibril formation clearly limited the dephosphorylation by CIP. Recently, evidence has shown that fibrils can sequester different kinds of proteins and change their corresponding physiological properties (58, 59). Fibrils might also influence the enzymatic activities (60, 61). Therefore, dephosphorylation of pAβ monomers mixed with equal moles of Fpβ fibrils (native PAGE in Fig. 4D) was tested to exclude the possibility that fibrils might trap the CIP and lower the enzymatic activity. Only native PAGE was used to identify the new bands after dephosphorylation because the bands from disrupted fibrils in SDS-PAGE would influence the results. Comparison of Fig. 4, C and D, showed that the presence of Fpβ fibrils did not affect the enzymatic activity of CIP because the dephosphorylation effects of this enzyme on pAβ monomers were similar both in the absence (Fig. 4C) and presence (Fig. 4D) of fibrils. Collectively, these results suggest that the phosphorylation at Ser8 would directly induce the formation of certain structures in Fpβ fibril.
FIGURE 4.

Dephosphorylation of pAβ monomers (A and C), Fpβ fibrils (B), and pAβ monomers mixed with equal moles of Fpβ fibrils (D). The pAβ monomers (A and C), Fpβ fibrils (B), or pAβ monomers mixed with equal moles of fibrils (D) were incubated with a series of CIP concentrations in 20 mm Tris·HCl buffer, 150 mm NaCl, pH 7.4, and traced using 12% Tricine-SDS-PAGE (A and B) and native PAGE (C and D), respectively. Enzymatic activity of CIP is shown at the bottom of the gel where U is the unit of CIP enzymatic activity. Dephosphorylation of pAβ monomers or Fpβ fibrils could generate Aβ in a concentration-dependent manner. Comparison of the dephosphorylation of Fpβ fibrils (B) with pAβ monomers (A) showed that fibril formation could clearly limit dephosphorylation partially. The same dephosphorylation trends of pAβ monomers in the absence (C) or presence (D) of Fpβ fibrils were observed. These results showed that the presence of Fpβ fibrils did not influence the enzymatic activity of CIP. The upper and lower protein bands in A and B (Tricine-SDS-PAGE) denote pAβ and Aβ, respectively. The upper and lower protein bands in C and D native PAGE) denote Aβ and pAβ, respectively.
Fpβ and Fβ Fibrils Show Distinct Propagation Activities and Cellular Toxicities
Distinct fibril morphologies have been considered to affect the symmetries and lateral association propensities of protofilaments (8). Consequently, the morphological differences between Fpβ and Fβ fibrils might imply their structural diversities, such as the surface exposure of different amino acid side chains, which might further be correlated with prion-like propagation (30, 48) and cellular toxicities (8, 9), etc.
First, the prion-like propagation of Fpβ and Fβ fibrils in vitro was monitored by a ThT fluorescence assay on the seeded fibrillation (62), and TEM was used to examine the structural natures of aggregates after seeding. Both Fpβ and Fβ fibrils were sonicated in an ice bath to produce small fragments (Fig. 5). The wild-type Aβ40, Aβ42, and pyroglutamated Aβ3–40, which is an important post-translationally modified form of Aβ (named Aβ(3pE)40 hereinafter) (63), were tested on the cross-seeding efficiencies for the seeds of Fpβ and Fβ fibrils. Fig. 6A plots the ThT kinetics curves of Aβ40 in the presence of 10% Fpβ and Fβ seeds. Both seeds greatly shortened the lag phase, and Fpβ initiated the fibrillation more rapidly compared with the Fβ-seeded fibrillation. The significant difference in the lag time between the Fpβ- (almost no lag time) and Fβ-seeded fibrillation (296 min) is shown in Fig. 6D. We emphasize that the results in Fig. 6A suggested that the wild-type Aβ40 formed fibrils more rapidly in the presence of Fpβ seeds in comparison with its own seeds, meaning that the cross-seeding between pAβ and wild-type Aβ40 was more efficient than the self-seeding of wild-type Aβ40. Interestingly, we found that the ThT intensities of seeded plateaus in Fig. 6A were lower than that of unseeded. The concentrations of Aβ40 in both the parental and seeded fibrils were 40 μm. Therefore, the intensity difference was not likely to originate from the quantities of fibrils. We postulated that the ThT fluorescence intensity was reduced in seeded fibrils because certain fibril structures with low ThT fluorescence emission were selectively amplified during the seeding process as has been suggested by a previous work from Tycko and co-workers (64). Fig. 6, B and C, further show that, for the Aβ(3pE)40 and wild-type Aβ42, the Fpβ induced more efficient cross-seeding than the wild-type Fβ. Overall, our results suggested that the pAβ (i.e. Ser8-phosphorylated Aβ40) might serve as an intrinsic trigger to promote the fibrillation of a variety of Aβ species. As revealed by TEM, both the Aβ40 and Aβ(3pE)40 fibrils seeded from Fpβ showed similar twist-like morphologies (Fig. 6, G and H, right panel), whereas the Aβ40 and Aβ(3pE)40 fibrils seeded from Fβ showed ribbon-like morphologies (Fig. 6, G and H, left panel). These results indicated that both Fpβ and Fβ fibrils could propagate their structures to Aβ40 and Aβ(3pE)40. However, under the same seeding conditions, although the fibrillation times were significantly shortened, a similar morphology was obtained for both Fpβ- and Fβ-seeded Aβ42 fibrils (Fig. 6I) that was different from either the parental Fpβ or Fβ fibrils. It was proposed previously that the seeded fibrillation process involves mainly chain elongation from the existing structural patterns of the seeds (65). For Aβ40 and Aβ(3pE)40, it was interesting that the Fpβ seeds served as a better structural template for the Aβ chain elongation compared with Fβ seeds, which would reflect the differences in the intrinsic architectures of the two fibrils. The conformational transition from partially folded Aβ to β-sheet-enriched state is an essential process for Aβ aggregation, and the seeded fibrillation might follow the “docking conformational change” mechanism in which the existence of fibril seeds as templates would lower the “activation energy” for the conformational switch (66–68). The differences of the fibrillation process in the presence of the Fβ and Fpβ seeds revealed that phosphorylated fibrils possibly contained the core structures that would decrease the energy barrier and promote the efficiency of chain elongation much more easily. Surprisingly, despite the high similarity of the Aβ40 and Aβ42 sequences, monomeric Aβ42 was incompatible with the structural nature of either Fβ or Fpβ seeds. Although the shortened lag time was detected when Aβ42 was seeded by both Fpβ and Fβ fibrils, the resulting Aβ42 fibrils did not inherit the parental structural information. This implies that Aβ42 cannot be directly converted to fibrils at the termini of the seeds of Fpβ and/or Fβ fibrils by using the structural template from seeds. A similar phenomenon was also observed in a previous study of α-synuclein strains (9). One possible explanation was that the Fpβ and Fβ seeds only served as a “surface” to catalyze the formation of fibrils but not as structural templates.
FIGURE 5.

TEM images of sonicated seeds of Fβ and Fpβ. Both Aβ (left) and pAβ (right) seeds were produced by sonicating the Fβ and Fpβ fibrils, respectively, in the ice bath three times (for 1.5 min each time).
