

Abstract
Traumatic brain injury (TBI), advanced age, and cerebral vascular disease are factors conferring increased risk for late onset Alzheimer’s disease (AD). These conditions are also related pathologically through multiple interacting mechanisms. The hallmark pathology of AD consists of pathological aggregates of amyloid-β (Aβ) peptides and tau proteins. These molecules are also involved in neuropathology of several other chronic neurodegenerative diseases, and are under intense investigation in the aftermath of TBI as potential contributors to the risk for developing AD and chronic traumatic encephalopathy (CTE). The pathology of TBI is complex and dependent on injury severity, age-at-injury, and length of time between injury and neuropathological evaluation. In addition, the mechanisms influencing pathology and recovery after TBI likely involve genetic/epigenetic factors as well as additional disorders or comorbid states related to age and central and peripheral vascular health. In this regard, dysfunction of the aging neurovascular system could be an important link between TBI and chronic neurodegenerative diseases, either as a precipitating event or related to accumulation of AD-like pathology which is amplified in the context of aging. Thus with advanced age and vascular dysfunction, TBI can trigger self-propagating cycles of neuronal injury, pathological protein aggregation, and synaptic loss resulting in chronic neurodegenerative disease. In this review we discuss evidence supporting TBI and aging as dual, interacting risk factors for AD, and the role of Aβ and cerebral vascular dysfunction in this relationship. Evidence is discussed that Aβ is involved in cyto- and synapto-toxicity after severe TBI, and that its chronic effects are potentiated by aging and impaired cerebral vascular function. From a therapeutic perspective, we emphasize that in the fields of TBI- and aging-related neurodegeneration protective strategies should include preservation of neurovascular function.
Keywords: Aging, Alzheimer’s disease, Amyloid-beta, Brain trauma, Neurodegeneration, Neurovascular unit
1. Goal of review
To discuss concepts and published research relating to disordered APP metabolism in the framework of traumatic brain injury (TBI) and aging as dual risk factors for development of late-onset Alzheimer’s disease (AD). The pathology of TBI is complex and dependent on multiple factors including injury severity, age-at-injury, and time between TBI and neuropathological evaluation. In this regard, the main pathological culprit underlying the link between TBI, aging, and chronic neurodegenerative disease is not well defined. Accumulations of amyloid-β (Aβ) peptide and hyper-phosphorylated tau (p-tau) protein occur in AD, and their presence is required for neuropathological diagnosis of the disease. However, similar aggregates are also detected in many aged cognitively normal people (Mufson et al., 2016; Price and Morris, 1999) and after TBI. Extracellular plaques of Aβ are documented acutely and chronically after severe TBI (Ikonomovic et al., 2004; Johnson et al., 2012; Roberts et al., 1991, 1994; Smith et al., 2003) as well as repetitive mild TBI (rmTBI) (Roberts et al., 1990; Stein et al., 2015). In the aging injured brain, Aβ may participate in a vicious cycle involving neuronal, synaptic, and cerebral vascular dysfunction and neuroinflammatory reaction (Cotman et al., 1996; Ramlackhansingh et al., 2011). Intracellular aggregates of p-tau are also detected after TBI and are currently designated as the defining pathology of chronic traumatic encephalopathy (CTE) (DeKosky et al., 2013; Gandy et al., 2014; Geddes et al., 1999; McKee et al., 2009; Omalu et al., 2005; Schmidt et al., 2001). Because brain accumulation of Aβ is the earliest biomarker change seen in the preclinical phase of AD (Jack et al., 2012), and is central to the amyloid cascade hypothesis of AD (Hardy and Selkoe, 2002), in this review we focus mainly on Aβ as a neuropathological link between TBI, aging, and AD.
2. Evidence linking TBI and AD
2.1. TBI as a risk factor for AD
TBI is considered a risk factor for AD, either as a precipitating event or by accelerating development of the disease (reviewed in (Lye and Shores, 2000; Van Den Heuvel et al., 2007; Vincent et al., 2014)). A link has been established between history of moderate or severe TBI with loss of consciousness (LOC) and AD (Mortimer et al., 1991) but the effect of outcomes varied among reports and some studies did not confirm this finding. This could be due to differences in TBI severity, age at the time of injury, and length of the injury-to-assessment interval, factors that are difficult to confirm in self-reported TBI (undocumented in medical records). For example, a study of 6645 subjects 55 yo and older with self-reported head trauma found no significant association between TBI with LOC and risk of dementia (Mehta et al., 1999). Similarly, TBI with LOC was not associated with dementia or AD neuropathology in three cohorts of patients segregated by duration of LOC, although an association was observed between history of TBI and Lewy body pathology (Crane et al., 2016). The use of self-reported, undocumented TBI, inclusion of a unique case cohort not representative of general population, and missing neuropathology data in some cohorts (e,g., diffuse plaques which are of principal interest in the pathology of severe TBI) complicates the interpretation of these reports. In contrast, self-reported TBI was associated with greater frequency of Aβ plaques in injured compared to age-matched uninjured subjects (Abner et al., 2014). A study of 79 healthy controls and 69 mild-moderate TBI patients with a short (1–2 years) follow up reported no association between TBI and amnestic mild cognitive impairment (MCI) or dementia (Rapoport et al., 2008). However, in a longer (5–7 years) follow up study of 164,661 trauma (TBI and non-TBI) patients older than 55 years and without baseline dementia during hospitalization, dementia was more prevalent after TBI (8.4%) compared to non-TBI trauma (5.9%) (Gardner et al., 2014). Moreover, risk of dementia was significantly associated with both age-at-injury and severity of injury, underscoring the importance of inclusion and accuracy of this information in these types of studies. After moderate-severe TBI, risk of dementia was increased in patients with age-at-TBI > 55, while mild TBI was a greater predictor of dementia in subjects with age-at-TBI > 65 (Gardner et al., 2014). Future studies should assess more precisely the influence of age at injury, time interval between TBI and assessment, and severity of injury on risk of dementia. Of importance, many studies fail to factor comorbid states such as peripheral or central vascular disease in the analyses. Does a history of hypertension or hyperlipidemia, stroke and ischemia influence risk for dementia after TBI? Insight into this question could prove invaluable for long term treatment of TBI patients if such a connection is made.
2.2. The spectrum of Aβ and tau pathology after TBI
2.2.1. Human studies
Aβ is the main component of amyloid plaques which are morphologically complex lesions (Dickson, 1997; Thal et al., 2015). In the earliest (preclinical) stages of AD, Aβ plaques are diffuse (Mufson et al., 2016) and composed predominantly of the longer Aβ42 form, comparable to Aβ plaques detected acutely in about 30% of people with severe TBI (Horsburgh et al., 2000; Ikonomovic et al., 2004; Roberts et al., 1994) (Fig. 1). In contrast, many chronic survivors of severe TBI, especially at advanced age, show widespread distribution of diffuse and neuritic Aβ plaques that are similar to those in pathologically confirmed AD (Johnson et al., 2012). The Aβ precursor protein (APP) also accumulates after TBI (Gentleman et al., 1993a; Graham et al., 1995; Ikonomovic et al., 2004; McKenzie et al., 1994; Roberts et al., 1991, 1994; Smith et al., 2003), and increased amyloidogenic (Aβ-producing) processing of APP has been postulated as a major cause of brain accumulation of Aβ (Thinakaran and Koo, 2008; Turner et al., 2003). Other factors contributing to Aβ deposition include impaired enzymatic degradation of Aβ (Johnson et al., 2009; Miners et al., 2008), alterations in brain efflux systems (Hawkes et al., 2014; Iliff et al., 2012), apolipoproteins (Castellano et al., 2011), and receptors involved in Aβ trafficking and clearance (Deane et al., 2004a, 2004b). Accordingly, in TBI patients with Aβ plaques, there is increased brain concentration of physiologically soluble (monomeric and oligomeric) Aβ1–42 peptide (DeKosky et al., 2007), which could be related to reduced cerebral spinal fluid Aβ concentrations detected after TBI (Kay et al., 2003), similar to what occurs in AD patients (Blennow et al., 2015) (Fig. 1E). Aβ may be acutely neurotoxic (Emre et al., 1992; Smith et al., 1998) and could initiate a cascade of pathophysiological events leading to more advanced AD pathology and dementia later in life (Roberts et al., 1994). The outcome of these pathological changes, particularly in the aging brain, is progressive impairment of neuronal function, inefficient repair of damaged synapses, and cognitive dysfunction.
Fig. 1.
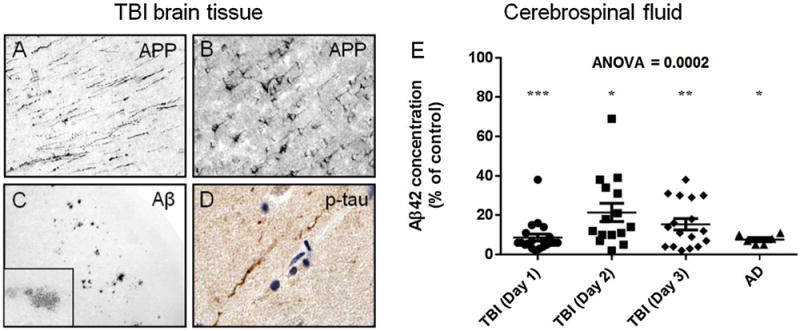
A–D: Brain tissue sections from a temporal cortex biopsy resected 12 h after severe TBI in a 39-year old subject from the University of Pittsburgh Brain Trauma Research Center (BTRC) were processed for immunohistochemistry using antibodies against the Aβ precursor protein (APP; polyclonal antibody anti-6, Athena), Aβ (antibody clone 10D5, Elan), and p-tau (antibody clone PHF-1, P. Davies, Albert Einstein College of Medicine). After severe TBI, APP accumulates in axons in the white matter (A), in cell bodies of pyramidal neurons in the grey matter (B), and Aβ deposits in diffuse Aβ plaques (C and inset). Rare profiles of phosphorylated tau (p-tau) immunoreactive fibers are detected in the gray matter (D; brown color is p-tau immunoreactivity, blue color is hematoxylin histological counterstain). E: Aβ1–42 peptide (Aβ42) concentrations (ELISA, Biosource) in cerebral spinal fluid (csf) from severe TBI patients (from the University of Pittsburgh BTRC; average age = 35.8 ± 15.7, range 17–65) at one, two, and three days after injury and from end-stage AD patients (from the University of Pittsburgh Alzheimer’s Disease Research Center; average age = 76.3 ± 10.2, range 63–91) are similarly reduced relative to levels in csf from cognitively normal control subjects (average age = 56.8 ± 14.5, range 25–78). *p < 0.05, **p < 0.01, ***p < 0.001 (Bonferroni multiple comparison post hoc test). Abbreviations: Aβ, amyloid-β; APP, Aβprecursor protein; p-tau, phosphorylated tau.
