

INTRODUCTION
Scope of the Review
Herein, we provide an up-to-date overview of the field of nanopore sensing with emphases placed on the advances made in the most recent two years (2014–2016). We will cover traditional sensing platforms including biological nanopores and solid-state nanopores with a focus on the recent demonstrations of new applications. Additionally, we will discuss novel nanopore sensing platforms such as plasmonic nanopores and hybrid nanopores. An outlook of nanopore sensing will be provided for future developments and directions in the field. We specifically do not consider applications from industry, where details related to specific components and operating conditions are often closely guarded.
Due to the great diversity of nanopore platforms and applications, no single review can include all published work in this rapidly growing field. Because of space limitations, we focus our attention primarily on work published between 2014 and 2016. We attempt to choose papers that showcase the newest and most exciting developments in this young yet already successful field. We selectively cite older literature prior to 2014 to provide the reader a more comprehensive view of the topic at hand. Throughout this Review, we will include previously published reviews dedicated for specific topics for interested readers to gain more information. Reviews that focus on comprehensive nanopore sensing platforms1–5 or biological nanopores,6, 7 solid-state nanopores,8–12 and plasmonic nano-pores13 have been published previously. Interested readers also can learn more about the history and development of nanopore DNA sensing in previous reviews.14–24
Overview of Nanopore Platforms
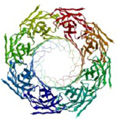
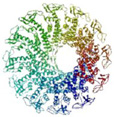
In the past two decades, the field of nanopore sensing has received wide attention due to the high sensitivity and versatility of this technique. Driven by technical advances in the fields of nanotechnology and molecular biology and improved fundamental understandings of nanoscale molecular transport mechanisms, a diversity of developments have been reported recently. Since the landmark demonstration of nucleotide detection with alpha hemolysin (α-HL) embedded within a lipid bilayer,25 a variety of biological nanopores with various characteristics have been developed and utilized in many sensing applications. Reported applications of these protein pores include detection of metal ions,26 small molecules,27, 28 nucleotides,29–36 and proteins,37–48 as well as differentiation between classes of nucleotide conformations.49, 50 Proof-of-principle studies of DNA sequencing have been realized by a combination of engineered biological nanopores with polymer-ase-based positional control.51 More recently, nanopore-based sequencing-by-synthesis (SBS) has been developed.52 In Table 1, six widely employed biological nanopores are shown along with corresponding critical dimensions and the analytes that have been detected.
Table 1.
Details of Commonly Utilized Biological Nanoporesa
| Biological Nanopore |
Structure | Critical Dimension |
Types of Analytes |
References | |
|---|---|---|---|---|---|
| Side View | Top View | ||||
| α-HL | 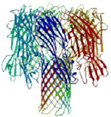 |
 |
1.4 nm108 | small molecules, RNA, ssDNA, dsDNA, proteins |
25, 28, 33, 37, 38, 41, 45, 52, 128, 129, 134– 143, 148–150, 152, 153, 157 |
| OmpG | 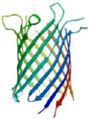 |
 |
1.3 nm118 | small molecules, proteins |
43, 44, 125, 126 |
| MspA |  |
 |
1.2 nm110 | ssDNA, dsDNA |
31, 51, 120–122, 154–156 |
| AeL |  |
 |
1.0 nm111 | ssDNA, proteins |
39, 46, 49, 127, 144, 145, 151 |
|
Phi29 Motor |
 |
 |
3.6 nm114 | small molecules, ssDNA, dsDNA, proteins |
48, 50, 130, 131, 146 |
| ClyA |  |
 |
3.3 nm116 | dsDNA, proteins |
40, 42, 47, 127, 132, 133, 147 |
Molecular graphics images were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081).249
Stimulated by the desire for nanopores with easier manufacturability and better stability, solid-state nanopores with diameters comparable to biological protein pores have been successfully fabricated within synthetic membranes such as silicon nitride (SiNx) and silica (SiO2) via ion beam or electron beam sculpting.53, 54 Solid-state nanopores in polymer membranes,55 alumina,56 and block copolymer membranes57 have also been reported. More recently, fabrication of ultrathin solid-state nanopores that might provide improved sensitivity and spatial resolution in sensing applications has attracted broad interest. Efforts to achieve ultrathin nanopores include electron irradiation-based thinning,58 plasma thinning,59, 60 and focused helium ion beam thinning.61 Another popular route is the direct creation of pores within ultrathin 2-dimensional (2D) materials, including graphene,62–65 boron nitride (BN),66 hafnium oxide (HfO2),67 and molybdenum disulfide (MoS2).68
Synthetic nanopores have tunable properties such as size, geometry, and surface chemistry as well as enhanced mechanical, thermal, and chemical stability in comparison to their biological counterparts. Nonetheless, synthetic nanopores usually lack the specificity of protein pores found in nature. To impart specificity or to eliminate unwanted surface properties from synthetic pores, a wide selection of surface functionalization approaches can be used. For instance, surface properties of nanopore walls can be altered through covalent binding of organosilane molecules,69 atomic layer deposition (ALD) of alumina,70–72 or electrostatic adsorption of molecules that have a charge opposite to the nanopore wall surface.73, 74 Bioinspired nanopore platforms, such as lipid bilayer coated nanopores,75, 76 biomimetic nuclear pore complexes,77 and glass pipettes with ion channels trapped at the tip opening78–80 also have been developed. Of note, modification of solid-state pores is usually applied to the entire nanopore surface rather than simple placement of desired functional groups at precise locations within the pore, as is commonly performed with protein pores, and atomically precise modification of synthetic pores remains to be achieved.
Increasing efforts have been undertaken to develop hybrid nanopore platforms that hold promise to combine merits from both biological nanopores and solid-state nanopores and further expand the scope of nanopore sensing.78–88 Moreover, integration of scanning probe techniques with nanopore platforms results in mobile nanopore sensors that can achieve localized analysis of a variety of surfaces and interfaces and, thus, provide numerous exciting opportunities in biophysics, cell biology, and material sciences.
DETECTION METHODS
To date, the majority of nanopore sensing studies have employed electrical signals for molecule detection/identification. The two techniques utilized to achieve sensing, resistive pulse sensing, and current–voltage (I–V) measurements are discussed below.
The precedent of resistive pulse sensing, the Coulter counter, was developed in the 1950s for the use of counting erythrocyte translocation through a micrometer-sized hole.89 Shrinking the aperture size to the nanoscale has led to the birth of modern nanopore sensors from nanofabrication techniques. Nanopore sensors that use resistive pulse sensing as the detection scheme usually consist of a nanoscale hole within a biological or synthetic membrane that separates two electrolyte-filled reservoirs and individual electrodes placed in the bath solution on either side of the membrane. When a constant potential difference is applied across the two electrodes, a steady-state ion current is established due to the flow of ions across the nanopore within the membrane. As individual analyte molecules pass through or interact with the pore, short-lived obstructions of the pore create detectable modulations in the ionic current. The amplitude, duration, and frequency of these transient current modulations depend on characteristics of the analyte molecule such as size, shape, and charge as well as interactions that take place between the molecule and pore.
To utilize I–V measurements as the sensing modality, interaction of analyte molecules with the nanopore wall (via either physical adsorption or covalent binding) leads to modulations in the surface charge of the nanopore wall that are identified by interpreting rectification changes in resistance as changes in the I–V responses. As an example, conical glass nanopores under basic pH will possess a negative surface charge due to deprotonation of the surface silanol groups. The negatively charged nanopore thus is selective for cations, which leads to a concentration polarization effect under applied potentials and results in rectified I–V curves. Introduction of analyte molecules bearing positively charged functional groups (i.e., amines) to the nanopore surface will render the pore positively charged, and an inversion of the rectified response will be observed.90–93 Comparison of I–V responses prior to and following introduction of analyte molecules provides a means for molecular detection. Through manipulation of nanopore dimensions, surface properties, applied potentials, and electrolyte conditions, nanopore platforms can be tailored to suit different sensor applications.
Although the capabilities of nanopore sensors are powerful, hurdles such as high-bandwidth electrical noise and low throughput restrain the development of electrical signal-based nanopore sensing platforms. To address these limitations, integration of additional sensing modalities, such as tunnelling detectors,94–96 optics,97–99 or plasmonic structures,100–104 along with nanopore platforms has been developed.
In comparison to detection schemes that use tunnelling current or surface plasmons, which require nanoscale features for successful signal production, optical detection does not necessarily require an aperture in the nanoregime, and thus, we will not cover optical-based detection in this Review. Interested readers can refer to an excellent review by Gilboa and Meller for more discussion about the recent advancements in optical sensing with solid-state nanopores.105 The above review covered detection methods that use total internal reflection fluorescence and confocal fluorescence microscopy, as well as a variety of background suppression approaches used for optical noise mitigation.
BIOLOGICAL NANOPORES
Protein channels in nature regulate molecular transport across the cell membrane and play imperative roles in a variety of biological processes. For nanopore sensing, biological nanopores are particularly appealing due to several advantages, such as atomic-precision structural reproducibility and pore sizes on a similar scale as many biologically important analyte molecules. In addition, biological nanopores can be engineered in a wide variety of approaches like site-directed mutagenesis or incorporation of specific adaptors.106 As a result, well-defined local changes can be made to protein nanopores to better adapt the pores for particular applications. Interested reader is directed to an excellent review by Ayub and Bayley for more discussion about recent advances in engineered biological pores.107 Herein, we will survey several well-studied biological nanopore platforms and discuss both native protein pores and engineered forms, with a particular emphasis on applications.
Assortment of Biological Pores
α-HL
Staphylococcal alpha hemolysin (α-HL) is a mushroom-shaped, heptameric transmembrane pore formed by self-assembly of seven monomers in the lipid bilayer.108 After insertion into the lipid bilayer, the protein pore has a cap domain with a large vestibule (with 2.6 nm opening and a wider 4.6 nm interior diameter) that faces the cis side of the membrane and a transmembrane β-barrel (with 5 nm length) that faces the trans side of the membrane. The vestibule and the β-barrel are connected by a constriction that gives the pore a critical dimension of 1.4 nm.108
MspA
Mycobacterium smegmatis porin A (MspA) is a funnel-shaped octameric channel pore that allows diffusional transport of water-soluble molecules in M. smegmatis.109 Geometrically, MspA consists of a single constriction with a diameter of 1.2 nm and a length of 0.6 nm.110
AeL
Aerolysin (AeL) is a heptameric pore with a narrow bottleneck-shaped structure at the extracellular entrance and a small extracellular mouth approximately 1 nm in diame-ter.111, 112 AeL pores in nature are responsible for the pathogenicity of Aeromonas hydrophila.113
Phi29 Motor
The bacteriophage phi29 motor mediates the entrance/exit of dsDNA into viruses during maturation and infection and has a truncated cone structure.114 The central channel consists of 12 copies of gp10 protein subunits that complex to form a dodecamer channel. The inner channel has a length of 7.5 nm and cross-sectional diameters of 3.6 nm at the narrow end and 6 nm at the wider end.115
ClyA
Cytolysin A (ClyA) from Salmonella typhi is a dodecameric transmembrane pore comprised of a central channel (13 nm in length) with a narrower opening at one end (~3.3 nm) and a wider opening at the other (~6.4 nm).116 ClyA exhibits cytolytic toxicity toward erythrocytes and other cultured mammalian cells.117
OmpG
Outer membrane protein G (OmpG) is a monomeric β-barrel protein nanopore with seven flexible loops at the extracellular side. The pore has a central cylindrical channel that is comprised of 14 β-strands. The central cylinder is slightly wider at the periplasmic exit (~2.2 nm) and gradually tapers to a 1.3 nm constriction near the extracellular side.118
Comparison of Biological Pores
In terms of pore size (here, we refer to pore size as the narrowest constriction diameter of the protein pores in their native forms), α-HL (~1.4 nm), MspA (~1.2 nm), OmpG (~1.3 nm), and AeL (~1.0 nm) are suitable for ssDNA translocation studies but not for larger analytes such as dsDNA and proteins in their native, folded confirmations. ClyA (~3.3 nm) and phi29 motor (~3.6 nm), on the other hand, are large enough to allow the passage of dsDNA molecules.
Another important factor is the length of the constriction region of the pore, as this length is vital to sensitivity in current measurements. Within a strand of DNA, the ion current signal from a given base as it translocates a nanopore is influenced by neighboring bases, and signal interpretation becomes extremely difficult unless the current signal is better isolated to decrease these contributions from nearby molecules. For example, as the distance between two bases is ~0.34 nm along ssDNA,119 a total number of ~15 bases will coexist within an α-HL pore that has a 5 nm long β-barrel. The most prominent advantage of the MspA pore is the 0.6 nm long constriction region, in which the ion current modulation is dominated by 3–4 bases that are in and around this area. In theory, MspA should provide improved sensitivity and spatial resolution to α-HL, and the Gundlach group has demonstrated the use of engineered forms of MspA pore in DNA sequencing31, 51, 120–122.
α-HL protein pores remain indefinitely open under a wide range of experimental conditions, which provides a stable baseline for molecular sensing experiments. OmpG pores, on the other hand, experience pH, voltage, and spontaneous gating caused by mobile loops at the extracellular side entrance.123 Site-directed mutagenesis has been used on OmpG pores to minimize gating behaviors, and the resultant “quieter” OmpG pore has been used for traditional stochastic current sensing experiments.124 Another interesting approach demonstrated by Chen and co-workers directly exploits the gating behavior of OmpG and uses the characteristic signal generated from specific interactions between the flexible loops and analyte molecules (i.e., loop dynamics) for analyte identification43, 44, 125, 126.
In terms of native surface properties, AeL pores contain seven unpaired residues that are positively charged within the pore lumen, which have been demonstrated to enhance the interaction of the pore with negatively charged analytes. MspA pores and ClyA pores, on the other hand, have negatively charged residues within the pore constriction. Mutagenesis has been used to remove negatively charged amino acids or add positively charged residues to the pore to better facilitate analyte translocations.31, 127 A similar approach has been taken to insert positively charged residues within α-HL pores.128
A particularly appealing feature exclusive to biological nanopores is the atomic precision with which they can be engineered as the availability of high resolution crystal structures of membrane proteins becomes more common. Though modifications of multimeric protein pores such as heptameric α-HL and octameric MspA have been studied and utilized extensively, the monomeric property of OmpG has the potential to simplify genetic/chemical modification procedures.
In summary, each proteinaceous pore has corresponding characteristics in terms of size, gating behaviors, and surface properties that can be individually exploited for different applications. A brief overview of the analytes that have been studied with these protein pores is shown in Table 1, and we will discuss these applications in more detail in the following text.
Applications in Detection of Nucleic Acids
The first demonstration of nucleic acid detection with protein pores was reported 20 years ago by Kasianowicz et al.,25 in which single-strand DNA (ssDNA) and RNA molecules were electrophoretically driven through an α-HL pore, and the trans-location of these molecules led to transient current modulations. The frequency of translocation events was directly correlated to analyte concentration, while the duration of the blockade was proportional to the length of the molecules. This seminal work sparked the field of nanopore sensing and set the foundation for further advancements.
Recently, the Bayley group has developed a truncated-barrel mutant (TBM) α-HL and incorporated an adaptor molecule within the truncated pore for nucleotide sensing applications. In comparison to WT α-HL with a 5 nm-long β-barrel, the TBM α-HL pores with shortened β-barrels have proven to be more suitable for high-resolution nucleotide sensing. Identification of four mononucleotides has been achieved with the engineered TBM α-HL pores with CD adaptors.33 Another interesting finding in this work is that the CD binds irreversibly to one member of the TBM α-HL family (TMBΔ6) to significantly increase the recording stability such that no breaks due to displacement of the adaptor are observed in current measurements. In addition, engineered forms of MspA pores with negatively charged amino acids substituted with neutral or positively charged residues have been used for nucleotide translocation studies.31
In work by Long and co-workers, detection of oligonucleotides was demonstrated with a wide-type AeL pore.49 The use of wild-type pores here greatly simplifies the experimental design through elimination of genetic modification procedures. Individual short polydeoxyadenines (dAn, with n ranging from 2 to 10) were readily resolved with aerolysin nanopores. In Figure 1a, an illustration is shown of the translocation of a short oligonucleotide through an AeL pore with the residual positive charge in the β-barrel labeled with red shading. Recordings of dAn oligonucleotides are shown in Figure 1b, with blockade current characteristics observed for dA2, dA3, dA4, dA5, and dA10. The red triangle under each current trace denotes one representative blockade event that has been expanded to the right for better visualization. The well-resolved current blockades resulted in distinguishable Gaussian distributions for all five oligonucleotides. In Figure 1c, the total count of current blockade events is plotted versus the residual current depth I/I0 (residual current magnitude I divided by open pore current Io; residual current refers to the remaining current after blockades). dA10 has the smallest residual current depth, meaning the pore is blocked to the greatest extent with dA10, whereas dA2 has the largest residual current depth, or the pore is least blocked upon translocation of dA2. This difference in current is consistent with respect to the oligonucleotide lengths. Moreover, discrimination of individual oligonucleotides within a mixture was demonstrated from the characteristic current signature displayed by each analyte. Finally, a stepwise-cleavage reaction of an oligonucleotide via the restriction enzyme exonuclease I was performed and monitored in real-time. The cleaved products were clearly detected from both continuous current traces and current histograms. The authors further examined the shorter oligonucleotides (dAn, n < 5) with α-HL, but no blockade events were seen likely because of fast translocation speeds caused by weak interactions between the α-HL pore surface and analytes. In contrast, the slow translocation speed of dAn in the AeL pore (~2–10 nt/ms) is approximately 3 orders of magnitude slower compared to that of the most efficient mutant of α-HL.129 The authors attribute the significantly decreased translocation speed to enhanced nucleotide–pore interaction due to the pore’s small constriction and highly charged β-barrel wall.
