

Abstract
Mechanistic target of rapamycin complex 1 (mTORC1) controls biosynthesis and has been implicated in uncontrolled cell growth in cancer. Although many details of mTORC1 regulation are well understood, a systems-level, predictive framework synthesizing those details is currently lacking. We constructed various mathematical models of mTORC1 activation mediated by Akt and aligned the model outputs to kinetic data acquired for growth factor-stimulated cells. A model based on a putative feedforward loop orchestrated by Akt consistently predicted how the pathway was altered by depletion of key regulatory proteins. Analysis of the successful model also elucidates two dynamical motifs: neutralization of a negative regulator, which characterizes how Akt indirectly activates mTORC1, and seesaw enzyme regulation, which describes how activated and inhibited states of mTORC1 are controlled in concert to produce a nonlinear, ultrasensitive response. Such insights lend quantitative understanding of signaling networks and their precise manipulation in various contexts.
Keywords: Akt PKB, mammalian target of rapamycin (mTOR), mathematical modeling, protein kinase, signal transduction, feedforward loop
Introduction
Composed of the protein kinase mechanistic target of rapamycin (mTOR)2 along with four other proteins that modulate substrate recognition, mTORC1 controls diverse functions in mammalian cells. Notably, mTORC1 is activated in response to growth factor stimulation and nutrient availability, promoting the growth of normal and transformed cells at the level of protein and lipid synthesis (1, 2). Activation of mTORC1 is mediated by the small GTPase, Ras homology enriched in brain (Rheb), which is negatively regulated by the GTPase-activating protein activity of tuberous sclerosis complex (TSC1/2) (3–6). The active, GTP-bound form of Rheb accumulates when TSC1/2 is deactivated by phosphorylation of TSC2 on regulatory sites. Activation of phosphoinositide 3-kinase (PI3K) and Akt, leading to TSC2 phosphorylation, is the canonical upstream pathway (7, 8). Studies also link extracellular signal-regulated kinase (ERK) signaling to inactivating phosphorylation of TSC2 (by ERK1/2 and by the ERK substrate, ribosomal s6 kinase (RSK)) (9–13). Another complication is that there is a second mechanism by which Akt positively influences mTORC1 activity. Akt phosphorylates proline-rich Akt substrate of 40 kDa (PRAS40), an obligate member of mTORC1 that blocks mTOR kinase activity, leading to sequestration of PRAS40 by 14-3-3 proteins (14–16). Akt-mediated neutralization of either TSC1/2 or PRAS40 might be sufficient for activation of mTORC1, and one or the other mechanism might prove dominant in a certain cell/environmental context; however, we considered and tested the alternate possibility that both mechanisms orchestrate mTORC1 activation, as suggested by an important study using purified proteins (14). In the parlance of systems biology, this is recognized as a coherent feedforward loop. To dissect the complexities of mTORC1 regulation, we adopted a quantitative approach, integrating experimental measurements and perturbations together with computational modeling and analysis (17–19).
Materials and Methods
Cell Culture
NIH 3T3 mouse fibroblasts were acquired from the American Type Culture Collection (Manassas, VA). The cells were cultured at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, and the antibiotics penicillin and streptomycin. All tissue culture reagents were purchased from Invitrogen.
Antibodies and Other Reagents
Human recombinant platelet-derived growth factor BB (PDGF-BB) was purchased from PeproTech (catalog number 100-14B). Antibodies acquired from Cell Signaling Technology are against ERK1/2 (catalog number 9107), MEK1/2 (catalog number 9126), Akt (catalog number 9272), ribosomal S6 kinase 1 (S6K1) (catalog number 2708), TSC2 (catalog number 4308), PRAS40 (catalog number 2691), β-actin (catalog number 4970), ERK Thr(P)202/Tyr(P)204 (catalog number 9101), MEK Ser(P)217/Ser(P)221 (catalog number 9121), Akt Ser(P)473 (catalog number 9271), S6K1 Thr(P)389 (catalog number 9234), TSC2 Thr(P)1462 (catalog number 3617), PRAS40 Thr(P)246 (catalog number 2997), and PRAS40 Ser(P)183 (catalog number 5936). Antibodies against Rheb were purchased from Santa Cruz Biotechnology (catalog number sc-271509). The pharmacological inhibitors U0126 and Akt inhibitor VIII were from Calbiochem (catalog numbers 662005 and 124017), and rapamycin was purchased from Sigma-Aldrich (catalog number R8781). Cells were preincubated with the indicated inhibitor or DMSO control for 40 min prior to growth factor stimulation. The concentrations of the inhibitors used were 10 μm U0126, 10 μm Akt inhibitor VIII, and 100 nm rapamycin. Lipofectamine and PLUS transfection reagents were purchased from Invitrogen (catalog numbers 18324012 and 11514-015). Except where noted otherwise, all other reagents were purchased from Sigma-Aldrich.