In contrast to severe TBI, rmTBI has been connected more closely to tau than Aβ pathology. For example, a histological study of autopsy brains from boxers with dementia pugilistica (DP) reported tau pathology in the absence of Aβ plaques (Corsellis et al., 1973). However, Roberts and colleagues re-examined the same cases using antigen retrieval methods and Aβ immunohistochemistry and demonstrated widespread Aβ plaques in all of the DP cases (Roberts et al., 1990). Importantly, both Aβ and tau pathology in DP cases differ from the classic Aβ plaques and neurofibrillary tangles (NFT) of AD. In DP, Aβ plaques are mainly diffuse and difficult to detect using standard histological techniques (Roberts et al., 1990), as in severe TBI (see Section 3.2). In addition, tau-immunoreactive “NFT” in DP have different laminar distribution patterns than those in AD (Hof et al., 1992). Geddes and colleagues suggested that after rmTBI tau pathology may precede Aβ plaques, based on their findings of intracellular tau aggregates with a perivascular distribution and absence of Aβ plaques in four autopsy brains (Geddes et al., 1999). This finding was replicated by others (McKee et al., 2009; Omalu et al., 2005; Schmidt et al., 2001) and deep sulcal and perivascular distribution of tau-immunoreactive neurons and astrocytes has been designated the key pathological characteristic of CTE, providing the basis for its current neuropathological staging (McKee et al., 2015). Accordingly, CTE was defined as a “tauopathy”, however new research demonstrated Aβ plaques in 52% of CTE cases (Stein et al., 2015), which exceeds the reported 30% incidence of Aβ plaques in severe TBI. The incidence of Aβ plaques after rmTBI may be greater, because not all who suffer rmTBI develop CTE. Collectively, these observations demonstrate overlap in the pathology of severe TBI, rmTBI (including CTE), and AD, and emphasize that polypathology, rather than just tauopathy or amyloidopathy, more accurately underlies the chronic neuropathological course of TBI (Turner et al., 2016; Washington et al., 2016). In this regard, evidence is emerging that other proteins, such as alpha-synuclein and TDP-43, can accumulate in the brain after TBI (Crane et al., 2016; Johnson et al., 2011; McKee et al., 2010).
2.2.2. Animal studies
Experimental studies of TBI-induced Aβ accumulation/toxicity as a risk of AD pathology have been methodologically diverse and often have produced conflicting results (Bird et al., 2016). These studies have been challenging, mainly because the amino acid sequence of the Aβ peptide differs between humans and commonly used experimental animals such as rats and mice. However, several animals produce Aβ with an amino acid sequence identical to that in humans and have utility in studies of the TBI-AD connection. After non-impact head rotational acceleration injury in pigs, Aβ accumulation was observed in injured axons and plaque-like structures (Smith et al., 1999). In aged guinea pigs, fluid percussion injury (FPI) resulted in diffuse Aβ plaques, and immunoreactivity to APP and tau increased with more advanced age over longer survival periods in this model (Bates et al., 2014). To study human Aβ response in brain injured mice, many investigations focused on transgenic mouse models of amyloidosis where APP is continually over-expressed, producing supra-physiologic brain concentrations of APP and Aβ. Brain injury experiments using these models produced varying outcomes. In Tg2576 mice (Hsiao et al., 1996), rmTBI at 9 months of age (prior to Aβ plaque formation in this model) increased brain Aβ concentrations and Aβ plaque deposition at survival intervals up to 16 weeks (Uryu et al., 2002). In contrast, PDAPP mice (Games et al., 1995) with cortical impact brain injury at 4 months of age (prior to Aβ plaque formation in this model) had acute, transiently increased brain Aβ concentration (Smith et al., 1998). In aged PDAPP mice (24 months old, with robust plaque pathology), severe CCI injury resulted in fewer Aβ plaques, particularly in the hippocampus ipsilateral to injury compared to the contralateral hippocampus (Nakagawa et al., 2000). This “regression” of Aβ plaques could be due to extensive neuropil loss in the hippocampus, because unlike the rmTBI model, severe CCI injury induces a profound cortical cavitation lesion. Using the PSAPP transgenic mouse model of AD (Holcomb et al., 1998) and moderate-severe controlled cortical impact (CCI) injury at three months of age (time of Aβ plaque pathology onset in this transgenic model), Tajiri and colleagues observed greater Aβ plaque load six weeks after injury which also produced significant cell loss in the hippocampus (Tajiri et al., 2013). Using 5–7 month old 3× transgenic mice which over-produce both human Aβ and human tau (Oddo et al., 2003), Brody and colleagues (Tran et al., 2011) reported that severe CCI resulted in axonal accumulation of Aβ and tau. Interestingly, blockade of Aβ accumulation in this model did not affect tau immunoreactivity, leading the authors to suggest independent courses of these two AD pathologies after TBI (Tran et al., 2011). Another study used 3×Tg mice at ages prior to plaque deposition and reported that CCI injury produces acute increases in both insoluble (fibrillar) and soluble (oligomeric) Aβ, while changes in tau were not examined (Washington et al., 2014). The use of animal models in research of tau changes after TBI has been reviewed extensively by Abisambra and Scheff (Abisambra and Scheff, 2014). Collectively these animal studies demonstrate that formation of AD-like pathology after TBI is dependent on animal model, type and severity of injury, age at injury, and length of survival period. Furthermore, transgenic models over-expressing APP/Aβand/or tau may be more suitable for studies of individuals with genetic predisposition for neurodegenerative diseases. To avoid confounds associated with transgene-driven APP overexpression, TBI studies by our group (Abrahamson et al., 2013, 2006, 2009) and others (Webster et al., 2015) utilized a human Aβ knock-in mouse model (hAβ KI mice) where APP expression is under control of the endogenous promoter. CCI injury in hAβ KI mice consistently produced significant increases and accumulation of Aβ and phosphorylated tau (Fig. 2).
Fig. 2.
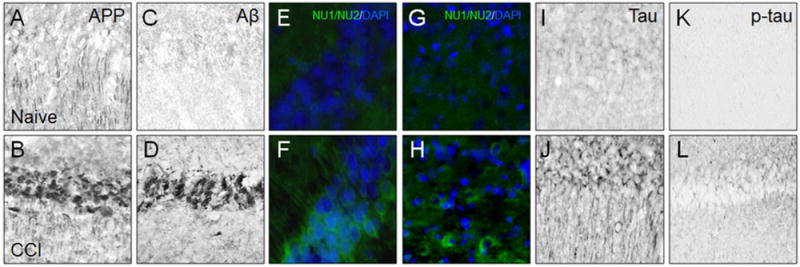
Immunohistochemical analysis of hippocampus CA1 (A–F; I–L) and pericontusional somatosensory cortex (G, H) in human Aβ knock-in mice free of TBI (Naïve; A, C, E, G, I, K) and two weeks after severe controlled cortical impact injury (CCI; B, D, F, H, J, L). Tissue sections were immunoreacted with antibodies recognizing amyloid precursor protein (polyclonal anti-APP antibody CT695, Biosource; A, B), Aβ peptide (polyclonal anti-Aβ42 antibody, Millipore; C, D), Aβ oligomers (antibodies clones NU1/NU2, generous gift from W. Klein, Northwestern University; E, F and G, H), total tau (polyclonal anti-Tau antibody, Dako; I, J), and phosphorylated tau (biotinylated antibody clone AT8, Thermo, p-tau; K, L). Each marker is detected at low levels in naïve mice and prominently after CCI injury.
3. Effect of aging on Aβ-induced risk for developing AD after TBI
3.1. The “two-hit” hypothesis of Aβ toxicity
The two-hit hypothesis has been formulated separately in the fields of TBI and AD, to explain the pathogenic role of Aβ. Related to TBI, Smith and colleagues (Smith et al., 1998) argued that neurons have a potential or latent vulnerability to Aβ toxicity at a given brain concentration (“first hit”) and that only after a “second hit”, such as TBI, is this potential manifested. Accordingly, “Aβ is necessary but not sufficient to cause neuronal death until a second pathological process potentiates its neurotoxicity” (Smith et al., 1998). In the context of AD as a chronic clinical-neuropathological sequela of TBI, amyloidogenic APP metabolism plays a role in both immediate (primary) and delayed (secondary) effects of TBI, while brain aging-related dysfunction (and non-Aβ-related secondary injury processes resulting from TBI) could be a second insult exacerbating these pathological pathways and contributing to other pathologies, such as tau, and clinical dementia onset.
3.2. Aging as a modifying factor in the relationship between TBI and AD
In younger subjects with severe TBI, accumulation of Aβ does not fully replicate the pathology seen in AD dementia patients. Diffuse Aβ plaques are detected shortly after injury, when neuritic Aβ plaques are absent or rare (Gentleman et al., 1997; Graham et al., 1995; Ikonomovic et al., 2004; Roberts et al., 1991). In addition, the acute phase after severe TBI is not associated with prominent cortical tau pathology (Ikonomovic et al., 2004). However in aged subjects with TBI and longer survival periods, widespread Aβ plaques (both diffuse and neuritic) and classic NFT can be detected (Johnson et al., 2012). Aβ and tau changes are also seen in some professional athletes with rmTBI, however these two pathologies can present with different proportions and time of onset. For example, rmTBI can produce a pathological feature of CTE which is characterized primarily by tau changes (McKee et al., 2015), while in aged people with same type of injury tau pathology frequently co-presents with Aβ plaques (Stein et al., 2015), more typical of AD. Thus, aging can modify the chronic post-TBI course by providing a second insult that drives the neuropathological outcome to more closely resemble the neuropathology of AD. Differential time course of induction and progression of Aβ and tau pathologies after severe TBI or rmTBI may be also influenced by genetic susceptibility and epigenetic mechanisms. The effect of these multiple factors on pathological mechanisms contributing to patient’s outcome after TBI is beginning to be understood. Advanced age is also a significant factor contributing to impaired recovery after TBI (Coronado et al., 2005; Czosnyka et al., 2005; Hukkelhoven et al., 2003) including increased risk for developing AD/dementia. This is particularly important in the context of growing aged populations of civilians and Veterans (Ortman et al., 2014), greater numbers of geriatric patients with chronic survival after head trauma (Adams and Holcomb, 2015), and increasing burden of elderly TBI patients on the health care system (Thompson et al., 2006). Recently, there is an indication of reduced inpatient mortality after TBI (Maxwell et al., 2015). Though encouraging from the standpoint of acute medical care, this will increase the number of aging TBI survivors at risk for developing chronic neurodegenerative diseases and dementia towards the end of their life. Notably, age alone is not a significant predictor of TBI outcome (Joseph et al., 2014), and aging-related comorbid states such as cerebrovascular disease may be superimposed on or work in synergy with TBI to worsen neurological outcomes (Plassman and Grafman, 2015).
