

Abstract
Fast-scan cyclic voltammetry (FCV) is an established method to monitor increases in extracellular dopamine (DA) concentration ([DA]o) in the striatum, which is densely innervated by DA axons. Ex vivo brain slice preparations provide an opportunity to identify endogenous modulators of DA release. For these experiments, local electrical stimulation is often used to elicit release of DA, as well as other transmitters, in the striatal microcircuitry; changes in evoked increases in [DA]o after application of a pharmacological agent (e.g., a receptor antagonist) indicate a regulatory role for the transmitter system interrogated. Optogenetic methods that allow specific stimulation of DA axons provide a complementary, bottom-up approach for elucidating factors that regulate DA release. To this end, we have characterized DA release evoked by local electrical and optical stimulation in striatal slices from mice that genetically express a variant of channelrhodopsin-2 (ChR2). Evoked increases in [DA]o in the dorsal and ventral striatum (dStr and vStr) were examined in a cross of a Cre-dependent ChR2 line (“Ai32” mice) with a DAT::Cre mouse line. In dStr, repeated optical pulse-train stimulation at the same recording site resulted in rundown of evoked [DA]o using heterozygous mice, which contrasted with the stability seen with electrical stimulation. Similar rundown was seen in the presence of a nicotinic acetylcholine receptor (nAChR) antagonist, implicating the absence of concurrent nAChR activation in DA release instability in slices. Rundown with optical stimulation in dStr could be circumvented by recording from a population of sites, each stimulated only once. Same-site rundown was less pronounced with single-pulse stimulation, and a stable baseline could be attained. In vStr, stable optically evoked increases in [DA]o at single sites could be achieved using heterozygous mice, although with relatively low peak [DA]o. Low release could be overcome by using mice with a second copy of the Ai32 allele, which doubled ChR2 expression. The characteristics reported here should help future practitioners decide which Ai32;DAT::Cre genotype and recording protocol is optimal for the striatal subregion to be examined.
Keywords: Ai32, Cre-loxp, fast-scan cyclic voltammetry, local stimulation, neurotransmitter uptake, optogenetics
Introduction
Fast-scan cyclic voltammetry (FCV) has been used to monitor spontaneous and evoked increases in extracellular concentrations of readily oxidizable substances, both in vivo(1) and ex vivo in brain slices.2 Much of the work has focused on dopamine (DA) in the striatum, which has been facilitated by the robust and largely contaminant-free signal from dense innervation by DA axons.3 The most common method used to evoke DA release in brain slices has been local electrical stimulation (eStim). Notably, local eStim activates the release of not only DA, but also other transmitters and modulators from the striatal microcircuitry. This has been used advantageously to identify local factors that modulate evoked increases in extracellular dopamine concentration ([DA]o).4 In such studies, modulation by an endogenously released transmitter is inferred by comparing responses evoked by relatively long pulse-trains consistently delivered to the same recording site, first in the absence and then in the presence of a selective receptor antagonist. Depending on the conditions, the modulation revealed might be either direct or indirect via polysynaptic interactions within a local circuit.
More recently, specific activation of DA axons using light-sensitive ion channels and optical stimulation (oStim) has become available to help elucidate local factors that regulate striatal DA release and determine whether observed regulation is direct or indirect. This is achieved through a simple deductive comparison of results obtained by classical eStim to those obtained by oStim, keeping in mind that although glutamate5,6 and GABA7,8 can be co-released from DA axons with oStim, other potential modulatory substances, like acetylcholine (ACh), are not.9 Optogenetic studies have primarily used vector-based methods10 for genetic access to the cell type of interest. For specific activation of DA release, there are many choices of optogenetic reagents for cell-specific targeting, but usually a Cre-dependent channelrhodopsin-2 (ChR2) encoding construct is delivered by injection of an adeno-associated virus (AAV) into midbrain DA cell body regions.11 The primary target sites for AAV injection are the substantia nigra pars compacta (SNc) projecting to the dorsal striatum (dStr) and the ventral tegmental area (VTA) projecting to the ventral striatum (vStr, including nucleus accumbens shell and core). Viral injection must be done in mice (or rats12) expressing Cre-recombinase in specific cell types, in this case by using a promoter selective for DA neurons. In these systems, optimization and standardization of experimental procedures for oStim are time-consuming and lab-specific, but also very necessary because of the variability in virus titer and other parameters affecting ChR2 expression level after injection. The virus-based approaches, however, do have the advantage of high expression levels and the ability to target anatomically distinct subsets of a given cell type.
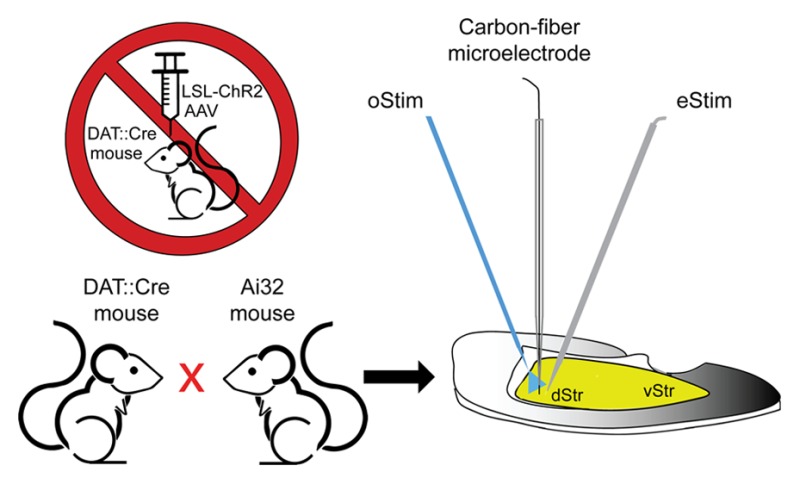
If anatomical dissociation is not necessary, an alternative to viral injection is to cross two transgenic/knock-in mouse lines; one for Cre expression and one for Cre-dependent ChR2 expression. This approach eliminates the need for surgeries and avoids potential side-effects of AAVs.13 One Cre-dependent mouse line introduced for such crosses is called “Ai32” (developed at the Allen Institute14); this line expresses the H134R variant of ChR2 from the otherwise ubiquitously and constitutively expressed ROSA26 locus after removal of an upstream loxP-flanked stop codon (LSL) in Cre-expressing cells. The crossing procedure theoretically should yield more homogeneous expression levels of ChR2 throughout the entire projection field than is possible with viral injection and thus greater reproducibility across laboratories using similar oStim protocols in various experimental contexts. Here, we characterize the suitability of using the progeny from a cross of Ai32 mice with a DA cell-specifying Cre-expressing line that is driven by the DA transporter (DAT) promoter (DAT::Cre mice)15 for oStim of striatal DA axons in FCV studies.
Results and Discussion
One aspect of the DAT::Cre-dependent mouse cross that demanded quantitation was their DAT function, as previous characterization of DAT::Cre mice showed a decrease in the expression level of DAT protein as an apparent side-effect of the genetic manipulation.15 The extent of functional consequences on DAT activity was not examined, however. We therefore first assessed DA uptake characteristics in ex vivo striatal slices from the DAT::Cre heterozygous mice used in the cross. By analyzing the falling phase of the [DA]o release profile evoked by single-pulse eStim recorded at multiple sites in a given slice (multiple-site recording), we found a significant decrease in the maximum DA uptake velocity (Vmax) in DAT::Cre+/– versus littermate control (DAT::Cre–/−) mice in both dStr and vStr core (Figure 1A,B). This was accompanied by a significant increase in peak eStim-evoked [DA]o reached in DAT::Cre+/– mice compared to control mice, within each region (Figure 1A,C). Given that evoked increases in [DA]o reflect the balance between release and uptake at a given site, higher [DA]o would be consistent with the predicted effect of decreased DA uptake.
Figure 1.