Figure 1.

Applications of biological nanopores for the detection of nucleotides. (a–c) Detection of oligonucleotides (polydeoxyadenines (dAn), n = 2, 3, 4, 5, 10) with an aerolysin pore. (a) Schematic illustration of a short oligonucleotide passing through an aerolysin nanopore embedded in a lipid bilayer. (b) From top to bottom, representative current traces recorded without dAn and with the addition of dA2, dA3, dA4, dA5, and dA10. The red triangles denote typical blockades shown in the insets. (c) Histogram for the blockade events associated with dAn with Gaussian fits. (a–c) Reproduced from Cao, C.; Ying, Y.-L.; Hu, Z.-L.; Liao, D.-F.; Tian, H.; Long, Y.-T. Nat. Nanotechnol 2016 11, 713–718 (ref 49), with permission from Nature Publishing Group. (d, e) Indirect detection of dsDNA with an α-HL pore embedded in a lipid bilayer. Laser light is focused onto a lipid bilayer membrane that contains a single α-HL nanopore and initiates unzipping of dsDNA. (d) Illustration of dsDNA trapped within a α-HL pore and the corresponding current–time trace. (e) Illustration of unzipped DNA translocation through the α-HL pore and the corresponding current-time trace. (d, e) Reproduced from Angevine, C. E.; Seashols-Williams, S. J.; Reiner, J. E. Anal. Chem 2016, 88, 2645–2651 (ref 135). Copyright 2016 American Chemical Society. (f–h) Identification of a mismatched base pair in a dsDNA sequence by the latch constriction zone of α-HL. (f) Schematic illustration of a dsDNA trapped within the vestibule and three dsDNA duplex designs with a mismatched CC pair at different locations within the strand. Representative current–time traces (g) and histograms (h) of the current states for the different duplexes. (f–h) Reproduced from Johnson, R. P.; Fleming, A. M.; Beuth, L. R.; Burrows, C. J.; White, H. S. J. Am. Chem. Soc. 2016, 138, 594–603 (ref 140). Copyright 2016 American Chemical Society.
In comparison to other well-studied nanopores, the phi29 motor has a larger diameter (~3.6 nm) and allows direct translocation of dsDNA, ssDNA, and RNA under an applied electric field.50, 130, 131 The crystal structure of the phi29 motor has been reported previously,115 and a variety of engineering modifications can be performed to tune the functionality of the pore for specific applications. For example, an engineered phi29 motor pore with reduced channel size has been used to discriminate between translocation of ssDNA and RNA.131 Recently, Guo and co-workers studied the translocation dynamics of dsDNA with respect to different sequence lengths and conformational properties.50 In this work, the dsDNA molecules used were short strand linear dsDNA (12, 20, 35, and 141 bp), long strand linear dsDNA (500–2000 bp), and folded dsDNA (tetra-stranded DNA). Characterization of translocation dynamics was based on the current blockade amplitude as well as the dwell time. In the ionic current traces, blockade events were readily detected after addition of dsDNA molecules. For linear dsDNA, the current blockade percentage (calculated by [(Iopen – IDNA)/Iopen] × 100%) was deduced to be 32%. This ~30% current blockade is consistent with the ratio of dsDNA cross-sectional area (~3.1 nm2) and phi29 constriction area (~10.2 nm2). Next, dwell time of linear dsDNA with different lengths was investigated. For shorter strands of dsDNA, a relatively long dwell time relative to their length was observed, whereas dwell time increased linearly with length for longer dsDNA strands. Furthermore, translocation experiments of folded dsDNA and tetra-strand dsDNA both gave rise to a ~64% blockade percentage. In the experiments with folded dsDNA, current signatures with distinct features for linear dsDNA (straight), folded dsDNA (Type I), and partially folded dsDNA (Type II and Type III) were observed. Quantitative analysis for all events was performed, and the event percentage was calculated as a ratio of the number of events for a specific type to the total event count. From the statistical data, the combined event percentage of Type I, Type II, and Type III was ~15%, meaning only 15% of the dsDNA translocated through the pore in a folded fashion while the remaining 85% translocated in a linear fashion (defined as straight here). These results suggest that a bigger energy barrier must be overcome for dsDNA to translocate the pore in a folded fashion. Moreover, the dwell time for folded dsDNA was measured to be ~2 times longer than that of linear dsDNA. In the folded dsDNA configuration, the electrostatic repulsion between the two strands will displace both strands from the pore axis and lead to stronger DNA–pore interactions, which might reduce the overall force on the molecule and result in increased translocation time. This work is the first demonstration of the use of protein nanopores to distinguish folded dsDNA conformations.
Although the ~3.3 nm ClyA pore is larger than the hydrodynamic diameter of dsDNA, double-stranded molecules do not enter the nanopore under physiological salt conditions, likely due to the large electrostatic repulsion from the negatively charged residues within the pore lumen.132 Initial dsDNA translocation experiments with ClyA pores were performed at high ionic strength (i.e., 2.5 M NaCl) at which the negative charges of the nanopore are effectively screened.133 In Franceschini et al.,127 an engineered ClyA pore (ClyARR) was constructed via substitution of neutral residues with two ring structures of positively charged arginine residues at the cis entrance and the midsection of the pore. Successful dsDNA translocation experiments under physiological conditions were performed with this engineered ClyA nanopore.
Direct translocation of dsDNA through α-HL is in fact unobtainable due to the size limitation of the pore. At room temperature, dsDNA usually blocks the pore for an extended period of time, and an electrical potential with opposite polarity (i.e., negative potential on trans electrode) is needed to electrostatically repel the dsDNA molecule out of the pore and back to the cis chamber. However, at elevated temperatures, the free energy barrier to unzip dsDNA will be lowered and dsDNA can decompose into ssDNA molecules able to pass through the pore.134 Such temperature-dependent melting properties of dsDNA molecules can be exploited to control the dsDNA residence time by adjusting the solution temperature within the pore. Angevine et al. demonstrated the use of short-wavelength infrared (SWIR) laser heating to control the temperature of an α-HL pore and further applied the method to study melting properties of short dsDNA homopolymers (<50 bp).135 A representative current trace at 22 °C is shown in Figure 1d, in which dsDNA remained in the larger double helix form and, thus, could not translocate through the pore due to size exclusion. In the current trace (Figure 1d), extended residence time was observed, and the majority of the time, i/i0 was close to 0, which indicates the pore was blocked by the dsDNA molecules trapped at the opening of the pore. Of note, i is the residual current, and i0 is the open pore current; thus, i/i0 = 1 represents open pore, and i/i0 = 0 represents blocked pore. Illumination of the sample with a 1444 nm laser induced local heating of the solution and initiated unzipping of dsDNA molecules; subsequent translocation of the resultant ssDNA molecules was then observed. In the current trace measured at 45 °C (Figure 1e), the dsDNA residence time significantly decreased, and short-lived blockades were observed. DNA residence time can be tuned from indefinite binding at room temperature to milliseconds at elevated temperatures, and the authors discovered that the mean residence time of dsDNA could be adjusted over a wide temperature range via IR heating control. In addition, the mean residence time depends on both the dsDNA size and the temperature. Using the residence time distributions, the relative composition of similarly sized dsDNA (20-mer and 30-mer homopolymers dsDNA) in a binary dsDNA mixture was obtained.
Another progression in dsDNA detection with α-HL has been demonstrated by the White group with a system that can detect abasic sites and monitor the kinetics of enzymatic DNA repair. For this nanopore sensor, the ~2.6 nm constriction at the top of the vestibule of α-HL (known as the “latch”) is utilized as a dsDNA-specific sensing zone.136–138 In brief, a ssDNA tail fused with the dsDNA molecule-of-interest is driven into the α-HL pore. While the ssDNA is small enough to pass through the central 1.4 nm constriction and thread into the β-barrel section of the pore, the wider dsDNA strand remains temporarily lodged within the α-HL vestibule. This confinement of dsDNA within the initial section of the pore provides sufficient time to resolve highly sensitive modulations in current signal that relate to structure. This system has been utilized to study both the abasic sites within damaged DNA at which neither the typical purine nor the pyrimidine base is present as well as furanic nucleotide analogs incorporated within the dsDNA sequence. When structural variations are located in close proximity to the latch constriction, distinct changes in ionic current occur within a two-state modulating current. The frequency of current modulation and amplitude of the two states is unique to abasic sites to allow molecular recognition. Further investigations were performed to determine the effects of temperature, salt concentration, and electrolyte cations on both current modulation and overall residence stability of analyte dsDNA in an attempt to optimize conditions for base-modification detection.136, 137 Detection of both single base-pair replacement (e.g., GC pair replaced by AT pair) and epigenetic markers (e.g., 5-methylcytosine) has also been achieved with α-HL latch sensors.139
In Jin et al., identification of the latch region as a dsDNA-specific sensing zone was reported for the first time.138 The authors prepared a series of DNA duplexes each containing a single analogous abasic site, tetrahydrofuran (THF or F), at different positions in the duplex sequence while maintaining a guanine (G) opposite to the furan. The current modulations were studied for these duplexes relative to the different positions of FG pair along the duplex. The maximum current modulation, or the highest sensitivity, was observed when the FG pair was in line with the latch constriction as the duplex resided within the vestibule. The striking discovery of the dsDNA-specific sensing zone at the latch constriction has set the foundation for various new applications.
The high sensitivity of the latch zone has been further demonstrated by another recent report, in which mismatched base pairs CC and CA were readily discriminated from canonical Watson–Crick CG pairing from distinct current modulation signatures.140 Identification of such dsDNA sequence differences is only achievable when the mismatch is in close proximity to the latch constriction zone, as visualized in the schematic of Figure 1f. Current–time traces (Figure 1g) and histograms of the current states (Figure 1h) are shown for the analytes CC6, CC9, and CC13. The subtitle number in the sample name indicates the position along the dsDNA strand at which the CC mismatch occurs relative to the 3′ end at which the ssDNA tail is fused. When the mismatched CC base pair occurred at the 6th or 13th position, a uniform single-state current signature was observed; for CC9, the mismatched pair was aligned with the latch constriction and a distinct two-state modulating current (I1 and I2, indicated by arrows in the CC9 current trace for better visualization) was clearly observed. The strong dependence of current modulation on mismatch location within the duplex demonstrates that the latch sensing zone is highly localized to give good spatial resolution along DNA chains. The authors attribute the distinct two-state modulations in current to cytosine base flipping in and out of the helical structure and subsequent interactions between the mismatched base pair and amino acid residues at the α-HL latch constriction. This hypothesis is supported by prolonged residence time of dsDNA with a mismatch in comparison to fully complementary dsDNA. More recently, the White group further studied the temperature dependence of the base flipping effect at a CC mismatch site situated at the latch constriction of α-HL and derived the corresponding kinetic and thermodynamic parameters from the current measurements. The obtained activation energy for flipping a cytosine located at the latch constriction of α-HL is in good agreement with previously reported values for base flipping.141
Applications in Detection of Proteins
Direct detection of protein molecules with biological nanopores is challenging because the diameter of most biological nanopores, including α-HL, is smaller than the size of most protein molecules in folded and functional forms. Indirect detection of proteins via α-HL pores has been reported previously.38, 41, 45, 142 In one approach, the activity of a protein enzyme is measured by monitoring the ion current modulations that occur due to protease–substrate reactions. In the absence of protease, translocation of intact peptide substrate molecules through a nanopore will produce one type of event. In the presence of the protease, proteolytic cleavage of the peptide substrate creates multiple fragments, which give rise to new blockade events. The fragments produced from protease digestion have shorter length when compared to the original substrate and, thus, they have distinct current features with shorter residence time and smaller blockade amplitude in comparison to the original peptide substrate. Analysis of the frequency and amplitude of events can be performed to determine overall enzyme activity. This method has been applied to study HIV-1 protease activity.41
In Zhou et al.,142 indirect detection of trypsin was similarly accomplished via characteristic blockade current signals of cleaved products from proteolytic cleavage of a lysine-containing peptide substrate. The substrate was selected because catalytic cleavage of peptides by trypsin takes place within the sequence directly after positively charged lysine amino acid residues. In the absence of trypsin, translocation of the peptide substrate provided one distinct current signature. The presence of trypsin resulted in cleavage of the peptide substrate into two fragments. Therefore, addition of trypsin led to two types of new events, with shorter dwell times and smaller blockade amplitudes. Figure 2a,b shows the basic principle for indirect trypsin detection with α-HL as well as 3D plots of event counts vs residence time vs blockade amplitude. From the plotted data, differences between current signatures associated with the intact peptide substrate (red), fragment 1 (blue), and fragment 2 (yellow) can be observed. Additionally, the frequency of new events by fragments 1 and 2 increased with prolonged measurement times as well as increased trypsin concentration, which confirms the origin of the new current signatures indeed were the degradation products of the trypsin cleavage reaction. Finally, to obtain the trypsin activity, one of the cleaved products (hereafter referred to as P1) from the trypsin cleavage reaction of a peptide substrate was first synthesized and then subjected to nanopore recording experiments. A calibration curve of event counts versus P1 concentration was constructed. Next, through comparison of the number of events of P1 generated from the trypsin digestion reaction with the calibration curve, the concentration of P1 in the enzymatic reaction mixture solution can be obtained, and the activity of trypsin can be determined in terms of units, calculated from micromoles of P1 fragment released from the peptide substrate per minute.
Figure 2.

Applications of biological nanopores in the detection of proteins. (Top) Indirect detection of protein molecules with α-HL. Comparison of current modulations measured for the initial peptide substrate (a) and its cleaved products (b) can be performed to determine trypsin activities. 3D plots of event counts vs residence time vs blockade amplitude show a clear difference between whole peptide substrate (red), fragment 1 (blue), and fragment 2 (yellow). (a, b) Reproduced from Zhou, S.; Wang, L.; Chen, X.; Guan, X. ACS Sensors 2016, 1, 607–613 (ref 142). Copyright 2016 American Chemical Society. (Bottom) Detection of protein with an OmpG pore. (c) OmpG-PEG2-biotin pore without bound streptavidin and corresponding characteristic current trace; (d) OmpG-PEG2-biotin pore with bound streptavidin and corresponding characteristic current trace. (c, d) Reproduced from Fahie, M.; Chisholm, C.; Chen, M. ACS Nano 2015, 9, 1089–1098 (ref 43). Copyright 2015 American Chemical Society.
In another study, a similar approach was used to detect the activity of a diagnostically relevant hydrolytic enzyme, renin protease, within a matrix of serum proteins.45 The advancement achieved by this work is the ability to perform both the protease cleavage reaction and the purification of the cleaved products within a single spin-column. In brief, peptide substrates derived from angiotensinogen are tethered to microscale beads, and after addition of renin-containing blood serum, protease cleavage of the peptide substrate will produce peptide fragments. The cleaved products, Ang-His6, then bind with a second type of bead present within the column, Ni2+-NTA-beads. Meanwhile, the interference serum proteins are washed away. Finally, the purified Ang-His6 peptides are eluted from the spin-column and then measured with nanopore current recordings. Ionic current recordings of the enzymatically cleaved products were performed with an α-HL pore, and current signatures for the cleaved products were clearly observed, from which the protease activity was determined.