4. Neurovascular unit and the TBI-AD link
The relationship between altered vascular responses after TBI and risk for developing chronic neurodegenerative diseases is not well understood (Logsdon et al., 2015). Defining the molecular mechanisms or vulnerable cell types which drive vascular impairment in TBI patients could facilitate treatment interventions aimed at preventing or ameliorating chronic pathology and neuropsychological impairment. Specifically, maintaining the functional integrity of cerebral neurovascular unit (NVU) after TBI is critical, and perhaps equally important as current neurocentric “neuroprotection” strategies (Logsdon et al., 2015), for preventing chronic accumulation of toxic proteins and for promoting the restoration of neuronal homeostasis and functional recovery. Cerebrovascular consequences of TBI are in part determined by the type of TBI and include cerebral vasospasm, hemorrhage (epidural, subdural, subarachnoid, and intraparenchymal), and vasogenic edema all of which can result in brain ischemia. TBI involves complex changes in cerebral blood flow (CBF), with some areas (typically proximal to the site of TBI) showing profound hypoperfusion and other areas (typically distal or contralateral to the site of TBI) showing hyperemia (Abrahamson et al., 2013; Akbik et al., 2016). As discussed below, accumulations of Aβ peptides (both oligomeric and fibrillar assemblies) affect cellular components of the NVU and can foster secondary injury processes including activation of cell death pathways (apoptosis), oxidative stress, and inflammation. These effects are likely exacerbated by aging, contributing to the risk of AD.
4.1. Endothelial cells
Tight junctions between endothelial cells of blood vessels form the blood brain barrier (BBB) at the interface between the vasculature and brain parenchyma. Endothelial cells also function as a conduit for molecules to access the brain through receptor mediated transport. In the context of Aβ, the receptor for advanced glycation end products (RAGE) is an important mediator of Aβ transport from the vasculature into brain parenchyma (Zhao et al., 2015a; Zlokovic, 2011). Dysfunction of endothelial cells in TBI and aging results in BBB breakdown and increased brain penetrance of toxic molecules (Price et al., 2016). Similarly, receptor-mediated transport of toxic molecules such as Aβ (via low density lipoprotein receptors) out of the brain is dependent on endothelial cells, disruption of which leads to accumulation of Aβ. Zlokovic and colleagues (Zhao et al., 2015b) demonstrated that phosphatidylinositol-binding clathrin assembly protein (PICALM, a genetic factor linked to AD (Harold et al., 2009) expressed in endothelial cells (Baig et al., 2010)) is an important mediator of LRP-bound Aβ transcytosis and clearance from the brain. Because enhancement or restoration of PICALM function could counteract detrimental effects of Aβ accumulation at the NVU, it will be important to define changes in PICALM expression after TBI and during aging.
Endothelial cells can be affected by high concentrations of Aβ in the brain, such as in AD (Kelleher and Soiza, 2013) but also after TBI (DeKosky et al., 2007). Moreover, endothelial cell aging likely increases their susceptibility to toxic effects of Aβ (Brandes et al., 2005). This toxicity occurs via stimulation of cell death pathways involving caspase-3 and caspase-8, TNF-related-apoptosis-inducing-ligand (Ghiso et al., 2014; Xu et al., 2001), ASK1-JNK/p38 apoptotic signaling pathways (Hsu et al., 2007), death receptors DR4 and DR5 (Ghiso et al., 2014), oxidative stress pathways, and intracellular calcium ion overload (Koizumi et al., 2016). Additional pathways involve altered endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) production (Suhara et al., 2003), and disruption of vascular proliferation (Grammas et al., 1995), potentially through effects on vascular endothelial growth receptor (Patel et al., 2010). Specifically, age-related changes in eNOS and NO production (Barton et al., 1997) could potentiate Aβ-induced impairment in endothelium-dependent vasodilatation after TBI.
Endothelial cells produce endothelin-1 (ET-1), a potent vasoconstrictor that is upregulated by Aβ (Palmer et al., 2012) and could contribute to vascular impairment in aging and AD (Love and Miners, 2016), but also after TBI. Aβ can upregulate at least one of the enzymes that regulate production of ET-1, endothelin-converting enzymes 1 and 2 (ECE-1 and ECE-2; (Palmer et al., 2009)), which interestingly also can degrade Aβ (Eckman et al., 2001). Love and colleagues (Palmer et al., 2009) demonstrated upregulation of ECE-2 mRNA and protein as well as increased activity of ECE-1 in AD brain tissue. This could be detrimental, through enhanced production and vasoconstrictor activity of ET-1, but also protective through degradation of Aβ. ET-1 is also upregulated acutely after TBI (Armstead and Kreipke, 2011), however its suppression through receptor pharmacology (Kreipke et al., 2010) or through mRNA silencing (Petrov, 2009) can restore CBF in TBI models. Since both endothelial cells and pericytes express ET-1 receptors, ET-1-induced changes in the NVU function might be mediated by both cell types (Dore-Duffy et al., 2011).
In addition to enhancing ET-1 induced vasoconstriction (Paris et al., 2003), Aβ could also impair CBF via loss or uncoupling of NO signaling (Austin et al., 2013). In an aged rat model of chronic brain hypoperfusion, de la Torre and colleagues reported that eNOS inhibition impaired behavioral outcome, leading these authors to conclude that eNOS (and NO production) might be a critical factor in maintaining at least a certain degree of cerebral perfusion even in the compromised state (hypoperfusion) (de la Torre and Aliev, 2005). This is relevant to aging and AD, where cerebrovascular impairment with hypoperfusion is related to brain accumulation of Aβ which is both toxic to endothelial cells (Thomas et al., 1996) and produces vasoactive effects (Iadecola, 2004; Townsend et al., 2002). Our work in the hAβ KI mouse model, which has little if any fibrillar Aβ but produces increased concentrations of physiologically soluble (monomeric and oligomeric) human Aβ after TBI, provides additional evidence for vasoactive effects of Aβ. CCI injury produced significantly greater impairment of CBF in hAβ KI mice compared to their wild type counterparts (C57Bl/6) (Abrahamson et al., 2013). This suggests that compared to murine Aβ, soluble human Aβ has stronger vasoconstriction effects after TBI.
4.2. Pericytes
Pericytes are components of the NVU (Winkler et al., 2014) and important regulators of BBB formation and maintenance, neurovascular coupling, and clearance of molecules, including soluble Aβ, from the brain (Bell et al., 2010; Winkler et al., 2014). Pericyte cell death, migration, and altered phagocytic and metabolic activity is reported in a wide variety of pathological conditions, including TBI and AD (for reviews see (Castejon, 2011; Winkler et al., 2014)). The effects of experimental TBI on pericytes is complex and dependent on the TBI model used and length of survival after injury. An ultrastructural study in adult rats with closed head injury demonstrated that within hours after injury pericytes migrated away from vessel cell walls in the vicinity of the lesion (Dore-Duffy et al., 2000). It is unclear whether this is a protective response or if the loss of capillary-associated pericytes further contributes to BBB breakdown and subsequent vasoactive edema after TBI. In mice subjected to a more severe, penetrating CCI injury model, pericyte cell death was observed at three days post injury in the ipsilateral hippocampus and perilesional cortex (Choi et al., 2016) whereas at five days post injury pericyte proliferation was detected in perilesional cortex (Zehendner et al., 2015). The complex response of pericytes to TBI requires further characterization, particularly in aging brain where these cells are involved in BBB disruption. Using an in vitro model of accelerated senescence, Yamazaki and colleagues (Yamazaki et al., 2016) observed an association of endothelial cell and pericyte senescence with breakdown of the BBB. This idea is supported by Zlokovic and colleagues (Montagne et al., 2015) who demonstrated BBB breakdown in the hippocampus of aged people. Moreover, pericyte senescence could potentiate TBI-associated BBB breakdown which can persist lengthily after injury (Hay et al., 2015). Pericytes undergo constriction after ischemia (Hall et al., 2014), and under pathological conditions such as TBI and AD, their dysfunction could contribute to chronic changes in neurovascular coupling. In AD, molecular signaling between pericytes and other cell components of the NVU is compromised (Winkler et al., 2014) and pericyte loss is accelerated, likely related to disrupted Aβ clearance (Halliday et al., 2016). In support of this, an association between pericyte loss and accumulation of soluble Aβ was demonstrated in transgenic APPswe mice (Sagare et al., 2013). Whether a similar relationship between pericyte changes and Aβ accumulation exists in chronic TBI needs to be investigated and may identify novel mechanisms contributing to risk for AD after TBI.
4.3. Astrocytes
Astrocytes exhibit changes in gene expression and phenotypic changes in the presence of oligomeric and fibrillar Aβ (Hu et al., 1998; Mulder et al., 2012). This is exemplified in their association with classic Aβ plaques in AD (Dickson, 1997; Thal, 2012; Wisniewski and Wegiel, 1991) where they may have a role in Aβ degradation (Wegiel et al., 2001) and glial scarring (Nagele et al., 2004). The relation between astrocytes and TBI-induced changes in Aβ is not well understood, particularly in the acute stages after severe TBI in young adults when diffuse Aβ plaques are predominant. A study examining pathology in frontal cortex biopsy samples from a subject with DP demonstrates a close correspondence between the laminar distribution of Aβ plaques, tangles, and GFAP-immunoreactive astrocytosis (Saing et al., 2012). A particularly striking feature of this case study is the conspicuously greater association of astrocytes with blood vessels in the DP case compared to cases with either AD of frontal temporal dementia, suggesting interplay between Aβ, astrocyte activation, and cerebral vasculature in brain injury. In addition, perivascu-lar clustering of tau-immunoreactive astrocytes (and neurons) is a pathological feature of CTE. Astrocytes play critical role in the maintenance of the BBB through vasoactive endothelial growth factor signaling mechanisms (Argaw et al., 2012) and interactions with extracellular matrix proteins. Astrocytic regulation of the BBB responds to, and could mediate, leukocyte trafficking (Lecuyer et al., 2016). Astrocytes are also involved in neurovascular coupling and functional hyperemia, in part through glutamate or ATP signaling-induced release of vasoactive substances, including arachidonic acid metabolites such as 20-HETE (Howarth, 2014). With age, astrocytes increase expression of glial fibrillary acidic mRNA and shift to a pro-inflammatory phenotype (reviewed in (Garwood et al., 2016)), and exhibit signs of oxidative damage and expression of the senescence associated secretory phenotype (Garwood et al., 2016; Simpson et al., 2010). Astrocytic dysfunction could result in impaired Aβ clearance, increased release of pro-inflammatory molecules, and enhanced leukocyte trafficking at the BBB which all relate to neuroinflammatory processes (discussed in Section 5.6) and could contribute to AD pathology with aging and after TBI. The relation of these factors to functional changes of the NVU in aging and TBI needs to be investigated.