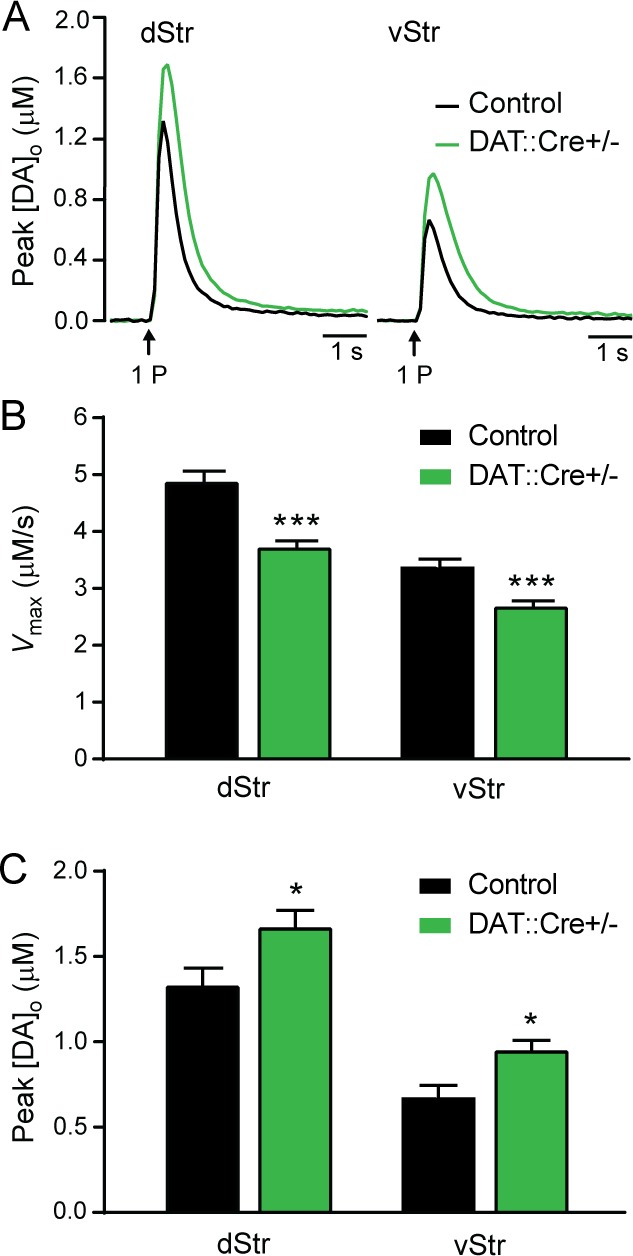
Evaluation of Vmax for DA uptake in control versus DAT::Cre+/– mice. (A) Single-pulse (1P) eStim-evoked [DA]o from multiple-site recording (n = 45–49 records in slices from 4 mice per group) in dStr and vStr (nucleus accumbens core) in control (DAT::Cre–/−) and DAT::Cre+/– mice. (B) Average Vmax for DA uptake computed by a scripted MATLAB analysis of the first 4 data points (separated by 100 ms) after the peak from panel A, fitted to a Michaelis–Menten model that assumes a Km of 0.9 μM for DA at the DAT for each region and genotype (***p < 0.001 vs control). (C) Summary of data in panel A, showing average peak [DA]o evoked in dStr and vStr from control and DAT::Cre+/– mice (*p < 0.05 vs control). Data are means + SEM and comparisons were made using one-way ANOVA followed by Bonferroni’s post hoc tests.
We next set out to characterize ChR2 expression in the Ai32+/–;DAT::Cre+/– mouse cross (referred to hereafter as HET/HET) mice. These mice express a ChR2 fused to the yellow fluorescent protein (YFP). Visual inspection of coronal striatal slices using a fluorescent stereomicroscope showed clear YFP fluorescence with a decreasing dorsal to ventral gradient (Figure 2A). Quantitative Western blot analysis of ChR2 expression in tissue samples from dStr and vStr from HET/HET mice confirmed a regional difference in protein levels, indicated by a significant main effect of region in a two-way ANOVA (F1,20 = 14.09; p < 0.01; Figure 2C). Moreover, these data revealed an age dependent increase in ChR2 expression in the dStr of 10–17 week old mice versus 18–25 week old mice (Figure 2B,C). Expression levels in samples from mice aged 3–5 weeks were significantly lower than the 18–25 week group as well but did not differ from 10–17 week old mice. In contrast, ChR2 expression in the vStr showed no significant changes with age (one-way ANOVA: F2,7 = 0.96, p = 0.43; Figure 2B,C).
Figure 2.
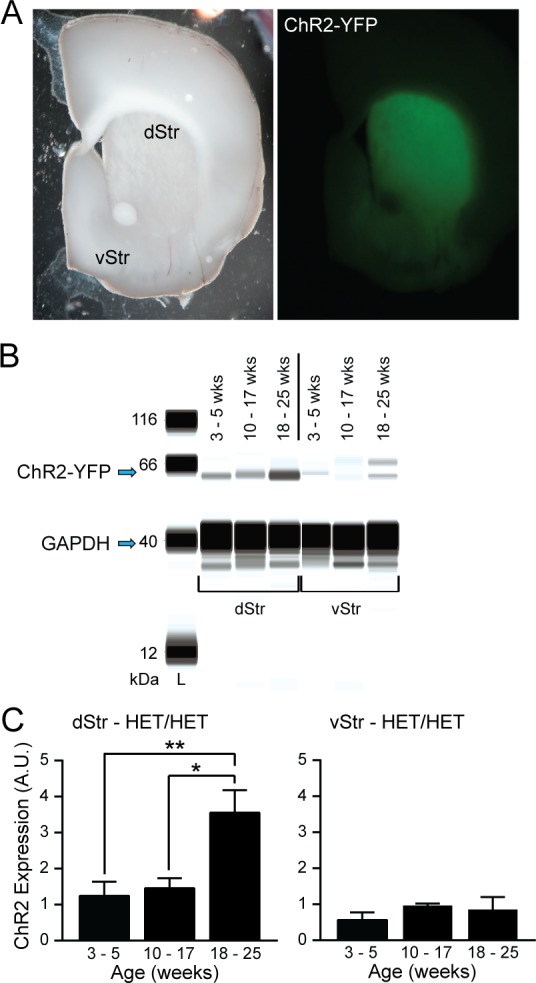
ChR2–YFP expression with age in HET/HET dStr and vStr. (A) Coronal striatal slices viewed through a fluorescent stereomicroscope with either brightfield illumination (left) or a green filter cube to reveal YFP-fused ChR2 expression (right). (B) Example capillary electropherograms from immunoassays (SimpleWestern) for YFP and GAPDH (a loading control protein) for dStr and vStr from 3–5 week old, 10–17 week old, and 18–25 week old HET/HET mice. (C) Summary of YFP expression (as percent of GAPDH) in dStr (left, n = 5–6 samples from 3–4 mice per age group) and vStr (right, n = 3–4 samples from 3–4 mice per age group). Data are means ± SEM and comparisons were made using one-way ANOVA followed by Tukey’s post hoc tests (*p < 0.05, **p < 0.01).
We next compared the characteristics of DA release with pulse-train eStim and oStim in evoked [DA]o as well as the stability of this measure in striatal slices from HET/HET mice. Relatively long trains of eStim pulses (e.g., 30 pulses at 10 Hz) have been used to reveal local circuit-mediated regulation of striatal DA release, because this allows sufficient time for initially released transmitters to act on DA release evoked by subsequent pulses in a train.16,17 We were therefore interested in establishing a similar pulse-train procedure for oStim. In preliminary studies, we determined that the minimal duration and light intensity for oStim that generated a robust but modulatable increase in [DA]o was 250 μs and 400 μW, which was used in most subsequent experiments, including all those using pulse-train stimulation. Increases in [DA]o evoked using these parameters was action potential dependent, as indicated by complete loss of a release response in the presence of tetrodotoxin (TTX; 0.1 μM), a voltage-gated Na+ channel blocker (normalized to peak control response: 100% ± 21% for control vs −0.7% ± 1.2% in TTX; n = 4; t-test, p < 0.001). Indeed, increases in [DA]o evoked by oStim pulses of 5 ms (longest duration tested) are also blocked by TTX.6
We then examined the stability of evoked [DA]o in the dStr using repeated pulse-train stimulation in the same recording site (same-site recording) at 10 min intervals in slices from 8–17 week old HET/HET mice (Figure 3A,B). Consistent with previous results in DAT::Cre mice after viral transfection,18 pulse-train eStim produced a fast-rising increase in [DA]o that was stable with repeated stimulation in the dStr (3 consecutive stimulations with no significant difference in peak; Figure 3A,C). Notably, pulse-train oStim produced release profiles in the dStr that were similarly shaped to those produced by eStim (Figure 3A,B). This preservation of shape suggests that the factors governing the response are intrinsic to DA axons, including DAT-dependent uptake,19,20 D2 DA receptor-dependent autoregulation, and axonal release probability. In contrast to the stability of eStim-evoked increases in [DA]o (Figure 3A,C), however, oStim-evoked [DA]o in this age group diminished progressively (oStim1 vs oStim3, p < 0.01; oStim1 vs oStim4, p < 0.001; repeated-measures ANOVA with Sidak’s multiple comparison test between stimulations) with repeated pulse-train stimulation at the same recording site (Figure 3B,D). Given that much longer stimulus pulses (1–5 ms) have been used for both eStim and oStim,6,7,21 it is not likely that the small difference in oStim and eStim pulse duration used here contributed to this rundown. Because Western blot analysis revealed an age dependent increase in ChR2 expression in the dStr of older (18–42 week) HET/HET mice, we examined whether this might increase stability of evoked [DA]o in this age group. As with younger HET/HET mice, eStim-evoked [DA]o reached stability after the second stimulation (Figure 3C). With oStim, however, evoked [DA]o was still not stable by the fourth stimulation, although the rate of rundown was less than in slices from younger (8–17 week) mice (Figure 3D, Sidak’s test between ages; oStim2, p < 0.05; oStim3, p < 0.01). Indeed there was no difference in evoked [DA]o between the groups by the fourth stimulation (8–17 week oStim4 vs 18–42 week oStim4, p = 0.38), with both showing a significant decline from oStim1 (oStim4 vs oStim1, p < 0.001).