Lysate Preparation and Immunoblotting
Cells were grown to ∼80% confluence in 60-mm dishes and serum-starved for 4 h prior to stimulation. Cells were treated with various pharmacological inhibitors or DMSO vehicle control for ∼40 min prior to growth factor stimulation. Cells were lysed in 50 mm HEPES, 100 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm Na3VO4, 50 mm β-glycerophosphate, 5 mm NaF, 1 mm EGTA, 10 mm sodium pyrophosphate, and 10 μg/ml each of aprotinin, leupeptin, pepstatin A, and chymostatin. An aliquot of each clarified lysate was prepared for standard SDS-PAGE, transfer to polyvinylidene fluoride membrane (Immobilon, EMD Millipore), and immunoblotting with primary and horseradish peroxidase (HRP)-conjugated secondary antibodies and SuperSignal West Femto substrate (Thermo Fisher). Multiple blots comparing lysates prepared on the same day, representing either different inhibitor treatments or different cell variants and respective control conditions, were performed in parallel and exposed at the same time. Chemiluminescence was measured using a SYNGENE G:BOX Chemi XRQ digital imaging system. Densitometry analysis was performed with local background subtraction using Quantity One software (Bio-Rad Laboratories Inc.). All blots yielded single bands with the mobilities expected, and the linearity of the densitometry measurements with respect to analyte amounts was confirmed. Immunoblotting data were first normalized by appropriate loading controls measured for the same lysates. The phospho-protein amount was normalized by the total amount of the same protein, except in the case of phospho-TSC2; blotting of total TSC2 yielded unacceptable variability, and hence total ERK1/2 was used instead. To trend-normalize the data across independent experiments, the data were further scaled by the mean of a small subset of the samples (see Fig. 1B: 1 nm PDGF time course (0, 5, 15, 30, 60, and 120 min); see Fig. 3A: shNEG time course for each comparison).
FIGURE 1.
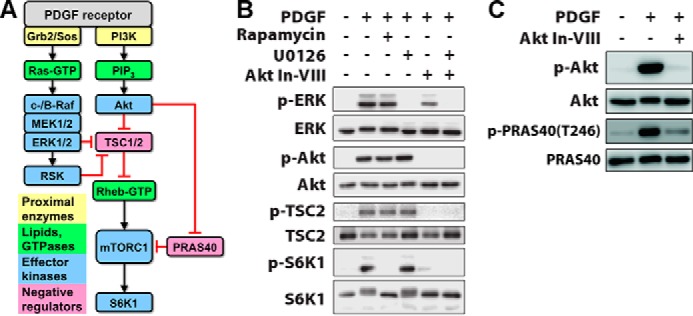
Delineation of signaling pathways that mediate growth factor-stimulated mTORC1 activation. A, mTORC1 is activated via Akt phosphorylation and neutralization of negative regulators, TSC2 and PRAS40. ERK-dependent neutralization of TSC2 has also been reported. The black arrows and red bars signify positive and negative regulation, respectively. PIP3, phosphoinositide 3,4,5-trisphosphate. B, in NIH 3T3 cells, PDGF-BB stimulation (1 nm, 15 min) elicits phosphorylation (p) of ERK, Akt, TSC2, and S6K1; the latter is blocked by rapamycin (100 nm). The MEK inhibitor U0126 (10 μm) blocks ERK activation but has no discernible effect on mTORC1-dependent S6K1 phosphorylation. Akt inhibitor VIII (Akt In-VII, 10 μm) blocks the phosphorylation of Akt and TSC2 as expected and ablates S6K1 phosphorylation. C, Akt inhibitor VIII (10 μm) also blocks phosphorylation of PRAS40 Thr246 as expected.
FIGURE 3.