5. Role of Aβ and aging in primary and secondary effects of TBI and risk of AD
Primary pathology of TBI results from mechanical forces. Severe and penetrating brain injury causes immediate cell lysis with release of intracellular ions, neurotransmitters, and peptides/proteins, axonal damage and disconnection. Dysfunction of neurovasculature, including disruption of the BBB and consequent influx of peripheral proteins and immune cells into brain parenchyma is another important consequence of severe TBI. In rmTBI, the effects of mechanical forces are more subtle but nevertheless result in axonal and vascular pathology. Secondary injury processes occur following primary injury and develop over a longer timescale, and include oxidative stress, excitotoxicity, activation of cell death pathways, and neuroinflammation. In the aging injured brain, these secondary injury processes are exacerbated, and recovery processes (synaptogenesis and angiogenesis) are compromised. Thus, secondary injury effects are likely important mechanistic connections between TBI and AD, particularly in the aging brain (see schematic in Fig. 3), and they can be modified by genetic and epigenetic factors. In the following sections we discuss pathological consequences of primary and secondary TBI effects in the context of altered Aβ metabolism and the aging brain, and how this can contribute to increased risk for AD later in life.
Fig. 3.
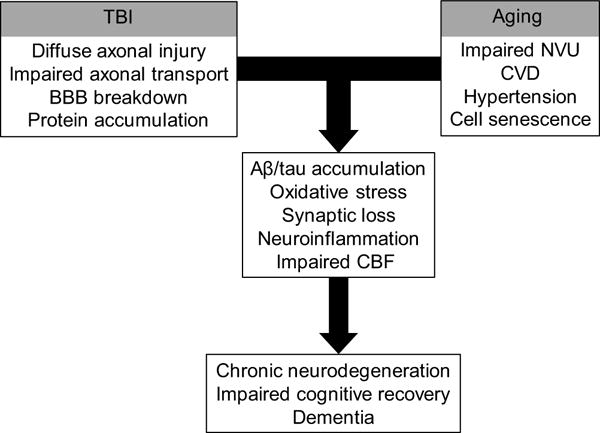
Flow diagram illustrating concepts discussed in the current review, including the interaction between TBI and aging in relation to neurovascular dysfunction as precursors to, or risk factors for AD. In addition, genetic predisposition and epigenetic factors can influence pathways toward recovery or chronic pathology. Abbreviations: Aβ, amyloid-β; BBB, blood brain barrier; CBF, cerebral blood flow; CVD, cerebrovascular disease; NVU, neurovascular unit; TBI, traumatic brain injury.
5.1. Diffuse axonal injury
Mechanical forces of TBI disrupt intra-axonal cytoskeletal structure and increase axolemmal permeability with subsequent pathological influx of Ca2+ ions and activation of caspases, calpain-mediated spectrin proteolysis, and mitochondrial damage (Buki et al., 2000; Buki and Povlishock, 2006). Axonal swellings, a pathological hallmark of diffuse axonal injury (DAI) visualized histologically as axonal bulbs (retraction balls), accumulate cell organelles, vesicles, and proteins. Among the proteins normally undergoing fast axonal transport, APP accumulates within hours after TBI in axonal swellings, where it co-localizes with Aβ (Smith et al., 2003). Accordingly, axonal APP immunoreactivity is considered a reliable marker of DAI (Gentleman et al., 1993b; Stone et al., 2000) and precedes the appearance of silver staining positivity, a marker of neuronal/axonal degeneration (McKenzie et al., 1996; Sherriff et al., 1994). APP immunoreactive axonopathy is also seen in long-term survivors of blast TBI (months to years), in a pattern described by Koliatsos and colleagues as spheroids and varicosities with perivascular distribution in cortical white matter (Ryu et al., 2014). Thus, axonal injury results from multiple forms of TBI and involves an interplay between brain injury and APP/Aβ that are likely amplified, or at least persist, with aging (Chen et al., 2009).
The relationship between axonal accumulation of APP/Aβ and parenchymal Aβ plaque deposition after TBI is not clear. APP-immunoreactive deposits are prevalent in areas of axonal injury (Graham et al., 1996; Ikonomovic et al., 2004) and APP-immunoreactive swollen axons associate with Aβ plaques at least acutely after TBI (Ikonomovic et al., 2004; Smith et al., 2003, 1999) (see Fig. 1). This suggests that injured axons may be the source of Aβ in plaques, however in an autopsy study of subjects with remote brain injuries (three years prior to death), lack of widespread plaque pathology despite axonal accumulation of Aβ led Smith and colleagues to suggest that TBI-induced plaques may regress over time (Chen et al., 2009). Interestingly, APP/Aβ deposition is more frequently seen in aged people who suffer a TBI, most typically as a consequence of a fall, the leading cause of head injury in the elderly (Faul and Coronado, 2015), thus more advanced age may exacerbate AD pathological processes after TBI.
There have been significant recent advances in detecting and monitoring axonal injury and Aβ deposition in living TBI patients, using imaging techniques typically applied in clinical studies of aging, MCI, and AD: MRI of axonal injury and connectivity (diffuse tensor imaging (DTI) and high definition fiber tractography) (MacDonald et al., 2007, 2011; Presson et al., 2015) as well as amyloid PET (Eisenmenger et al., 2016; Gatson et al., 2016; Hong et al., 2014; Kawai et al., 2013; Mitsis et al., 2014; Scott et al., 2016). Several of these clinical analyses replicate data from histopathology studies. Kawai and colleagues (Kawai et al., 2013) reported [C-11]PiB retention in 27% (3/11) of imaged patients, reminiscent of the percent of severe TBI cases with histological evidence of Aβ plaques. Jack and colleagues (Mielke et al., 2014) found that among the MCI subjects undergoing [C-11]PiB PET imaging in the Mayo Clinic Study of Aging, those with self-reported TBI had greater PiB-PET retention. In a study of long-term survivors of moderate-severe TBI, increased PiB-PET was associated with DTI evidence of altered connectivity involving the posterior cingulate cortex (Scott et al., 2016), a cortical area vulnerable to Aβ accumulation in AD. It will be important to use these imaging modalities in conjunction with imaging measures of vascular dysfunction to assess the relation between amyloid lesions and CVD after TBI, particularly in the aging brain as has been investigated in non-TBI subjects (Marchant et al., 2013).
5.2. Synapse loss
As discussed above, DAI disrupts axonal transport (both retrograde and anterograde) which, in addition to accumulation of proteins in axonal swellings, results in disconnection/deafferentation (see (Povlishock and Katz, 2005) for review). The time course of this process differs between experimental animals and humans and is associated with alterations in synapses (Povlishock et al., 1992). While axonal disruption can be extensive after TBI, Povlishock and colleagues pointed out that “the amount of degenerating nerve terminals far exceeds the number of identified damaged fibers” (Povlishock and Katz, 2005) underlining the importance of synapse degeneration in TBI. Detection of synapse degeneration/loss in TBI can be confounded by ongoing restorative processes including synapse regeneration, a phenomenon that could reflect brain compensatory response or an aberrant response to loss of synapses. Experimental studies by Cotman and colleagues revealed that this process is active in the hippocampus after brain injury (Cotman et al., 1981) and can be influenced by aging. This is most clearly demonstrated in the entorhinal cortex lesion (ECx) model. After complete unilateral ECx, greater loss of synapse density in the outer molecular layer of the dentate gyrus and slower synapse replacement was observed in aged compared to young rats (Hoff et al., 1982). Loss of synapses is a structural correlate of cognitive impairment in AD (Scheff and Price, 2006), and it could contribute to increased risk for dementia after TBI, particularly chronically with aging. When synapses are quantified in clinical groups with no cognitive impairment, MCI and mild AD, synapse loss occurs in a stepwise fashion in some neocortical areas (inferior temporal gyrus and posterior cingulate cortex (Scheff et al., 2015, 2011)) while in others (e.g., precuneus) synapse loss is seen only in mild AD (Scheff et al., 2013). Synapse loss chronically after experimental TBI might recapitulate regional vulnerability patterns seen in AD. For example, cortical impact injury produces secondary injury with extensive synapse loss in the underlying hippocampus, a region vulnerable to synapse loss in AD (Scheff et al., 2005). The role of Aβ in synapse loss after experimental TBI remains to be investigated. We demonstrated that CCI injury produced greater loss of pre- and post-synaptic markers in hAβ KI mice compared to wild type counterparts expressing endogenous (mouse) Aβ (Fig. 4). This suggests that human Aβ oligomers play a role in synapse degeneration after TBI, similar to what occurs in AD (Walsh et al., 2002). Advanced age, particularly >65 years involves synapse loss (Masliah et al., 2006, 1993), and this could contribute to late-onset AD dementia in some aging survivors of TBI since any significant synapse loss could lower the threshold for clinical manifestation of AD.
Fig. 4.
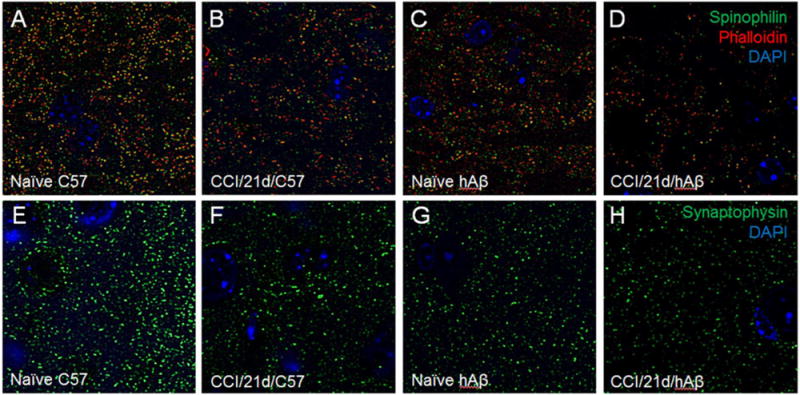
Confocal microscopy analyses of dendritic spines double immunolabeled using antibodies against spinophilin (polyclonal anti-spinophilin antibody, Millipore; green, A–D) and phalloidin (f-actin probe, Thermo; red, A–D) and presynaptic terminals immunolabeled with anti-synaptophysin antibody (polyclonal anti-synaptophysin antibody, Thermo; green, E–H) in the hippocampus of naïve (uninjured) wild type mice (line C57Bl/6, A, E) and naïve (uninjured) human Aβ (hAβ) knock-in mice (C, G) compared to CCI injured wild type mice (B, F) and hAβ mice (D,H) with 21 days survival. Blue fluorescence is DAPI counterstain. CCI injury results in loss of both spinophilin/phalloidin positive dendritic spines and synaptophysin positive presynaptic terminals, and the extent of this loss is greater in hAβ mice when compared to wild type mice.