Figure 3.
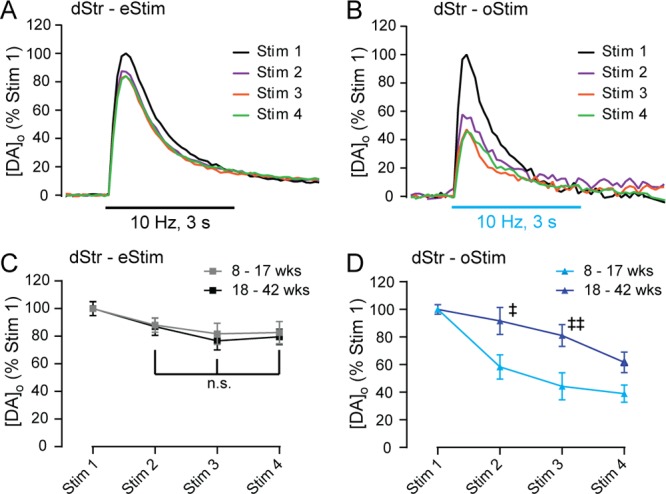
Attenuation of evoked [DA]o in dStr with repeated pulse-train oStim but not eStim. (A) Representative eStim-evoked increases in [DA]o from the first four consecutive pulse-train stimulations (30 pulses, 10 Hz) at 10 min interstimulus intervals at a single recording site in the dStr of a HET/HET mouse, demonstrating relative stability. Records were normalized to peak evoked [DA]o for the first stimulation at each site (Stim 1) and are color coded by stimulation order. (B) Same as in panel A, except that increases in [DA]o were evoked by repeated same-site oStim (30 pulses, 10 Hz; 10 min interstimulus interval) and show marked rundown with successive same-site stimulations. (C) Summary of data in panel A, showing average peak [DA]o evoked by eStim in 8–17 week old mice (gray line, n = 9 sites) versus 18–42 week old mice (black line, n = 7 sites) within single recording sites. (D) Summary of data in panel B, showing average peak [DA]o evoked by oStim in 8–17 week old mice (light blue line, n = 5 sites) versus 18–42 week old mice (dark blue line, n = 9 sites) within single recording sites. Points denote peak responses from four successive stimulations, normalized to Stim 1 of a given stimulus type. Data are means ± SEM and comparisons were made using a two-way repeated-measures ANOVA followed by Sidak’s tests between (‡p < 0.05 8–17 week oStim2 vs 18–42 week oStim1; ‡‡p < 0.01 for a similar between-age comparison at oStim3) and Dunnett’s tests within age groups (n.s., p > 0.05 for all comparisons of eStim2/3/4 with one another).
A possible contributing factor to this limited stability might be impaired maintenance of the readily releasable pool22 because of decreased DAT function (Figure 1) and consequently decreased recycling of DA after release.23 The stability of peak evoked [DA]o with eStim in HET/HET dStr (Figure 3A,C) argues that this is not the primary cause. However, to determine whether rundown with oStim might reflect insufficient time to replenish releasable DA pools during a 10 min stimulus interval, we applied an additional fifth stimulation 20 min after oStim4 in a subset of experiments. We found that there was only a modest 10% recovery in [DA]o evoked by oStim after this longer interstimulus period, which was not statistically different from oStim4 (7 sites, t-test, p > 0.05). Another possible cause of rundown with oStim could be tissue damage from the laser light used to activate ChR2. However, this is unlikely, given that oStim-evoked [DA]o is stable in the vStr (see below). Instead, these data suggest the alternative hypothesis that there may be a neurochemical depression of evoked DA release in dStr in the absence of other factors normally evoked by eStim, particularly ACh, as discussed further below.
The lack of stability of pulse-train oStim-evoked [DA]o within a reasonable time frame or age range precludes use of same-site controls for pharmacological studies of DA release regulation in the dStr. To avoid the confounding influence of rundown with repeated oStim in dStr, we tested the alternative strategy of examining evoked [DA]o from multiple recording sites within a discrete subregion of the striatum. This multiple-site recording minimizes any variance due to heterogeneity in release sites.24,25 We used this strategy in our studies of DA uptake (Figure 1) and in earlier studies that compared single-pulse eStim-evoked [DA]o between slices from control and mutant mice24 or before and after drug application in rat striatal slices.25 We compared peak evoked [DA]o between same-site and multiple-site recording for eStim and oStim in HET/HET dStr (Figure 4). Average peak [DA]o evoked by same-site pulse-train eStim (3–4 stimulations at 7 sites; Figure 4A,D) did not differ significantly from the average multiple-site recordings (n = 14 sites; two-way ANOVA followed by Tukey’s multiple comparison test, p = 0.19), which reflects the relative stability of eStim-evoked peak [DA]o at a given site. In contrast, average peak [DA]o evoked by same-site pulse-train oStim (last 3 stimulations in 13 sites) was significantly lower than the average from multiple-site recording (n = 23 sites; p < 0.001; Figure 4B,D) reflecting the marked rundown with same-site oStim. Thus, multiple-site recording provides a useful alternative to the same-site design when a reliable baseline value prior to pharmacological manipulation is necessary in the dStr of HET/HET mice. Notably, the peak evoked increases in [DA]o were significantly lower with oStim than with eStim (p < 0.05; Figure 4D) with the multiple-site recording protocol and were in fact lower throughout oStim versus eStim trains (two-way repeated-measures ANOVA, stimulus type × time interaction: F29,1044 = 8.49, p < 0.001; Figure 4C).
Figure 4.
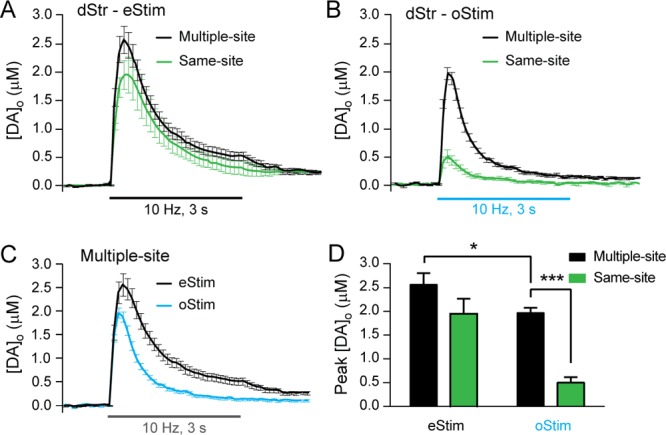
Peak [DA]o evoked by same-site and multiple-site sampling in dStr. (A) Average increases in [DA]o evoked by pulse-train eStim (30 pulses, 10 Hz) from 3–4 stable records per site with repetitive same-site stimulation at 10 min intervals (n = 7 stimulations in slices from 2 mice) or from multiple-site recording (n = 15 stimulations in slices from 4 mice). (B) Average increases in [DA]o evoked by pulse-train oStim (30 pulses, 10 Hz) from the last 3 reliably detectable responses with repetitive same-site stimulation (n = 13 stimulations in slices from 4 mice) or from multiple-site recording (n = 23 sites in slices from 4 mice). (C) Comparison of peak [DA]o evoked by multiple-site recording in HET/HET dStr with eStim (from panel A) and oStim (from panel B). (D) Summary of the data illustrated in panels A–C, showing comparisons of peak evoked [DA]o among various conditions. The multiple-site recording protocol allowed consistent and reliable control data to be obtained with oStim, whereas same-site recording did not. Significantly higher release was also seen with eStim than oStim (see text). Data are means ± SEM, and comparisons were made using a two-way ANOVA followed by Tukey’s post hoc tests (*p < 0.05; ***p < 0.001).