Another approach for protein detection is through the use of an enzyme motor to induce unfolding of the protein molecules and prompt translocation. A good example of such an enzyme motor is a protein unfoldase, ClpX, which can unfold substrate proteins by ATP-fueled mechanical pulling. Co-occurrence of such pulling and stochastic protein stability reduction can result in protein denaturation to prompt translocation. In nanopore sensing experiments, a model protein molecule is coupled to a polyanion linker with a ClpX-binding tag; then, the protein-linker-tag is added to the cis chamber (in which the wider opening of α-HL faces). Under applied potentials, the charged linker along with the tag is threaded through the α-HL pore into the trans side. The folded protein can then be captured in the α-HL pore lumen because the size of the protein is too large to pass through the constriction. Next, ClpX unfoldase is added to the trans chamber (in which the β-barrel opening of α-HL faces), and then, the unfoldase is able to bind to the affinity tag. In this way, unfoldase-assisted translocation is initiated and can then be monitored by ion current recordings.143 Later, Nivala et al.38 further advanced this approach in two major aspects, namely, addition of an ATP-regeneration mixture to the trans chamber to maintain a constant supply of ATP to the unfoldase and the use of a more robust/well-behaved unfoldase mutant (ClpXP). They then investigated the ability of the unfoldase-coupled nanopore to differentiate individual protein domains within a protein strand. In these experiments, an engineered protein molecule with four well-characterized folded domains was constructed and subjected to the unfoldase-coupled nanopore for measurements. Signals from the unfoldase-assisted translocation of the designed protein molecules could be used to discriminate among distinct protein domains. However, this approach could not be used to predict a theoretical current pattern for a given protein, and experimentally established patterns were needed for specific protein discrimination.
The aerolysin pore has also been used for unfolded protein translocation studies. Electrical stability tests of aerolysin in the presence of denaturing reagent were performed through the measurements of I–V curves as a function of guanidium chloride concentrations, and channel conductance remained unchanged up to 1 M guanidium chloride (a commonly used protein denaturing agent), which demonstrates that aerolysin can be used to study unfolded protein transport. The authors also studied unfolded protein translocation as a function of applied voltage and protein concentration. The blockade frequency and amplitude was found to vary exponentially with applied voltage and linearly with protein concentration. The dwell time of unfolded proteins decreased exponentially as applied voltage increased.144
Recently, the use of aerolysin to detect botulinum neurotoxins (BoNTs) has been reported.46 Indirect detection of the enzymatic activity of BoNT was accomplished via analysis of the current modulation caused by BoNT-digested fragments. The fragments were generated by the BoNT-digestion reaction of a recombinant synaptic protein substrate; only one cationic peptide from the digestion reaction leads to detectable current modulations likely due to the ion selectivity of the aerolysin pore, whereas the other fragments in the reaction mixture and serum cannot produce measurable blockade events. Therefore, by analysis of the current modulations generated by this cationic peptide, the presence of BoNT at subnanomolar concentration was determined.
In another study, translocation of a chimera protein molecule in the presence of a denaturing reagent was demonstrated through an aerolysin nanopore.39 The chimera consisted of a recombinant protein molecule covalently bound to a ssDNA oligonucleotide to allow amplification of the protein molecule via polymerase chain reaction (PCR). First, distinct current signatures for the translocation of the chimera, the protein alone, and then ssDNA alone were obtained. Next, trans-location experiments of the chimera molecules in the presence of denaturing reagent were performed. The unfolded protein molecules with ssDNA tags translocated from the cis to trans chamber were then subjected to quantitative reverse transcription PCR (qRT-PCR) to verify that the unfolded chimera protein molecules indeed translocated through the pore. Later, a systematic study was undertaken by Clark and co-workers to obtain more in-depth information about the translocation dynamics of unfolded protein molecules.145 They found that the translocation of unfolded protein molecules is mainly an enthalpically driven process due to the electrostatic interaction between the protein molecules and the pore, with a minor entropy component from the confinement of the proteins within the pore.
The phi29 motor channel represents an attractive alternative for protein detection because of its larger size that allows direct passage of proteins. In a recent study, translocation experiments of three different peptides and one unfolded peptide were performed with a phi29 pore.146 The parameter that was used for blockade event characterization is the current blockade percentage, defined as [(Ip − I0)/I0 × 100%], where Ip refers to the residual current after peptide blockade, and I0 refers to the open pore current. From the current blockade profiles, the four analytes were found to display a common peak with a blockade percentage of 31%, which is attributed to generic unfolded peptides approximately 2 nm in diameter. Additionally, all three peptides displayed their own unique blockade peaks with larger blockade percentages compared to the common peak, which serve as fingerprints for each individual peptide and could be used for differentiation. In addition, an engineered form of the phi29 motor has been used to probe a specific antibody.48 The engineered pore was constructed with the addition of Epithelial Cell Adhesion Molecule (EpCAM) peptides into all 12 subunits at the wider opening (C-terminal). Next, the binding of EpCAM antibodies (Abs) to the peptide-modified pore was studied. Stepwise blockades in current were observed due to the sequential binding of EpCAM antibodies to each peptide. Of note, each stepwise blockade event corresponds to one antibody binding event. Analysis of the dwell time was used to determine the docking and undocking kinetics of antibody–peptide interactions. Finally, discrimination of EpCAM antibody in the presence of other nonspecific antibodies or diluted serum was obtained, which further validated the specificity of this engineered platform.
To date, ClyA represents the largest-sized protein pore for sensing applications, which makes it particularly powerful in the studies of larger molecules such as proteins and protein–DNA complexes. A number of mutagenesis modifications have been applied to engineer ClyA pores for a wide variety of applications. In Soskine et al.,147 three different types of ClyA nanopores corresponding to 12-mer (opening diameter ~3.3 nm), 13-mer (~3.7 nm), and 14-mer (~4.2 nm) were engineered. These engineered ClyA nanopores have identical chemical compositions but different diameters. Their interactions with protein molecules such as human thrombin (HT) were studied, and ion current blockades of HT to the three pores were readily discriminated. Analysis of the dwell time suggested that larger pores impose less steric hindrance to HT, while HT can lodge within smaller pores for an extended period of time.
In addition, immobilization of proteins within the ClyA nanopore has been performed. The nanopore-confined proteins have been used to study ligand–protein interactions, as binding of ligand analytes to the proteins was reflected by nanopore conductance modulations.40, 42, 47 In a recent study, incorporation of two model proteins within ClyA pores has been carried out. Ion current recordings demonstrated that binding of analyte molecules to these immobilized proteins results in measurable conductance changes, which prove the proteins retain their functionality when trapped within the ClyA pore.47 ClyA nanopores have also been applied to study protein–DNA interaction using model systems such as the binding between HT and a thrombin binding aptamer (TBA).40 Upon addition of HT into the cis chamber, current blockade events were observed as a result of HT molecules entering the ClyA pore vestibule. After the addition of TBA molecules, binding of TBA to HT led to a decrease in the frequency of HT blockade, while a new event type from the HT–TBA complex was observed. Also, with increased TBA concentration, the event frequency of HT-only blockade further decreased. The HT–TBA complex could be trapped within the nanopore under proper applied potentials, which allowed a longer time for ion current recordings. A closer examination of the blockade current from the HT–TBA complex revealed the existence of two distinct current signatures, which were attributed to the formation of two isomeric HT–TBA complexes. In another study, proteinaceous rotaxane, a molecular structure in which a polypeptide thread is locked between two protein stoppers in the shape of a dumbbell, was assembled within a ClyA pore.42 Under low negative applied potentials, one of the two protein caps resided inside the pore indefinitely to prevent transport of protein molecules across the lipid membrane. Application of large negative potentials, on the other hand, was able to dislodge the interlocked rotaxane system and permit passage of protein molecules through the pore. Such rotaxane systems might be used as switches in molecular electronics due the control afforded by applied voltage to allow or block translocation of protein analytes through the pore.
In comparison to other well-studied protein pores, the sensing principle of OmpG is slightly different. The interaction of analyte protein molecules with the flexible loops leads to distinct characteristic signals for each protein molecule, with different frequency, duration, and open pore current. By analyzing the current patterns, molecular identity of the bound protein molecules can be achieved. For example, a specific ligand (i.e., PEGn-biotin) can be tethered to the most mobile loop (L6) of the OmpG pore to be used as a “fishing rod” for analyte proteins. To tether the ligand to the pore lumen, first a single cysteine mutation was introduced to the D224 residue of OmpG via mutagenesis. Next, the purified OmpG D224C proteins were labeled with maleimide-(PEG)n-biotin followed by a return of OmpG-PEGn-biotin construct to its native, folded structure. The tethered biotin group extends out from the OmpG pore to interact with and attract analyte proteins. Ionic current measurements reveal no measurable changes in the gating patterns of OmpG-PEGn-biotin relative to native OmpG pores. This modified pore has been utilized for the detection of streptavidin;43 the schematic of the detection strategy is shown in Figure 2c,d. Initially, in the absence of streptavidin, a baseline of gating events is measured as a control (Figure 2c). Once streptavidin binds with the biotinylated tether, both the frequency and amplitude of gating events reduce significantly (Figure 2d) such that analysis of the current response can clearly differentiate the presence or absence of streptavidin. The authors attribute the reduced gating frequency to a more rigid structure with bound streptavidin that restricts the bending of the L6 mobile loop. Control experiments, in which addition of streptavidin to native OmpG pores and addition of BSA to OmpG-PEGn-biotin pores exhibited no changes in gating activity, confirmed the altered gating was caused by the specific interaction between the streptavidin and the tethered biotin. To fully characterize the current patterns, analysis of five parameters was performed to generate a “fingerprint” for each protein analyte: (i) open probability; (ii) gating event frequency; (iii) intergating event duration; (iv) duration of gating events; (v) the open state conductance. A systematic study of the sensitivity and selectivity of this method (based on factors such as PEG linker length, applied voltage, salt concentration) has been performed to better understand the OmpG-linker-ligand system.125 Mouse monoclonal antibiotin antibodies (mAb), structural homologues, and isoforms of biotin-binding proteins (i.e., glycosylated and deglycosylated isoforms of avidin) were identified and differentiated according to the unique gating patterns displayed by each in a complex mixture.43, 44, 125, 126
Applications in Detection of Small Molecules and Polymers
Recently, wild-type (WT) α-HL has been used as a stochastic sensor to identify metallic nanoclusters (i.e., polyoxometalate (POM)) in solution. Decomposition of 12-phosphotungstic acid (PTA) into [PW11O39]7− and [P2W5O23]6− was investigated at various pH values (5.5 to 9.0). Ionic current measurements from α-HL were used to track the decomposition process, and the proposed decomposition products deduced from α-HL measurements were validated by 31P NMR.28 In another study, a stochastic sensor of a WT α-HL or mutant α-HL equipped with a noncovalently bound γ-cyclodextrin (γCD) was employed for the detection of sulfonate (MPSA)-labeled gold nanoparticles (NP).148 Recent work by Harrington et al. has demonstrated the use of an engineered α-HL with a pseudosubstrate sensor to detect protein kinase inhibitors.37 The sensor, comprised of a protease recognition site, a pseudosubstrate, and a peptide linker, which fuses the recognition site to the trans entrance of the α-HL lumen, accomplished detection of kinase protein from ionic current measurements.
In another study, a combination of aptamers and host–guest interactions has been used to develop a universal approach for nanopore sensing.149 In the proposed strategy, the first step is hybridization of an aptamer with a DNA probe which either is a complete match to the aptamer or contains several deliberately positioned mismatches. The DNA probe is modified with a host–guest complex in the middle of the strand which will produce a distinct current response as it moves through the α-HL. However, the hybridized DNA duplex is too large to pass through the α-HL pore, and therefore, no translocation event will occur. In the presence of an analyte molecule with greater affinity for the aptamer, however, the aptamer–DNA probe duplex can unwind to free the DNA probe from the host–guest complex. The DNA probe is now of sufficient size to pass through the pore, during which the characteristic signal will be observed. With this method, successful detection of vascular endothelial growth factor (VEGF), thrombin, and cocaine has been demonstrated.149
Nanopore-based single molecule mass spectrometry (Np-SMMS) with α-HL pores was first reported by Krasilnikov et al. in 2006.150 Strong interactions between PEG oligomers and the inner surface of the protein pore result in current modulations with prolonged dwell time and signal amplitude that is dependent on the size or mass of the PEG analyte. From the ion current signal, the molecular mass of the PEG molecules can be determined directly. More recently, Np-SMMS has been accomplished with AeL.151 A steeper mass-conductance relation and increased analyte dwell time within the AeL pore results in a significantly enhanced signal-to-noise ratio. With this improved Np-SMMS system, detection of PEG molecules with fewer than 25 repeat units has been possible.
Applications in Dynamic Process Studies
The high sensitivity in current recordings afforded by protein pores has also been exploited to study dynamic processes in real-time. A good example is the use of an engineered α-HL pore with a β-barrel that contains an unnatural amino acid with a terminal alkyne group to monitor the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction.152 From ionic current measurements, a long-lived intermediate was observed as the reaction proceeded.
In another report by Bayley and co-workers, continuous realtime tracking of the movement of a “single-molecule walker” at ambient conditions was accomplished.153 The stepwise motion of the walker molecule (an organoarsenic species) was observed across a one-dimensional “track” of thiols (five cysteine residues) within the β-barrel of an α-HL pore. As the loosely bound organoarsenic species moves stochastically along the linear track, As–S bonds are formed and broken, and individual steps can be observed by changes in the conductance state of the pore with millisecond time resolution and high precision.
Recently, a technique termed as single-molecule picometer resolution nanopore tweezer (SPRNT) was developed by the Gundlach group to monitor the motion of DNA as well as the conformational changes of the motor protein (phi29) as a DNA molecule passed through a nanopore (MspA).154 A key advance for the realization of the SPRNT technique is that the ionic current observed during ssDNA translocation can be correlated to the precise longitudinal position of the DNA within the pore. In SPRNT, the motion of nucleic acid processing enzymes (e.g., DNA polymerase phi29 or helicase/translocase Hel308) along DNA molecules has been studied, and detection of DNA displacements as small as 40 pm has been demonstrated.
Applications in DNA Sequencing
While protein nanopores have been used in a variety of ways to study different analytes, their most highly desired application is DNA sequencing. A significant roadblock toward this goal is the fast translocation speed of ssDNA molecules. A variety of novel approaches have been taken to address this problem.
In the seminal work reported by Manrao et al.,120 the combination of high current sensitivity offered by an engineered MspA nanopore and the reduced DNA trans-location speed obtained with a DNA polymerase phi29 (DNAP) enabled clear differentiation of single nucleotides as DNA molecules are threaded through the pore. In this system, the stepwise activity of the phi29 motor DNA polymerase (the same bacteriophage often utilized as a nanopore sensor itself) is employed to control the speed of DNA translocation through MspA. In studies without the presence of the polymerase, single-nucleotide resolution is not possible due to the velocity at which DNA translocation occurs (faster than 1 nucleotide/µs). By using the activity of the phi29 polymerase to “ratchet” the DNA through the pore, each signal step lasted ~28 ms before the polymerase moved the DNA strand forward through the pore by the distance of a single nucleotide position. On the basis of the sequencing results, it has been proposed that four nucleotides along the ssDNA strand in and around the MspA pore constriction contribute to the current signal. Thus, 44 = 256 possible base sequence combinations exist, each with a unique current signature akin to a molecular fingerprint.
Recently, Gundlach and co-workers measured the ionic current signals that correspond to each of the 256 individual quadromers and developed a quadromer map.155 The authors also investigated if the quadromer map would be predictive of current signatures for a previously unmeasured DNA sequence. A genomic DNA sequencing library of the bacteriophage was constructed from the quadromer map to attain theoretical current values and then experimentally nanopore-sequenced. A >90% correlation was observed between the predicted and the measured current levels, which further validates the accuracy of the quadromer map. A remaining limitation of this system is the lack of ability for direct de novo sequencing, meaning that without a reference for alignment, direct conversion of ion current signals to meaningful sequencing information has not yet been realized. However, with the development of the highly predictive quadromer map, this work still represents a historical milestone in the field of nanopore-based DNA sequencing.
The MspA–phi29 system has also been used to map DNA methylation. ssDNA molecules containing unmethylated, methylated, or hydromethylated cytosine sites within the strand were subjected to sequencing experiments. By comparing current obtained with ssDNA containing methylated cytosine sites to current obtained from unmethylated copies of the same ssDNA, precise location of the methylated sites was identified.156 Moreover, detection of unnatural nucleotides dNaM and d5SICS has been enabled by the MspA–phi29 system.121
Ju and co-workers first reported the use of a nanopore-based sequencing by synthesis (SBS) strategy to detect and differentiate between the four nucleotide base molecules in 2012.157 The idea of SBS is to attach identifiable tags to each of the nucleotides, such that the labels can be detected via ionic current recordings in nanopores during enzyme-catalyzed DNA synthesis. In this initial study, PEG tags with high salt concentrations were employed that have been proven less than ideal for polymerase activity. Recently, the use of oligonucleotide-based polymer tags bound to the terminal phosphate of each nucleotide has been reported.52 The two major criteria necessary in label design include: (i) distinct current modulations generated upon translocation of each base and (ii) proper activity of DNA polymerase still achieved upon addition of the tag. In Figure 3a, the sensor design is shown in which the phi29 DNA polymerase is covalently conjugated with the α-HL pore. Polymerase functions in nature to synthesize new copies of DNA through formation of a strand complementary to the original template; in this system, the DNA sequence-of-interest serves as a template and phi29 adds the necessary tagged nucleotide that pairs with the analyte sequence (i.e., A:T). During this synthesis reaction, the oligonucleotide tag is cleaved so that only the native nucleotide is added to the growing strand. Upon release, the polymer tag moves out of the pore and creates a signature current signal that is unique to the nucleotide incorporated within that position. Figure 3b shows a cartoon representation of the SBS process for the addition of each nucleotide. When polymerase adds a labeled base to the primer strand, the tag is released and a distinct transient current level is observed. Each of the four bases are labeled such that a unique current reduction occurs as the tag moves through the nanopore. Representative sequencing data is shown in Figure 3c.