FIGURE 6.

Propagation of Fpβand Fβ fibrils in vitro was monitored by ThT kinetics assay and TEM images. Propagation of preformed 10% Fpβ (red curves) and Fβ fibrils (blue curves) in 40 μm Aβ40 (A), 10 μm Aβ(3pE)40 (B), and 10 μm Aβ42 (C) in vitro at pH 7.4 was monitored by ThT fluorescence kinetics assay. The lag phase for Aβ40 (D), Aβ(3pE)40 (E), and Aβ42 (F) showed that both Fpβ (red column) and Fβ (blue column) can shorten the lag phase for amyloid formation. The structural natures of Aβ40, Aβ(3pE)40, and Aβ42 aggregates after seeding were examined using TEM (G, H, and I, respectively); left and right panels are structural images of Fβ-seeded fibrils and Fpβ-seeded fibrils, respectively. The TEM samples were prepared after 24-h incubation. For Aβ40 (G) and Aβ(3pE)40 (H), the structural information of parental Fβ fibrils and Fpβ fibrils can be inherited. The black and white arrowheads point to ribbon-like and twisted cylindrical morphologies, respectively. However, Aβ42 cannot sample the structural information from either Fβ or Fpβ fibrils (I). ThT fluorescence kinetics of different soluble Aβ species without any seeds and Fβ or Fpβ seeds only were also conducted as controls. Error bars represent S.E. (n = 3 independent measurements).
To explore whether the Fpβ fibrils possessed different levels of cytotoxicity compared with the wild-type Fβ fibrils, both fibrils were applied extracellularly to mouse neuroblastoma N2a cells and mouse microglia BV-2 cells. The cell viabilities were investigated using 3-(4-,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 7). After 24-h treatment, both fibrils showed concentration-dependent cytotoxicities to these two mammalian cells. The twisted Fpβ fibrils showed significantly higher cytotoxicity than the flat ribbon Fβ fibrils. Ribbon-like and twisted fibrils are two major types of fibril morphologies formed by amyloid proteins, including Aβ (8, 69, 70), amylin (71), and α-synuclein (9), depending on the growth conditions. Different fibril structures have been shown to correlate with distinct morphologies and different levels of concentration-dependent cytotoxicities (6, 69). We observed higher toxicity for the twisted Fpβ fibrils compared with the ribbon-like Fβ fibrils, which was consistent with the potential correlation between morphology and cytotoxicity observed in other Aβ and α-synuclein fibrils (8, 9). We suspect that different lateral association and symmetry of twisted and ribbon-like morphologies might lead to distinct packing geometries and different exposures of residues on the fibril surface and therefore might induce different levels of toxicity to cells.
FIGURE 7.
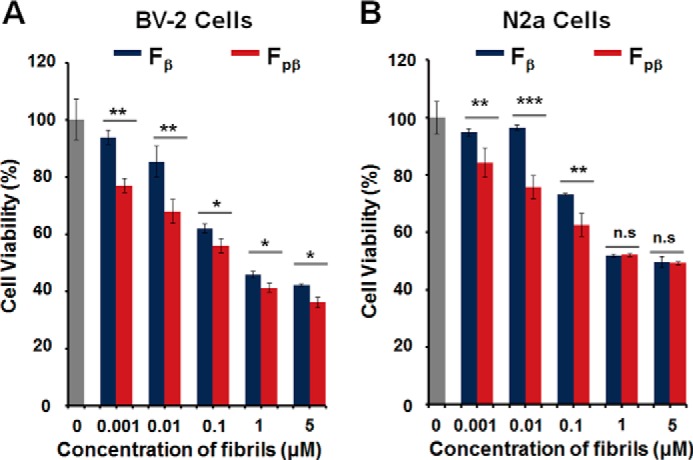
Cytotoxicity of Fpβ and Fβ fibrils were measured using MTT assay. Cell viability of BV-2 cells (A) and N2a cells (B) treated with Fβ and Fpβ fibrils, respectively, for 24 h was measured using an MTT assay. The blank control is shown in gray columns. Fpβ fibrils shows higher toxicity compared with Fβ fibrils in a concentration-dependent manner. n.s., not significant. S.E. is shown as error bars (n = 3 independent measurements; *, p < 0.05; **, p < 0.01; ***, p < 0.001 as evaluated with independent Student's t test).
Molecular Structural Differences between the Fpβ- and Fβ-seeded Aβ40 Fibrils
Given the differences in the fibril morphologies, biophysical characterizations, and cytotoxicities, we were interested in whether there are molecular level structural variations between the Fpβ and Fβ fibrils. This was done by MAS NMR studies on the Fpβ- and Fβ-seeded Aβ fibrils. The seeded fibrils were prepared by adding 10% Fpβ or Fβ seeds to freshly dissolved 40 μm Aβ40 monomers followed by quiescent incubation for 48 h under physiological temperature and pH. This protocol minimized the manipulations on fibrillar structures and was likely to produce seeded fibrils that were similar to their parents (8, 72). We verified the structural similarity using TEM and CD spectroscopy and examined the cytotoxicities of daughter fibrils using the MTT assay (Fig. 8). The CD spectra showed minimum peaks at 216 and 222 nm for the Fpβ- and Fβ-seeded fibrils, respectively (Fig. 8A), that were identical to their parent fibrils. In Fig. 8B, the TEM images show that the features for the Fpβ and Fβ fibrils, which were the twisted cylindrical and flat ribbon-like morphologies, respectively, were propagated to their daughter fibrils. In Fig. 8C, we found that daughter fibrils from both Fβ and Fpβ seeds showed concentration-dependent cytotoxicities to the two mammalian cells. Daughter fibrils seeded from Fpβ also showed higher cytotoxicity where the trend was similar to that of their parental fibrils. Therefore, we concluded that Fpβ- and Fβ-seeded fibrils would retain structural characteristics from their parental fibrils.
FIGURE 8.