A common factor in the lower stability and decreased peak amplitude of pulse-train oStim- versus eStim-evoked [DA]o is the absence of concurrently released local transmitters with selective oStim of DA axons. The most obvious candidate is ACh, which is well-known to facilitate DA release.9,26 Indeed, low-frequency eStim-evoked [DA]o is markedly decreased in the presence of nicotinic ACh receptor (nAChR) antagonists4 or after genetic deletion of ACh synthesis.27 We examined the influence of the absence of concurrently released ACh with oStim of DA axons using mecamylamine, a nAChR antagonist4,27 (Figure 5). We first examined the influence of nAChR antagonism on peak [DA]o evoked by eStim or oStim in the dStr of HET/HET mice using multiple-site recording. Consistent with previous reports,7,9 averaged peak eStim-evoked [DA]o showed a significant decrease (p < 0.001) in mecamylamine (10 μM), whereas oStim-evoked release was unaltered (p = 0.88; Figure 5A,B). Moreover, the decrease in peak eStim-evoked [DA]o with nAChR antagonism was statistically indistinguishable (t-test on % decrease, p = 0.43) from the difference between peak [DA]o evoked by eStim and oStim in HET/HET dStr (Figure 4C,D), presumably reflecting the lack of ACh facilitation of DA release via nAChRs9,26 with oStim pulses. In contrast to these results, Melchior et al.7 reported higher oStim- versus eStim-evoked DA release in the vStr core after viral transfection. In addition to possible regional differences between that study and the present one, another contributing factor might be the 20 Hz train stimulation frequency used by Melchior and colleagues, given that DA release evoked by higher frequency pulse-trains is amplified in the absence of concurrent ACh release with oStim.9 Their observation that single-pulse oStim evoked lower [DA]o than eStim in the core argues against, but does not exclude, the alternative explanation of higher ChR2 levels with viral transfection than in our mouse cross.
Figure 5.
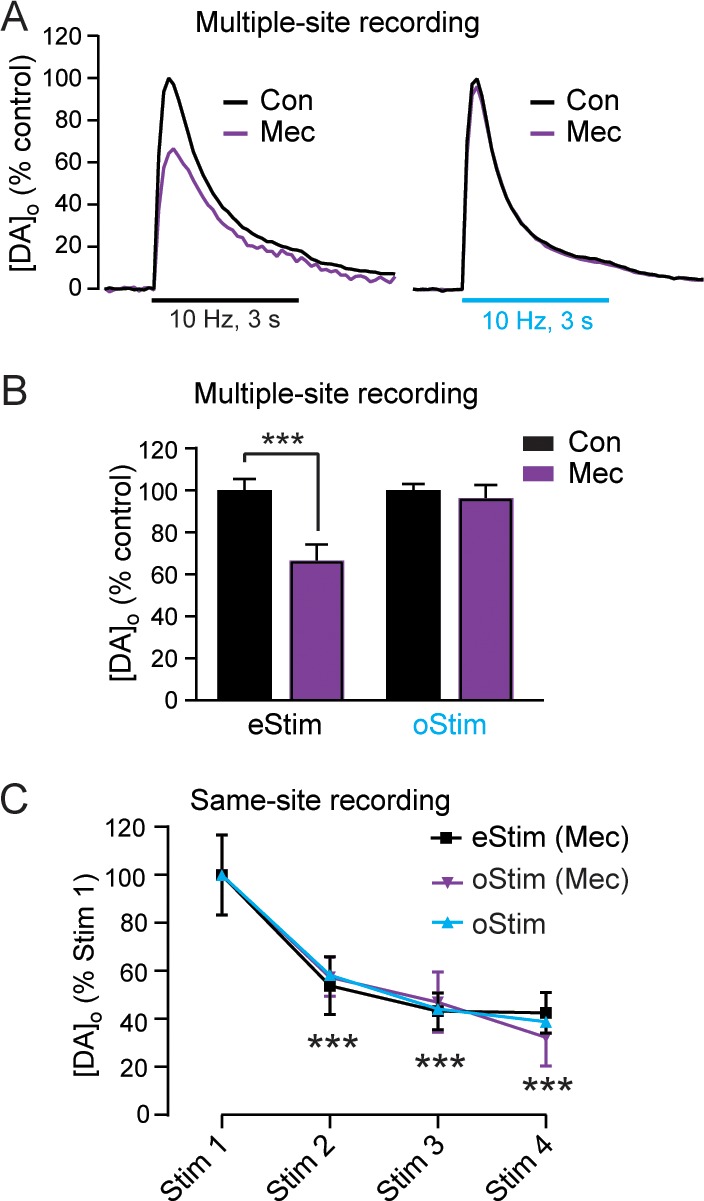
Acetylcholine facilitates evoked [DA]o increases with pulse-train eStim, but not oStim, in dStr. (A) Average increase in [DA]o evoked by multiple-site pulse-train (30 pulses, 10 Hz) eStim (left) or oStim (right) in the dStr from HET/HET mice before (black trace) and after (purple trace) application of mecamylamine (Mec, 10 μM); error bars are omitted for clarity. (B) Summary of the average peak [DA]o in panel A evoked by pulse-train eStim (left; n = 20 sites in slices from 3 mice) and oStim pulse-trains (right; n = 20 sites in slices from 3 mice), before and after Mec application (two-way ANOVA followed by Sidak’s tests *p < 0.05 vs control). (C) Effect of nAChR antagonism by Mec (10 μM) on same-site increases in [DA]o evoked by pulse-train eStim or oStim in the dStr of slices from HET/HET mice. Points denote average peak [DA]o evoked from four successive same-site stimulations after application of Mec (n = 5–6 sites per stimulus type), normalized as a percentage of [DA]o evoked by Stim 1. The rundown trace obtained for oStim (from Figure 3C) in the absence of Mec is also shown for comparison (with error bars omitted for clarity). Comparisons are made using a two-way repeated-measures ANOVA followed by Dunnett’s tests (***p < 0.001 for the Stim1 vs Stim2/3/4 comparisons within any of the three conditions). Data are means ± SEM.
Antagonism of nAChRs by mecamylamine also impaired the stability of same-site eStim-evoked DA release in the dStr of 8–17 week HET/HET mice. After stable pulse-train eStim-evoked [DA]o was established, mecamylamine was applied. This caused a progressive decrease in peak eStim-evoked [DA]o (Figure 5C) that closely mirrored the pattern of rundown in oStim-evoked [DA]o in the dStr of 8–17 week HET/HET mice (Figure 5C). This suggests that rundown with oStim is due primarily to lack of ACh facilitation of DA release when DA axons are stimulated in isolation. The similarity of the rundown patterns for eStim in mecamylamine and oStim alone also argues against a role for tonic ACh release that might boost DA releasability. Indeed, the pattern of rundown seen in [DA]o evoked with eStim was also similar to that evoked with oStim when nAChRs were blocked (Figure 5C).
Given that pulse-train stimulation could cause greater depletion of DA in the absence of concurrently released ACh, thereby enhancing rundown, we hypothesized that stable peak evoked [DA]o might be achieved with single-pulse instead of pulse-train oStim. A previous set of studies in a similar mouse cross assessed optimal single-pulse stimulation parameters for oStim in the vStr,28 but other factors like release stability and TTX sensitivity were not reported. In other recent studies, Foster and colleagues21 achieved stable single-pulse oStim-evoked release in Ai32+/–;DAT::Cre+/– mice when oStim was alternated with single pulse eStim. Here we examined single-pulse oStim and eStim in separate experiments in dStr from HET/HET mice. Initial (Stim 1) single-pulse evoked increases in [DA]o evoked by oStim were lower than those evoked by eStim (1.08 ± 0.11 μM for oStim, n = 16 sites from 8 mice, vs 1.72 ± 0.38 μM for eStim, n = 6 sites from 6 mice), which was also noted in previous optogenetic studies in virally transfected mice.7 For oStim, 0.25 and 2 ms pulse durations were tested; the pattern of evoked [DA]o with consecutive stimulations was similar for both (66% ± 4% of Stim 1 by the fourth stimulation for 0.25 ms and 68% ± 3% of Stim 1 for 2 ms), so the data were pooled (Figure 6). We found that although peak [DA]o in dStr evoked at 5 min intervals by single-pulse oStim showed greater rundown during the first four simulations than the minimal change seen with single-pulse eStim (Stim2, p < 0.05 between stimulus types; Stim3 and Stim4, p < 0.001; Figure 6A,B,D), this was less profound than the rundown seen with pulse-train oStim (compare with Figure 3; unpaired t-test on 30P vs 1P oStim4, **p < 0.01). Indeed, in five experiments in which the recording period was extended, four stable consecutive same-site single-pulse evoked [DA]o release events could be achieved (Figure 6D,E), with oStim1′ typically occurring within 30 min and oStim4′ within 45 min of oStim1 (i.e., a maximum of 10 same-site stimulations). Testing ended once stability was reached with average peak [DA]o evoked for oStim4′ that was 62% ± 7% of initial oStim1 (n = 5; Figure 6E).