Figure 3.

α-HL–polymerase sensor for DNA sequencing. (a) Schematic of the sensor design with phi29 DNA polymerase molecule conjugated to an α-HL nanopore. (b) Sequencing-by-synthesis (SBS) schematic that shows the sequential capture and detection of tagged nucleotides as they are incorporated into the growing DNA strand to produce nucleotide-specific current blockades. (c) Example of the homopolymer sequence reads. Reproduced from Fuller, C. W.; Kumar, S.; Porel, M.; Chien, M.; Bibillo, A.; Stranges, P. B.; Dorwart, M.; Tao, C.; Li, Z.; Guo, W.; et al. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 5233–5238 (ref 52). Copyright 2016 National Academy of Sciences.
Despite the previously discussed achievements, techniques that utilize biological pores for DNA sequencing do have their limitations. For instance, the fragile lipid bilayer tends to rupture and break at high salt concentrations, a condition that is often necessary in most DNA sequencing experiments to minimize electroosmotic drag on the DNA molecules and provide sufficient ionic current for proper read-out. Also, the stochastic skipping and backstepping manners of enzyme motors (i.e., phi29) can result in deletion and insertion errors. To implement nanopore sequencing in practical applications, parallel detection is needed to maximize throughput and obtain sequencing information in a reasonable time scale. However, mass production of lipid bilayer arrays remains a difficult challenge.
SOLID-STATE NANOPORES
Though the use of biological nanopores as sensors has proven successful, there remains a number of limitations with those systems, such as lack of freedom to tune the protein pore size/geometry as well as limited stability of the protein nanopore– lipid bilayer system under harsh conditions of pH, temperature, and ionic concentration. Thus, solid-state nanopores have been introduced to circumvent these limitations of biological nanopore setups. Solid-state nanopore platforms possess several unique advantages, including tunable pore size and shape with subnanometer resolution, mechanical robustness, superior thermal and chemical stability over a wide range of conditions (i.e., pH, temperature, concentration), parallel fabrication techniques that can easily produce many identical nanopore setups, and integration compatibility with sophisticated electronics and optical readout systems.
Advances in Fabrications
To date, a variety of solid-state nanopore fabrication approaches along with a broad collection of supporting membrane materials have been exploited. Examples include porous polymer membranes obtained via the track-etch technique,55 block copolymers (BCPs) produced by self-assembly of two or more chemically distinct polymer blocks,57 alumina fabricated through the anodic oxidation method,56 and nanopores within silicon or 2D materials fabricated by ion beam sculpting53 or electron beam construction with TEM,54 as well as glass nanopores created by laser/heat assisted manipulation with a commercially available pipet puller or reactive ion etching of glass-encapsulated Pt wires.158 For readers looking for more specific discussions regarding these nanopore platforms and details of their fabrication, several recent books and reviews are available that cover the following topics: nanoporous alumina,159 glass nanopipettes in the analytical sciences,160, 161 and fabrication techniques as well as fundamental mechanisms about nanoscale platforms.162
The use of ion and electron beams has been widely employed to fabricate solid-state nanopores in the sub-5 nm range since their inception. These methods offer a high degree of control over the final pore dimensions to promote a wide range of studies. However, the expensive instrumentation, low sample throughput, and complicated fabrication procedure that requires experienced personnel restrict the productivity and reproducibility of these techniques. Focused-ion-beam (FIB) milling has proven valuable for direct patterning of multiple chips with high resolution, though typically results in nanopores with diameters of 10 to 20 nm with a single, direct exposure.
A viable alternative for nanopore fabrication in commercially available SiNx membranes has recently been developed that involves controlled dielectric breakdown.163 In this method, a constant potential is applied to the SiNx membrane immersed in solution to induce leakage current. It is important that the potential applied results in an electric field strength close to the membrane’s dielectric breakdown strength, i.e., 0.4–1 V/nm. Creation of a single nanopore is indicated by an abrupt increase in the leakage current signal. Through careful feedback control, sub-2 nm pores can be fabricated with high reproducibility. If larger sizes are desired, an AC square wave electric pulse can be applied to further expand the initial nanopore. Automated fabrication of nanopores with a specific size has been developed through autotermination of the applied voltage once the leakage current exceeds a predefined threshold current.164 The relative symmetric geometry and surface charge distribution of pores fabricated in this fashion have been confirmed by ohmic I–V responses. The size of the nanopore can be deduced from the measured conductance with a known equation165 or can be directly visualized from electron microscopy, although the latter might lead to variations in the pore size from interaction with the electron beam. More recently, a similar electrical pulse method has been used to fabricate nanopores within graphene films.166 In this approach, brief voltage pulses of 250 ns duration were used to induce pore formation to minimize the amount of material removed during each pulse and thus gain better control over the final pore size. Despite the simple fabrication procedure and extensively studied pore formation mechanism, the unpredictability of the pore position remains the major limitation of this approach. Recently, plasmonicassisted dielectric breakdown has been utilized to create nanopores at directed locations through focusing the electric field to a nanoscale spot via integrated metallic nanostructures.
In Feng et al., a novel approach for individual nanopore fabrication in single-layer molybdenum disulfide (MoS2) with subnanometer resolution that uses an electrochemical reaction has been developed.167 Briefly, a transmembrane potential is applied to the MoS2 membrane while the ionic current is monitored. Surface defects serve as active sites to facilitate local electrochemical dissolution of MoS2 when a transmembrane potential higher than the oxidation potential of MoS2 is applied. Nanopore size can be controlled via an automated ion current feedback system. To stop the formed pore from further expanding, the applied potential is simply stopped once the desired current threshold, which is directly correlated with the pore size, is reached.
Another recent trend in the solid-state nanopore community is the fabrication of ultrathin nanopores. Drndić and co-workers have developed an electron-irradiation-based thinning method of SiNx membranes.58 In this method, the target membrane area is raster-scanned with the electron beam from a scanning transmission electron microscope (STEM), and real-time acquisition of both a high-angle annular dark-field (HAADF) STEM image and an energy electron-loss spectrum (EELS) of the same area is achieved. By monitoring and quantifying the mass loss with the HAADF STEM images and EELS, the film thickness can be controlled. Electron irradiation sputters N and Si atoms, with the rate of N sputtering significantly higher compared to that of Si, such that an amorphous silicon (a-Si) membrane is produced. The final drilling step is also performed with the STEM electron beam, and subnanometer resolution in the pore formation step is ensured by the small probe size and accurate lateral movement of the electron beam. With this approach, fabrication of nanopores with a thickness of 1.4 nm and diameter of 2.5 nm has been obtained.
2D Materials for Solid-State Nanopore Fabrication
Direct creation of ultrathin nanopores within atomically thin membranes made from 2-dimensional (2D) materials, including graphene,62–64, 168 boron nitride (BN),66 hafnium oxide (HfO2),67 and molybdenum disulfide (MoS2),68 is presently under intense study. Monolayers of 2D materials hold promise of superb spatial resolution as they represent the thinnest materials with thickness comparable to the spacing between DNA bases. When a DNA molecule passes through a nanopore in such a platform, the resultant current modulation may be the result of a single base interaction with the pore and enable high resolution DNA sequencing in comparison to a combined blockade effect from several bases within the sensing constriction of a conventional nanoporous system.
Graphene is a single layer of carbon atoms arranged in a hexagonal lattice with a nominal thickness of 0.34 nm. Since initial proof-of-principle demonstrations of dsDNA detection in graphene nanopores with resistive pulse sensing techniques,62–65 intense research efforts have been focused on this material, and unique characteristics of graphene have been exploited for exciting new applications. In a study by Dekker and co-workers,169 a hydrophilic layer applied to the graphene nanopore wall was able to minimize the attractive hydrophobic interactions between the nucleotide molecules and the pore surface. These nanopores exhibit enhanced durability and have been used for ssDNA translocation studies. In another study, the hydrophobic interaction between the DNA and the graphene was exploited to slow down the translocation speed.170 A graphene/Al2O3/graphene multilayer pore was fabricated, and ssDNA translocation speed was indeed reduced. Despite these exciting developments, drawbacks such as high noise levels and positional fluctuations during DNA trans-location associated with graphene nanopores still stand in the way of single-base resolution, and research focus has shifted to exploit the conductivity properties of graphene for sequencing applications. Ideas such as measurement of tunnelling current modulations during DNA translocation via a graphene nanogap171 or monitoring in-plane current variations caused by traversal of the DNA molecule via a graphene nanoribbon172 have been proposed. However, the majority of studies about such ideas published to date are theoretical studies and experimental demonstrations remain limited.
While intense efforts have been undertaken to improve the sensing performance of graphene nanopores, researchers are actively seeking alternative 2D materials for ultrathin nanopore fabrication. For instance, better control of DNA translocation dynamics through MoS2 nanopores has been achieved through establishment of an ionic liquid/water viscosity gradient system. The high viscosity of ionic liquid effectively reduces DNA translocation speed and enables single-nucleotide identification.173
Interested readers are directed to an excellent review article by Schneider and co-workers, in which the present status and future prospective for integration of 2D materials with nanopore platforms for single molecule sensing are discussed in great detail.11 For more discussion on the specific use of graphene for DNA sequencing applications, users are directed to another recently published review.174 A comprehensive overview of graphene and graphene related materials covering both fundamental research challenges and practical applications can also be found in a recent published review.175
Applications in Detection of Nucleic Acids
Since the first demonstration of dsDNA detection with a SiNx nanopore with resistive pulse sensing,53 significant research interest has further advanced solid-state nanopore applications. To depict a better physical picture of dsDNA translocation, several important factors and their effect on dsDNA translocation have been studied, including spatial parameters such as the length of the dsDNA molecule and the diameter/thickness of the nanopore, as well as physical parameters such as the applied voltage, surface charge of the nanopore, electrolyte concentration, and the environmental temperature.30, 176–186
An example is the study of DNA translocation dynamics performed by Dekker and co-workers.187 In this work, linear dsDNA molecules with markers (i.e., short oligonucleotide protrusions) positioned at known locations were subjected to translocation experiments. By monitoring the relative position of the markers as the DNA moves through the pore, the translocation velocity has been determined.
Another important application is the discrimination of different DNA conformations from unique current signatures upon nanopore translocation. For instance, one major limitation in nucleic acid synthesis reactions is deleterious products formed during synthesis, such as structural misassemblies and small local defects. Carson et al. demonstrated the use of a solid-state nanopore platform to differentiate between deleterious products and desired synthesis products.188 First, detection of small sequence defects such as single base mismatches was demonstrated. Ionic current modulation caused by a single base mismatch was relatively small and therefore difficult to detect, but by binding the single mismatch to a mismatch-binding protein, the resultant signal from the mismatch was amplified to enable successful identification. Four representative misassembled structures were studied, and clear differences in ionic current blockade shape, duration, and frequency were observed for these molecules in comparison to the signal of correctly assembled DNA. In Figure 4a, schematic illustrations (left), representative current traces (center), and log-dwell time histograms (right) are shown for properly assembled dsDNA molecules (top) and one of the misassembled conformations termed the Holiday junction (bottom). The two types of events are well-differentiated in both different current signatures and histograms.
Figure 4.

Different applications of solid-state nanopores. (a) Detection of various dsDNA conformations with a 2.5 nm SiNx nanopore. Schematic illustration, representative current trace, and log-dwell time histogram of DNA product (top) and one type of DNA misassembly (bottom). Reproduced from Carson, S.; Wick, S. T.; Carr, P. A.; Wanunu, M.; Aguilar, C. A. ACS Nano 2015, 9, 12417–12424 (ref 188). Copyright 2015 American Chemical Society. (b) Detection of short dsDNA molecules with a 7 nm SiNx nanopore. Translocation experiments of monovalent streptavidin (MS) only, dsDNA (bio90) only, and MS incubated with bio90 at a molar ratio of 8:1. (Left) Current traces for MS only; (center) bio90 only; (right) MS–bio90 mixture over a range of potentials. Reproduced from Carlsen, A. T.; Zahid, O. K.; Ruzicka, J. A.; Taylor, E. W.; Hall, A. R. Nano Lett 2014 14, 5488–5492 (ref 191). Copyright 2014 American Chemical Society. (c) Detection of zinc finger proteins (ZFPs) along a long strand DNA molecule with SiNx nanopores. (Top) Illustrations and representative current characteristics of two types of translocation events for a DNA–protein complex with ZFP bound in the center. (Bottom) Illustrations and representative current characteristics of two types of translocation events for a DNA–protein complex with ZFP bound at the 2-to-5 position. Reproduced from Yu, J.-S.; Lim, M.-C.; Huynh, D. T. N.; Kim, H.-J.; Kim, H.-M.; Kim, Y.-R.; Kim, K.-B. ACS Nano 2015 9, 5289–5298 (ref 200). Copyright 2015 American Chemical Society. (d) Detection of bound proteins with designed DNA carriers. (Top) Schematic illustration of the detection scheme. (Middle) Two DNA carrier designs, with three biotin sites (3B) and no biotin sites (0B). (Bottom) Plot of percentage of positive translocations with different protein mixture–DNA carrier combinations. Reproduced from Bell, N. A. W.; Keyser, U. F. J. Am. Chem. Soc 2015 137, 2035–2041 (ref 202). Copyright 2015 American Chemical Society.
In a recent report, detection of DNA knots along a long strand DNA molecule has been demonstrated through the use of solid-state nanopores.189 In this work, instead of a typical 1 M KCl electrolyte solution, bath solutions of 2 or 4 M LiCl were used to decrease the translocation speed of DNA molecules. As a result, a higher signal-to-noise ratio was obtained, which allowed higher measurement bandwidth and enabled detection of otherwise poorly resolved DNA knot configurations. When a dsDNA molecule passes through the pore in linear fashion, a current blockade I1 is induced. If this molecule translocates while in a folded conformation, a current blockade twice the amplitude as I1 (2I1) is produced. The presence of a DNA knot is indicated by additional sharp blockade spikes with high amplitude and short duration observed in addition to the usual I1 or 2I1 blockades that represent linear or folded molecules, respectively. These distinct current signatures were further utilized to study knotting probability, size, and position along the DNA chain.
Recently, Dekker and co-workers reported another approach to slow down DNA translocation by the use of glutamate (Glu) solutions.190 In comparison to conventionally used Cl−-based solutions, glutamate-based solutions have lower conductivity and greater viscosity. Increased viscosity of the electrolyte solution leads to higher dragging force on the DNA molecules and, thus, results in prolonged translocation time, albeit the price of reduction in current blockade amplitude is paid at the same time owing to the decreased conductivity of glutamate salt solutions.
Another recent advancement in unambiguous discrimination and quantification of modified short dsDNA molecules has been demonstrated by Hall and co-workers.191 In this work, individual monovalent streptavidin (MS) proteins were employed as high-affinity tags to label biotinylated 90-bp dsDNA (bio90). Translocation experiments were performed with only MS as a sample, only bio90 as a sample, and finally a mixture (molar ratio 8:1) of MS and bio90. As seen in Figure 4b, in the cases of only MS (Figure 4b, left) or only bio90 (Figure 4b, center), translocation frequency was relatively low. The low translocation frequency of MS can be explained both by the fast translocation velocity of MS molecules due to a strong electrophoretic driving force from the significant negative charge of the protein and the bandwidth limitation of the measurement electronics. Absence of observed bio90 events is attributed to infrequent translocation caused by the electroosmotic dragging force. When the two bind to form a complex, a significant increase in the event frequency was observed (Figure 4b, left) as a result of the reduced translocation speed of MS–bio90 complex. The authors hypothesize the slower translocation for the complex occurs because attachment of the nucleic acid chain to the MS adds an additional electroosmotic force to the electrophoretic force experienced by MS alone, and this drag decreases the velocity of the otherwise rapid movement of the protein. Event frequency was demonstrated to be directly correlated with DNA–protein complex concentration, and direct molecular quantification was enabled. Using this method, the concentration of biotinylated DNA within a mixture of both biotinylated and unmodified DNA could be obtained by interpreting the translocation frequency.