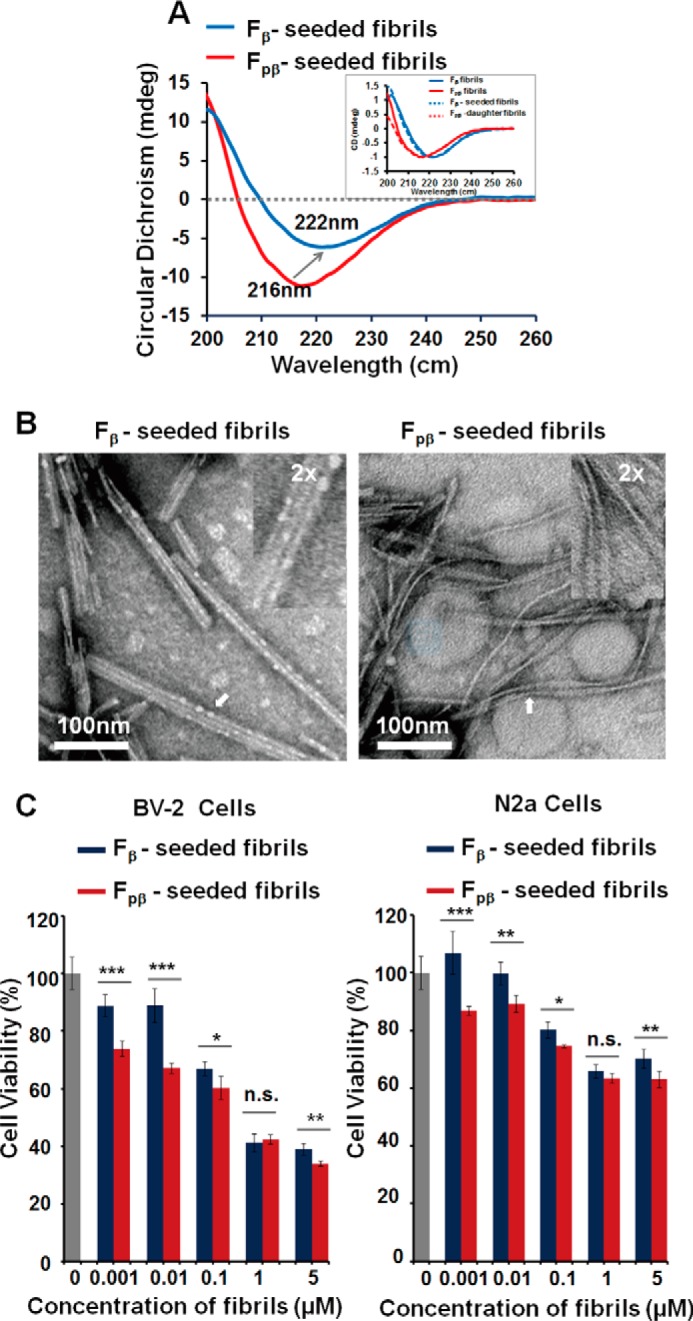
CD spectroscopy, TEM images, and MTT assay indicated that structural features of Fpβ and Fβ fibrils could be propagated to their daughter fibrils. 40 μm Aβ40 was used to examine the structural natures after seeding. A, CD spectroscopy showed that the minimum peak of Fpβ- and Fβ-seeded fibrils was the same as their parent fibrils, respectively. The inset is normalized CD spectroscopy curves of parent and daughter fibrils. B, TEM images of Fpβ- and Fβ-seeded fibrils were identical to their parent fibrils. The samples were prepared after 48-h incubation. C, MTT assay showed that Fpβ-seeded fibrils had higher cytotoxicity of which the trend was the same as for their parental fibrils. S.E. is shown as error bars (n = 3 independent measurements; *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant; as evaluated with independent Student's t test). mdeg, millidegrees.
Extensive MAS NMR measurements were performed to probe the residue-specific secondary structures of both seeded fibrils. We used six scattering uniformly isotope-labeled Aβ peptides to cover the majority of the primary sequence, especially the typical β-strands and interstrand loop segments from the known Aβ fibril structures (24, 27, 69, 73–75). Representative NMR spectra and the summary of chemical shift deviations are plotted in Fig. 9. The chemical shift assignments for both seeded fibrils are provided in Tables 1 and 2. Overall, both fibrils showed typical β-loop-β structures like other known 40-residue Aβ fibrils. However, detailed structural variations were observed. First, the local secondary structure of Gly9, which is next to the phosphorylated Ser8, changed significantly. The wild-type Fβ fibrils showed multiple Gly9 C′-Cα cross-peaks, whereas the phosphorylated Fpβ fibrils had a predominant cross-peak (Fig. 10A). This observation was consistent with the 31P NMR (Fig. 3) and confirmed that the local conformation around Ser8 became better defined due to the phosphorylation. Second, the site-specific phosphorylation affected not only the local conformation but also the overall secondary structures of the resulting fibrils. As shown in Fig. 9, we identified significant chemical shift deviations between the Fpβ and Fβ fibrils, and the largest chemical shift differences were observed for residues Gly9, the segments between Leu17 and Ile32, and the residue Val39. Particularly, the segments Asp23–Ser26 and Gly29–Ile32 showed the most significant chemical shift deviations between the two fibrillar species. The C-terminal β-strand region from Gly33 to Gly38, in contrast, seemed to have little structural variation. Additionally, it seemed that the Fpβ fibrils had more ordered C termini compared with the Fβ fibrils because the Val39 cross-peaks were much stronger in the Fpβ-seeded fibrils then in the Fβ-seeded fibrils (Fig. 10, B and C). Importantly, the segment between Ala21 and Ala30 is generally considered as the interstrand loop region based on the known 40-residue Aβ fibril structures and might be involved in the early stages of fibrillation according to previous studies on Aβ and other similar types of amyloid peptides (76, 77). Because the seeded fibrils were likely to have the same structures as their parents, our results suggested that the phosphorylation on Ser8 might affect the intermediate Aβ structures formed during the nucleation steps through specific molecular interactions between the phosphorylated Ser8 side chains and residues located in the loop segment between Ala21 and Gly29. Interestingly, recent work (46) showed that the phosphorylation of Ser8 stabilized the Aβ fibrils through a potential inter-residue interaction among Asp7 and the phosphorylated Ser8 and Ser26, which was consistent with our NMR data.
FIGURE 9.

MAS NMR spectra and chemical shift deviations of Fβ- and Fpβ-seeded 40-residue Aβ fibrils. The Fpβ-seeded fibrils (A, C, and E) and Fβ-seeded fibrils (B, D, and F) were isotope-labeled at different residues. Intraresidual Cα-Cβ cross-peaks are highlighted in colored lines. The differences between dashed and solid lines in B, D, and F indicate shifts in cross-peaks. G shows the residue-specific chemical shift deviations (δ(Fpβ-seeded) − δ(Fβ-seeded)) for C′ (black squares), Cα (red circles), and Cβ (blue triangles). The solid orange lines highlight the ±1.0-ppm threshold for significant chemical shift deviation based on the estimation of 13C NMR line widths. Residues with at least one significant chemical shift deviation among C′, Cα, and Cβ are highlighted in green on the x axis.
TABLE 1.
13C chemical shifts for the Fpβ-seeded fibrils
| C′ | Cα | Cβ | Cγ | Cδ | Cϵ | |
|---|---|---|---|---|---|---|
| ppm | ppm | ppm | ppm | ppm | ppm | |
| Gly9 | 172.4 | 45.4 | ||||
| Leu17 | 174.2 | 54.1 | 45.8 | 28.8 | 24.6 | |
| Val18 | 172.9 | 61.1 | 35.3 | 21.2 | ||
| Phe19 | 172.4 | 56.2 | 42.7 | |||
| Phe20 | 172.8 | 56.8 | 39.8 | |||
| Ala21 | 175.4 | 50.1 | 22.9 | |||
| Asp23 | 173.9 | 56.5 | 43.7 | 176.2 | ||
| Val24 | 173.8 | 62.9 | 32.5 | 20.7 | ||
| Gly25 | 171.9 | 44.8 | ||||
| Ser26 | 172.3 | 56.2 | 65.8 | |||
| Lys28 | 173.6 | 55.8 | 35.2 | 25.5 | 31 | 42.6 |
| Gly29 | 172.7 | 45.8 | ||||
| Ala30 | 176.6 | 53.4 | 20.9 | |||
| Ile31 | 174.5 | 62.2 | 38.3 | 27.7 | 15.6 | 12.9 |
| Ile32 | 174.4 | 59.1 | 42.3 | 27 | 18.9 | 14.1 |
| Gly33 | 171 | 44.9 | ||||
| Leu34 | 173.6 | 53.6 | 46 | 27.5 | 24.6 | |
| Val36 | 174.4 | 60.4 | 34.6 | 21.6 | ||
| Gly37 | 171.7 | 47.5 | ||||
| Gly38 | 173.5 | 49.8 | ||||
| Val39 | 176 | 60.4 | 35.3 | 21.7 |
TABLE 2.