Figure 6.

Stability of single-pulse evoked increases in [DA]o with eStim and oStim in dStr. (A) Representative single-pulse (1P) evoked increases in [DA]o from four consecutive stimulations (at 5 min intervals) at a single recording site in HET/HET dStr with eStim. (B) Same as panel A, except that same-site single-pulse oStim was used. (C) Average peak [DA]o evoked by the first four consecutive single-pulse eStim (black line; n = 6 sites in slices from 6 mice) and oStim (blue line; n = 16 sites in slices from 8 mice) indicating a rundown of ∼30% with oStim. (D) In a subset of oStim experiments (n = 5 sites in slices from 5 mice), in which the recording time was increased, stable evoked increases in [DA]o could be achieved, after a total rundown of approximately 40% relative to Stim 1 (C). (E) Average evoked peak responses [DA]o for four consecutive 1P oStim after stability was reached, typically by the 6th stimulation, designated Stim 1′–4′ (one-way repeated-measures ANOVA followed by Dunnett’s test: p > 0.05 for oStim1′ vs oStim2/3/4′). Data are means ± SEM and comparisons are made by a two-way repeated-measures ANOVA followed by Dunnett’s tests (***p < 0.001; oStim1 vs oStim2/3/4).
Like eStim in dStr, pulse-train eStim in the nucleus accumbens shell in the vStr of HET/HET mice evoked stable increases in [DA]o that typically remained elevated throughout the pulse train (Figure 7A). The shape of pulse-train eStim-evoked [DA]o in vStr differed markedly from that in dStr, which shows an early peak in [DA]o then a return toward baseline (e.g., Figure 3A). The differences in release profile shapes from dStr and vStr are typical of those seen in wild-type rodents and have been suggested to reflect different patterns of modulation that regulate release throughout the pulse-train.29,30 In contrast to eStim, pulse-train oStim failed to evoke DA release in almost all recording sites in the medial shell of the vStr in age-matched HET/HET mice. In the one recording site in which oStim-evoked [DA]o was detected, in common with pulse-train oStim in the dStr, peak [DA]o was also unstable in this vStr recording site. In contrast, reliable evoked increases in [DA]o could be evoked using pulse-train oStim in the ventral part of the shell, although the shape of the concentration–time responses appeared to differ somewhat from those evoked using pulse-train eStim (Figure 7A,B). Moreover, unlike in the medial shell or dStr, oStim-evoked increases in [DA]o in the ventral shell were remarkably stable (Figure 7B,D).
Figure 7.
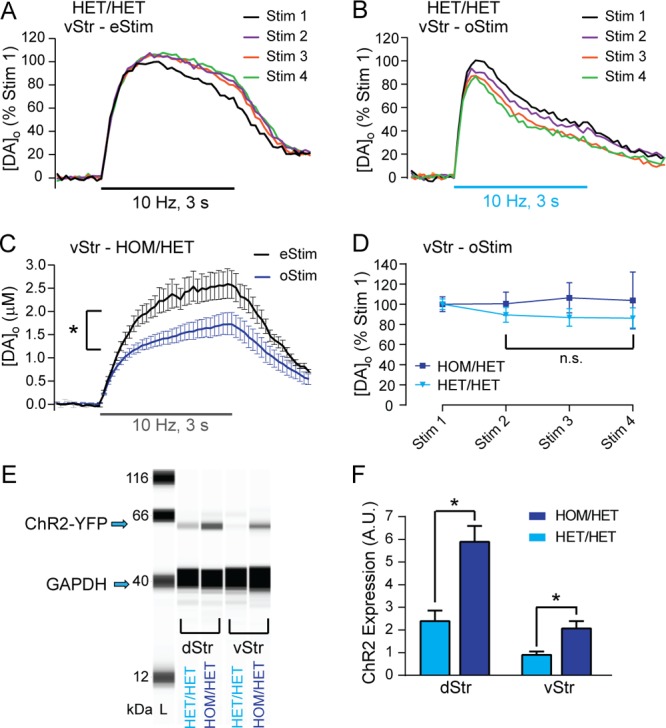
Pulse-train oStim-evoked increases in [DA]o are stable in the vStr of HOM/HET mice that express higher striatal ChR2 than seen in HET/HET mice. (A) Representative records from the first four consecutive pulse-train (30 pulses, 10 Hz) eStim-evoked increases in [DA]o in the vStr of a HET/HET mouse. (B) Same as in panel A, except that increases in [DA]o were evoked by same-site oStim. (C) Average pulse-train evoked responses in vStr of HOM/HET mice with repeated same-site eStim (black trace; n = 6 stimulations in slices from 2 mice) or oStim (dark blue trace; n = 15 stimulations in slices from 5 mice). (D) Summary of peak evoked oStim responses during four consecutive pulse-train stimulations in HET/HET mice (light blue triangles; from panel B) and HOM/HET mice (dark blue squares) normalized to Stim 1. (E) Example capillary electropherograms from immunoassays (SimpleWestern) for YFP and GAPDH (a loading control protein) for HET/HET and HOM/HET dStr (left) and vStr (right). The YFP antibody labeled a band at ∼60 kDa, which is the molecular weight (MW) of YFP plus ChR2 to which it is fused. (F) Summary of YFP expression (as percent of GAPDH) in dStr (left, n = 11 samples from 4 mice) and vStr (right, n = 7 samples from 4 mice) of HET/HET (light blue bars) and HOM/HET mice (dark blue bars; n = 7 samples from 4 mice for dStr, and n = 5 samples from 4 mice for vStr. Data are means ± SEM and comparisons were made using a two-way ANOVA followed by Holm–Sidak tests (*p < 0.05).
Given the difficulty in obtaining reliable oStim-evoked release in medial nucleus accumbens shell and relatively low release levels in ventral shell compared to those in dStr (0.49 ± 0.60 μM; n = 14), we tested a strategy to amplify oStim-evoked release levels by using HOM/HET mice (i.e., mice carrying two copies of the ChR2 construct; see Methods). Strikingly, the medial shell of the vStr in slices from age-matched HOM/HET mice exhibited robust oStim-evoked increases in [DA]o (1.72 ± 0.24 μM; n = 15) with a similar release profile to that with eStim (Figure 7C). Even with the expected increase in ChR2 in HOM/HET mice, however, average peak evoked [DA]o with oStim in HOM/HET vStr was significantly lower than for eStim (Figure 7C) and, indeed, was lower throughout the stimulus train than with eStim (two-way repeated-measures ANOVA, main effect of stimulus type, F1,19 = 7.27, p < 0.05; stimulus type × time interaction F29,551 = 4.59, p < 0.001). As in dStr, this may predominantly reflect the absence of ACh facilitation of DA release during low frequency oStim. Stable increases in [DA]o with repeated pulse-train oStim at the same site were achieved in HOM/HET mice (Figure 7D).
To determine the extent to which a theoretical doubling of gene dose in HOM/HET compared to HET/HET mice actually altered ChR2 expression, we compared ChR2 levels in tissue samples from dStr and vStr from HET/HET and HOM/HET mice using Western blots (Figure 7E). As predicted, doubling the gene dose roughly doubled ChR2 expression in both regions, with significantly higher levels in HOM/HET versus HET/HET in dStr and in vStr (two-way ANOVA, main effect of genotype, F1,26 = 22.21, p < 0.001; Figure 7F). ChR2 expression was significantly higher in dStr overall compared to vStr from the same mice (main effect of region, F1,26 = 28.72, p < 0.001).
The use of HOM/HET mice for vStr experiments (Figure 7) provides an additional strategy for achieving stable oStim same-site recordings with comparable evoked [DA]o release profiles to those evoked by eStim. The finding that ChR2 levels were higher in the dStr, as well as vStr, suggested that this genotype could be valuable for pulse-train oStim in dStr, as well. However, in HOM/HET dStr, oStim-evoked [DA]o showed an unusually protracted release profile that was initially very high (Figure 8A), then still ran down with subsequent pulse-trains, as seen in age-matched HET/HET mice (Figure 8A,B). Using the area under the curve during the 3 s stimulation period as an index of DA release throughout a stimulus train in dStr, we found that oStim-evoked release was significantly higher in HOM/HET compared to HET/HET (Figure 8C,D). Although we cannot rule out the possibility that oStim-evoked release in HOM/HET is actually normal and reflects restoration of releasability that is absent even with eStim, the similarity in the shapes of oStim and eStim responses in HET/HET dStr (Figure 3A,B) argues against this.