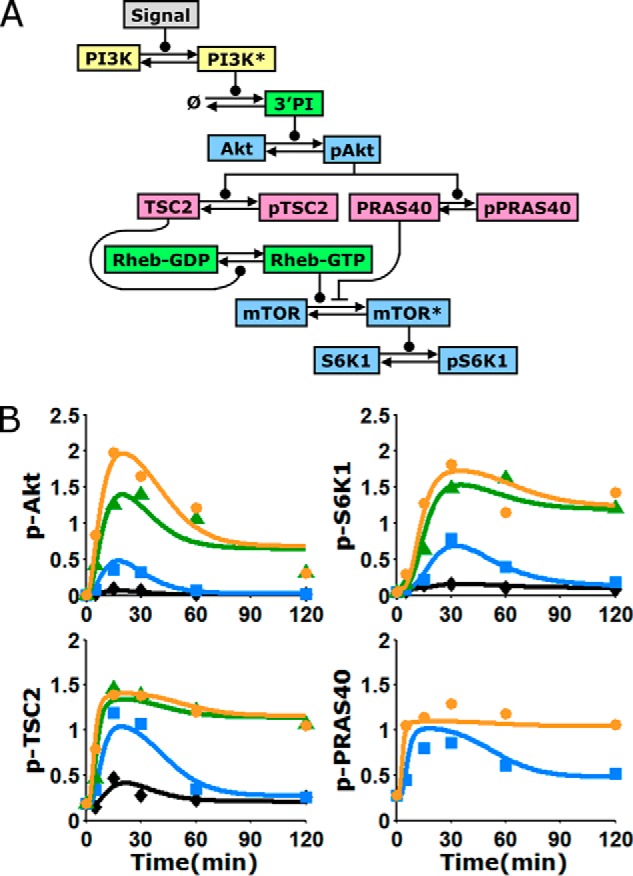
Kinetic model of the Akt/mTORC1 signaling axis, trained on quantitative data. A, schematic of the kinetic model, composed of ordinary differential equations in time. Arrows indicate transformations; connectors ending in circles and bars signify positive and negative regulation of the transformation, respectively. Asterisks signify active kinases; PI, phosphoinositides. B, alignment of the model to the data means presented in Fig. 2B (symbols). The curves represent the mean of the fit to the data, averaging over a large ensemble of parameter sets (n = 10,000; see supplemental Fig. S1 for more detail). See legend in Fig. 2 for explanations of colors and symbols.
Plasmids and Lentiviral Infections
pLKO-puro vectors containing short hairpin sequences targeting Mus musculus TSC2 (5′-ATATCAAGTTTAAGAGAGAGG-3′ and 5′-AATGAGGCTCTCATACACTCG-3′), Rheb (5′-AAGACTTTCCTTGTGAAGCTG-3′ and 5′-AAATTGGCCTTCAACAAACTG-3′), and PRAS40 (5′-TTCTGGAAGTCGCTGGTATTG-3′ and 5′-TAATATTTCCGCTTCAGCTTC-3′) were acquired from the UNC Lenti-shRNA Core Facility (UNC Chapel Hill, Chapel Hill, NC). Lentivirus was produced by lipofection of 293T cells with shRNA-containing vector and the packaging plasmids pCMV-VSVG (8454; Addgene) and pCMV-DR8.91. The control plasmid containing a nontargeting shRNA sequence (5′-TTATCGCGCATATCACGCG-3′), pLKO-shNEG-puro (Everett (31)), and pCMV-DR8.91 were gifts from R. Everett (Medical Research Council–University of Glasgow Centre for Virus Research, Glasgow, Scotland, UK). Virus was harvested 24, 48, and 72 h after transfection, and the pooled, conditioned medium was supplemented with 8 μg/ml Polybrene before adding to target cells. After incubating for 24 h, the infected cells were selected in growth medium supplemented with 2 μg/ml puromycin (Fisher Scientific).
Computational Modeling and Analysis
Formulation of the model equations, the algorithm for acquiring parameter set ensembles, the variations of the model that were tested, generation of model predictions, and mathematical analysis of the model are described in detail in supplemental Text S1.