13C chemical shifts for the Fβ-seeded fibrils
| C′ | Cα | Cβ | Cγ | Cδ | Cϵ | |
|---|---|---|---|---|---|---|
| ppm | ppm | ppm | ppm | ppm | ppm | |
| Gly9 | 170.8 | 44.5 | ||||
| Leu17 | 173.8 | 52.1 | 43 | 26.2 | ||
| Val18 | 173 | 60.6 | 35.8 | 21.2 | ||
| Phe19 | 172 | 55.6 | 41.6 | |||
| Phe20 | 174.2 | 57.1 | 39.2 | |||
| Ala21 | 174.4 | 50 | 23.3 | |||
| Asp23 | 173.8 | 55.9 | 42.5 | |||
| Val24 | 175 | 64.2 | 33.2 | 21.6 | ||
| Gly25 | 172.7 | 46.2 | ||||
| Ser26 | 174.2 | 55.5 | 65.7 | |||
| Lys28 | 173.9 | 55.4 | 35.2 | 27.4 | 30.2 | 42.5 |
| Gly29 | 171 | 44.3 | ||||
| Ala30 | 174 | 50.6 | 23 | |||
| Ile31 | 174.6 | 61.7 | 39.5 | 28.7 | 17.4 | 13.6 |
| Ile32 | 174.6 | 58.5 | 40.5 | 26.2 | 16.2 | 12.1 |
| Gly33 | 170.8 | 44.7 | ||||
| Leu34 | 173.3 | 53.4 | 45.9 | 27.4 | 24.8 | |
| Val36 | 174.1 | 59.7 | 35.6 | 20.7 | ||
| Gly37 | 171.4 | 47.2 | ||||
| Gly38 | 173.1 | 49.3 | ||||
| Val39 | 175.4 | 61.5 | 33.7 | 22.3 |
FIGURE 10.
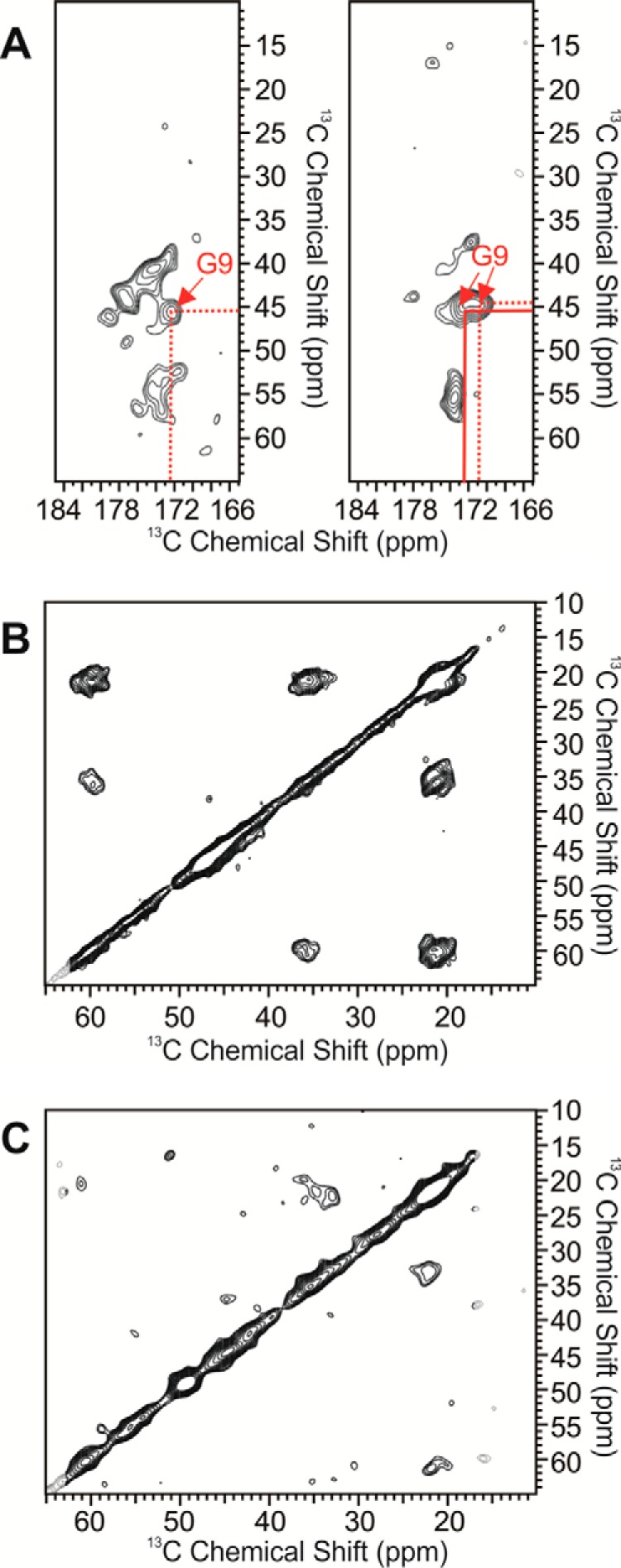
2D 13C-13C MAS NMR spectra. A, cross-peaks for Gly9 in Fpβ-seeded (left) and Fβ-seeded (right) fibrils. B and C, cross-peaks for Val39 in Fpβ-seeded (B) and Fβ-seeded (C) fibrils.
Discussion
In this decade, polymorphic Aβ fibrils with different neuronal cytotoxicities and propagation efficiencies have attracted intense attention (6). Small variations of microenvironments in vitro will change the kinetics and thermodynamics for aggregation (8, 70). This may be induced by the coexistence of multiple nucleation processes, each of which favors its own fibril structures (78). Morphological differences induced by variations in fibrillation conditions imply that experimentally based microenvironments (agitation, pH, ionic strength, etc.) may lead to a favorable nucleation process (8, 9, 50, 69, 70, 78). For Aβ40 fibrils, two major types of fibrillar morphologies have been reported. One is the twisted filament with a 3-fold molecular symmetry, and the other is the ribbon-like filament with a 2-fold molecular symmetry (8, 69, 70). Herein, we report the effects of phosphorylation at Ser8, which is an existing post-translational modification of Aβ in vivo (42, 43), on the fibrillation of Aβ. Similar to the effects of microenvironment variation on amyloid formation, phosphorylation at Ser8 changes the nucleation process of Aβ and finally leads to fibrils with distinct morphologies. Under the same fibrillation conditions, the pAβ folds into a twisted fibril, and Aβ folds into a flat ribbon-like fibril. The N-terminal post-translational modifications of Aβ, such as phosphorylation (31), pyroglutamation (63), and nitration (79), have been reported to control the kinetic process for Aβ fibrillation, accelerate plaque formation, and alter the prion-like propagation efficiency. We first revealed that phosphorylation at Ser8, acting as an intrinsic molecular trigger beyond the variations of fibrillation conditions, can induce pAβ folding into different fibril morphologies.