Figure 8.
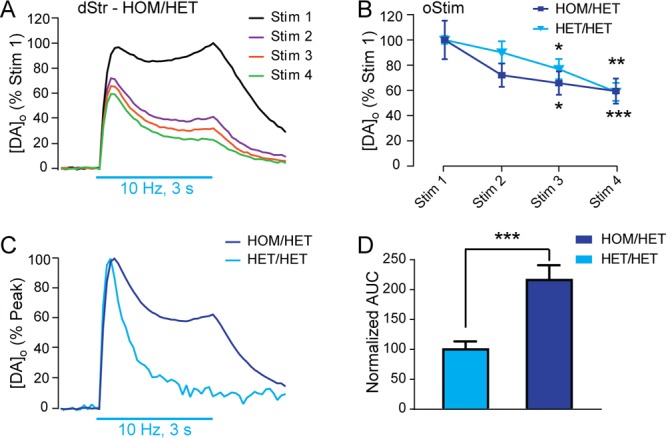
Pulse-train oStim-evoked increases in [DA]o are broadened in HOM/HET dStr. (A) Average pulse-train oStim-evoked increases in [DA]o from four consecutive stimulations in dStr of HOM/HET mice (n = 4 sites in slices from 4 mice) demonstrates a broadened response, especially in Stim 1; error bars are omitted for clarity. (B) Summary of panel A showing the average peak evoked [DA]o during oStim pulse-trains in HOM/HET mice as well as in HET/HET mice; in both genotypes there was a significant difference between Stims 3 and 4 vs Stim 1 at the peak of the response (*p < 0.05, **p < 0.01, **p < 0.001; upper symbols pertain to comparisons within HET/HET data, lower symbols for HOM/HET data). (C) Average of Stims 1–4 in same-site experiments in HOM/HET (dark blue trace; n = 16 stimulations in slices from 4 mice) and HET/HET mice (light blue trace; n = 11 stimulations in slices from 4 mice), each normalized to their own peak. (D) Quantitation of the area under the curve (AUC) during the 3 s stimulus train for the records in panel C. The AUC was normalized to that in HET/HET mice to quantify the protracted increase in evoked [DA]o vs time in HOM/HET dStr compared to the response in HET/HET mice (***p < 0.001, unpaired t-test with Welch’s correction). All data are means ± SEM and comparisons for panel B were made using a two-way repeated-measures ANOVA with Dunnett’s tests within genotypes (oStim1 vs oStim2/3/4).
The strategy of doubling the gene dose for ChR2 by using HOM/HET mice therefore apparently leads to appropriate expression levels of ChR2 in the vStr but excessively high expression levels and an aberrant release profile in the dStr. This regional pattern of ChR2 expression and oStim responses is consistent with the idea that Cre expression levels, which are driven by the DAT promoter, are not in excess, and are limiting ChR2 expression in this particular digenic system, especially in the vStr. This hypothesis seems reasonable given that endogenous DAT expression is known to be approximately 2-fold lower in vStr compared to dStr,19,20 which corresponds with weaker oStim-evoked release in vStr vs dStr in the HET/HET mice. qPCR experiments examining the proportion of completed Cre-mediated recombination events over time would need to be conducted to confirm this.
The data reported here are consistent with another previously reported advantage of Ai32 knock-in crosses, which is that the Cre allele and Ai32 allele can be kept heterozygous (HET) yet oStim will still be efficacious in many cell-types14 (DA neurons were not among those tested). The prospect of using knock-in mice with only one copy of the transgene integrated into a specific location of the genome is attractive for its simplicity, as well as for decreasing the likelihood of genetic side-effects. This is in contrast to a typical characteristic of conventional transgenic lines, in which a high copy number must be achieved to ensure the line is widely useful. For vStr, however, we also needed a higher copy number with our knock-in line and achieved this by breeding HOM/HET mice that exhibited stable same-site oStim-evoked increases in [DA]o. The Allen Institute has recently deposited an improved generation of various optogenetic knock-in mice (e.g.., the Ai90D line) with The Jackson Laboratory, which express from the TIGRE locus instead of the ROSA26 locus and have a tetracycline responsive (TRE) promoter.31 These improvements result in apparently higher opsin expression levels than are expressed in the Ai32 mice used here. There is additionally the option to titer expression lower by administering the mice doxycycline, which could be useful for oStim in the dStr, for example.
It should be noted that ongoing improvements in transgenic mouse lines could yield a net advantage in using them over using knock-in mouse lines in the near future. For example, using a creative method to achieve multiple copies, Ting and Feng recently designed an improved DAT-containing bacterial artificial chromosome (BAC) that can be used to make transgenic mice with modified DA systems.32 This improved DAT-BAC could achieve specific targeting without the need for a foreign viral IRES sequence insertion into an endogenous UTR. This is important because IRES insertions into the 3′-UTR, such as the one present in DAT::Cre knock-in mice, have been shown to decrease bicistronic mRNA transcript stability (e.g., decreased stability of the single mRNA strand that encodes both DAT and ChR2) through increased nonsense-mediated decay. The result is knock-down of the endogenous gene used to drive Cre expression33 (the DAT in our case), which may underlie the decreased DAT protein levels reported previously and decreased DAT function reported here for DAT::Cre mice.
Overall, the data reported here demonstrate that the Ai32;DAT::Cre line can yield reproducible, DA-specific optical stimulation throughout the striatum. A number of other laboratories have used virally transfected ChR2 successfully for oStim-evoked DA release. In those reports, DA release evoked by single pulses or short pulse trains (≤5 pulses) appear to be stable.9,18,21,26 However, as far as we are aware the stability of longer pulse-trains has not been tested. Given that the rundown seen in the dStr in slices from HET/HET mice reflects the absence of ACh contributions to DA release stability (Figure 5C), similar rundown might be expected after viral transfection, as well.
The genetic approaches used here are arguably less labor intensive than a viral-based approach. Furthermore, the expression level of ChR2 is sufficiently high to facilitate robust light-activated axonal DA release with moderate oStim parameters as an index of functional sensitivity. This opens the possibility of using these mice for studies of local microcircuitry in striatum. We recommend using such a mouse cross for studies that do not require projection-specific targeting of the transgene (e.g., nigrostriatal), recognizing that a viral-based approach would allow better anatomical dissociation and potentially higher ChR2 expression. Based on the results reported here, we recommend using the HET/HET genotype of Ai32;DAT::Cre mice with multiple-site pulse-train recording for studies in the dStr, and the HOM/HET genotype for studies in the vStr and other regions where endogenous DAT expression levels are low.19,20 These strategies successfully overcame potential limiting factors in the use of these mice, including DA release rundown with same-site pulse-train stimulation in the dStr, and low oStim evoked DA release in the vStr. Together with a better understanding of the consequences of the absence of concurrent ACh release, these approaches should facilitate more effective use of these viral-free mice to study the regulation and roles of oStim-evoked DA release.
Methods
Animals
Male and female mutant mice were tested; no obvious gender differences were noted in the parameters examined, so data from both were pooled. Mice used in the course of these studies were were tested at 3−5 weeks, 8−17 weeks, or 18−42 weeks of age; however, most data reported here were obtained using mice between 8 and 17 weeks. Mutant mice expressing the H134R variant of ChR2 selectively in DA neurons were generated by the following strategy: B6.SJL-Slc6a3tm1.1(cre)Bkmn/J (DAT::Cre) heterozygous mice were bred with B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze (Ai32) homozygous mice. Both knock-in mouse lines were purchased from The Jackson Laboratory (Bar Harbor, ME). The F1 progeny of the genotype Ai32+/–;DAT::Cre+/– were self-crossed to generate the production line of breeders used to generate mutant mice for voltammetry. Control mice examined in DA uptake studies were DAT::Cre–/– littermates of DAT::Cre+/– mice, which have a C57Bl6/J background.
All mutant mice used for voltammetric recordings were heterozygous for a bicistronic (IRES-containing) allele encoding Cre recombinase under control of the DAT promoter, and most were additionally either heterozygous or homozygous carriers of the Cre-dependent LSL-ChR2(H134R)–EYFP fusion cassette from the Ai32 line. This leads to constitutive but restricted expression of a blue light-sensitive ion channel from the otherwise ubiquitously expressed ROSA26 locus. The terms for the mutant genotypes used are Ai32+/–;DAT::Cre+/– (HET/HET) and Ai32+/+;DAT::Cre+/– (HOM/HET) mice. The mice were genotyped by standard PCR procedures using a Veriti thermal cycler (model 9902 from Applied Biosystems; Waltham, MA) according to the protocols published by Jackson.