5.3. Oxidative stress
Detrimental effects of reactive oxygen species (ROS) due to mitochondrial leakage or accumulation of Aβ have been posited to underlie oxidative damage and cellular dysfunction in aging (Indo et al., 2015) and AD (Swomley and Butterfield, 2015), respectively. Studies by Scheff and colleagues demonstrate an association between oxidative stress and loss of synaptic proteins in the presence of Aβ pathology in preclinical and prodromal AD (Scheff et al., 2016). Although the cause and effect relationship between ROS and A in cellular/synaptic dysfunction is still a matter of debate in the field of neurodegenerative disorders (Mattson, 2011), both variables likely influence outcome, including risk for AD, after TBI. ROS production increases after TBI and could contribute to DNA damage, protein and lipid peroxidation, mitochondrial dysfunction and cell death (Hall, 2015). At the level of the NVU, pathological levels of ROS can induce cell death and disrupt the BBB (von Leden et al., 2016). In aging, mitochondria dysfunction and reductions in antioxidant enzymes increase oxidative stress at the NVU, in part through peroxinitrite formation (van der Loo et al., 2000) resulting from the interaction of endothelial cell derived NO and superoxide. These mechanisms could be exacerbated in the presence of Aβ or, conversely, promote brain accumulation of Aβ. The connection between Aβ and oxidative stress after TBI was examined in nine month old Tg2576 mice (prior to plaque deposition in this model) and wild type littermates subjected to rmTBI. In this study, Trojanowski and colleagues (Uryu et al., 2002) detected increased Aβ concentrations and deposition, increased levels of lipid peroxidation, and cognitive impairment in transgenic but not wild type mice (Uryu et al., 2002) and suggested that brain Aβ accumulation and oxidative stress could work synergistically after TBI to increase the risk for AD (Uryu et al., 2002). Collectively, ROS appear to be pathologically active in TBI, aging, and AD and should be targeted by therapies aiming to break the link between brain injury and AD.
5.4. Apoptosis
Apoptosis is an important cell death mechanism in TBI (reviewed in (Bramlett and Dietrich, 2015)). Pro-apoptotic molecules such as cytochrome-c and apoptosis inducing factor are released from damaged mitochondria and activate caspases in injured axonal segments, contributing to cytoskeletal damage. Increased Aβ production after TBI could contribute to the induction of apoptosis through caspase activation (Allen et al., 2001; Nishimura et al., 2002; Uetsuki et al., 1999) which could, in turn, promote amyloidogenic processing of APP (Gervais et al., 1999). Enhanced caspase cleavage of APP after TBI or in aging leads to Aβ over-production and could contribute to the pathological link between TBI and AD (Stone et al., 2002; Zhao et al., 2003). Interestingly, Koliatsos and colleagues reported that in subjects with no cognitive impairment (NCI), there is little if any evidence of apoptosis in the absence of Aβ pathology, however apoptosis is present in NCI cases with Aβ plaques (a sign of pre-clinical AD) and is markedly increased in AD brains, (Troncoso et al., 1996). Although unexplored, the implications of these observations are that TBI patients who develop Aβ plaques could exhibit greater neuronal loss, and with aging this could lower the threshold for clinical manifestation of AD. We investigated, albeit indirectly, the effects of caspase-cleaved APP and human Aβ on outcome after TBI (Abrahamson et al., 2006), and observed increased levels of APP, caspase-cleaved APP, and Aβ monomers/oligomers after severe CCI injury in hAβ KI mice. Pan-caspase inhibition immediately after CCI injury reduced levels of both caspase-cleaved APP and Aβ and improved histological outcome (Abrahamson et al., 2006), suggesting that targeting caspases after TBI could reduce injury-induced AD-like pathology. Aging, when superimposed on TBI, could enhance apoptotic response to brain injury and this could impair neurological outcome. In support of the notion that aging can exacerbate injury-induced neuronal degeneration, FPI produced higher number of TUNEL positive neurons in aged rats when compared to juvenile rats (Sun et al., 2013).
5.5. Neuroinflammation
Aging is associated with a shift to a pro-inflammatory state (Godbout and Johnson, 2009; Sandhir et al., 2008), enhanced microglia reactivity (Yu et al., 2002), and microglia senescence (Neumann et al., 2009; Streit et al., 2008). Microgliosis and reactive astrocytosis accompany amyloid lesions in AD, and they are potentially involved in Aβ-related neurodegeneration after TBI. Astrocyte signaling mechanisms are activated after TBI, and are associated with heterogeneous morphological changes determined in part by injury severity (reviewed in (Burda et al., 2016)). Van Eldik and colleagues observed that closed head injury produced a delayed but enhanced and persistent astrocytic response in hAβ KI mice with the PS1 mutation compared to wild type mice (Webster et al., 2015). Moreover, greater plaque load was detected in hAβ KI/PS1 mice after TBI compared to sham-operates, and the degree of astrocyte activation was also greater in injured transgenic mice compared to transgenic sham operates and injured wild type mice. These results suggest a relation between injury-induced Aβ changes and astrocyte activation after TBI. These processes could contribute to clearance of cell debris and neurovascular remodeling (Burda et al., 2016) and involve several receptors including toll-like receptors (TLRs) and RAGE. Astrocytes are capable of internalizing and metabolizing Aβ (Wyss-Coray et al., 2003), possibly through Aβ binding to the low density lipoprotein receptor-related protein 1 (Thal, 2012). After TBI astrocytes release inflammatory mediators including cyclo-oxygenase-2 and metalloproteinases, also active in AD (Phillips et al., 2014), and could play a role in sequestering toxic Aβ.
Microglia have the potential to be beneficial or detrimental after TBI, depending on their polarization/characterization as M1 (pro-inflammatory) or M2 (immunosuppressive), reviewed in (Loane and Kumar, 2016). Different phenotypes of microglial activation and related neuroreparatory versus neurotoxic processes need to be explored in relation to Aβ changes after TBI. In adult hAβ KI mice subjected to CCI injury we observed a rapid (hours) and sustained (weeks) increase in microglia immunoreactive for ionized calcium binding adaptor molecule 1, and the course of this reactive process corresponded to increased accumulation of human Aβ and loss of synaptophysin immunoreactivity in the cortex and hippocampus (Abrahamson et al., 2009). Similarly, more pronounced microglia reaction and more sustained memory impairment after closed head injury were reported in hAβ KI/PS1 mice compared to wild-type mice (Webster et al., 2015). Recent research supports combined effects of TBI and aging on microglia function. Aging can affect microglia function after TBI (Lourbopoulos et al., 2015), possibly reflected by morphological changes (Harry, 2013), which could influence outcome at different stages of recovery (Hefendehl et al., 2014; Norden et al., 2015). For example, in the aging brain the microglia profile changes into a primed (inflammatory) state which involves an exaggerated cytokine (IL-1β) response and increased expression of inflammatory (IL-1β, TNF-α) as well as immune (MHC II) markers, similar to what occurs after TBI and in AD (reviewed in (Norden et al., 2015)). Furthermore, aging impairs the ability of microglia to internalize Aβ, despite their preserved ability to adhere to Aβ fibrils both in situ (plaques) and isolated in vitro (Floden and Combs, 2011). Aging-related impairment in Aβ clearance from brain may explain greater vulnerability to neurodegenerative processes after injury in the aging brain (Harry, 2013). Animal studies also demonstrate that after TBI, peripheral monocyte infiltration and activation is greater in aged mice (Morganti et al., 2016). Thus, inflammatory processes activated after TBI may be exacerbated by aging-related pathophysiology and this could result in chronic neuroinflammation conferring greater risk of developing AD later in life.
6. Summary and conclusions
There is substantial evidence linking TBI to increased risk for AD and supporting a role for disordered APP metabolism and aging in this relationship. It is also becoming clear that both severe TBI and rmTBI involve complex polypathologies, with Aβ and tau changes occurring in different proportions at different time points after injury, and can also include aggregates of other proteins linked to chronic neurodegenerative diseases (e.g., TDP-43, alpha-synuclein). In this regard, future studies should strive to explore a wider array of aggregation-prone molecules, and employ more diverse assessment approaches to investigate pathological molecular changes not readily detectable in routine diagnostic neuropathology workups (e.g., soluble Aβ and tau oligomers). Studies of TBI should also define the time window of opportunity for treatments prior to advanced age when risk is greater for sustained accumulation of pathological protein aggregates, neuronal degeneration, and cognitive and behavioral changes. These treatments should aim to suppress prolonged accumulation of Aβ and tau, and also prevent or attenuate pathological effects of these molecules on cellular components of the NVU. Ideally such strategies would supplement neuroprotection-focused therapies and advance the efforts to break the link between TBI and AD.
Acknowledgments
This work was supported by Veterans Affairs grants 1I01RX000511, 1I01RX000952, 1I01RX001778, NIA grants AG014449 and AG05133, and NINDS grant NS30318.