The prion-like strain phenomenon where a single amyloid protein gives rise to multiple distinct phenotypes has been correlated with the ability of some polypeptides or proteins to fold into distinct fibril structures in neurodegenerative disease (16, 17). Aβ seeds have been found to possess a transmission risk like prion protein aggregates in vivo (22). Our study herein indicates that phosphorylation results in Aβ fibril morphology distinct from the wild-type Aβ, which proposes a possible intrinsic origin of polymorphous fibrils of Aβ. Neurotoxic studies and seeding experiments show that Fpβ and Fβ fibrils from pAβ and wild-type Aβ, respectively, have different cytotoxicities and seeding efficiencies, which probably arise from strain-specific structural variation. The results suggest that phosphorylation may act as a possible factor to induce the formation of strains of Aβ. A recent study by Rijal Upadhaya et al. (43) showed that phosphorylation of Aβ occurred in the Alzheimer's brain in a hierarchical sequence and promoted the formation of final toxic aggregates, and this phosphorylation was assumed to be indicative for biochemical Aβ stage 3 and associated with symptomatic AD. We suggest that the occurrence of phosphorylation was not just an inducer for the further accumulation of total aggregates but might be an initiator for the formation of certain amyloid morphologies with specific biological activities.
Recently, the structures and stabilities of Aβ40 phosphorylated at Ser8 have been studied by Rezaei-Ghaleh et al. (46, 55) using solution NMR spectroscopy and molecular dynamics simulations. These studies mainly concluded that the phosphorylated Ser8 was not included in the fibril core, which typically consisted of residues 15–40. In the monomeric state in solution, the phosphorylation at Ser8 mainly affected the local conformation from residues 4 to 16. However, it also promoted the inter-residual interactions between segments Asp23–Gly25 and Gly29–Ala30, which are located in the inter-β-strand loop region in Aβ fibrils (55). Combined with our solid-state NMR measurements on the chemical shift differences between Fpβ- and Fβ-seeded fibrils, we propose that the Ser8 phosphorylation might affect the initial nucleation of Aβ fibrillation. This site-specific phosphorylation might stabilize certain conformations within the segment Asp23–Ala30, presumably through a transient hydrogen bonding interaction based on the previous molecular dynamics simulation (46). The fact that we observed the most significant chemical shift differences within the segments Asp23–Ser26 and Gly29–Ile32 between the two types of fibrils further supports the hypothesis. The residues located between two typical β-strands in amyloid fibrils have been proposed as general initial nucleation sites in the fibrillation process of Aβ and other types of amyloid peptides (76, 77). Therefore, stabilization of local conformations in this region might accelerate the rates of nucleation and may lead to different fibril morphology.
In summary, amyloid fibrils together with small aggregates, such as oligomers and protofibrils, are considered important pathological agents in AD. Our study raises the possibility that different Aβ strains would occur in a phosphorylation-dependent manner. Compared with the wild-type Aβ fibrils, phosphorylation at Ser8 leads to an overall fibril structural change with significant chemical shift deviations in the segments Asp23–Ser26 and Gly29–Ile32. Along with the recent study from Rezaei-Ghaleh et al. (46, 55), we propose that the effects of phosphorylation at Ser8 on the local conformation of residues 4–16 and the inter-residual interactions in the interstrand loop region from residues 21 to 30 would cooperatively promote pAβ to fold with a distinct nucleation process and finally lead to a fibril morphology different from that of wild-type Aβ. Combined with the potential link between phosphorylation and symptomatic AD (43), we propose that pAβ might serve as a specific therapeutic target for AD. The specific link between Aβ strains and phosphorylation raises the possibility of designing structure-based imaging agents and inhibitors to diagnose or prevent AD at different stages.
Experimental Procedures
Sample Preparation
All Aβ peptides with the same primary sequence (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV) were prepared using Fmoc chemistry with Fmoc-Val-Wang resin (0.18 mmol/g; GL Biochem (Shanghai) Ltd.). Fmoc-Ser(PO3Bzl)-OH (where Bzl is benzyl) (GL Biochem (shanghai) Ltd.) and uniformly 13C- and 15N-labeled amino acids (Cambridge Isotope Laboratories, Inc.) were incorporated into the peptides to achieve phosphorylated and isotope-labeled Aβ peptide sequences. The Glu3-pyroglutamated Aβ40 (Aβ(3pE)40; pEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV) was also synthesized using Fmoc chemistry. Aβ42 peptide was purchased from Your Bio-Tech Partner, Shanghai, China, and used directly. All peptides were purified by reverse-phase HPLC (C18 column, Waters 600) and identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/TOF MS, ABI Research). All fibril samples (including the Fβ and Fpβ fibrils) were prepared following the method described previously (80). Briefly, the Aβ or pAβ peptides were dissolved in HFIP (1 mg/ml, Sigma) for at least 4 h at ambient temperature to remove any preformed aggregates. Then the HFIP was removed by blow drying with N2 and further vacuum drying for at least 3 h using an oil pump. The thin film of peptides was then resuspended in 20 mm Tris·HCl buffer, 150 mm NaCl, pH 7.4, and incubated at 37 °C with 200-rpm continuous shaking for 2 days to produce mature fibrils.
ThT Fluorescence Kinetics Assay
The ThT fluorescence kinetics assay was conducted on the Synergy 4 (BioTek) plate reader using a 96-well plate (λex = 440 nm and λem = 480 nm). For monitoring the fibrillation process of Aβ and pAβ (40 μm), ThT signals (20 μm) were recorded at 37 °C under continuous shaking (medium) with a time interval of ∼10 min between each recording. For tracking the aggregation process of different Aβ species (40 μm Aβ40, 10 μm Aβ(3pE)40, and 10 μm Aβ42) in the presence of Fβ and Fpβ seeds (10%), the seeds of Fβ and Fpβ were prepared by sonicating the resulted fibrils three times (1.5 min each time) in an ice bath. ThT signals were recorded at 37 °C under quiescent conditions with a time interval of 10 min between each recording. The fibrillation ThT kinetics curves of Aβ and pAβ were fitted using a sigmoidal function, F − F0 = 1/(1 + exp(γmax(τ1/2 − t))), in which τ1/2 is the half-completion time of aggregation and γmax is the maximum growth rate (50). The tlag (lag time) was determined as the time intercept of the line best fitted to the linear portion of the F to t coordinate. All the experiments were replicated at least three times.
TEM
Fibrils samples (8 μl) were directly incubated on a carbon-coated copper grid for 2 min and then stained with 2% sodium phosphotungstic acid, pH 6.5, for 1 min. TEM images were recorded using a Hitachi-7650B electron microscope at 80 kV.
CD
CD spectroscopy in the 200–260-nm region (far-UV region) was performed with a Chirascan Plus CD spectrophotometer (Applied Photophysics). All samples (40 μm; 200 μl) were directly loaded in a 1-mm width CD cuvette, and signals were recorded at room temperature with three-scan signal averaging.