More recently, the Hall group applied this approach to detect microRNA (miRNA) and short nucleic acids.32 In translocation experiments for short nucleotide sequences (34-nt, termed as ssBio34 or dsBio34), only formation of dsBio34–protein complex caused increased translocation frequency, whereas for dsBio34 only, ssBio34 only, or ssBio34–protein complex, translocation frequency remained very low. This method then was used to recognize a specific target sequence (i.e., complementary to ssBio34) within a heterogeneous mixture of both the target sequence and other nonspecific decoy sequences. Increased translocation frequency was only observed when the target sequence, the ssBio34, and streptavidin were present in the sample, as the hybridization of the target sequence with ssBio34 resulted in dsBio34 and then prompted the formation of dsBio34–protein complex, which finally led to the increased translocation frequency. Furthermore, detection of a specific miRNA was demonstrated. To do so, a biotinylated 23-nt ssDNA (ssBio23) complementary to the target miRNA was constructed. Similarly, only a DNA–RNA–MS mixture yielded increased event frequency, whereas for miRNA only, ssBio23 only, and MS only, the event frequency was low. This work represents an important advance in miRNA analysis with solid-state nanopore platforms. In comparison to previous work that used ultrathin nanopores for miRNA detection,59 in which intensive sample purification and preconcentration was required due to the nonselective nature of conventional solid-state nanopore detection, the work present here has shown the ability to unambiguously resolve target sequences within a heterogeneous background, which has the potential to simplify the sample pretreatment procedure.
Applications in Direct Detection of Proteins
Measurements of protein translocation through solid-state nanopores has been challenging, owing to the fast translocation speed of protein molecules (i.e., ~1 protein/µs), poor signal-to-noise ratio during translocations, and variable interactions between the protein molecules and the nanopore wall.192 According to simulation results,192, 193 for the translocation of sub-100 kDa protein molecules through solid-state nanopores with diameters >10 nm, only the slowest ~0.1% of the translocations are observed when using current amplifiers with 10 kHz bandwidth. Therefore, direct analysis of protein translocation events through solid-state nanopores suffers from insufficient temporal resolution, which results in a substantial fraction of undetected signals. To date, a variety of novel approaches have been developed for protein detection with solid-state nano-pores.
Direct detection of a wide range of protein molecules (14 to 465 kDa), such as lysozyme, avidin, and IgG, via a glass nanopore has been reported.194 Since glass nanopores provide lower electrical noise, instead of the traditionally used 10 kHz filter, a 100 kHz filter was used for experiments. Further advanced electronics capable of performing high-bandwidth (1 MHz) electrical measurements have enabled direct analysis of translocation of sub-30 kDa protein molecules through nanopores in SiNx membranes and HfO2 membranes.195
An alternative way to directly detect protein translocation is to slow down the motion of protein molecules and prolong their residence time within the nanopore, such that time-resolvable signals can be obtained. Meller and co-workers have found that, by fine-tuning the electrolyte pH to the isoelectric point (pI) of the analyte protein, translocation speed of the analyte protein can be slowed down because of the diminished electrophoretic mobility when the net charge of the protein molecules approaches zero.196 Therefore, at a pH near the pI, the protein translocation speed will decrease, leading to longer residence time and providing better temporal resolution. Influence of electroosmotic flow has also been considered, and a pH value at which the SiNx membrane will still maintain a slight negative charge is selected, such that the cation-based electroosmotic flow can further slow down the protein translocation. The decreased translocation speed enabled detection of a small protein, ubiquitin (Ub). In addition, two Ub dimers with the same molecular weight, but different molecular structures, were readily discriminated. Furthermore, the authors used the method to monitor deubiquitination reactions of di-Ub. The cleaved products at different reaction durations were analyzed by both gel electrophoresis and nanopore ionic current recordings. Differentiation of uncleaved di-Ub, cleaved mono Ub, and a mixture of the two was demonstrated. This work presents a novel approach to facilitate detection of proteins with solid-state nanopores by fine-tuning the electrolyte pH. Albeit the decreased translocation speed is beneficial and enabled detection of sub-10-kDa proteins, the event frequency decreased as well, likely due to the change of protein–pore interactions as a result of varying pH, which needs further investigation.
In addition, by fine-tuning the nanopore surface properties, enhanced interaction between the protein molecules and the pore wall can be obtained, facilitating the detection of protein molecules. For instance, direct detection of bovine serum albumin (BSA) has been reported with Al2O3 modified nanopore.72 Atomic layer deposition of Al2O3 was applied to the surface of PET conical nanopores to (i) shrink the pore size and (ii) render the surface positively charged. Bare PET membranes contain negative surface charge due to the presence of carboxylate groups. The Al2O3 surface, on the other hand, is positively charged at neutral pH, as the nominal isoelectric point of Al2O3 is pH = 9. Therefore, the Al2O3-coated nanopore wall contains positive surface charge, which poses a weak attraction to the negatively charged BSA molecules. This weak electrostatic attraction effectively reduced the trans-location speed of BSA molecules, and well-defined time-resolvable signals of BSA translocation were observed. Interestingly, a biphasic pattern was observed for BSA translocations, which elucidated a unique mechanism for protein translocation that involved transient redistributions of ions within the pore as a result of BSA molecules adsorption on or desorption at the nanopore wall.
Lipid bilayer coating has been applied to SiNx membranes as well as glass nanopipettes, to prevent nonspecific interactions of protein molecules with the pore wall, enabling the detection of protein molecules.197 In addition, modifications of the solid-state nanopore with specific receptors (i.e., nitrilotriacetic acid receptor) for target protein detection (i.e., His-tagged proteins) are another route for direct detection of protein molecules.198
Applications in Detection of DNA-Bound Proteins
A novel approach to detect biomolecules bound to a long dsDNA strand has been developed. The basic principle is simple: DNA passes through a nanopore in a linear head-to-tail fashion that causes a transient change in ionic current from the temporal blockage of the pore. As a result, a characteristic blockade current will be observed from the DNA molecule itself during translocation. Any biomolecules bound to the DNA strand will result in a secondary level current change in addition to the characteristic blockade current for the bare DNA molecules. Consequently, by monitoring the current blockage pattern during the translocation of a long strand of DNA, local properties of the biopolymer can be obtained as well as information about the bound biomolecules. In addition to the study of the binding process between DNA and proteins, this interaction has been applied to enable detection of small proteins as analytes; unbound proteins typically translocate a nanopore at ultrafast speed, but the DNA molecule acts as an anchor to slow down the protein upon complexation. Recently, several groups have demonstrated the realization of this concept in various interesting applications.
Dekker and co-workers have used an antibody that binds randomly to DNA molecules as the model protein molecule and demonstrated the detection of individual bound antibodies on the lambda phage 48.5 kbp DNA (λ-DNA) strand.199 These anti-DNA antibodies are positively charged, meaning free antibodies in the cis chamber of a resistive pulse setup are repulsed and cannot translocate the nanopore unless bound to negatively charged DNA molecules. Solutions that contained DNA and anti-DNA antibodies were first incubated to ensure DNA–antibody binding. Next, the DNA–antibody mixtures were measured with glass nanopore sensors and, indeed, current spikes with short durations and large amplitudes were observed in addition to signal of blockade events by only the DNA strand. Analysis of the second-level current spike position supported the random binding process of anti-DNA antibodies, as the spikes were found to distribute randomly over the duration of the first blockade current level associated with DNA translocation. This work represents a realization of DNA-bound protein detection with solid-state nanopores and provides new inspirations for further development.
In Yu et al.,200 a similar method was employed to detect zinc finger proteins (ZFPs) along a dsDNA molecule. Each zinc finger is able to recognize a specific DNA sequence in the dsDNA major groove via a single α-helix. ZFPs can be engineered to have enhanced affinity to specific desired sequences through the addition of structural linkers. In the work described above, anti-DNA antibodies exhibited random binding all along the dsDNA molecule; within this study, the binding locus of a ZFP along the dsDNA strand can be precisely controlled. From the known position of the ZFP binding site within the dsDNA molecule, one can predict when the additional current spike that corresponds to bound ZFP will occur within the initial current blockade. Analysis of the ratio of DNA translocation time before (t1) and after (t2) the ZFP peak allows the position of ZFP binding site on the dsDNA to be deduced with a simple calculation: t1/(t1 + t2). To investigate identification of the location of bound protein along the dsDNA molecules from the current measurements, the authors designed two different DNA–protein complexes: one as a 520 bp DNA chain with a ZFP binding site at the center of the molecule and the other as a 5605 bp DNA strand with a ZFP binding site at the asymmetric position of 2-to-5 within the dsDNA strand. Figure 4c, top, shows a schematic illustration of the sensor as well as ionic current recordings of the symmetric 520 bp dsDNA with ZFP protein bound in the center of the strand. Figure 4c, bottom, shows a similar depiction of the asymmetric 5605 bp dsDNA with ZFP protein bound at the 2-to-5 position. Two types of events were observed in the current–time measurements, one that corresponds to passage of bare dsDNA as a single level current drop and another that corresponds to translocation of ZFP-bound DNA with the additional current reduction to the first level due to the extra volume added by the ZFP to further block the pore. The relative position of the additional current loss caused by the bound ZFP–DNA complex with respect to the overall dsDNA blockade current match well with the physical position of the bound ZFP to the dsDNA molecule in both scenarios.
Meller and co-workers demonstrated the detection of a transcription factor (TF) protein bound to DNA molecules.201 The protein used in this study was the DNA-binding domain of Early Growth Response 1 (EGR1), a zinc-finger protein named zif268. The protein zif268 adopts one of two configurations when bound to DNA molecules depending on whether the binding was specific or not. Interpretation of current traces during translocation experiments enabled the differentiation between specific and nonspecific zif268-DNA binding conformations simply from the distinct current blockade patterns for each scenario.
In a recent report,202 long-strand dsDNA molecules (~7.2 kbp) were designed and engineered to bind single or multiple proteins at tailored positions, termed as DNA carriers. The presence or absence of specific protein molecules then could be determined by monitoring the characteristic current signatures for specific DNA carrier–protein complexes. In the initial proof-of-principle experiment, assembly of the protein–DNA carrier complex was done in three steps (i) biotin groups were attached to the center of the dsDNA carrier (with 1B, 3B, and 5B corresponding to one, three, and five biotin sites, respectively); (ii) streptavidin was added in great excess; (iii) the protein–DNA mixture was incubated to ensure successful binding of streptavidin to the biotin site(s) on the DNA carrier. The protein–DNA carrier complexes were then measured via a glass conical nanopore ~15 nm in diameter. In the ionic current traces, the protein signal was clearly observed in the middle of the DNA signal because the bound protein molecules were spatially positioned at the center of the DNA carrier. The schematic illustration of the experiment is shown in Figure 4d, top. Additionally, the amplitude of the protein signal increased along with the number of protein binding sites present to further confirm that the secondary signal in addition to the DNA signal was indeed from the bound protein(s). With this nanopore sensor, identification of specific proteins within a mixture was also demonstrated. Two protein mixtures were prepared of four different protein molecules, such that one combination contained streptavidin (mixture 1) and the other substituted this protein for BSA (mixture 2). Each protein mixture was incubated with two different types of DNA carriers with either three biotin sites (3B DNA) or zero biotin sites (0B DNA), as visualized in Figure 4d, middle. Next, ion current measurements were performed on the different mixtures. Indeed, characteristic protein current signals were only observed when mixture 1 was incubated with 3B DNA and streptavidin was available to bind with the biotin sites (Figure 4d, bottom). The bar graph in this figure of percent positive translocation plotted versus type of DNA carrier refers to the number of events in which protein current signals were observed out of the overall total event counts. The generic nature of this method was demonstrated by design of different DNA carriers.
In another report, the fraction of DNA carriers that displayed the additional current reduction within the overall DNA current blockade (caused by the DNA–protein complex) was utilized to determine protein concentration.203 In the ionic current recordings for the streptavidin–DNA carrier complex, a comparison between the histogram of secondary signal (from streptavidin) and an all-point histogram (from all linear translocation events) was made. The two peaks were found to be overlapping, which indicates the change in current from streptavidin translocation is similar to that caused by dsDNA. Due to this peak overlap, some of the signal from streptavidin would be buried in the dsDNA signal and, thus, impair the sensitivity and accuracy of the method. Next, another protein pair, digoxigenin–antidigoxigenin, was tested. The larger size of digoxigenin compared to streptavidin resulted in a larger secondary current amplitude. With the increased difference in current signal, the unbound dsDNA and protein-bound DNA signals were well-separated and the sensitivity and accuracy of the sensor was greatly improved.
Recently, Bell and Keyser further advanced this “DNA carrier–protein” detection approach and demonstrated highly multiplexed sensing with digitally encoded nanostructures.204 In this work, a library of DNA nanostructures was created such that each could be identified by a unique barcode. Barcode regions were constructed along a long strand dsDNA molecule and consisted of five basic units, or bits, each containing 11 protruding DNA dumbbell hairpins. Each bit in the barcode was used to define a “yes” or “no” answer for the presence of dumbbell hairpins. Sufficient spacing was necessary to ensure each bit could be measured individually, so each bit of 220 bp was separated by 312 bp. In each barcode, the first and fifth bit were used to signify the start and end of barcode reading. The second, third, and fourth bit were used to signal “on/1” in the presence of the hairpin or “off/0” in the absence of the hairpin. As a result, a total of 23 = 8 different barcodes (000, 001, 010, 011, 100, 101, 110, and 111) were generated from the designed DNA nanostructures. Translocation experiments of all eight barcode designs were performed via a glass conical nanopore. The authors demonstrated that the 3-bit barcode can be assigned with a 94% accuracy when the DNA nanostructures were electrophoretically driven through a solid-state nanopore. With the developed barcode strategy, one member of the 8 barcodes, 011, was selected and further equipped with a binding site for a specific antibody at a directed location. Incubation of the modified 011 barcode with target antibodies was performed, and the mixture was then analyzed by the nanopore sensor. Positive antibody signals were observed for ~97% of the 011 barcode translocations, whereas only 4% of the signals in the control experiment (no added antibody) were positive. Finally, multiplex measurements of four antibodies were demonstrated. In this experiment, four barcodes from the library were selected, and each barcode was engineered with a specific binding site for one of the four target antibodies. The remaining four unmodified barcodes were used as controls. Next, the four target antibodies, four barcodes modified with binding sites, and four unmodified barcodes were mixed and then examined with nanopore ionic current measurements. Detection and differentiation of the four antibodies were readily obtained by the clear difference between the barcodes. The fascinating part of this experiment is that the basic design of the 3-bit code can be scaled up to allow more barcode variations and thus further increase the number of analyte molecules that can be studied simultaneously.
Applications in Detection of Protein–DNA Interactions
Nanopores can be used to monitor chemical and biochemical reactions of specific analytes as measurable structural changes are often observed alongside these reactions, and such changes in structure will usually lead to detectable current modulations during translocation experiments. However, to study a chemical reaction for an extended period of time is difficult because of the fast translocation speed of certain analyte molecules such as DNA. Stein and co-workers constructed a novel nanopore entropic cage structure on which one side contained a nanopore (~10 nm in diameter) and the other side contained a larger opening (~200–300 nm); the two openings were then connected through a micrometer-scale inner cavity of ~2–3 µm. The structure was designed such that a single DNA molecule could be captured and entropically trapped for a prolonged duration.205 Another unique feature of this cage structure is the permeability to molecules such as restriction enzymes, which can enter the cage and cleave the stable, trapped DNA molecule in a sequence-specific manner. After the enzyme cleavage reaction, the fragments then can be controllably driven out of the nanopore orifice and the number and sizes of the cleaved products analyzed upon exit. Entropic traps such as this cage structure can be used as a nanoreactors and will likely find widespread use for numerous applications.
Hall and co-workers examined the interaction between ssDNA molecules and E. coli single-strand binding proteins (SSBs) with translocation experiments via SiNx nanopores.206, 207 Their studies demonstrated that the SSB– ssDNA complex has a characteristic current signature that is distinct from either SSB alone or ssDNA alone. Of note, SSB only binds to ssDNA but not dsDNA. When a mixture of dsDNA and ssDNA are exposed to a large excess of SSB, dsDNA molecules remain unbound while nearly all ssDNA molecules will complex with SSB. The unique current signature of the complex can be clearly identified from the current signal for unbound dsDNA such that one can differentiate dsDNA from ssDNA. Furthermore, the interaction between both circular ssDNA and linear ssDNA with SSB was studied, and the translocation characteristics share similar trends, which indicates that the formation of the SSB–ssDNA complex is independent of ssDNA confirmation.