References
- Abisambra JF, Scheff S. Brain injury in the context of tauopathies. J Alzheimers Dis. 2014;40:495–518. doi: 10.3233/JAD-131019. [DOI] [PubMed] [Google Scholar]
- Abner EL, Nelson PT, Schmitt FA, Browning SR, Fardo DW, Wan L, Jicha GA, Cooper GE, Smith CD, Caban-Holt AM, Van Eldik LJ, Kryscio RJ. Self-reported head injury and risk of late-life impairment and AD pathology in an AD center cohort. Dement Geriatr Cogn Disord. 2014;37:294–306. doi: 10.1159/000355478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood DG, Clark RSB, DeKosky ST. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp Neurol. 2006;197:437–450. doi: 10.1016/j.expneurol.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Abrahamson EE, Ikonomovic MD, Dixon CE, DeKosky ST. Simvastatin therapy prevents brain trauma-induced increases in beta-amyloid peptide levels. Ann Neurol. 2009;66:407–414. doi: 10.1002/ana.21731. [DOI] [PubMed] [Google Scholar]
- Abrahamson EE, Foley LM, Dekosky ST, Hitchens TK, Ho C, Kochanek PM, Ikonomovic MD. Cerebral blood flow changes after brain injury in human amyloid-beta knock-in mice. J Cereb Blood Flow Metab. 2013;33:826–833. doi: 10.1038/jcbfm.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams SD, Holcomb JB. Geriatric trauma. Curr Opin Crit Care. 2015;21:520–526. doi: 10.1097/MCC.0000000000000246. [DOI] [PubMed] [Google Scholar]
- Akbik OS, Carlson AP, Krasberg M, Yonas H. The utility of cerebral blood flow assessment in TBI. Curr Neurol Neurosci Rep. 2016;16:72. doi: 10.1007/s11910-016-0672-3. [DOI] [PubMed] [Google Scholar]
- Allen JW, Eldadah BA, Huang X, Knoblach SM, Faden AI. Multiple caspases are involved in beta-amyloid-induced neuronal apoptosis. J Neurosci Res. 2001;65:45–53. doi: 10.1002/jnr.1126. [DOI] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead WM, Kreipke CW. Endothelin-1 is upregulated after traumatic brain injury: a cross-species, cross-model analysis. Neurol Res. 2011;33:133–136. doi: 10.1179/016164111X12881719352174. [DOI] [PubMed] [Google Scholar]
- Austin SA, Santhanam AV, Hinton DJ, Choi DS, Katusic ZS. Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J Neurochem. 2013;127:691–700. doi: 10.1111/jnc.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig S, Joseph SA, Tayler H, Abraham R, Owen MJ, Williams J, Kehoe PG, Love S. Distribution and expression of picalm in Alzheimer disease. J Neuropathol Exp Neurol. 2010;69:1071–1077. doi: 10.1097/NEN.0b013e3181f52e01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton M, Cosentino F, Brandes RP, Moreau P, Shaw S, Luscher TF. Anatomic heterogeneity of vascular aging: role of nitric oxide and endothelin. Hypertension. 1997;30:817–824. doi: 10.1161/01.hyp.30.4.817. [DOI] [PubMed] [Google Scholar]
- Bates K, Vink R, Martins R, Harvey A. Aging, cortical injury and Alzheimer’s disease-like pathology in the guinea pig brain. Neurobiol Aging. 2014;35:1345–1351. doi: 10.1016/j.neurobiolaging.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird SM, Sohrabi HR, Sutton TA, Weinborn M, Rainey-Smith SR, Brown B, Patterson L, Taddei K, Gupta V, Carruthers M, Lenzo N, Knuckey N, Bucks RS, Verdile G, Martins RN. Cerebral amyloid-beta accumulation and deposition following traumatic brain injury—a narrative review and meta-analysis of animal studies. Neurosci Biobehav Rev. 2016;64:215–228. doi: 10.1016/j.neubiorev.2016.01.004. [DOI] [PubMed] [Google Scholar]
- Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36:297–309. doi: 10.1016/j.tips.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J Neurotrauma. 2015;32:1834–1848. doi: 10.1089/neu.2014.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Buki A, Povlishock JT. All roads lead to disconnection?—traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–193. doi: 10.1007/s00701-005-0674-4. discussion 193–194. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2016;275:305–315. doi: 10.1016/j.expneurol.2015.03.020. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castejon OJ. Ultrastructural pathology of cortical capillary pericytes in human traumatic brain oedema. Folia Neuropathol. 2011;49:162–173. [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Maki T, Mandeville ET, Koh SH, Hayakawa K, Arai K, Kim YM, Whalen MJ, Xing C, Wang X, Kim KW, Lo EH. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat Med. 2016 doi: 10.1038/nm.4188. http://dx.doi.org/10.1038/nm.4188 (Epub ahead of print) [DOI] [PubMed]
- Coronado VG, Thomas KE, Sattin RW, Johnson RL. The CDC traumatic brain injury surveillance system: characteristics of persons aged 65 years and older hospitalized with a TBI. J Head Trauma Rehabil. 2005;20:215–228. doi: 10.1097/00001199-200505000-00005. [DOI] [PubMed] [Google Scholar]
- Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Nieto-Sampedro M, Harris EW. Synapse replacement in the nervous system of adult vertebrates. Physiol Rev. 1981;61:684–784. doi: 10.1152/physrev.1981.61.3.684. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Tenner AJ, Cummings BJ. Beta-amyloid converts an acute phase injury response to chronic injury responses. Neurobiol Aging. 1996;17:723–731. doi: 10.1016/0197-4580(96)00117-0. [DOI] [PubMed] [Google Scholar]
- Crane PK, Gibbons LE, Dams-O’Connor K, Trittschuh E, Leverenz JB, Keene CD, Sonnen J, Montine TJ, Bennett DA, Leurgans S, Schneider JA, Larson EB. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. 2016;73:1062–1069. doi: 10.1001/jamaneurol.2016.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czosnyka M, Balestreri M, Steiner L, Smielewski P, Hutchinson PJ, Matta B, Pickard JD. Age, intracranial pressure, autoregulation, and outcome after brain trauma. J Neurosurg. 2005;102:450–454. doi: 10.3171/jns.2005.102.3.0450. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS, Ikonomovic MD. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64:541–544. doi: 10.1001/archneur.64.4.541. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Blennow K, Ikonomovic MD, Gandy S. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol. 2013;9:192–200. doi: 10.1038/nrneurol.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004a;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004b;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60:55–69. doi: 10.1006/mvre.2000.2244. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Wang S, Mehedi A, Katyshev V, Cleary K, Tapper A, Reynolds C, Ding Y, Zhan P, Rafols J, Kreipke CW. Pericyte-mediated vasoconstriction underlies TBI-induced hypoperfusion. Neurol Res. 2011;33:176–186. doi: 10.1179/016164111X12881719352372. [DOI] [PubMed] [Google Scholar]
- Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer’s amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- Eisenmenger LB, Huo EJ, Hoffman JM, Minoshima S, Matesan MC, Lewis DH, Lopresti BJ, Mathis CA, Okonkwo DO, Mountz JM. Advances in PET imaging of degenerative, cerebrovascular, and traumatic causes of dementia. Semin Nucl Med. 2016;46:57–87. doi: 10.1053/j.semnuclmed.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Emre M, Geula C, Ransil BJ, Mesulam MM. The acute neurotoxicity and effects upon cholinergic axons of intracerebrally injected beta-amyloid in the rat brain. Neurobiol Aging. 1992;13:553–559. doi: 10.1016/0197-4580(92)90055-3. [DOI] [PubMed] [Google Scholar]
- Faul M, Coronado V. Epidemiology of traumatic brain injury. Handb Clin Neurol. 2015;127:3–13. doi: 10.1016/B978-0-444-52892-6.00001-5. [DOI] [PubMed] [Google Scholar]
- Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J Alzheimers Dis. 2011;25:279–293. doi: 10.3233/JAD-2011-101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gandy S, Ikonomovic MD, Mitsis E, Elder G, Ahlers ST, Barth J, Stone JR, DeKosky ST. Chronic traumatic encephalopathy: clinical-biomarker correlations and current concepts in pathogenesis. Mol Neurodegener. 2014;9:37. doi: 10.1186/1750-1326-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014;71:1490–1497. doi: 10.1001/jamaneurol.2014.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwood CJ, Ratcliffe LE, Simpson JE, Heath PR, Ince PG, Wharton SB. Review: astrocytes in Alzheimer’s disease and other age-associated dementias; a supporting player with a central role. Neuropathol Appl Neurobiol. 2016 doi: 10.1111/nan.12338. [DOI] [PubMed] [Google Scholar]
- Gatson JW, Stebbins C, Mathews D, Harris TS, Madden C, Batjer H, Diaz-Arrastia R, Minei JP. Evidence of increased brain amyloid in severe TBI survivors at 1, 12, and 24 months after injury: report of 2 cases. J Neurosurg. 2016;124:1646–1653. doi: 10.3171/2015.6.JNS15639. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Graham DI, Roberts GW. Molecular pathology of head trauma: altered beta APP metabolism and the aetiology of Alzheimer’s disease. Prog Brain Res. 1993a;96:237–246. doi: 10.1016/s0079-6123(08)63270-7. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993b;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW, Griffin WS, Graham DI. A beta 42 is the predominant form of amyloid beta-protein in the brains of short-term survivors of head injury. Neuroreport. 1997;8:1519–1522. doi: 10.1097/00001756-199704140-00039. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Ghiso J, Fossati S, Rostagno A. Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J Alzheimers Dis. 2014;42(Suppl 3):S167–176. doi: 10.3233/JAD-140027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin North Am. 2009;29:321–337. doi: 10.1016/j.iac.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Graham DI, Gentleman SM, Lynch A, Roberts GW. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol. 1995;21:27–34. doi: 10.1111/j.1365-2990.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- Graham DI, Gentleman SM, Nicoll JA, Royston MC, McKenzie JE, Roberts GW, Griffin WS. Altered beta-APP metabolism after head injury and its relationship to the aetiology of Alzheimer’s disease. Acta Neurochir. 1996;(Suppl 66):96–102. doi: 10.1007/978-3-7091-9465-2_17. [DOI] [PubMed] [Google Scholar]
- Grammas P, Botchlet T, Fugate R, Ball MJ, Roher AE. Alzheimer disease amyloid proteins inhibit brain endothelial cell proliferation in vitro. Dementia. 1995;6:126–130. doi: 10.1159/000106934. [DOI] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED. The contributing role of lipid peroxidation and protein oxidation in the course of CNS injury neurodegeneration and neuroprotection: an overview. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. CRC Press/Taylor & Francis; Boca Raton (FL): 2015. Chapter 6. [PubMed] [Google Scholar]
- Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;36:216–227. doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Gen. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry GJ. Microglia during development and aging. Pharmacol Ther. 2013;139:313–326. doi: 10.1016/j.pharmthera.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO. Failure of perivascular drainage of beta-amyloid in cerebral amyloid angiopathy. Brain Pathol. 2014;24:396–403. doi: 10.1111/bpa.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AM, Smith DH, Stewart W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol. 2015;74:1147–1157. doi: 10.1097/NEN.0000000000000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. 2014;13:60–69. doi: 10.1111/acel.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- Hoff SF, Scheff SW, Benardo LS, Cotman CW. Lesion-induced synaptogenesis in the dentate gyrus of aged rats: I. Loss and reacquisition of normal synaptic density. J Comp Neurol. 1982;205:246–252. doi: 10.1002/cne.902050304. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hong YT, Veenith T, Dewar D, Outtrim JG, Mani V, Williams C, Pimlott S, Hutchinson PJ, Tavares A, Canales R, Mathis CA, Klunk WE, Aigbirhio FI, Coles JP, Baron JC, Pickard JD, Fryer TD, Stewart W, Menon DK. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014;71:23–31. doi: 10.1001/jamaneurol.2013.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh K, Cole GM, Yang F, Savage MJ, Greenberg BD, Gentleman SM, Graham DI, Nicoll JA. Beta-amyloid (abeta)42(43), abeta42, abeta40 and apoE immunostaining of plaques in fatal head injury. Neuropathol Appl Neurobiol. 2000;26:124–132. doi: 10.1046/j.1365-2990.2000.026002124.x. [DOI] [PubMed] [Google Scholar]
- Howarth C. The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci. 2014;8:103. doi: 10.3389/fnins.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hsu MJ, Hsu CY, Chen BC, Chen MC, Ou G, Lin CH. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J Neurosci. 2007;27:5719–5729. doi: 10.1523/JNEUROSCI.1874-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ. Amyloid-beta peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998;785:195–206. doi: 10.1016/s0006-8993(97)01318-8. [DOI] [PubMed] [Google Scholar]
- Hukkelhoven CW, Steyerberg EW, Rampen AJ, Farace E, Habbema JD, Marshall LF, Murray GD, Maas AI. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99:666–673. doi: 10.3171/jns.2003.99.4.0666. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo HP, Yen HC, Nakanishi I, Matsumoto K, Tamura M, Nagano Y, Matsui H, Gusev O, Cornette R, Okuda T, Minamiyama Y, Ichikawa H, Suenaga S, Oki M, Sato T, Ozawa T, Clair DK, Majima HJ. A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr. 2015;56:1–7. doi: 10.3164/jcbn.14-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Lowe V, Kantarci K, Bernstein MA, Senjem ML, Gunter JL, Boeve BF, Trojanowski JQ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Knopman DS. Alzheimer’s disease neuroimaging, I., shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch Neurol. 2012;69:856–867. doi: 10.1001/archneurol.2011.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Graham DI, Stewart JE, Praestgaard AH, Smith DH. A neprilysin polymorphism and amyloid-beta plaques after traumatic brain injury. J Neurotrauma. 2009;26:1197–1202. doi: 10.1089/neu.2008.0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Trojanowski JQ, Smith DH. Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 2011;122:715–726. doi: 10.1007/s00401-011-0909-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B, Pandit V, Rhee P, Aziz H, Sadoun M, Wynne J, Tang A, Kulvatunyou N, O’Keeffe T, Fain MJ, Friese RS. Predicting hospital discharge disposition in geriatric trauma patients: is frailty the answer? J Trauma Acute Care Surg. 2014;76:196–200. doi: 10.1097/TA.0b013e3182a833ac. [DOI] [PubMed] [Google Scholar]
- Kawai N, Kawanishi M, Kudomi N, Maeda Y, Yamamoto Y, Nishiyama Y, Tamiya T. Detection of brain amyloid beta deposition in patients with neuropsychological impairment after traumatic brain injury: PET evaluation using Pittsburgh compound-B. Brain Inj. 2013;27:1026–1031. doi: 10.3109/02699052.2013.794963. [DOI] [PubMed] [Google Scholar]
- Kay AD, Petzold A, Kerr M, Keir G, Thompson EJ, Nicoll JA. Cerebrospinal fluid apolipoprotein E concentration decreases after traumatic brain injury. J Neurotrauma. 2003;20:243–250. doi: 10.1089/089771503321532824. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, Soiza RL. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis. 2013;3:197–226. [PMC free article] [PubMed] [Google Scholar]
- Koizumi K, Wang G, Park L. Endothelial dysfunction and amyloid-beta-induced neurovascular alterations. Cell Mol Neurobiol. 2016;36:155–165. doi: 10.1007/s10571-015-0256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreipke CW, Schafer PC, Rossi NF, Rafols JA. Differential effects of endothelin receptor A and B antagonism on cerebral hypoperfusion following traumatic brain injury. Neurol Res. 2010;32:209–214. doi: 10.1179/174313209X414515. [DOI] [PubMed] [Google Scholar]
- Lecuyer MA, Kebir H, Prat A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochim Biophys Acta. 2016;1862:472–482. doi: 10.1016/j.bbadis.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol. 2016;275:316–327. doi: 10.1016/j.expneurol.2015.08.018. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW. Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol. 2015;5:1147–1160. doi: 10.1002/cphy.c140057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourbopoulos A, Erturk A, Hellal F. Microglia in action: how aging and injury can change the brain’s guardians. Front Cell Neurosci. 2015;9:54. doi: 10.3389/fncel.2015.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Miners JS. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016;131:645–658. doi: 10.1007/s00401-015-1522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- MacDonald CL, Dikranian K, Bayly P, Holtzman D, Brody D. Diffusion tensor imaging reliably detects experimental traumatic axonal injury and indicates approximate time of injury. J Neurosci. 2007;27:11869–11876. doi: 10.1523/JNEUROSCI.3647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, Snyder AZ, Raichle ME, Witherow JR, Fang R, Flaherty SF, Brody DL. Detection of blast-related traumatic brain injury in U.S. military personnel. N Engl J Med. 2011;364:2091–2100. doi: 10.1056/NEJMoa1008069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, Mack WJ, DeCarli C, Weiner MW, Mungas DM, Chui HC, Jagust WJ. The aging brain and cognition: contribution of vascular injury and abeta to mild cognitive dysfunction. JAMA Neurol. 2013;70:488–495. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–197. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- Masliah E, Crews L, Hansen L. Synaptic remodeling during aging and in Alzheimer’s disease. J Alzheimers Dis. 2006;9:91–99. doi: 10.3233/jad-2006-9s311. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Commentary: proteooxidotoxic process of aggregation. Neuromol Med. 2011;13:91–92. doi: 10.1007/s12017-011-8146-x. [DOI] [PubMed] [Google Scholar]
- Maxwell CA, Miller RS, Dietrich MS, Mion LC, Minnick A. The aging of America: a comprehensive look at over 25,000 geriatric trauma admissions to United States hospitals. Am Surg. 2015;81:630–636. doi: 10.1177/000313481508100630. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M, Budson AE. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015;25:350–364. doi: 10.1111/bpa.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie JE, Gentleman SM, Roberts GW, Graham DI, Royston MC. Increased numbers of beta APP-immunoreactive neurones in the entorhinal cortex after head injury. Neuroreport. 1994;6:161–164. doi: 10.1097/00001756-199412300-00041. [DOI] [PubMed] [Google Scholar]
- McKenzie KJ, McLellan DR, Gentleman SM, Maxwell WL, Gennarelli TA, Graham DI. Is beta-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathol. 1996;92:608–613. doi: 10.1007/s004010050568. [DOI] [PubMed] [Google Scholar]
- Mehta KM, Ott A, Kalmijn S, Slooter AJ, van Duijn CM, Hofman A, Breteler MM. Head trauma and risk of dementia and Alzheimer’s disease: the Rotterdam study. Neurology. 1999;53:1959–1962. doi: 10.1212/wnl.53.9.1959. [DOI] [PubMed] [Google Scholar]
- Mielke MM, Savica R, Wiste HJ, Weigand SD, Vemuri P, Knopman DS, Lowe VJ, Roberts RO, Machulda MM, Geda YE, Petersen RC, Jack CR., Jr Head trauma and in vivo measures of amyloid and neurodegeneration in a population-based study. Neurology. 2014;82:70–76. doi: 10.1212/01.wnl.0000438229.56094.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsis EM, Riggio S, Kostakoglu L, Dickstein DL, Machac J, Delman B, Goldstein M, Jennings D, D’Antonio E, Martin J, Naidich TP, Aloysi A, Fernandez C, Seibyl J, DeKosky ST, Elder GA, Marek K, Gordon W, Hof PR, Sano M, Gandy S. Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatry. 2014;4:e441. doi: 10.1038/tp.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Riparip LK, Chou A, Liu S, Gupta N, Rosi S. Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J Neuroinflammation. 2016;13:80. doi: 10.1186/s12974-016-0547-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies: EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ikonomovic MD, Counts SE, Perez SE, Malek-Ahmadi M, Scheff SW, Ginsberg SD. Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav Brain Res. 2016;311:54–69. doi: 10.1016/j.bbr.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder SD, Veerhuis R, Blankenstein MA, Nielsen HM. The effect of amyloid associated proteins on the expression of genes involved in amyloid-beta clearance by adult human astrocytes. Exp Neurol. 2012;233:373–379. doi: 10.1016/j.expneurol.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging. 2004;25:663–674. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura I, Uetsuki T, Kuwako K, Hara T, Kawakami T, Aimoto S, Yoshikawa K. Cell death induced by a caspase-cleaved transmembrane fragment of the Alzheimer amyloid precursor protein. Cell Death Diff. 2002;9:199–208. doi: 10.1038/sj.cdd.4400931. [DOI] [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96:29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. discussion 128–134. [DOI] [PubMed] [Google Scholar]
- Ortman JM, Velkoff VA, Hogan H. Current Population Reports. U.S. CensusBureau; Washington, DC: 2014. An Aging Nation: The Older Population in the United States. Population Estimates and Projections; pp. 25–1140. https://www.census.gov/prod/2014pubs/p25-1140.pdf. [Google Scholar]
- Palmer JC, Baig S, Kehoe PG, Love S. Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by abeta. Am J Pathol. 2009;175:262–270. doi: 10.2353/ajpath.2009.081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JC, Barker R, Kehoe PG, Love S. Endothelin-1 is elevated in Alzheimer’s disease and upregulated by amyloid-beta. J Alzheimers Dis. 2012;29:853–861. doi: 10.3233/JAD-2012-111760. [DOI] [PubMed] [Google Scholar]
- Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: role of inflammation. Neurol Res. 2003;25:642–651. doi: 10.1179/016164103101201940. [DOI] [PubMed] [Google Scholar]
- Patel NS, Mathura VS, Bachmeier C, Beaulieu-Abdelahad D, Laporte V, Weeks O, Mullan M, Paris D. Alzheimer’s beta-amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. J Neurochem. 2010;112:66–76. doi: 10.1111/j.1471-4159.2009.06426.x. [DOI] [PubMed] [Google Scholar]
- Petrov T. Amelioration of hypoperfusion after traumatic brain injury by in vivo endothelin-1 knockout. Can J Physiol Pharmacol. 2009;87:379–386. doi: 10.1139/y09-022. [DOI] [PubMed] [Google Scholar]