Results
In Growth Factor-stimulated Cells with Strong Activation of Both Akt and ERK, mTORC1 Activation Is Predominantly Akt-dependent
We studied mTORC1 signaling dynamics in mouse fibroblasts stimulated with PDGF (Fig. 1A). In this system, both the Ras/ERK and PI3K/Akt pathways are strongly activated, as we have characterized in quantitative detail (20–22). Moreover, PDGF receptors mediate activation of type IA PI3Ks via direct, high-avidity recruitment of the p85 regulatory subunits (23), thus avoiding the complication of negative feedback affecting the adaptor protein, insulin receptor substrate 1 (IRS)-1 (24, 25). Chemical inhibitors were used to assess the relative contributions of Ras/ERK and PI3K/Akt pathways to mTORC1 (Fig. 1B). PDGF stimulated robust phosphorylation of S6K1 Thr389, a quintessential substrate of mTORC1, whereas this response was completely blocked by the mTORC1-specific inhibitor, rapamycin. This treatment had no discernible effect on ERK or Akt phosphorylation (even for different PDGF doses and stimulation times; data not shown), consistent with the expected lack of negative feedback. An inhibitor that antagonizes the pleckstrin homology domain of Akt blocked PDGF-stimulated phosphorylation of Akt, TSC2 Thr1462, and PRAS40 Thr246 as expected (Fig. 1, B and C), and this treatment ablated S6K1 phosphorylation as well; in contrast, a MEK inhibitor that blocks activation of ERK had no discernible effect on phospho-S6K1 (Fig. 1B). These results establish PI3K/Akt signaling as the predominant pathway to mTORC1 in our system.
Quantification of Akt/mTORC1 Signaling Dynamics Defines Distinct Kinetic Features
This pathway was probed further at the level of system dynamics, considering phosphorylated Akt as the input, phosphorylation of S6K1 as the output, and Akt-dependent phosphorylation of TSC2 and of PRAS40 as key, intermediate steps (Fig. 2). The PDGF dose and stimulation time were systematically varied, and immunoblotting was carefully performed (Fig. 2A) to acquire normalized, quantitative data that reveal a number of characteristic features. Consistent with previous work, Akt phosphorylation is transient, which has been attributed to PDGF depletion and down-regulation of PDGF receptors, showing dose saturation at ∼0.3 nm PDGF-BB (21). S6K1 phosphorylation shows a similar pattern, except with a noticeable kinetic delay and a less dramatic adaptation of the maximal response when compared with Akt (Fig. 2B). By comparison, it is curious that Akt-dependent regulation of TSC2 (Thr(P)1462) and PRAS40 (Thr(P)246) exhibits greater sensitivity to PDGF dose than either the input or the output of the pathway, judging from the relative saturation of the responses observed with 0.03 nm versus 1 nm PDGF-BB; we also examined phosphorylation of PRAS40 on another regulatory site, Ser183 (26), which increased only modestly in response to PDGF stimulation (Fig. 1B). These distinct kinetics and dose responses suggest that Akt controls mTORC1 activation via mechanisms with complex or counterintuitive properties.
FIGURE 2.

Dynamics of growth factor-stimulated Akt/mTORC1 signaling. A and B, representative immunoblots (A) and quantification (B) of Akt, TSC2, PRAS40, and S6K1 phosphorylation (p) as a function of time and PDGF-BB dose as indicated. The data are normalized by loading controls and then by the mean value of the 1 nm time course for each phospho-protein. The normalized data are reported as mean ± S.E. (n = 5 independent experiments).
A Mechanistic Model of mTORC1 Regulation Captures the Dynamics of the Pathway and Offers a Platform for Quantitative Predictions
To evaluate whether or not the kinetics of the Akt/mTORC1 signaling axis are consistent with the current knowledge of mTORC1 regulation, a simplified mass action model composed of differential equations in time was formulated. The model describes, with a minimal number of adjustable parameters, the recruitment of PI3K, accumulation of phosphoinositides, activation of Akt, parallel phosphorylation of TSC2 and PRAS40 by Akt, and effect of TSC2 phosphorylation on Rheb-GTP (Fig. 3A). To model the activity of mTORC1, leading to phosphorylation of S6K1, we considered mTOR to be in pseudo-equilibrium with two entities. One is unphosphorylated PRAS40, an obligate member of the mTORC1 complex. The other represents either the physical binding of Rheb-GTP or, because the mechanism is currently unclear, an indirect yet proportional effect of Rheb exerted on the complex, e.g. by affecting substrate binding. Importantly, the model allows for these interactions to range from mutually exclusive to synergistic, and thus it can be used to predict the nature of mTORC1 regulation.
A Monte Carlo algorithm was used to directly and globally align the model and data set (20, 21). The goal of this exercise was not to identify a single parameter set that fits the data best (arguably, a fruitless task for models with even modest complexity). Rather, the algorithm collects a large ensemble of parameter sets that fit the data near optimally. This approach allows one to analyze the distributions of the parameter values (supplemental Text S1), but more importantly, it can be used to generate a corresponding ensemble of predictions for various hypothetical scenarios. Before applying the model in this manner, we judged the quality of fit and deemed that the ensemble average of the aligned model output captures all of the aforementioned features of the training data (Fig. 3B). Hence, we assert that the data are indeed consistent with the biochemical mechanisms encoded by the model equations. Alternative models, in which it is assumed that mTORC1 is regulated solely by TSC1/2 or by PRAS40, did not yield a better fit to the data (supplemental Text S1).