Applications in DNA Sequencing
An innovative proposal for next-generation DNA sequencing with nanopores utilizes detection via tunneling current. The setup consists of a nanopore and integrated metallic nanogap electrodes such that the nanopore is used for analyte translocation and the electrodes measure the tunnelling current with respect to analyte translocation. Tunnelling current is generated when an electrical potential is applied between the metallic electrodes. As analyte molecules pass through the nanopore, characteristic tunnelling current modulations can be observed, through which molecular information can be concluded. It has been theoretically predicted that characteristic tunnelling current signatures can be observed for each individual nucleotide because of their distinct chemical composition, molecular size, and local electron density.171
Single molecule detection of thymidine 5′-monophosphate (TMP), guanosine 5′-monophosphate (GMP), and cytidine 5′-monophosphate (CMP) has been demonstrated via tunneling current.95 Simultaneous detection of DNA molecules with both the tunneling current method and resistive pulse sensing has also been reported.208
Integration with Scanning Probe Microscopy Techniques
Incorporation of conventional solid-state nanopore platforms with scanning probe microscopy techniques, such as scanning ion conductance microscopy (SICM)209 and scanning electrochemical microscopy (SECM),210 leads to exciting new systems we describe here, the mobile nanopore sensors. With these techniques, qualitative and quantitative analysis of a variety of surfaces and interfaces can be performed with superior spatial resolution for numerous exciting opportunities in biophysics, cell biology, and material sciences.
Merits of Glass Nanopipettes
Nanopipettes have been the most widely used platform in the development of nonstationary sensors because of some unique advantages. One of the most attractive features for nanopipettes is the facile and low-cost fabrication process. Unlike nanopores fabricated from SiNx, Si, or graphene, which require elaborate clean room facilities, expensive instruments, and special nanofabrication expertise, a nanopipette can be readily obtained in a few seconds by pulling a quartz or borosilicate capillary with a benchtop puller instrument. Additionally, the size and geometry of nanopipettes can be fine-tuned by adjustment of fabrication parameters, and integration of nanopipettes with positioning systems such as stepper motors or piezoelectric stages can easily be attained. Thus, opposed to nanopores drilled in membrane materials that have fixed positions, nanopipettes can be coupled with high-precision positioning systems for subnanometer manipulation in x, y, and z dimensions.
Scanning Probe Microscopy Techniques
For scanning probe sensors, there must exist some type of distance-dependent relationship between the nanopore and the sample surface that can be exploited for accurate control of sensor position with nanometer precision. With the accurate probe position control, SICM is suitable for such applications.
The operation of SICM relies on an electrolyte-filled nanopipette as a scanning probe to scan over a sample immersed within an electrolyte bath. A distance-dependent ion current is generated as a potential is applied between the working electrode inserted in the nanopipette and the reference electrode in the bulk bath solution, and this current signal serves as feedback to maintain a constant probe–sample distance. As the pipet is moved across a sample surface, the z-position of the pipet is adjusted to maintain a constant ion current and, thus, distance from the surface to create a topographical image of the sample. Concurrently, an ion– current image is generated. Although SICM is known as simply a surface imaging technique,209 we must emphasize that use of SICM reaches beyond high-resolution imaging or nano-positioning; this technique has developed into a powerful platform for nanoscale investigation of cellular activities, local ion fluxes, and other intriguing surface properties. We refer interested readers to a review by Baker and co-workers for the basics of this technique as well as applications reported prior to 2012.211 In addition to the typical high-resolution images of live cells and sample surfaces attained with traditional SICM, a variety of novel sensing applications have been incorporated, including electrochemical activity mapping, local cellular conductance sensing, surface charge mapping, and localized delivery/sampling.
SECM is another scanning probe microscopy technique used to measure local electrochemical activities.210 In a typical SECM experiment, a micro- or nanoelectrode is moved close to a surface of interest, and spatially resolved electrochemical information is obtained through measurement of faradaic current produced from the redox reaction of electrochemical active mediators. Readers are directed to earlier literature for more detailed discussion about SECM basics and previous applications.212, 213
Topographical Sensors
The use of nanopipettes and ion-current feedback in SICM results in a noncontact imaging technique with nanometer resolution and the ability to operate in physiological buffers for investigation of soft and responsive cell surfaces. A direct comparison of atomic force microscopy (AFM) and SICM has been performed in which microvilli and whole-cell geometry were studied.214 Living microvilli structures were resolved by both AFM and SICM, and though AFM provides higher spatial resolution in theory, it is less advantageous for biological samples. Because force interactions are used for feedback during scanning, AFM probes can contact the soft cell surface and physically distort the sample to degrade the overall image quality. Imaging quality of AFM measurements significantly improved after cell fixation whereas the quality of images produced with SICM was unaffected by the fixation process. Additionally, the cantilever force in AFM scans strongly influenced the measured height and width of a cell; with SICM, the contact-free imaging mechanism resulted in constant cell dimensions observed for a wide range of probe– sample distances. Long-term microvilli dynamics were also studied with both AFM and SICM. AFM image quality worsened over time likely due to probe–sample interactions, whereas SICM images demonstrated consistent imaging quality. These studies demonstrate the superior suitability of SICM for delicate biological samples.
The use of SICM as a topographical sensor to gain information about exocytosis of a target protein (von Willebrand factor (vWF)) after phorbol-12-myristate-13-acetate (PMA) stimulation in living endothelial cells (human umbilical vein endothelial cells (HUVECs) also has been reported.215 In this study, exocytosis of vWF was observed from sequential topographic images with SICM (Figure 5a). Before stimulation (at 0 min), the boundary of the live cell was clearly resolved; after stimulation (from 6 to 14 min), secretion materials with protrusion-shapes were observed from the SICM topography images. Additionally, vWF strings and pore formations as well as dynamic changes of the cell membrane during exocytosis were also captured by SICM.
Figure 5.

Representative applications of solid-state nanopore integration with scanning probe techniques. (a) SICM sequential topographical images of the protrusion formation process in living HUVECs after PMA stimulation. Reproduced from Nashimoto, Y.; Takahashi, Y.; Ida, H.; Matsumae, Y.; Ino, K.; Shiku, H.; Matsue, T. Anal. Chem 2015 87, 2542–2545 (ref 215). Copyright 2015 American Chemical Society. (b) (Left) Illustration of potentiometric scanning ion conductance microscopy (P-SICM) setup for measurement at a tricellular tight junction (tTJ). (Right) Apparent local conductance of tricellulin overexpression MDCKII cells in Ringer solution and in NMDG+ replacement solution. Reproduced from Zhou, L.; Gong, Y.; Sunq, A.; Hou, J.; Baker, L. A. Anal. Chem 2016, 88, 9630–9637 (ref 225). Copyright 2016 American Chemical Society. (c) Simultaneous topographical sensing and surface-charge sensing of a nonuniform polystyrene film on glass. (Left) SICM topography image of substrate with hopping mode at ΔV = 0 and AFM image of a similar area within the substrate. Normalized DC component and AC phase shift at negative tip bias (middle) and positive tip bias (right). Reproduced from Perry, D.; Al Botros, R.; Momotenko, D.; Kinnear, S. L.; Unwin, P. R. ACS Nano 2015 9, 7266–7276 (ref 230). Copyright 2015 American Chemical Society.
Electrochemical Activity Sensors
In-depth understanding of nanoscale heterogeneities at electroactive surfaces/interfaces is key for advancement in a variety of energy conversion devices like fuel cells and batteries. Hybrid SICM–SECM techniques, which combine the robust probe–surface distance control of SICM and the superior electrochemical sensitivity of SECM, have been developed and proven useful to probe nanoscale electrochemical information.216, 217
Recently, O’Connell and Wain demonstrated the use of a hybrid SICM–SECM sensor to image surface features as small as 100–150 nm.218 The sensor used in this study was a dual-barrel probe with one open barrel filled with electrolyte for probe position control and one barrel filled with Pt-deposited carbon for electrochemical information collection. Electrochemical activity of individual Pt nanospheres was imaged via oxygen reduction reaction (ORR). Later, they reported use of a similar approach to detect localized H2O2 generation at individual catalytic gold nanoparticles (AuNPs).219 High resolution SICM topographical and SECM electrochemical images revealed electrochemical heterogeneity of the surface. In another work, a similar Pt-deposited double-barrel nanoprobe was employed to collect electrochemical images of immunocytochemically stained EGFR proteins on A431 cells.220
Different probe configurations have also been reported. For example, Baker and co-workers demonstrated the use of a gold-crescent-nanopore sensor for the investigation of degraded Nafion membrane performance, in which heterogeneous membrane permeability was probed with SICM–SECM.221 An elegant approach reported by Unwin and co-workers has demonstrated use of a bare nanopipette for dynamic visualization of interfacial reactivity without further electrode fabrication procedures.222 The basic principle is simple yet intriguing: the ion current recorded by the pipet sensor is extremely sensitive to local conductivity change, and local conductivity depends on the ion composition in the solution. Therefore, as the probe is positioned in close proximity to an electrochemical reaction center that produces or consumes specific ions, the dynamic process can be sensed by the probe in the form of ion current as the conductivity changes. In this study, voltammetric mapping of ion-flux spatial distribution from a local electrochemical reaction at a substrate was obtained.
Cellular Conductance Sensors
Potentiometric-SICM (P-SICM) is another advanced form of SICM.223 In P-SICM, the sensor is a dual-barrel probe, with one barrel used for nanoscale electrode positioning and the other barrel used to measure local conductance in the form of potential deflection. A schematic of P-SICM is shown in Figure 5b, left. Potentiometric measurement in P-SICM provides an enhanced signal-to-noise ratio. Apparent local conductance of trancellular transport pathways (measured at cell body, CB) and paracellular transport pathways (measured at bicellular tight junctions, bTJs) within epithelial cell monolayers has been measured with P- SICM.223, 224
Recently, Zhou et al. reported the use of P-SICM to resolve heterogeneity in epithelial paracellular transport pathways for the first time and explored the functions of a tight junction protein, tricellulin, in the regulation of ion-transport properties of bTJs and tricellular TJs (tTJs).225 Single tTJ pores were found to be 10 nm in diameter and tTJs (as determined from topography images) only take up less than 1% of the total epithelial surface area. The nanometer resolution of nanopipette sensors is critical to resolve and quantify such unique and discrete transport pathways within an epithelial cell layer. Further, a large organic molecule (N-methyl-d-glucamine (NMDG+)), which can only pass through tTJs, but not bTJs, was used to study the size-selective transport at the different cell junctions through substitution of the major cation (Na+) in the basolateral bath solution with isomolar NMDG+. Subsequent P-SICM measurements were carried out, and from the conductance histogram (Figure 5b, right), a clear difference between the conductance of bTJs and tTJs was observed.
Surface Charge Sensors
In work by Sa and Baker, the use of a nanopipette to probe substrate surface charge was reported for the first time.226 The asymmetric I–V response of a quartz nanopipette under applied potentials was found to be strongly dependent on the surface charge of the substrate when the probe was in close proximity to the substrate. Taking cue of the changes in rectifying characteristics of the I–V responses at different probe–substrate distances, the surface charge polarity of the substrate has been determined. In addition, simulation results provide a more thorough understanding of this substrate-induced current rectification and mass transport phenomena.227 Recently, similar methods were employed to sense the surface-charge behavior of polycarbonate membranes in different electrolyte solutions such as propylene carbonate, which is widely used in lithium-ion batteries.228
A novel technique termed bias-modulated SICM (BM-SICM) has been developed229 and used as both a topographical and surface-charge sensor.230 In BM-SICM, a modulated bias (AC) instead of a constant bias is applied to the nanopipette electrode. The phase shift of the resultant AC ion current is sensitive to probe–substrate distance and hence is used as the feedback signal to provide topography information on the sample. The AC ion current induced by small AC bias (zero net bias) on the probe electrode was found to be insensitive to surface charge. Therefore, BM-SICM provides advantages in accurate topographic imaging in buffers with low ionic strength, in which surface charges affect the DC ion current in conventional SICM feedback signal. To implement surface charge mapping based on BM-SICM, a DC bias in addition to the AC bias is applied to the nanopipette electrode and a hopping voltammetric scan method is used. The nanopipette sensor is approached to the surface with zero DC bias in order to obtain accurate topographical information; the probe is then held steady at both a close position and far position as a DC bias is swept from −0.4 to +0.4 V to record two cyclic voltammgrams to extract the surface charge-induced ICR change. Surface charge mapping was carried out for a glass substrate with a randomly distributed polystyrene film. This surface contained both negatively charged glass and neutral polystyrene. In Figure 5c, an SICM topographical image recorded at zero net bias and an AFM image of the film are shown. In addition, images of DC current and AC phase shift at both negative tip bias and positive tip bias are shown (with both normalized by the bulk responses), and surface-charge distribution is clearly resolved. For example, when the negatively charged probe (with DC current at negative tip bias) scanned over a negatively charged glass region, an enhancement in ionic concentration near the tip led to a higher conductance state. Therefore, the normalized current magnitude ratio was over 1, as seen in Figure 5c (negative tip bias). For AC phase behavior, a stronger contrast was observed with respect to the surface charge compared to the DC current. In a recent work in Perry et al., this elegant approach has been further applied to sense surface charge on live cells and reveal new features that otherwise cannot be accessed with other techniques.231
An alternative design for a surface-charge sensor has been reported by Mirkin and co-workers, namely, plasmonic-based electrochemical impedance imaging (P-EIM).232 An oscillating potential is applied to a micropipette positioned near a gold substrate electrode, which results in local surface-charge redistribution (electric field) on the substrate electrode. Simultaneously, light is directed to the gold substrate with proper incident angle to excite surface plasmons. As the surface plasmon resonance condition is sensitive to surface charge, it becomes possible to sense the surface charge distribution via plasmonic imaging. The surface charge distribution dependence on (i) pipet size and (ii) distance between pipet tip and gold substrate was investigated.
Localized Delivery/Sampling Tool
The ability of nanopipettes to manipulate ultrasmall volumes and to act as molecular reservoirs to deliver/sample biomolecules is of great interest. Controllable voltage-driven delivery/sampling of biomolecules by a nanopipette has been demonstrated in a number of different experiments.74, 233, 234 The deposition/ capture of molecules is controlled by amplitude and polarity of the applied potential to the pipet electrode.
Recently, Mirkin and co-workers reported the use of nanopipettes to both eject and capture individual AuNPs. Negatively charged 10 nm AuNPs were used in the experiments. Translocation of AuNPs into/out of the nanopipette orifice resulted in distinct current modulations which enabled quantification of the number of particles delivered or collected. Upon application of negative potential to the pipet electrode, ejection of AuNP occurred, whereas at positive applied potentials, AuNPs were captured by the probe. Incorporation of SICM with this miniaturized delivery system allowed localized delivery of single particles.235 Moreover, the use of nanopipettes to directly sample native cellular environments and then evaluate local distribution of biomolecules such as mRNA, DNA, and lipids has been reported.236–238
In another report, deposition of free-standing three-dimensional (3D) nanostructures was accomplished with a dual-barrel nanopipette probe, where one barrel was used for distance control (as in conventional SICM) and the other barrel was used as a local source for patterning precursors (Cu2+).239 For patterning, the probe is brought into close proximity with the underlying Au substrate surface and a delivery potential is applied to the precursor-containing barrel to drive out copper ions. Next, as the Cu deposition process starts (prompted by the substrate potential) and the deposited feature grows in the confined region under the nanopore orifice of the probe, a slow retraction of the nanopipette takes place due to the positional feedback of SICM. With this approach, pillars, zigzags, and Γ-like structures were created. Another advance presented here is the capability for the same probe to first produce the nanostructure and then subsequently image the resultant feature.
BIOLOGICAL AND SOLID-STATE NANOPORE COMPARISON
In this section, we provide a comparison of solid-state and biological nanopores to provide readers a more comprehensive idea of the merits and/or drawbacks of both platforms. Although the majority of widely employed biological nanopores exhibit excellent tolerance in a surprisingly wide range of experimental conditions, the stability of the fragile lipid bilayer which supports these pores is far less than ideal. The durability of solid-state nanopores prepared in silicon supports, polymer membranes, and glass is unarguably better compared to their biological counterparts.
Biological nanopores remain unrivaled for their atomic-level reproducibility. Every α-HL pore has the same size/geometry/ surface properties unless purposely altered by modification. State-of-the-art fabrication techniques for synthetic nanopores, however, still do not have such precision.
The diameter, thickness, and shape of solid-state nanopores can be tuned to better suit the desired sensing application. Nature has preset the geometry and dimensions of protein pores, and though alterations in biological pore sizes have been developed,33, 147 they require intensive engineering efforts. Additionally, protein pores have much less flexibility in tuning the size compared to solid-state nanopores.
With respect to surface modification adaptability, biological nanopores are presently better suited than solid-state nanopores. Thanks to high-resolution crystal structures of commonly used protein pores, site-directed mutagenesis can be performed to precisely place functional groups at exact target locations with relative ease. Specific point modifications within synthetic nanopores have yet to be realized despite the wide selection of applicable surface modification techniques.