XRD
Fβ and Fpβ fibrils were centrifuged (12,000 rpm) at 4 °C for 30 min, and the resulting pellets were washed with ddH2O three times to remove salts. The pellets were then resuspended in 10 μl of ddH2O, and the final suspension was aligned between two fire-polished glass rods at 4 °C overnight. XRD images were recorded on a Rigaku Micromax-007 X-ray generator equipped with an R-Axis IV++ area detector.
FT-IR Spectroscopy
FT-IR spectroscopy was performed on a Frontier FT-IR (PerkinElmer Life Sciences) equipped with an attenuated total reflectance accessory at ambient temperature. 1 ml of Fβ and Fpβ fibrils was centrifuged (12,000 rpm) at 4 °C for 30 min. The pellets were then washed with ddH2O three times to remove salts. The pellets were then frozen and dried into powders. The monomeric samples were prepared by dissolving Aβ and pAβ (1 mg/ml) in HFIP overnight, and then the solutions were frozen and lyophilized into powders. The final powders were directly loaded on the attenuated total reflectance accessory for acquisition of data.
Dephosphorylation by CIP
All samples with or without CIP (Sigma) were incubated at 37 °C for 30 min. The final mixtures were then directly loaded and analyzed using Tricine-SDS-PAGE and native PAGE, respectively. The PAGE images were recorded using VersaDoc 3000 (Bio-Rad). For all the experiments, 20 μl of 40 μm Aβ monomers or fibrils was used.
Neuronal Cell Toxicity Assay
To detect the toxicities of the two fibrils, the fibrils were centrifuged at 12,000 rpm at 4 °C for 1 h, and the final pellets were resuspended in the cell culture medium, sonicated in an ice bath three times (1.5 min each time). MTT reduction assays were used to determine the effects of Fβ and Fpβ fibrils on viability of neuronal cells. Mouse neuroblastoma N2a cells and murine microglia BV-2 cells were plated at a density of 5000 cells/well on 96-well plates in 150 μl of culture medium (50% α-minimum Eagle's medium and 50% DMEM with 10% fetal bovine serum for N2a cells and 100% DMEM with 10% fetal bovine serum for BV-2 Cells). After 24-h incubation at 37 °C, the medium was exchanged for fresh medium (100% DMEM without fetal bovine serum) with Fβ and Fpβ fibrils. After 24-h incubation, 20 μl of MTT (5 mg/ml) was added into each well for a further 4-h incubation. The culture medium was then discarded, and 150 μl of DMSO was added to dissolving formazan completely. Absorbance at 490 nm was measured using Synergy 4 plate reader to calculate the cell viability.
MAS NMR
The seeded fibrils for MAS NMR measurements were prepared by incubating the isotope-labeled monomeric Aβ40 with 10% Fβ and Fpβ seeds for 2 days quiescently. The fibrils were then centrifuged at 100,000 × g for 1 h at 4 °C (Beckman Coulter). The final pellets were freeze-dried and packed into 2.5-mm MAS rotors with rehydration using deionized water (1 μl/mg). All MAS NMR measurements were performed on a 600-MHz Bruker solid-state NMR spectrometer equipped with a 2.5-mm TriGamma MAS probe. The MAS frequency was set to 10 kHz for all experiments. The sample temperature was kept at ∼ 5 °C using a 270 K N2 cooling line. The 31P spectra were recorded using direct excitation with a 50-kHz 31P 90° radiofrequency (rf) pulse and a 100-kHz continuous wave 1H decoupling during the acquisition time. The two-dimensional (2D) 13C-13C spin diffusion spectra were recorded using a pulse sequence composed of a 60-kHz 1H excitation 90° pulse, a linearly ramped 1H-13C cross-polarization period, a 10-kHz rf-assisted diffusion 1H field during the 10-ms mixing period, and a 100-kHz 1H two-pulse phase modulation decoupling. The 31P and 2D spectra were processed using Topspin and NMRPipe software, respectively.
Author Contributions
Z.-W. H. synthesized the peptides, designed and conducted all experiments, and wrote the paper. M.-R. M. contributed to the XRD and CD experiments. Y.-X. C. and Y.-F. Z. analyzed the results and revised the paper intellectually. W. Q. designed and conducted the NMR experiments, analyzed the results, and wrote the paper. Y.-M. L. conceived the project, designed all the experiments, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We sincerely acknowledge Dr. Cong Liu (Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China) for kind help with XRD measurement and very useful discussions.
This work was supported by Major State Basic Research Development Program of China Grant 2013CB910700, National Natural Science Foundation of China Grants 21472109 and 81661148047, a start-up fund from Binghamton University (to W. Q.), and National Science Foundation Major Research Instrumentation Grant 0922815. The authors declare that they have no conflicts of interest with the contents of this article.
- AD
- Alzheimer's disease
- Aβ
- amyloid-β
- Aβ(3pE)40
- Glu3-pyroglutamated Aβ40
- pAβ
- Ser8-phosphorylated Aβ
- MAS
- magic angle spinning
- TEM
- transmission electron microscopy
- XRD
- X-ray diffraction
- CIP
- calf intestinal alkaline phosphatase
- MTT
- 3-(4-,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ThT
- thioflavin T
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- HFIP
- hexafluoroisopropanol
- ddH2O
- double distilled H2O.