Perturbations of Feedforward mTORC1 Regulation Are Consistent with Model Predictions
With a consistent model of the pathway, we sought to analyze the contributions of TSC1/2 and PRAS40 in the putative feedforward loop from Akt to mTORC1. To do so, we designed experiments to perturb those contributions, and in parallel, we used the model and its established ensemble of parameter sets to predict the effects of those perturbations. Seven cell lines were established, each with stable expression of a distinct shRNA: two targeting sequences for each of TSC2, PRAS40, and Rheb, and a non-targeting control. For each targeting shRNA, the extent of protein depletion/knockdown was quantified by immunoblotting, as was the attendant effect on PDGF-stimulated S6K1 phosphorylation kinetics. Depletion of either TSC2 or PRAS40 significantly enhanced S6K1 phosphorylation in cells stimulated with a moderate dose (0.03 nm) of PDGF, whereas depletion of Rheb diminished the response in cells stimulated with a saturating dose (Fig. 4A). In all cases, the effect was magnified according to the extent of protein depletion. We conclude that both modes of mTORC1 regulation, TSC1/2 regulation of Rheb and direct antagonism by PRAS40, contribute substantially in this system.
FIGURE 4.

Perturbation of mTORC1 regulation alters system dynamics, in accordance with model predictions. A, stable shRNA-expressing cell lines were established to perturb key regulatory proteins in the Akt/mTORC1 axis: TSC2, PRAS40, and Rheb. For each target, two hairpins targeting different sequences were selected, based on the extent of protein knockdown (% KD, as indicated), along with a non-targeting control line (shNEG). Representative immunoblots and quantification of target protein knockdown and S6K1 phosphorylation (p) for the indicated PDGF stimulation conditions are shown. Phospho-S6K1 data are normalized by total S6K1 and then by the mean value of each shNEG time course. The normalized data are reported as mean ± S.E. (n = 3 independent experiments). B, the corresponding model predictions are the means of the model ensemble (n = 10,000) for each condition, normalized in the same manner as the data. C, model predictions as in B, using an alternative parameter set ensemble acquired by fitting the data as in Fig. 3B but with the parameter constraints α = β = 1.
Turning to the model, we accounted for the estimated degree of protein knockdown in each cell line as a corresponding fractional change in the associated kinetic parameter. Adjusted thus, each parameter set in the ensemble was used to predict the phospho-S6K1 time course for each scenario relative to the unperturbed control (the latter time course being used to normalize both the data and the model predictions) (Fig. 4B). The a priori predictions stand in semi-quantitative agreement with the experimental data, validating the modeling approach. Apparently, the dynamic response of the pathway to changing Akt activity, affected by the PDGF dose in the training data set, consistently reflects the sensitivities of mTORC1 to regulation by TSC1/2/Rheb and PRAS40, as probed by the shRNA depletion studies.
Two key parameters of the model, α and β, describe how PRAS40 binding regulates mTORC1. The parameter α describes the activity of Rheb-activated mTORC1 with PRAS40 absent relative to a hypothetical state in which mTORC1 is both Rheb-activated and PRAS40-bound. A value of α > 0.5 indicates that PRAS40 binding reduces mTORC1 activity, representing a non-competitive mode of regulation. The parameter β represents the cooperativity of Rheb activation and PRAS40 binding. If β < 1, the two processes are negatively cooperative, representing a competitive mode of regulation by PRAS40 (whereas the processes are independent if β = 1). As discussed in more depth below, the algorithm used to acquire parameter sets consistently selected low values of β (all 104 values are < 0.01), implying that PRAS40 and Rheb-GTP interactions with mTORC1 are mutually exclusive. A separate fit to the training data set was performed with the constraints α = β = 1, considering the non-competitive regulation mechanism. Although the fit to the training data set was only marginally worse with these constraints (supplemental Text S1 and supplemental Fig. S1), the resulting ensemble of parameter sets yielded predictions with a visibly poorer match to the shRNA-mediated depletion experiments (Fig. 4C).