- Phillips EC, Croft CL, Kurbatskaya K, O’Neill MJ, Hutton ML, Hanger DP, Garwood CJ, Noble W. Astrocytes and neuroinflammation in Alzheimer’s disease. Biochem Soc Trans. 2014;42:1321–1325. doi: 10.1042/BST20140155. [DOI] [PubMed] [Google Scholar]
- Plassman BL, Grafman J. Traumatic brain injury and late-life dementia. Handb Clin Neurol. 2015;128:711–722. doi: 10.1016/B978-0-444-63521-1.00044-3. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Erb DE, Astruc J. Axonal response to traumatic brain injury: reactive axonal change, deafferentation, and neuroplasticity. J Neurotrauma. 1992;9(Suppl 1):S189–200. [PubMed] [Google Scholar]
- Presson N, Beers SR, Morrow L, Wagener LM, Bird WA, Van Eman G, Krishnaswamy D, Penderville J, Borrasso AJ, Benso S, Puccio A, Fissell C, Okonkwo DO, Schneider W. An exploratory analysis linking neuropsychological testing to quantification of tractography using High Definition Fiber Tracking (HDFT) in military TBI. Brain Imaging Behav. 2015;9:484–499. doi: 10.1007/s11682-015-9386-4. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and preclinical Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Price L, Wilson C, Grant G. Blood-Brain Barrier Pathophysiology following Traumatic Brain Injury. In: Laskowitz D, Grant G, editors. Translational Research in Traumatic Brain Injury. CRC Press/Taylor & Francis; Boca Raton (FL): 2016. Chapter 4. [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Wolf U, Herrmann N, Kiss A, Shammi P, Reis M, Phillips A, Feinstein A. Traumatic brain injury, apolipoprotein E-epsilon4, and cognition in older adults: a two-year longitudinal study. J Neuropsychiatry Clin Neurosci. 2008;20:68–73. doi: 10.1176/jnp.2008.20.1.68. [DOI] [PubMed] [Google Scholar]
- Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–378. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Graham DI. Beta A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Horkayne-Szakaly I, Xu L, Pletnikova O, Leri F, Eberhart C, Troncoso JC, Koliatsos VE. The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol Commun. 2014;2:153. doi: 10.1186/s40478-014-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saing T, Dick M, Nelson PT, Kim RC, Cribbs DH, Head E. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma. 2012;29:1054–1070. doi: 10.1089/neu.2011.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol. 2008;213:372–380. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2011;24:547–557. doi: 10.3233/JAD-2011-101782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, Mufson EJ. Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J Alzheimers Dis. 2013;35:599–609. doi: 10.3233/JAD-122353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Ansari MA, Roberts KN, Schmitt FA, Ikonomovic MD, Mufson EJ. Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimers Dis. 2015;43:1073–1090. doi: 10.3233/JAD-141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Ansari MA, Mufson EJ. Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol Aging. 2016;42:1–12. doi: 10.1016/j.neurobiolaging.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, Leech R, Greenwood RJ, Turkheimer FE, Gentleman SM, Heckemann RA, Matthews PM, Brooks DJ, Sharp DJ. Amyloid pathology and axonal injury after brain trauma. Neurology. 2016;86:821–828. doi: 10.1212/WNL.0000000000002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Haynes LJ, Theaker R, Gelsthorpe C, Baxter L, Forster G, Lace GL, Shaw PJ, Matthews FE, Savva GM, Brayne C, Wharton SB, Function M.R.C.C, Ageing Neuropathology Study, G Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol Appl Neurobiol. 2010;36:25–40. doi: 10.1111/j.1365-2990.2009.01030.x. [DOI] [PubMed] [Google Scholar]
- Smith DH, Nakamura M, McIntosh TK, Wang J, Rodriguez A, Chen XH, Raghupathi R, Saatman KE, Clemens J, Schmidt ML, Lee VM, Trojanowski JQ. Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol. 1998;153:1005–1010. doi: 10.1016/s0002-9440(10)65643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, Daneshvar DH, Mez J, Solomon T, Meng G, Kubilus CA, Cormier KA, Meng S, Babcock K, Kiernan P, Murphy L, Nowinski CJ, Martin B, Dixon D, Stern RA, Cantu RC, Kowall NW, McKee AC. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015;130:21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Singleton RH, Povlishock JT. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 2000;871:288–302. doi: 10.1016/s0006-8993(00)02485-9. [DOI] [PubMed] [Google Scholar]
- Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J Neurotrauma. 2002;19:601–614. doi: 10.1089/089771502753754073. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Miller KR, Lopes KO, Njie E. Microglial degeneration in the aging brain–bad news for neurons? Front Biosci. 2008;13:3423–3438. doi: 10.2741/2937. [DOI] [PubMed] [Google Scholar]
- Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, McPhie DL, Walsh K, Querfurth H. Abeta42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK-3beta signaling-dependent mechanism. Neurobiol Aging. 2003;24:437–451. doi: 10.1016/s0197-4580(02)00135-5. [DOI] [PubMed] [Google Scholar]
- Sun D, McGinn M, Hankins JE, Mays KM, Rolfe A, Colello RJ. Aging-and injury-related differential apoptotic response in the dentate gyrus of the hippocampus in rats following brain trauma. Front Aging Neurosci. 2013;5:95. doi: 10.3389/fnagi.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swomley AM, Butterfield DA. Oxidative stress in Alzheimer disease and mild cognitive impairment: evidence from human data provided by redox proteomics. Arch Toxicol. 2015;89:1669–1680. doi: 10.1007/s00204-015-1556-z. [DOI] [PubMed] [Google Scholar]
- Tajiri N, Kellogg SL, Shimizu T, Arendash GW, Borlongan CV. Traumatic brain injury precipitates cognitive impairment and extracellular abeta aggregation in Alzheimer’s disease transgenic mice. PLoS One. 2013;8:e78851. doi: 10.1371/journal.pone.0078851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Walter J, Saido TC, Fandrich M. Neuropathology and biochemistry of abeta and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015;129:167–182. doi: 10.1007/s00401-014-1375-y. [DOI] [PubMed] [Google Scholar]
- Thal DR. The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp Neurol. 2012;236:1–5. doi: 10.1016/j.expneurol.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, McCormick WC, Kagan SH. Traumatic brain injury in older adults: epidemiology, outcomes, and future implications. J Am Geriatr Soc. 2006;54:1590–1595. doi: 10.1111/j.1532-5415.2006.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M. Proinflammatory and vasoactive effects of abeta in the cerebrovasculature. Ann N Y Acad Sci. 2002;977:65–76. doi: 10.1111/j.1749-6632.2002.tb04799.x. [DOI] [PubMed] [Google Scholar]
- Tran HT, LaFerla FM, Holtzman DM, Brody DL. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci. 2011;31:9513–9525. doi: 10.1523/JNEUROSCI.0858-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncoso JC, Martin LJ, Dal Forno G, Kawas CH. Neuropathology in controls and demented subjects from the Baltimore Longitudinal Study of Aging. Neurobiol Aging. 1996;17:365–371. doi: 10.1016/0197-4580(96)00028-0. [DOI] [PubMed] [Google Scholar]
- Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Turner RC, Lucke-Wold BP, Robson MJ, Lee JM, Bailes JE. Alzheimer’s disease and chronic traumatic encephalopathy: distinct but possibly overlapping disease entities. Brain Inj. 2016;11:1–14. doi: 10.1080/02699052.2016.1193631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetsuki T, Takemoto K, Nishimura I, Okamoto M, Niinobe M, Momoi T, Miura M, Yoshikawa K. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci. 1999;19:6955–6964. doi: 10.1523/JNEUROSCI.19-16-06955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Laurer H, McIntosh T, Pratico D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer’s disease: a review. Prog Brain Res. 2007;161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- Vincent AS, Roebuck-Spencer TM, Cernich A. Cognitive changes and dementia risk after traumatic brain injury: implications for aging military personnel. Alzheimers Dement. 2014;10:S174–187. doi: 10.1016/j.jalz.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- Washington PM, Morffy N, Parsadanian M, Zapple DN, Burns MP. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J Neurotrauma. 2014;31:125–134. doi: 10.1089/neu.2013.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington PM, Villapol S, Burns MP. Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol. 2016;275:381–388. doi: 10.1016/j.expneurol.2015.06.015. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Van Eldik LJ, Watterson DM, Bachstetter AD. Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J Neurosci. 2015;35:6554–6569. doi: 10.1523/JNEUROSCI.0291-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014;24:371–386. doi: 10.1111/bpa.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J. Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging. 1991;12:593–600. doi: 10.1016/0197-4580(91)90091-w. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen S, Ku G, Ahmed SH, Xu J, Chen H, Hsu CY. Amyloid beta peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J Cereb Blood Flow Metab. 2001;21:702–710. doi: 10.1097/00004647-200106000-00008. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, Kanekiyo T. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016;47:1068–1077. doi: 10.1161/STROKEAHA.115.010835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Go L, Guinn BA, Fraser PE, Westaway D, McLaurin J. Phenotypic and functional changes in glial cells as a function of age. Neurobiol Aging. 2002;23:105–115. doi: 10.1016/s0197-4580(01)00258-5. [DOI] [PubMed] [Google Scholar]
- Zehendner CM, Sebastiani A, Hugonnet A, Bischoff F, Luhmann HJ, Thal SC. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci Rep. 2015;5:13497. doi: 10.1038/srep13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Su J, Head E, Cotman CW. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer’s disease. Neurobiol Dis. 2003;14:391–403. doi: 10.1016/j.nbd.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015a;163:1064–1078. doi: 10.1016/j.cell.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, Owens NC, Rege SV, Si G, Ahuja A, Zhu D, Miller CA, Schneider JA, Maeda M, Maeda T, Sugawara T, Ichida JK, Zlokovic BV. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015b;18:978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC, Aliev G. Inhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changes. J Cereb Blood Flow Metab. 2005;25:663–672. doi: 10.1038/sj.jcbfm.9600057. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Leden RE, Yauger YJ, Khayrullina G, Byrnes KR. Central nervous system injury and nicotinamide adenine dinucleotide phosphate oxidase: oxidative stress and therapeutic targets. J Neurotrauma. 2016 doi: 10.1089/neu.2016.4486. http://dx.doi.org/10.1089/neu.2016.4486 (Epub ahead of print) [DOI] [PMC free article] [PubMed]