Fast-Scan Cyclic Voltammetry (FCV)
Evoked increases in [DA]o in striatal slices were monitored using FCV, as described previously.2 Briefly, mice were anesthetized with Euthasol (diluted with water to administer 120 mg/kg pentobarbital, ip) before decapitation and brain removal. Coronal slices (300 μm thick) were prepared on a Leica VT1200S vibroslicer in an ice-cold sucrose-based cutting solution containing the following (in mM): sucrose (225), NaHCO3 (28), d-glucose (7), MgCl2 (7), sodium pyruvate (3), KCl (2.5), NaH2PO4 (1.25), ascorbic acid (1), CaCl2 (0.5). The slices were then transferred to a chamber containing a HEPES-based solution for storage at room temperature until the experiment was started. The HEPES-based solution contained (in mM): NaCl (120), NaHCO3 (20), d-glucose (10), HEPES acid (6.6), KCl (5), HEPES sodium (3.3), CaCl2 (2), MgSO4 (2). After at least 1 h, a slice was transferred to a recording chamber where it was superfused with artificial cerebrospinal fluid (aCSF) at 1.5 mL/min at 32 °C. aCSF contained the following (in mM): NaCl (124.2), NaHCO3 (26), d-glucose (10), KCl (3.76), CaCl2 (2.4), MgSO4 (1.33), KH2PO4 (1.23). All physiological solutions were equilibrated with 95% O2/5% CO2. Stimulation of DA release was initiated typically 30 min after transfer to the chamber. Recordings were made in slices for up to 8 h after cutting (maximum of 3 slices/day). All experiments using mice conformed to the Guide for Care and Use of Laboratory Animals, and the procedures were approved by the NYUMC Institutional Care and Use Committee.
Recordings were made using carbon-fiber microelectrodes (7 μm fiber diameter) with a triangular potential waveform of −0.7 V|+1.3 V|−0.7 V (v.s. Ag/AgCl) applied at 100 ms intervals.2 Release was evoked either by light applied via a 200 μm diameter optical fiber or by a bipolar stimulating electrode placed within 200 μm of the recording electrode. The light source was a 473 nm blue laser (Laserglow Technologies; Toronto, Ontario), and a power meter equipped with a photodiode sensor was used to determine the light intensity delivered (PM100USB from Thorlabs; Newton, NJ). Light pulse duration and timing were controlled by a Master-8 (A.M.P.I., Jerusalem, Israel). Optical (oStim) or electrical (eStim) pulse-train stimulation at 10 Hz (30 pulses, 3 s train duration) was used for most experiments, although single-pulse oStim or eStim was used in some. For eStim, stimulus pulses were 300 μA in amplitude and 0.1 ms in duration, and for oStim, parameters were 400 μW in intensity with 0.25 ms duration for pulse trains and 0.25 or 2 ms duration for single pulses.
Two basic experimental designs were tested. For same-site recording, the oStim probe or bipolar eStim electrode was positioned near the recording electrode, and [DA]o was evoked for four consecutive stimulations. For multiple-site recording, oStim- or eStim-evoked DA release was assessed at a population of unique sites in one striatal subregion per slice. For all experiments, evoked increases in [DA]o were quantified by postexperimental calibration of the carbon fiber microelectrode in the recording chamber in aCSF.2
Evaluation of Vmax in Control versus DAT::Cre+/– Mice for Evoked [DA]o in Striatal Slices
To minimize recording or experimenter bias, DA uptake experiments were conducted blinded to genotype. Single-pulse multiple-site eStim was used to evoke increases in [DA]o in slices from one male DAT::Cre+/– mouse and a control (DAT::Cre–/−) littermate on a given day. The order of testing was alternated between days to balance any time-of-day influence. Single-pulse evoked [DA]o was recorded in 5–7 sites in the dorsolateral dStr and 5–6 sites in the nucleus accumbens core of the vStr per slice; 2 slices per mouse were used with 4 mice per group. The use of single pulses for Vmax analysis is essential to avoid contributions from autoreceptor regulation of evoked DA release.
To determine whether DAT-mediated DA uptake was compromised in DAT::Cre+/– mice, the initial portion of the falling phase (typically 4 sample points) of single-pulse evoked [DA]o curves were fitted to the Michaelis–Menten equation with a custom-coded MATLAB script, to extract Vmax (maximal uptake rate constant) as described previously.22Km (which is inversely related to the affinity of the DAT for DA) was fixed at 0.9 μM34 and assumed not to be altered in the DAT::Cre+/– knock-in line. Moreover, Km is known to be similar across striatal subregions.35 The goodness of fit (R2) for each release event was also determined; Vmax values that were associated with a R2 < 0.9 were excluded from the data set.
Determination of ChR2–YFP Expression in HET/HET and HOM/HET Mice
Expression of YFP was visualized qualitatively in brain slices using an Olympus SZX16 stereomicroscope; YFP was excited with 460−495 nm light and emission was collected with a GFP filter transmitting between 510 and 550 nm. Images were acquired at 3.5X magnification with a Canon Rebel T6i fitted to the microscope phototube. Expression levels of ChR2–YFP were determined by Nanobiotec (Whippany, NJ) using an automated capillary-based nanoproteomic immunoassay,36 Simple Western System (ProteinSimple). Four HET/HET mice from each age group and four HOM/HET mice were examined. After anesthetization and decapitation, as described above, the brain was removed, and tissue samples from dStr and vStr were dissected from each mouse on a glass plate over ice, then frozen immediately on dry ice in 1.5 mL microcentrifuge tubes. Samples were stored at −80 °C until processing. For analysis, tissue samples were sonicated in appropriate buffers with fluorescent molecular weight (MW) standards. After the samples were heated to 95 °C for 5 min, 4 μL aliquots were loaded into individual capillaries. Samples were passed through stacking and separation matrices for 30 min at 250 V. Proteins were immobilized on capillary walls using photoactivated capture chemistry, capillaries were incubated with a blocking reagent for 23 min, and target proteins were probed with primary antibodies (anti-YFP and anti-GAPDH antibodies, from Cell Signaling Technology, Inc., Beverly, MA), 1:200–400 diluted in blocking buffer and incubated for 200 min, and subsequently with horseradish peroxidase-conjugated anti-mouse secondary antibodies. A mixture of luminol and peroxide was added following manufacturer’s protocol. The resulting chemiluminescent signal was captured by a CCD camera, and the signal intensities were analyzed using Compass Software (ProteinSimple).
Chemicals
Mecamylamine hydrochloride and all chemicals for slice preparation, superfusion, and DA for microelectrode calibration were from Sigma (St. Louis, MO).
Data Analysis and Statistics
All statistical analyses were performed using GraphPad Prism 6.0, and most numerical analysis was performed using MS Excel. Unpaired Student’s t tests were used for two-group comparisons. Two-way repeated-measures ANOVA were performed for all line graphs (except Figure 6E, which was a one-way ANOVA) as well as for Figures 4C and 7C. Two-way ANOVA was performed for Figures 4D, 5B, and 7F. One-way ANOVA was performed for Figures 1, 2C, and 8D. All ANOVA was followed by Tukey’s, Dunnett’s, Bonferroni’s, or Sidak’s post hoc tests as indicated.
Acknowledgments
We thank Dr. Mitchell Chesler and his group for the use of equipment for mouse genotyping. We also thank Dr. Jennifer Arnold for her assistance with dissections for the SimpleWestern analysis. The MATLAB script for Vmax analysis was designed and provided by Dr. Charles Nicholson.
Author Contributions
B.O. conducted optical and electrical pulse-train stimulation experiments, analyzed and graphed data, and drafted the manuscript. J.C.P. conducted the Km/Vmax experiments and other experiments involving single-pulse stimulation and contributed to optical and electrical pulse-train stimulation experiments, analyzed and graphed data, and contributed to writing the manuscript. M.E.R. contributed to project design, supervised data collection and interpretation of results, and contributed to manuscript preparation.
This work was supported by NIH/NIDA Grants R01 DA038616 (M.E.R.) and T32 DA007254 (B.O.).
The authors declare no competing financial interest.