Finally, solid-state nanopores are more amenable to parallel fabrication of multiple identical devices and can be readily integrated to other nanodevices. The fragile lipid bilayer support for protein nanopores, however, limits the capability of mass production.
PRACTICAL CONCERNS AND CHALLENGES
The most widely used detection scheme for both biological and solid-state nanopores is the resistive pulse sensing method, in which detection of analyte molecules is dependent on ionic current blockades. This technique suffers in high frequency electrical noise in the measured ionic current signal as a result of the experimental setup: the insulative membrane sandwiched between electrolyte solutions with ions on either side resembles a parallel plate capacitor. Because ionic current signal measured with nanopore sensors is intrinsically small (pA to nA range), it is usually necessary to apply a low-pass filter to the signal. Temporal resolution is also an important factor to consider for analyte translocation experiments, and a temporal resolution of 100 µs is set when a 10 kHz filter is applied. This temporal resolution is sufficient for many studies, but sampling is far too slow for DNA sequencing applications. For example, with solid-state nanopore platforms, DNA molecules move at a speed of >10 nucleotides/µs;240 for sensors that employ biological nanopores such as α-HL, the translocation velocity is >1 nucleotide/µs.31, 34, 35 Insufficient temporal resolution may greatly distort the obtained results simply because the recording system is not fast enough to capture all the events. Such issues have been addressed by two main approaches: attempts to slow down the molecular translocation velocity as well as speed up the recording electronics. The former can be achieved through careful adjustment of experimental conditions, such as electrolyte pH, viscosity, and nanopore surface proper-ties.170, 196, 241 It should be noted that such modification might inherently alter the molecular flux, however. Improved electronics capable of measurements with bandwidth as quick as 1 MHz have been reported,194, 195 and the 1 µs temporal resolution offered by such electronics has already shown promise in a variety of exciting applications. Unfortunately, these newer current amplifiers capable of such high-bandwidth measurements are usually quite expensive. For more information about how to control DNA velocity during nanopore translocation as well as a detailed comparison of state-of-the-art DNA sequencing techniques, readers are directed to an excellent review published recently.242
Increasing efforts have been put into the development of: (i) new measurement modalities beyond ionic current signals and (ii) hybrid nanopore platforms which combine merits from both biological and solid-state nanopores to further expand the applications of nanopore sensing. In the following sections, we provide an overview of novel nanopore platforms and their exciting applications.
PLASMONIC NANOPORES
Plasmonic nanopores represent a novel class of solid-state sensor and usually consist of metallic nanostructures at the vicinity of nanopores. Upon light illumination, the metallic nanostructures in the pore focus the optical field to a nanoscale “hot spot”. This enhanced optical field has a variety of unique properties that can be applied to provide exciting new opportunities for molecular sensing.
Plasmonic Nanopore Configurations
Generally, a plasmonic nanopore platform contains a nanometer-sized aperture with integrated metallic nanostructures. The metallic nanostructure is used as a nanoantenna that has the ability to enhance and focus the optical field from an incoming light beam to a nanoscale volume, and the nanopore serves as the conduit for analyte translocation as in traditional sensing experiments. Representative illustrations and microscopy images of plasmonic nanopore configurations are shown in Figure 6.
Figure 6.

Representative configurations of plasmonic nanopores. (a) Schematic illustration of DNA molecule translocation through a gold bowtie antenna with a 10 nm nanopore at the gap center (left) and TEM image of the gold bowtie plasmonic nanopore device (right). Reproduced from Nicoli, F.; Verschueren, D.; Klein, M.; Dekker, C.; Jonsson, M. P. Nano Lett 2014 14, 6917–6925 (ref 103). Copyright 2014 American Chemical Society. (b) Schematic of a graphene nanopore with self-integrated optical antenna (left) and SEM image of a graphene nanopore with integrated nanoparticle (right). Reproduced from Nam, S.; Choi, I.; Fu, C.-c.; Kim, K.; Hong, S.; Choi, Y.; Zettl, A.; Lee, L. P. Nano Lett 2014 14, 5584–5589 (ref 245). Copyright 2014 American Chemical Society. (c) Schematic of a bullseye plasmonic nanopore with a top-down view of features milled into gold film (dimensions: (i) ring period 518 nm, (ii) pore diameter 80 nm, and (iii) ring width 80 nm) and a side-on cross-section of freestanding membrane (left). SEM image with a top-down view of the nanopore/ bullseye structure milled into the membrane. Reproduced from Crick, C. R.; Albella, P.; Ng, B.; Ivanov, A. P.; Roschuk, T.; Cecchini, M. P.; Bresme, F.; Maier, S. A.; Edel, J. B. Nano Lett 2014 15, 553–559 (ref 101). Copyright 2014 American Chemical Society. (d) Schematic of the “pore-in-cavity” plasmonic nanopore with a plasmonic cavity on top of a nanopore (left). TEM image of a silicon nitride membrane surrounded by the gold nanocavity with the designed boundary of the gold cavity indicated by the yellow dashed line (middle). Magnified TEM image of red dashed circle in middle panel, showing a typical 10 nm nanopore inside the gold nanocavity (right). Reproduced from Li, Y.; Nicoli, F.; Chen, C.; Lagae, L.; Groeseneken, G.; Stakenborg, T.; Zandbergen, H. W.; Dekker, C.; Van Dorpe, P.; Jonsson, M. P. Nano Lett 2015 15, 776–782 (ref 246). Copyright 2015 American Chemical Society.
Figure 6a shows a gold bowtie nanoantenna within a thin membrane (i.e., SiNx) that is comprised of two equilateral triangles separated by a small gap with a nanopore at the center.103 It is vital to ensure precise alignment of the nanopore with the optical hot spot such that molecules experience the greatest field enhancement as they move through the pore. Dekker and co-workers have developed a self-aligned fabrication method based on plasmonic-excitation-promoted dielectric breakdown.243 The gold bowtie nanoantenna is fabricated on top of a SiNx membrane via e-beam lithography. Illumination of the gold bowtie structure along the main longitudinal axis of the structure creates the “hot spot” of locally enhanced field strength confined within the center of the bowtie. Upon simultaneous application of a laser to excite the gold bowtie and induce plasmon formation and an electrical bias to the solid-state membrane close to the breakdown voltage of SiNx, a nanopore is created at the point of greatest field strength within the center of the bowtie. Pore formation is indicated by an abrupt increase in ionic current as ions become able to pass through the newly formed opening. Two approaches can be used to further confirm nanopore alignment within the plasmonic hot spot. First, the nanopore can be directly visualized with TEM. Another interesting approach uses a low-power laser beam to raster-scan the membrane while ionic current is monitored. As the laser illuminates the bowtie structure near the nanopore, an increase in the ionic current is observed due to the increased electrolyte conductivity caused by localized heating of the plasmonic structure.100 The alignment of the nanopore with the plasmonic hot spot is verified when the location of maximum current is found to be within the center of the plasmonic bowtie upon illumination. The plasmon-assisted dielectric breakdown approach presented in this work is particularly useful to fabricate nanopores within plasmonic systems in which the position of the hot spot is unknown or hard to predict244 as the approach herein ensures automatic nanopore alignment at the location of greatest field intensity.
Another method has been developed recently that uses a one-step fabrication procedure to form graphene nanopores with self-integrated/aligned optical nanoantennas.245 For this fabrication method, gold nanorods are first drop-cast on either a free-standing graphene film or graphene on an underlying carbon support. Illumination of the surface with a wavelength of light that corresponds to the peak absorbance of the gold nanorods is performed and causes nanometer-sized heated spots via photon-to-heat conversion. By heating the nanorods to a near melting point, the rods become mobile and create nanopores at the highly localized heat pockets. Figure 6b shows the schematic and TEM image of a graphene nanopore with a gold nanoparticle next to the pore opening as a self-integrated optical antenna. In addition, tunability of nanopore shape and size has been demonstrated through simple variation in illumination laser intensity and initial nanoparticle dimensions.
Edel and co-workers have developed a device with a solid-state nanopore surrounded by a bullseye-shaped plasmonic structure.101 This device, created on a free-standing Au-coated SiNx membrane with FIB milling/patterning, is shown by a schematic illustration and SEM image in Figure 6c.
A different system, termed “plasmonic pore-in-cavity”, was developed by Jonsson and co-workers and consists of a nanopore integrated in conjunction with a plasmonic nanoslit cavity.246 In Figure 6d, the schematic of the “pore-in-cavity” plasmonic system and TEM image of the cavity and nanopore are shown. In this method, the free-standing SiNx membrane at the bottom of a gold nanoslit was fabricated via alternative chemical vapor deposition (CVD) of SiNx, Au, and SiNx again; the nanopore is then drilled into the membrane by TEM.
Plasmonic Nanopore Applications
Local Heating
The highly localized heating effect upon laser illumination has been exploited by Jonsson and Dekker to generate high-resolution optical intensity profiles.100 With this technique, variations in local optical intensity modulate plasmonic heating to change the conductivity of the solution, which can be measured electrically via ionic current through the nanopore. By monitoring current as a 10 mW laser beam focused by a high numerical aperture microscope objective scanned over the focal plane of the nanopore, a three-dimensional intensity distribution can be generated. This study represents the first use of a nanopore to probe local heating effects in plasmonic nanostructures.
Further investigations have provided more insight into these plasmonic-induced heating effects. Simulation results by Nicoli et al.103 have demonstrated that, as the plasmonic nanopore sensor is excited along the longitudinal direction, a highly focused hot spot with a significantly enhanced optical intensity, or electric field, is observed directly at the gap in the bowtie at the same location of the nanopore. Though no excitation is found along the transverse direction, light absorption and local heating are still discernible, but to a much smaller extent. Overall, the localized heat effect for plasmonic nanopores occurs in both longitudinal and transverse directions, with a pronounced effect in the longitudinal direction.
Applications in Detection of Nucleotides
In Nicoli et al.,103 the influence of plasmonic excitation on dsDNA translocation was investigated. In Figure 7a, left, open pore current was observed to increase as plasmonic excitation was increased through adjustment of the illuminating laser power. The purple trace shows a control experiment of translocation of dsDNA molecules without any illumination and thus no plasmonic excitation; distinct downward spikes in current were observed as a result of the passage of dsDNA molecules through the nanopore. The green and orange traces show studies that utilized different levels of plasmonic excitation with laser powers of 5 and 10 mW, respectively. A clear increase in open pore current from ~6 nA (0 mW) to ~9 nA (5 mW) and then to ~12 nA (10 mW) was observed. Current signals of typical translocation events are highlighted in Figure 7a, center. Additionally, histograms of conductance blockade were constructed, as shown in Figure 7a, right, and conductance blockades were observed to increase with plasmonic excitation. These observations can, again, be explained by the presence of localized heating at the nanopore; local increases in temperature enhance electrolyte conductivity which leads to a measurably greater signal of ion current.
Figure 7.

Representative applications of plasmonic nanopores. (a) Application in DNA translocation studies. (Left) Examples of current traces without (green) and with plasmonic excitation using laser power of 5 mW (purple) and 10 mW (orange). (Center) Magnified current signals for DNA translocation events. (Right) Corresponding histogram of conductance blockades for DNA translocation. Reproduced from Nicoli, F.; Verschueren, D.; Klein, M.; Dekker, C.; Jonsson, M. P. Nano Lett 2014 14, 6917–6925 (ref 103). Copyright 2014 American Chemical Society. (b) Proposed approach for DNA sequencing with a plasmonic nanopore. (Top) Schematic of proposed plasmonic nanopore device to trap and sequence DNA. (Middle) Duty cycle of laser illumination and simulated displacement of the DNA molecule. (Bottom) Raman intensity signals recorded during translocation of ssDNA through a single plasmonic hot spot. Reproduced from Belkin, M.; Chao, S.-H.; Jonsson, M. P.; Dekker, C.; Aksimentiev, A. ACS Nano 2015 9, 10598–10611 (ref 104). Copyright 2015 American Chemical Society. (c) Photoresistance control of a plasmonic nanopore. (Top) Schematic illustration of experiment setup that uses a plasmonic “pore-in-cavity” structure. (Middle) Ionic current traces of the nanopore when illuminated with different laser powers. (Bottom) Current–voltage curves that highlight rectification effects of high powered laser illumination. Reproduced from Li, Y.; Nicoli, F.; Chen, C.; Lagae, L.; Groeseneken, G.; Stakenborg, T.; Zandbergen, H. W.; Dekker, C.; Van Dorpe, P.; Jonsson, M. P. Nano Lett 2015 15, 776–782 (ref 246). Copyright 2015 American Chemical Society.
Another finding in this work by Nicoli et al. is that the frequency of dsDNA translocation events in LiCl buffers increases significantly upon plasmonic excitation. The authors attribute this increased frequency to the localized nature of plasmonic heating that creates a large thermal gradient within the vicinity of the nanopore. Negative thermophoresis, or the movement of molecules from cold to warmer regions within solution, has been demonstrated to occur within LiCl buffer solutions. Upon plasmonic excitation, DNA molecules experience this phenomenon and move toward the locally heated nanopore region for a higher capture rate and increased event frequency. Measurements conducted in LiCl buffers typically suffer from very low event rates (owing to the partial reduction of DNA charge as a result of stronger transient binding between Li+ and DNA as compared to Na+ or K+) but are still widely used because of slower dsDNA translocation that results in enhanced signal-to-noise and better resolution.241 If greater event frequency occurs, a larger number of events will take place in a given time, which is beneficial for statistical analysis. Thus, plasmonic nanopore systems may find utility for the study of DNA under these experimental conditions.
More recently, theoretical studies have been undertaken to determine the feasibility of combining plasmonic systems (to directly trap and control DNA displacement through a solid-state nanopore) with surface-enhanced Raman spectroscopy (SERS) measurements to achieve a new method of sequencing.104 The proposed schematic for this setup uses a gold bowtie plasmonic nanopore and is shown in Figure 7b, top. Molecular dynamic simulations have revealed that the localized optical hot spots can trap the DNA molecules during translocation to reduce overall translocation velocity. As increased illumination laser power is applied to the system, the translocation speed of dsDNA is further reduced due to local plasmonic forces that pull the molecules toward the local maximum plasmonic field at the edge of the gold bowtie. With the highest illumination laser power investigated, movement of the DNA molecule could be fully stopped. Therefore, with periodic modulation of the plasmonic field, stepwise displacement of the DNA molecule to the pore and plasmonic maximum is achievable (Figure 7b, middle). Moreover, the highly focused optical field induced by plasmonic excitation greatly increases the possibility of Raman emission from the molecules, which is beneficial as each section of the DNA molecule produces characteristic Raman signals that correspond to nucleotide structure. Raman spectra of the four DNA nucleotides have been approximated by individual Gaussian distributions with distinct peak intensity frequencies (with cytosine (C) centered at 800 cm−1, thymine (T) centered at 780 cm−1, adenine (A) centered at 735 cm−1, and guanine (G) centered at 660 cm−1). The Raman spectrum of the entire DNA sequence under study can be viewed as a superposition of each Gaussian distribution scaled by a field enhancement factor. Modulation in Raman intensity can be measured for all four nucleotides as the DNA molecule translocates the pore and, with a deconvolution algorithm, DNA sequencing information can be obtained. With traditional nanopore sensors, the conversion of signal into a known sequence is nontrivial as all information is condensed into a single current value; in this SERS system, Raman intensity at four specific frequencies (each corresponding to a particular nucleotide) is measured simultaneously such that there is little to no interference from neighboring nucleotides. In Figure 7b, bottom, Raman signals measured upon ssDNA translocation through a plasmonic hot spot are shown, and peaks within the intensity traces correlate to individual nucleotides. The simulation results in this work suggest a novel approach of DNA sequencing with plasmonic nanopores. Practical implementation of this concept will require precise control over DNA–nanopore interactions to maintain accurate Raman signals.
Photoresistance Switching
Another recent finding with plasmonic nanopores is the ability to modulate resistance through adjustment of overall illumination power. Plasmonic nanopores with tunable resistances have been used as fluidic switches through control of laser illumination.246Figure 7c, top, shows the experimental setup of a nanopore integrated with a metal nanocavity. Current–time measurements with low-power laser illumination and high-power laser illumination are shown in Figure 7c, middle. Upon illumination by a 2 mW laser, the system current increased due to the plasmonic excitation. However, illumination of the nanopore with a higher power laser (i.e., 15 mW) demonstrated the opposite response with a decrease in nanopore current. A systematic study revealed that current increased monotonically with laser power up to 12 mW. Once the laser power was further increased over this 12 mW cutoff, a rapid decrease in current was observed such that the resistance of the nanopore exposed to 30 mW laser light exhibited a resistance of ~500% of the original. This resistance modulation is a reversible process as the system reset to initial nanopore current values after the laser was turned off. In addition, I–V curves recorded without laser illumination (black) and with 2 mW (blue) as well as 15 mW (brown) laser illumination are shown in Figure 7c, bottom. With no application of laser light as well as use of a lower power laser, linear I–V responses were observed with nominal pore resistances of 11.0 and 9.1 MΩ, respectively. High-power laser illumination, however, led to a rectified I–V curve. The authors attribute this interesting photoresistance switching effect to the reversible blockage of the nanopore by plasmonic-induced nanobubbles. The dynamic control of nanopore resistance exhibited by plasmonic excitation offers a new method for nanoscale fluid manipulation.