Analysis of the Model Reveals Neutralization of a Negative Regulator and Seesaw Regulation as Dynamical Motifs of the Akt/mTORC1 Signaling Axis
To gain further insight into the model predictions, we performed a computational analysis of the steady state, with the variable representing Akt activity artificially held at various fixed values. As anticipated based on inspection of the kinetic data, phosphorylation of TSC2 and of PRAS40 is predicted to be readily saturated with increasing Akt activity, whereas the predicted responses of Rheb-GTP and mTORC1 activity are far less so (Fig. 5A). This is attributed to the indirect mechanism of mTORC1 activation, whereby phosphorylation by Akt neutralizes the negative regulators TSC1/2 and PRAS40. We recently proposed deactivation/neutralization of a negative regulator as a common but as yet underappreciated motif in signaling networks (27); the mechanism yields a broader range of sensitivity to the input, beyond saturation of regulator phosphorylation, because it is the unphosphorylated state of the regulator that exerts influence on the pathway (Fig. 5A). This property of the motif is manifest in the model predictions and, accordingly, explains the ability of the model to capture complex features of the kinetic data.
FIGURE 5.
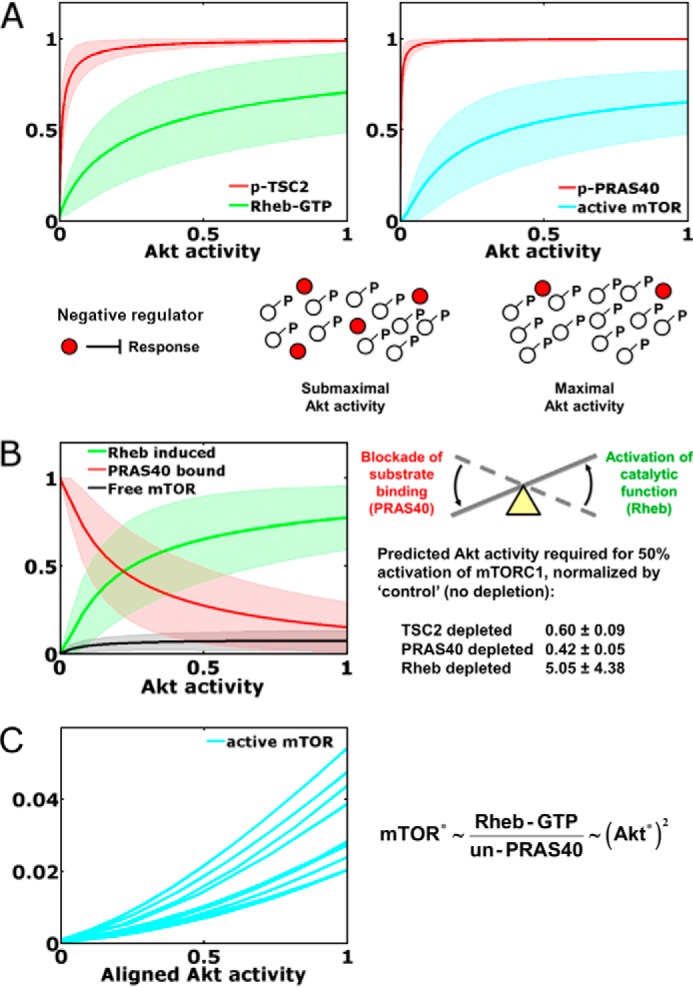
Dynamical motifs encoded in Akt/mTORC1 signaling dynamics. A, neutralization of negative regulators. Ensemble predictions of the model at steady state (mean ± S.D., n = 10,000) show that phosphorylation (p) of the negative regulators TSC2 and PRAS40 approach saturation at low stoichiometries of Akt activation. This offers maximal sensitivity of Rheb-GTP loading and mTORC1 activation, because TSC2 and PRAS40 regulate those responses in their unphosphorylated states. B, seesaw regulation of mTORC1. Steady-state analysis of the model predicts the status of the mTORC1 complex (mean ± S.D., n = 10,000), with a transition from largely PRAS-inhibited and largely Rheb-induced states. Also shown are the model-predicted Akt activity levels, normalized by that of the control, required to elicit half-maximal activation of mTORC1 in cells with TSC2, PRAS40, or Rheb depleted by a nominal 80% (mean ± S.D., n = 10,000). C, as shown for 10 randomly selected parameter sets, seesaw regulation of mTORC1 encodes an ultrasensitive, nonlinear input-output relationship over the relevant range of Akt activity (i.e. with Akt activity scaled as it was in the alignment to phospho-Akt data, where a value of 1 corresponds to the mean of the 1 nm PDGF time course). Asterisks indicate kinase activity. UN-PRAS40, unphosphorylated PRAS40.