References
- Robinson D. L.; Venton B. J.; Heien M. L.; Wightman R. M. (2003) Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin. Chem. 49, 1763–1773. 10.1373/49.10.1763. [DOI] [PubMed] [Google Scholar]
- Patel J. C.; Rice M. E. (2013) Monitoring axonal and somatodendritic dopamine release using fast-scan cyclic voltammetry in brain slices. Methods Mol. Biol. 964, 243–273. 10.1007/978-1-62703-251-3_15. [DOI] [PubMed] [Google Scholar]
- Matsuda W.; Furuta T.; Nakamura K. C.; Hioki H.; Fujiyama F.; Arai R.; Kaneko T. (2009) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453. 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice M. E.; Patel J. C.; Cragg S. J. (2011) Dopamine release in the basal ganglia. Neuroscience 198, 112–137. 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber G. D.; Hnasko T. S.; Britt J. P.; Edwards R. H.; Bonci A. (2010) Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J. Neurosci. 30, 8229–8233. 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F.; Patel J. C.; Xenias H.; English D.; Tadros I.; Shah F.; Berlin J.; Deisseroth K.; Rice M. E.; Tepper J. M.; Koos T. (2010) Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J. Neurosci. 30, 7105–7110. 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior J. R.; Ferris M. J.; Stuber G. D.; Riddle D. R.; Jones S. R. (2015) Optogenetic versus electrical stimulation of dopamine terminals in the nucleus accumbens reveals local modulation of presynaptic release. J. Neurochem. 134, 833–844. 10.1111/jnc.13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch N. X.; Ding J. B.; Sabatini B. L. (2012) Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 490, 262–266. 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S.; Lalic T.; Platt N. J.; Jennings K. A.; Deisseroth K.; Cragg S. J. (2012) Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64. 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Gompf H. S.; Budygin E. A.; Fuller P. M.; Bass C. E. (2015) Targeted genetic manipulations of neuronal subtypes using promoter-specific combinatorial AAVs in wild-type animals. Front. Behav. Neurosci. 9, 152. 10.3389/fnbeh.2015.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S.; Steinberg E. E.; Foldy C.; Wall N. R.; Beier K.; Luo L.; Malenka R. C. (2015) Diversity of transgenic mouse models for selective targeting of midbrain dopamine neurons. Neuron 85, 429–438. 10.1016/j.neuron.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten I. B.; Steinberg E. E.; Lee S. Y.; Davidson T. J.; Zalocusky K. A.; Brodsky M.; Yizhar O.; Cho S. L.; Gong S. C.; Ramakrishnan C.; Stuber G. D.; Tye K. M.; Janak P. H.; Deisseroth K. (2011) Recombinase-driver rat lines: tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron 72, 721–733. 10.1016/j.neuron.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman S. L.; Beneduce B. M.; Drew I. R.; Regehr W. G. (2014) Achieving high-frequency optical control of synaptic transmission. J. Neurosci. 34, 7704–7714. 10.1523/JNEUROSCI.4694-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L.; Mao T.; Koch H.; Zhuo J. M.; Berenyi A.; Fujisawa S.; Hsu Y. W.; Garcia A. J.; Gu X.; Zanella S.; Kidney J.; Gu H.; Mao Y.; Hooks B. M.; Boyden E. S.; Buzsáki G.; Ramirez J. M.; Jones A. R.; Svoboda K.; Han X.; Turner E. E.; Zeng H. (2012) A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802. 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckman C. M.; Malik N.; Zhang Y.; Shan L.; Grinberg A.; Hoffer B. J.; Westphal H.; Tomac A. C. (2006) Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Avshalumov M. V.; Rice M. E. (2003) Activation of ATP-sensitive K+ (KATP) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc. Natl. Acad. Sci. U. S. A. 100, 11729–11734. 10.1073/pnas.1834314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. C.; Witkovsky P.; Coetzee W. A.; Rice M. E. (2011) Subsecond regulation of striatal dopamine release by pre-synaptic K-ATP channels. J. Neurochem. 118, 721–736. 10.1111/j.1471-4159.2011.07358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrover M. F.; Shin J. H.; Alvarez V. A. (2014) Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J. Neurosci. 34, 3183–3192. 10.1523/JNEUROSCI.4958-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hernandez T.; Barroso-Chinea P.; De La Cruz Muros I.; Del Mar Perez-Delgado M.; Rodriguez M. (2004) Expression of dopamine and vesicular monoamine transporters and differential vulnerability of mesostriatal dopaminergic neurons. J. Comp. Neurol. 479, 198–215. 10.1002/cne.20323. [DOI] [PubMed] [Google Scholar]
- Ciliax B. J.; Heilman C.; Demchyshyn L. L.; Pristupa Z. B.; Ince E.; Hersch S. M.; Niznik H. B.; Levey A. I. (1995) The dopamine transporter: immunochemical characterization and localization in brain. J. Neurosci. 15, 1714–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D. J.; Wilson J. M.; Remke D. H.; Mahmood M. S.; Uddin M. J.; Wess J.; Patel S.; Marnett L. J.; Niswender C. M.; Jones C. K.; Xiang Z.; Lindsley C. W.; Rook J. M.; Conn P. J. (2016) Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release. Neuron 91, 1244–1252. 10.1016/j.neuron.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venton B. J.; Seipel A. T.; Phillips P. E.; Wetsel W. C.; Gitler D.; Greengard P.; Augustine G. J.; Wightman R. M. (2006) Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J. Neurosci. 26, 3206–3209. 10.1523/JNEUROSCI.4901-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace L. J.; Hughes R. M. (2008) Computational analysis of stimulated dopaminergic synapses suggests release largely occurs from a single pool of vesicles. Synapse 62, 909–919. 10.1002/syn.20572. [DOI] [PubMed] [Google Scholar]
- Li X.; Patel J. C.; Wang J.; Avshalumov M. V.; Nicholson C.; Buxbaum J. D.; Elder G. A.; Rice M. E.; Yue Z. (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 30, 1788–1797. 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer M. A.; Woods C. A.; Patel J. C.; Lee C. R.; Witkovsky P.; Bao L.; Machold R. P.; Jones K. T.; de Vaca S. C.; Reith M. E.; Carr K. D.; Rice M. E. (2015) Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun. 6, 8543. 10.1038/ncomms9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R.; Mateo Y.; Mathur B. N.; Irving J.; Wang H. L.; Morales M.; Lovinger D. M.; Cheer J. F. (2012) Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2, 33–41. 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. C.; Rossignol E.; Rice M. E.; Machold R. P. (2012) Opposing regulation of dopaminergic activity and exploratory motor behavior by forebrain and brainstem cholinergic circuits. Nat. Commun. 3, 1172. 10.1038/ncomms2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Driscoll N.; Ozden I.; Yu Z.; Nurmikko A. V. (2015) Modulating dopamine release by optogenetics in transgenic mice reveals terminal dopaminergic dynamics. Neurophotonics 2, 031207. 10.1117/1.NPh.2.3.031207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J.; Trout S. J.; Kruk Z. L. (1992) Regional differences in evoked dopamine efflux in brain slices of rat anterior and posterior caudate putamen. Naunyn-Schmiedeberg's Arch. Pharmacol. 346, 267–276. 10.1007/BF00173539. [DOI] [PubMed] [Google Scholar]
- Brimblecombe K. R.; Gracie C. J.; Platt N. J.; Cragg S. J. (2015) Gating of dopamine transmission by calcium and axonal N-, Q-, T- and L-type voltage-gated calcium channels differs between striatal domains. J. Physiol. 593, 929–946. 10.1113/jphysiol.2014.285890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L.; Garner A. R.; Shimaoka D.; Chuong A. S.; Klapoetke N. C.; Li L.; van der Bourg A.; Niino Y.; Egolf L.; Monetti C.; Gu H.; Mills M.; Cheng A.; Tasic B.; Nguyen T. N.; Sunkin S. M.; Benucci A.; Nagy A.; Miyawaki A.; Helmchen F.; Empson R. M.; Knopfel T.; Boyden E. S.; Reid R. C.; Carandini M.; Zeng H. (2015) Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting J. T.; Feng G. (2014) Recombineering strategies for developing next generation BAC transgenic tools for optogenetics and beyond. Front. Behav. Neurosci. 8, 111. 10.3389/fnbeh.2014.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikama Y.; Hu H.; Ohno M.; Matsuoka I.; Shichishima T.; Kimura J. (2010) Transcripts expressed using a bicistronic vector pIREShyg2 are sensitized to nonsense-mediated mRNA decay. BMC Mol. Biol. 11, 42. 10.1186/1471-2199-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y.; Lee C. J.; Schmauss C.; Gonon F.; Sulzer D. (2001) Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J. Neurosci. 21, 5916–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Reith M. E.; Wightman R. M.; Kawagoe K. T.; Garris P. A. (2001) Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J. Neurosci. Methods 112, 119–133. 10.1016/S0165-0270(01)00459-9. [DOI] [PubMed] [Google Scholar]
- Chen J. Q.; Wakefield L. M.; Goldstein D. J. (2015) Capillary nano-immunoassays: advancing quantitative proteomics analysis, biomarker assessment, and molecular diagnostics. J. Transl. Med. 13, 182. 10.1186/s12967-015-0537-6. [DOI] [PMC free article] [PubMed] [Google Scholar]