References
- 1. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guo J. L., and Lee V. M. (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knowles T. P., Vendruscolo M., and Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 [DOI] [PubMed] [Google Scholar]
- 5. Kodali R., and Wetzel R. (2007) Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 17, 48–57 [DOI] [PubMed] [Google Scholar]
- 6. Tycko R. (2015) Amyloid polymorphism: structural basis and neurobiological relevance. Neuron 86, 632–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T., and Tanaka M. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., and Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 9. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stöhr J., Condello C., Watts J. C., Bloch L., Oehler A., Nick M., DeArmond S. J., Giles K., DeGrado W. F., and Prusiner S. B. (2014) Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. U.S.A. 111, 10329–10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo J. L., Covell D. J., Daniels J. P., Iba M., Stieber A., Zhang B., Riddle D. M., Kwong L. K., Xu Y., Trojanowski J. Q., and Lee V. M. (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frost B., and Diamond M. I. (2010) Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goedert M., Clavaguera F., and Tolnay M. (2010) The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325 [DOI] [PubMed] [Google Scholar]
- 14. Polymenidou M., and Cleveland D. W. (2012) Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 209, 889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chien P., Weissman J. S., and DePace A. H. (2004) Emerging principles of conformation-based prion inheritance. Annu. Rev. Biochem. 73, 617–656 [DOI] [PubMed] [Google Scholar]
- 16. Brettschneider J., Del Tredici K., Lee V. M., and Trojanowski J. Q. (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanders D. W., Kaufman S. K., Holmes B. B., and Diamond M. I. (2016) Prions and protein assemblies that convey biological information in health and disease. Neuron 89, 433–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xue W.-F., Hellewell A. L., Gosal W. S., Homans S. W., Hewitt E. W., and Radford S. E. (2009) Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 284, 34272–34282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kalapothakis J. M., Morris R. J., Szavits-Nossan J., Eden K., Covill S., Tabor S., Gillam J., Barran P. E., Allen R. J., and MacPhee C. E. (2015) A kinetic study of ovalbumin fibril formation: the importance of fragmentation and end-joining. Biophys. J. 108, 2300–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cras P., Kawai M., Lowery D., Gonzalez-DeWhitt P., Greenberg B., and Perry G. (1991) Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 88, 7552–7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yin R. H., Tan L., Jiang T., and Yu J. T. (2014) Prion-like mechanisms in Alzheimer's disease. Curr. Alzheimer Res. 11, 755–764 [DOI] [PubMed] [Google Scholar]
- 22. Jaunmuktane Z., Mead S., Ellis M., Wadsworth J. D., Nicoll A. J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A. S., Rudge P., Collinge J., and Brandner S. (2015) Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250 [DOI] [PubMed] [Google Scholar]
- 23. Paravastu A. K., Qahwash I., Leapman R. D., Meredith S. C., and Tycko R. (2009) Seeded growth of β-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. U.S.A. 106, 7443–7448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watts J. C., Condello C., Stöhr J., Oehler A., Lee J., DeArmond S. J., Lannfelt L., Ingelsson M., Giles K., and Prusiner S. B. (2014) Serial propagation of distinct strains of Aβ prions from Alzheimer's disease patients. Proc. Natl. Acad. Sci. U.S.A. 111, 10323–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cohen M., Appleby B., and Safar J. G. (2016) Distinct prion-like strains of amyloid β implicated in phenotypic diversity of Alzheimer's disease. Prion 10, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., and Ishii Y. (2015) Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 22, 499–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A. L., Vigouret J. M., Paganetti P., Walsh D. M., Mathews P. M., Ghiso J., et al. (2006) Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 [DOI] [PubMed] [Google Scholar]
- 29. Eisele Y. S., Obermüller U., Heilbronner G., Baumann F., Kaeser S. A., Wolburg H., Walker L. C., Staufenbiel M., Heikenwalder M., and Jucker M. (2010) Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 330, 980–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stöhr J., Watts J. C., Mensinger Z. L., Oehler A., Grillo S. K., DeArmond S. J., Prusiner S. B., and Giles K. (2012) Purified and synthetic Alzheimer's amyloid β (Aβ) prions. Proc. Natl. Acad. Sci. U.S.A. 109, 11025–11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar S., and Walter J. (2011) Phosphorylation of amyloid β (Aβ) peptides—a trigger for formation of toxic aggregates in Alzheimer's disease. Aging 3, 803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jawhar S., Wirths O., and Bayer T. A. (2011) Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J. Biol. Chem. 286, 38825–38832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bah A., Vernon R. M., Siddiqui Z., Krzeminski M., Muhandiram R., Zhao C., Sonenberg N., Kay L. E., and Forman-Kay J. D. (2015) Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519, 106–109 [DOI] [PubMed] [Google Scholar]
- 34. Kardos J., Kiss B., Micsonai A., Rovó P., Menyhárd D. K., Kovács J., Váradi G., Tóth G. K., and Perczel A. (2015) Phosphorylation as conformational switch from the native to amyloid state: Trp-cage as a protein aggregation model. J. Phys. Chem. B 119, 2946–2955 [DOI] [PubMed] [Google Scholar]
- 35. Du J. T., Li Y. M., Wei W., Wu G. S., Zhao Y. F., Kanazawa K., Nemoto T., and Nakanishi H. (2005) Low-barrier hydrogen bond between phosphate and the amide group in phosphopeptide. J. Am. Chem. Soc. 127, 16350–16351 [DOI] [PubMed] [Google Scholar]
- 36. Gandhi N. S., Landrieu I., Byrne C., Kukic P., Amniai L., Cantrelle F.-X., Wieruszeski J.-M., Mancera R. L., Jacquot Y., and Lippens G. (2015) A phosphorylation-induced turn defines the Alzheimer's disease AT8 antibody epitope on the tau protein. Angew. Chem. Int. Ed. Engl. 54, 6819–6823 [DOI] [PubMed] [Google Scholar]
- 37. Rezaei-Ghaleh N., Amininasab M., Giller K., Kumar S., Stündl A., Schneider A., Becker S., Walter J., and Zweckstetter M. (2014) Turn plasticity distinguishes different modes of amyloid-β aggregation. J. Am. Chem. Soc. 136, 4913–4919 [DOI] [PubMed] [Google Scholar]
- 38. Zhou L.-X., Zeng Z.-Y., Du J.-T., Zhao Y.-F., and Li Y.-M. (2006) The self-assembly ability of the first microtubule-binding repeat from tau and its modulation by phosphorylation. Biochem. Biophys. Res. Commun. 348, 637–642 [DOI] [PubMed] [Google Scholar]
- 39. Du J.-T., Yu C.-H., Zhou L.-X., Wu W.-H., Lei P., Li Y., Zhao Y.-F., Nakanishi H., and Li Y.-M. (2007) Phosphorylation modulates the local conformation and self-aggregation ability of a peptide from the fourth tau microtubule-binding repeat. FEBS J. 274, 5012–5020 [DOI] [PubMed] [Google Scholar]
- 40. Ma M.-R., Hu Z.-W., Zhao Y.-F., Chen Y.-X., and Li Y.-M. (2016) Phosphorylation induces distinct α-synuclein strain formation. Sci. Rep. 6, 37130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Milton N. G. (2005) Phosphorylated amyloid-β: the toxic intermediate in Alzheimer's disease neurodegeneration. Subcell. Biochem. 38, 381–402 [DOI] [PubMed] [Google Scholar]
- 42. Kumar S., Wirths O., Theil S., Gerth J., Bayer T. A., and Walter J. (2013) Early intraneuronal accumulation and increased aggregation of phosphorylated Aβ in a mouse model of Alzheimer's disease. Acta Neuropathol. 125, 699–709 [DOI] [PubMed] [Google Scholar]
- 43. Rijal Upadhaya A., Kosterin I., Kumar S., von Arnim C. A., Yamaguchi H., Fändrich M., Walter J., and Thal D. R. (2014) Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer's disease. Brain 137, 887–903 [DOI] [PubMed] [Google Scholar]