We further used the steady-state analysis to explore the status of the mTORC1 complex, revealing another characteristic pattern (Fig. 5B). The model predicts that almost all of the mTORC1 is either PRAS40-bound or in the Rheb-induced state, rather than the so-called free state (neither PRAS-bound nor Rheb-induced). Thus, the implication is that Akt exerts feedforward control of mTORC1 activity via a “seesaw” regulation mechanism, which we define as the concomitant dissociation of PRAS40 and activation of mTORC1 catalytic function through Rheb. Accordingly, the interpretation of the model is that depletion of TSC2, PRAS40, or Rheb perturbs that balance to differing extents, with depletion of Rheb exerting the largest effect (Fig. 5B). What quantitative feature of this regulation mechanism is encoded in the kinetic data? A signature of this mode of regulation is a nonlinear, ultrasensitive response of mTORC1 activity to Akt activation (Fig. 5C). As shown through a mathematical analysis (supplemental Text S1), the maximum sensitivity of the pathway is achieved when mTORC1 activity is proportional to the Rheb-GTP level and inversely proportional to the concentration of unphosphorylated PRAS40; given that PRAS40 phosphorylation is close to saturation, mTORC1 activity is roughly proportional to the Akt activity squared (Fig. 5C), i.e. with a Hill coefficient approaching 2.
As mentioned in the previous section, the value of the parameter that determines competition versus synergy between the PRAS40-bound and Rheb-induced states of mTORC1 (β) was consistently chosen such that the two states are mutually exclusive. According to the same mathematical analysis noted above, this is not a necessary condition for ultrasensitivity; the scenario in which PRAS40 regulation of mTORC1 is non-competitive is also capable of ultrasensitivity and is therefore only subtly different from the situation favored by the fitting algorithm.
Discussion
The Akt/mTORC1 signaling axis has attracted great interest because it affects cell growth in normal and cancer cells. In the context of growth factor stimulation, integration of quantitative measurements and mechanistic modeling elucidated the dose-dependent kinetics of the Akt/mTORC1 signaling axis, and the computational model consistently predicted how those kinetics were perturbed experimentally, via shRNA depletion of key regulatory proteins. Thus, the model accounts for the parallel contributions of TSC1/2/Rheb and PRAS40; we have shown that, together, these mechanisms constitute feedforward regulation of mTORC1. Further, model-driven analysis of the pathway revealed two dynamical motifs. One is the neutralization of a negative regulator, which applies to both arms of the feedforward loop; this motif has the distinct property that it gains sensitivity as the neutralizing modification (in this case, phosphorylation by Akt) approaches saturation (27), explaining key features of the pathway kinetics. The other motif is seesaw regulation, which describes how indirect activation of Rheb and the neutralization of PRAS40 converge to modulate mTORC1 activity in an ultrasensitive manner. Taken together, these systems-level insights offer a framework for comparing mTORC1 regulation across various physiological contexts and in cancer cells with misregulated/rewired signaling networks.
One specific prediction of the model that warrants further discussion concerns the state of the mTORC1 complex. Although the precise mechanism by which Rheb activates mTORC1 is still debated (28), the evidence is consistent with enhanced binding/access by mTORC1 substrates (29, 30). This concept is consistent with the seesaw model, given that bound PRAS40 exerts the opposite effect on substrate binding (16). The implication that Rheb and PRAS40 interactions with mTORC1 are mutually exclusive is also supported by experimental findings using purified proteins; Rheb-GTP, when present in vast excess, can counteract the inhibitory effect of PRAS40 (14). Another distinct possibility is that the Rheb-induced state defined in the model represents mTORC1 in complex with its various substrates, which directly compete with PRAS40 for mTORC1 binding (16).
Author Contributions
J. M. H. conceived and coordinated the study. A. R. performed and analyzed all of the experiments and computations. Both authors wrote the paper.
Supplementary Material
This work was supported in part by National Institutes of Health Grant R01-GM088987 (to J. M. H.) and National Science Foundation Grant 1330746. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Text S1 and supplemental Fig. S1.
- mTOR
- mechanistic target of rapamycin.