- 44. Kumar S., Rezaei-Ghaleh N., Terwel D., Thal D. R., Richard M., Hoch M., McDonald J. M., Wüllner U., Glebov K., Heneka M. T., Walsh D. M., Zweckstetter M., and Walter J. (2011) Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer's disease. EMBO J. 30, 2255–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar S., Singh S., Hinze D., Josten M., Sahl H. G., Siepmann M., and Walter J. (2012) Phosphorylation of amyloid-β peptide at serine 8 attenuates its clearance via insulin-degrading and angiotensin-converting enzymes. J. Biol. Chem. 287, 8641–8651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rezaei-Ghaleh N., Amininasab M., Kumar S., Walter J., and Zweckstetter M. (2016) Phosphorylation modifies the molecular stability of β-amyloid deposits. Nat. Commun. 7, 11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A., Barker S. J., Foley A. C., Thorpe J. R., Serpell L. C., Miller T. M., Grinberg L. T., Seeley W. W., and Diamond M. I. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peelaerts W., Bousset L., Van der Perren A., Moskalyuk A., Pulizzi R., Giugliano M., Van den Haute C., Melki R., and Baekelandt V. (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 [DOI] [PubMed] [Google Scholar]
- 49. Serpell L. C., Berriman J., Jakes R., Goedert M., and Crowther R. A. (2000) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. U.S.A. 97, 4897–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abelein A., Jarvet J., Barth A., Gräslund A., and Danielsson J. (2016) Ionic strength modulation of the free energy landscape of Aβ40 peptide fibril formation. J. Am. Chem. Soc. 138, 6893–6902 [DOI] [PubMed] [Google Scholar]
- 51. Sarroukh R., Goormaghtigh E., Ruysschaert J.-M., and Raussens V. (2013) ATR-FTIR: a “rejuvenated” tool to investigate amyloid proteins. Biochim. Biophys. Acta 1828, 2328–2338 [DOI] [PubMed] [Google Scholar]
- 52. Matsuzaki K. (2014) How do membranes initiate Alzheimer's disease? Formation of toxic amyloid fibrils by the amyloid β-protein on ganglioside clusters. Acc. Chem. Res. 47, 2397–2404 [DOI] [PubMed] [Google Scholar]
- 53. Xu F., Fu Z., Dass S., Kotarba A. E., Davis J., Smith S. O., and Van Nostrand W. E. (2016) Cerebral vascular amyloid seeds drive amyloid β-protein fibril assembly with a distinct anti-parallel structure. Nat. Commun. 7, 13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Makin O. S., Atkins E., Sikorski P., Johansson J., and Serpell L. C. (2005) Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. U.S.A. 102, 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rezaei-Ghaleh N., Kumar S., Walter J., and Zweckstetter M. (2016) Phosphorylation interferes with maturation of amyloid-β fibrillar structure in the N-terminus. J. Biol. Chem. 291, 16059–16067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zalatan J. G., and Herschlag D. (2006) Alkaline phosphatase mono- and diesterase reactions: comparative transition state analysis. J. Am. Chem. Soc. 128, 1293–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chaudhuri G., Chatterjee S., Venu-Babu P., Ramasamy K., and Thilagaraj W. R. (2013) Kinetic behaviour of calf intestinal alkaline phosphatase with pNPP. Indian J. Biochem. Biophys. 50, 64–71 [PubMed] [Google Scholar]
- 58. Olzscha H., Schermann S. M., Woerner A. C., Pinkert S., Hecht M. H., Tartaglia G. G., Vendruscolo M., Hayer-Hartl M., Hartl F. U., and Vabulas R. M. (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 [DOI] [PubMed] [Google Scholar]
- 59. Rahman M. M., Zetterberg H., Lendel C., and Härd T. (2015) Binding of human proteins to amyloid-β protofibrils. ACS Chem. Biol. 10, 766–774 [DOI] [PubMed] [Google Scholar]
- 60. Zorn J. A., Wille H., Wolan D. W., and Wells J. A. (2011) Self-assembling small molecules form nanofibrils that bind procaspase-3 to promote activation. J. Am. Chem. Soc. 133, 19630–19633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Julien O., Kampmann M., Bassik M. C., Zorn J. A., Venditto V. J., Shimbo K., Agard N. J., Shimada K., Rheingold A. L., Stockwell B. R., Weissman J. S., and Wells J. A. (2014) Unraveling the mechanism of cell death induced by chemical fibrils. Nat. Chem. Biol. 10, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Colby D. W., Zhang Q., Wang S., Groth D., Legname G., Riesner D., and Prusiner S. B. (2007) Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. U.S.A. 104, 20914–20919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nussbaum J. M., Schilling S., Cynis H., Silva A., Swanson E., Wangsanut T., Tayler K., Wiltgen B., Hatami A., Rönicke R., Reymann K., Hutter-Paier B., Alexandru A., Jagla W., Graubner S., et al. (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Qiang W., Yau W. M., and Tycko R. (2011) Structural evolution of Iowa mutant β-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J. Am. Chem. Soc. 133, 4018–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cohen S. I., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., and Knowles T. P. (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Esler W. P., Stimson E. R., Jennings J. M., Vinters H. V., Ghilardi J. R., Lee J. P., Mantyh P. W., and Maggio J. E. (2000) Alzheimer's disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39, 6288–6295 [DOI] [PubMed] [Google Scholar]
- 67. Nguyen P. H., Li M. S., Stock G., Straub J. E., and Thirumalai D. (2007) Monomer adds to preformed structured oligomers of Aβ-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. U.S.A. 104, 111–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. O'Brien E. P., Okamoto Y., Straub J. E., Brooks B. R., and Thirumalai D. (2009) Thermodynamic perspective on the dock-lock growth mechanism of amyloid fibrils. J. Phys. Chem. B 113, 14421–14430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Paravastu A. K., Leapman R. D., Yau W. M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qiang W., Kelley K., and Tycko R. (2013) Polymorph-specific kinetics and thermodynamics of β-amyloid fibril growth. J. Am. Chem. Soc. 135, 6860–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang S., Andreasen M., Nielsen J. T., Liu L., Nielsen E. H., Song J., Ji G., Sun F., Skrydstrup T., Besenbacher F., Nielsen N. C., Otzen D. E., and Dong M. (2013) Coexistence of ribbon and helical fibrils originating from hIAPP(20–29) revealed by quantitative nanomechanical atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 2798–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Spirig T., Ovchinnikova O., Vagt T., and Glockshuber R. (2014) Direct evidence for self-propagation different amyloid-β fibril conformations. Neurodegener. Dis. 14, 151–159 [DOI] [PubMed] [Google Scholar]
- 73. Petkova A. T., Yau W. M., and Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry 45, 498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Qiang W., Yau W. M., Luo Y., Mattson M. P., and Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sgourakis N. G., Yau W. M., and Qiang W. (2015) Modeling an in-register, parallel “Iowa'' Aβ fibril structure using solid-state NMR data from labeled samples with Rosetta. Structure 23, 216–227 [DOI] [PubMed] [Google Scholar]
- 76. Akinlolu R. D., Nam M., and Qiang W. (2015) Competition between fibrillation and induction of vesicle fusion for the membrane-associated 40-residue β-amyloid peptides. Biochemistry 54, 3416–3419 [DOI] [PubMed] [Google Scholar]
- 77. Shim S. H., Gupta R., Ling Y. L., Strasfeld D. B., Raleigh D. P., and Zanni M. T. (2009) Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 6614–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tanaka M., and Komi Y. (2015) Layers of structure and function in protein aggregation. Nat. Chem. Biol. 11, 373–377 [DOI] [PubMed] [Google Scholar]
- 79. Kummer M. P., Hermes M., Delekarte A., Hammerschmidt T., Kumar S., Terwel D., Walter J., Pape H. C., König S., Roeber S., Jessen F., Klockgether T., Korte M., and Heneka M. T. (2011) Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 71, 833–844 [DOI] [PubMed] [Google Scholar]
- 80. Jan A., Hartley D. M., and Lashuel H. A. (2010) Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer's disease research. Nat. Protoc. 5, 1186–1209 [DOI] [PubMed] [Google Scholar]