References
- 1. Shaw R. J., and Cantley L. C. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430 [DOI] [PubMed] [Google Scholar]
- 2. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garami A., Zwartkruis F. J., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S. C., Hafen E., Bos J. L., and Thomas G. (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 [DOI] [PubMed] [Google Scholar]
- 4. Inoki K., Li Y., Xu T., and Guan K. L. (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tee A. R., Manning B. D., Roux P. P., Cantley L. C., and Blenis J. (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 6. Li Y., Inoki K., and Guan K. L. (2004) Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol. Cell. Biol. 24, 7965–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manning B. D., and Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dibble C. C., and Cantley L. C. (2015) Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ballif B. A., Roux P. P., Gerber S. A., MacKeigan J. P., Blenis J., and Gygi S. P. (2005) Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. U.S.A. 102, 667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rolfe M., McLeod L. E., Pratt P. F., and Proud C. G. (2005) Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2). Biochem. J. 388, 973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma L., Chen Z., Erdjument-Bromage H., Tempst P., and Pandolfi P. P. (2005) Phosphorylation and functional inactivation of TSC2 by Erk: implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 [DOI] [PubMed] [Google Scholar]
- 12. Ma L., Teruya-Feldstein J., Bonner P., Bernardi R., Franz D. N., Witte D., Cordon-Cardo C., and Pandolfi P. P. (2007) Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 67, 7106–7112 [DOI] [PubMed] [Google Scholar]
- 13. Huang J., and Manning B. D. (2008) The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., and Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 15. Vander Haar E., Lee S. I., Bandhakavi S., Griffin T. J., and Kim D. H. (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323 [DOI] [PubMed] [Google Scholar]
- 16. Wang L., Harris T. E., Roth R. A., and Lawrence J. C. Jr. (2007) PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 282, 20036–20044 [DOI] [PubMed] [Google Scholar]
- 17. Janes K. A., and Lauffenburger D. A. (2013) Models of signalling networks: what cell biologists can gain from them and give to them. J. Cell Sci. 126, 1913–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Purvis J. E., and Lahav G. (2013) Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kolch W., Halasz M., Granovskaya M., and Kholodenko B. N. (2015) The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 15, 515–527 [DOI] [PubMed] [Google Scholar]
- 20. Wang C.-C., Cirit M., and Haugh J. M. (2009) PI3K-dependent crosstalk interactions converge with Ras as quantifiable inputs integrated by Erk. Mol. Syst. Biol. 5, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cirit M., Wang C.-C., and Haugh J. M. (2010) Systematic quantification of negative feedback mechanisms in the extracellular signal-regulated kinase (ERK) signaling network. J. Biol. Chem. 285, 36736–36744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ahmed S., Grant K. G., Edwards L. E., Rahman A., Cirit M., Goshe M. B., and Haugh J. M. (2014) Data-driven modeling reconciles kinetics of ERK phosphorylation, localization, and activity states. Mol. Syst. Biol. 10, 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Escobedo J. A., Navankasattusas S., Kavanaugh W. M., Milfay D., Fried V. A., and Williams L. T. (1991) cDNA cloning of a novel 85 kd protein that has SH2 domains and regulates binding of PI3-kinase to the PDGF β-receptor. Cell 65, 75–82 [DOI] [PubMed] [Google Scholar]
- 24. Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., Gout I., Downes C. P., and Lamb R. F. (2004) The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manning B. D., Logsdon M. N., Lipovsky A. I., Abbott D., Kwiatkowski D. J., and Cantley L. C. (2005) Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 19, 1773–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nascimento E. B., Snel M., Guigas B., van der Zon G. C., Kriek J., Maassen J. A., Jazet I. M., Diamant M., and Ouwens D. M. (2010) Phosphorylation of PRAS40 on Thr246 by PKB/AKT facilitates efficient phosphorylation of Ser183 by mTORC1. Cell. Signal. 22, 961–967 [DOI] [PubMed] [Google Scholar]
- 27. Rahman A., and Haugh J. M. (2014) Deactivation of a negative regulator: a distinct signal transduction mechanism, pronounced in Akt signaling. Biophys. J. 107, L29–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X., and Proud C. G. (2011) mTORC1 signaling: what we still don't know. J. Mol. Cell. Biol. 3, 206–220 [DOI] [PubMed] [Google Scholar]
- 29. Avruch J., Long X., Lin Y., Ortiz-Vega S., Rapley J., Papageorgiou A., Oshiro N., and Kikkawa U. (2009) Activation of mTORC1 in two steps: Rheb-GTP activation of catalytic function and increased binding of substrates to raptor. Biochem. Soc. Trans. 37, 223–226 [DOI] [PubMed] [Google Scholar]
- 30. Sato T., Nakashima A., Guo L., and Tamanoi F. (2009) Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 284, 12783–12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Everett R. D. (2010) Depletion of CoREST does not improve the replication of ICP0 null mutant herpes simplex virus type 1. J. Virol. 84, 3695–3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.