HYBRID NANOPORES
The motivation of hybrid nanopore systems is to exploit the merits and circumvent the disadvantages of both biological and solid-state nanopore platforms. For instance, substitution of the membrane support for protein pores from lipid bilayer to synthetic materials such as SiNx can dramatically increase system stability. Such hybrid nanopore platforms possess the advantages of atomic-precision structural reproducibility and surface-modification adaptability of protein pores while also bypassing the instability of the lipid bilayer. In addition, the use of DNA origami as highly stable chemical building blocks for semisynthetic membrane porin constructions offers superior versatility in nanopore configurations for numerous exciting opportunities. In this section, we aim to highlight some recent advances in hybrid nanopore platforms and their applications.
The first example of a hybrid nanopore was created by insertion of a preassembled α-HL pore within a SiNx pore.81 To ensure coaxial arrangement of the protein and SiNx pores, a DNA oligomer tethered to the lumen of the α-HL molecule pulls the biological pore into proper alignment as it translocates the solid-state pore. Insertion of the dsDNA-conjugated α-HL pore was achieved through application of a potential across the SiNx membrane. Successful translocation experiments of ssDNA through the α-HL pore within the SiNx support further verified the functionality of α-HL was preserved. This hybrid α-HL-SiNx nanopore has bridged the gap between the two worlds of solid-state nanopores and biological nanopores. However, the atomically precise size of the biological pore in this platform is a double-edged sword, as α-HL exhibits superior reproducibility unsurpassed by present nanofabrication technologies but has a fixed size that limits the variety of analytes that can be studied.
Another novel design of a hybrid nanopore sensor has been reported of carbon nanotubes (CNTs) embedded within a lipid bilayer or live cell membrane.83 When the lengths of the CNTs are comparable to the thickness of the lipid bilayer, spontaneous insertion into the lipid membrane will lead to formation of CNT membrane channels. An illustration of ssDNA translocation through the CNT porin and the representative data are shown in Figure 8a,b, respectively. Insertion of CNT porins into the membranes of live cells was verified by patch clamp measurements.
Figure 8.

Representative examples of hybrid nanopore platforms. (Top) Carbon nanotubes (CNTs) within a lipid bilayer. (a) Schematic depiction of ssDNA translocation event through a CNT porin in the lipid bilayer. (b) Representative current traces in the absence (top) and presence (bottom) of ssDNA. (a, b) Reproduced from Geng, J.; Kim, K.; Zhang, J.; Escalada, A.; Tunuguntla, R.; Comolli, L. R.; Allen, F. I.; Shnyrova, A. V.; Cho, K. R.; Munoz, D. Nature 2014 514, 612–615 (ref 83), with permission from Nature Publishing Group. (Bottom) Membrane patch-ion channel probe. (c) Instrumental setup for dual-barrel ion channel probe-SICM measurements. (d) Current–time (I–T) traces recorded with the ICP barrel close (top) and far (bottom) from surface substrate. (c, d) Reproduced from Shi, W.; Zeng, Y.; Zhou, L.; Xiao, Y.; Cummins, T. R.; Baker, L. A. Faraday Discuss 2016, DOI: 10.1039/C6FD00133E (ref 80), with permission of The Royal Society of Chemistry.
A novel approach for hybrid nanopore sensors that can scan surfaces has also been reported. Ion-channel probes (ICPs), in which multiple α-HL pores are reconstituted within a lipid bilayer that covers the opening of a glass micropipette tip,78 have been developed and integrated with SICM to provide a new method for spatially resolved sensing. As a potential is applied between the electrode in the bath solution and the electrode in the pipet, an ion current passes through the α-HL protein channels that can be used as feedback to control probe position and maintain a constant probe–surface distance, just as in conventional SICM. Preliminary results also demonstrated utilization of ICPs for topographic imaging. A similar method then followed by Macazo and White in which simultaneous imaging and detection of cyclodextrin molecules was achieved, though the resolution was relatively low (compared to what is ultimately achievable with SICM) due to the large probe size and the topography was obtained in constant-height mode without any feedback enabled.79 Shi et al. further advanced the ICP-SICM technique through the use of a dual-barrel pipet, which consists of an open barrel and a barrel with a membrane patch (MP) directly excised from a donor cell that contains ion channels (schematic shown in Figure 8c).80 Upon integration with SICM, the open barrel (SICM barrel) serves to measure the distance-dependent ion current for noninvasive imaging and positioning of the probe by the same feedback mechanism as in traditional SICM. The second barrel in the probe that supports the MP (ICP barrel) was used to investigate the activities of the ion channels of interest that are present on the patch. With SICM’s superior feedback control, channel activities of ligand-gated ion channels were analyzed with respect to the distance between the channels and the ligand source. In Figure 8d, distinct distance-dependent channel activities were observed: when the ion-channel probe was far away from the ligand source, the channel remained mostly in the CLOSED (C) state; once the probe was held in close proximity to the ligand source, significantly increased channel activity was observed in the OPEN (O) state. This study highlights the importance of robust distance control in investigations of ion-channel activities. Virtually any kind of cells that contain any kind of native or engineered ion channels can be used to form the membrane patch on the MP-ICP probe, which makes this approach extremely versatile. Further development of this technique may provide a way to study cell-to-cell or even channel-to-channel communications.
With the development of DNA origami techniques,247 another revolutionary hybrid nanopore platform that involves the insertion of 3D DNA origami structures into SiNx nanopores has been achieved by the Keyser Group. Subsequent dsDNA translocation through theses SiNx-support DNA origami porins was carried out successfully to further demonstrate their potential for sensing applications.82 In comparison to the self-assembly formation process that occurs with protein pores, creation of DNA origami porins is a bottom-up scaffolding approach that uses ssDNA building blocks to create predefined desired shapes. Insertion of synthetic origami pores within a lipid bilayer has also been demonstrated. The flexibility of origami design leads to creations of completely new pore architectures with high degrees of freedom that can be exploited for different applications. Readers can find more information about the DNA origami pores in a recent review by Keyser and co-workers, in which the developments, challenges, and future prospectives of DNA origami are discussed.248 A variety of DNA origami porin structures have been reported, and we have selectively included some representative designs herein.84–88
In Figure 9a, the placement of one such DNA–duplex porin to form a toroidal DNA-lipid channel (DLC) is shown.84 The pore design consists of a 19-base long DNA duplex with six hydrophobic porphyrin tags attached to thymidine bases, such that the porphyrin protrusions act as membrane anchors when the duplex is inserted into a lipid bilayer (Figure 9b). Binding of the synthetic DNA–duplex with a bilayer was confirmed by the observation of bright rings around Giant Unilamellar Vesicles (GUVs) caused by the fluorescence of the porphyrin tags. Furthermore, the ion conductance capability of the DNA– duplex porin was investigated with measurements of ion current. Stepwise increases in current were observed after addition of the DNA duplex to a lipid bilayer, and the channel conductance was observed to be ~0.06 nS (Figure 9c). In comparison to previously reported synthetic DNA porins or biological pores, the DNA–duplex porin lacks a central channel but is still permeable to ions to provide a conductance pathway. Further experimental and simulation investigations of ion fluxes through this DNA duplex origami may provide fundamental understanding of ion-conduction mechanisms across lipid bilayer membranes.
Figure 9.

Representative DNA origami nanopores. (a) Envisioned placement and conductance mechanism of the DNA duplex (blue) with membrane anchors (red) that form a toroidal lipid pore (yellow). (b) Sequence of the nucleotide duplex, with the locations of tags within the sequence shown in red and their corresponding chemical structures. (c) Ionic current trace of duplex insertion in the lipid bilayer membrane. (a–c) Reproduced from Gopfrich, K.; Li, C.-Y.; Mames, I.; Bhamidimarri, S. P.; Ricci, M.; Yoo, J.; Mames, A.; Ohmann, A.; Winterhalter, M.; Stulz, E.; Aksimentiev, A.; Keyser, U. F. Nano Lett 2016 16, 4665–4669 (ref 84). Copyright 2016 American Chemical Society. (d) Envisioned positioning of the funnel-shaped DNA porin (red) in the lipid membrane (yellow) with each DNA duplex represented as a rod. (e) Side and top views of the DNA porin with 19 cholesterol tags (orange). (f) Ionic current trace of two consecutive DNA–origami porin insertions in the lipid bilayer membrane. (d–f) Reproduced from Gopfrich, K.; Li, C.-Y.; Ricci, M.; Bhamidimarri, S. P.; Yoo, J.; Gyenes, B.; Ohmann, A.; Winterhalter, M.; Aksimentiev, A.; Keyser, U. F. ACS Nano 2016 10, 8207–8214 (ref 85). Copyright 2016 American Chemical Society. (g) Schematic illustration of the ligand-trigger mechanism of a designed DNA porin. Conversion of the nanopore from CLOSED state (NP-C) to OPEN state (NP-O) is achieved through binding of the complementary “key” oligonucleotide to the “lock” oligonucleotide positioned at the lumen of the pore. (h) Flux histograms of CF and SRB through NP-C exposed to a mismatching key and NP-C after addition of a matching key to yield NP-O. (g, h) Reproduced from Burns, J. R.; Seifert, A.; Fertig, N.; Howorka, S. Nat. Nanotechnol 2016 11, 152–156 (ref 87), with permission from Nature Publishing Group.
The DNA duplex porin represents the smallest DNA-based ion channel to date. The largest synthetic pore within a lipid bilayer achieved at the time of this publication has also been achieved with DNA origami and has a size similar to the nuclear pore complex with a conductance value ~10-fold larger than previously reported man-made membrane channels.85 In Figure 9d, a schematic of a funnel-shaped DNA origami porin inserted within a lipid bilayer is shown. This porin was assembled with a 7249 base scaffold, 179 ssDNA staples, and 19 cholesterol anchors to assist insertion into the lipid bilayer. The total length of the porin is 54 nm with a smaller opening measured to be 6 nm and a larger orifice measured to be 22 nm (Figure 9e). Similar characterization was performed for this system as with the DNA–duplex porin, and binding between the DNA porin and GUVs was verified. Furthermore, the ion conductance was measured (shown in Figure 9f), and multiple high-conductance membrane-insertion steps were observed with a single channel conductance value of 40 nS. The large DNA porin described in this work can provide additional opportunities in sensing applications of larger biomolecules such as proteins.
Further development of DNA origami techniques has led to the creation of synthetic DNA porins with selective gating capabilities that control the selective passage of analyte molecules across the bilayer.87 For such a system, visualized via a cartoon schematic in Figure 9g, the binding event of a specific ligand to a DNA nanopore (NP) initially in a CLOSED state (NP-C) initiates a nanomechanical change in the pore conformation and causes the channel to change to the OPEN state (NP-O). The DNA porin in this work was comprised of six 50-nt oligonucleotides that assembled to form a pore ~9 nm in length and ~5 nm in width. The membrane-insertion stability and single channel conductance of the synthetic NP were measured and confirmed. Next, the authors incorporated the ligand-gating capability to the nanopore by adding an oligonucleotide sequence to one end of the pore lumen; the pore in this conformation without any bound ligand is referred to as NP-C. The conversion of NP-C to NP-O was facilitated by the introduction of the complementary oligonucleotide sequence that is able to bind to the DNA sequence present on the porin. This gating scheme can be thought of as a key and lock system: the oligonucleotide bound to the closed pore is the “lock,” and the pore only transitions to an open state once the complementary “key” sequence interacts with the bound “lock.” The selectivity of this DNA porin was demonstrated by transport experiments of two fluorophores with different molecular charges. The pore exhibited a 13-fold higher selectivity for passage of sulpho-rhodamine B (SRB), a molecule with one positive and two negative charges, over 6-carboxyfluorescein (CF), a molecule with three negative charges. The electrostatic selectivity of the DNA porin is due to the negatively charged pore wall. Finally, the authors demonstrated the use of the designed DNA pore to control transmembrane transport. Minimal fluxes were observed for both fluorophores after addition of mismatched oligonucleotides to the NP-C configurations; i.e., the wrong “key” does not unlock and open the pore. However, addition of the matching oligonucleotides converted NP-C to NP-O, and a significant increase in SRB flux was observed while CF flux remained low, as seen in Figure 9h. The two advances presented in this work with DNA porins, namely, the selectivity and the ligand-gating properties, signify another big step toward synthetic mimics of biological pores from nature.
FUTURE PROSPECTIVE
This Review has provided a summary of the recent advances in the field of nanopore sensing, including progress on the development of biological nanopores, solid-state nanopores, and plasmonic and hybrid systems for a wide variety of applications. In the long-run, expansion of the nanosensor toolbox beyond traditional resistive pulse sensing measurements will continue to push the boundaries of this field. A number of new, emerging detection methods have already been reported: integration of tunnelling current detectors will allow investigation with extremely high resolution at the submolecular level, whereas the integration of plasmonic structures to nanopore platforms will provide additional handles to suit the system for different applications. Development of hybrid nanopore platforms will bring the advantages of both the biological and solid-state worlds together for increased versatility toward various applications.
In addition, development of nanotechnology and DNA-engineering techniques provides promise to expand the limitations of nanopore dimensions and functionality. Presently, biological nanopores are most widely used due to their unprecedented reproducibility in both geometry and chemical-property modifications that can better tune the system for the desired application. Thanks to nature, fabrication of an a-HL pore with identical dimensions is trivial. The protein pore is produced by self-assembly of single/multiple subunit(s), and the position of modified functional groups within the lumen of the pore can be precisely controlled with atomic resolution. Though today s fabrication techniques for solid-state nanopores do not offer the reproducibility or adaptability of biological systems, new advances that enable more precise control over nanopore size, geometry, and chemical properties will significantly benefit application of solid-state pores for a variety of sensors.
Improved electronics have enabled higher quality, low-noise measurements through high-bandwidth recording capabilities. Slower translocation speeds of analytes have been achieved with optimized experimental conditions, such as pH as well as electrolyte composition and concentration. The goal of nanopore-based DNA sequencing envisioned 20 years ago has been realized with a variety of smart approaches, including the novel design of a protein nanopore coupled with a polymerase motor.
The evolution of this newer analytical detection technique is driven by a highly multidisciplinary community; the work of chemists, biologists, physicists, and engineers continues to advance the field of nanopore sensing and push the boundaries of possibility with lower limits of detection and exciting new applications. Rapid expansion in this field has led to a number of nanopore-themed conferences, such as the biyearly Gordon Research Conference on “Nanoporous Materials and their Applications” that was initiated in 2011. As the scientific community continues to focus on building smaller, faster, and better tools, nanoporous sensors will surely continue to find application and relevance in the future.
Acknowledgments
We gratefully thank the National Science Foundation, Directorate for Mathematical and Physical Sciences, Division of Chemistry Award #1507341 and the U.S. Department of Health and Human Services, National Institutes of Health, National Institute of Biomedical Imaging and Bioengineering for award R21EB02229701, as well as Indiana University for support of our efforts.
Biographies
Wenqing Shi received her B.S. in chemistry from Wuhan University (China) in 2011. She is currently a graduate student under Professor Lane A. Baker at Indiana University, Bloomington, with a research focus in the use of unconventional nanopipettes for local chemical analysis. She is interested in design and construction of novel nanopipette platforms to expand the applications of scanning ion conductance microscopy (SICM).
Alicia K. Friedman is presently a Ph.D. candidate under the guidance of Professor Lane A. Baker at Indiana University. She received her B.A. in chemistry from Vanderbilt University in 2009 and anticipates her Ph.D. in Analytical Chemistry in 2017. Her work in graduate school has focused on the development of nanoporous bioanalytical platforms, specifically biomimetic hydrogels of the Nuclear Pore Complex and ion-channel probes for scanning ion conductance microscopy (SICM).
Lane A. Baker is presently a James F. Jackson Associate Professor of Chemistry at Indiana University. He received a B.S. degree from Missouri State University in 1996. He was awarded his Ph.D. degree at Texas A&M University in 2001. He completed a National Research Council Postdoctoral Associateship at the Naval Research Laboratory in Washington, DC in addition to a postdoctoral appointment at the University of Florida. His current research interests include electrochemical methods for analysis and imaging, and work in his group is focused on applications of nanopores for the development of chemical and biochemically selective membranes, sensor development, and electrochemical imaging.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Venkatesan BM, Bashir R. Nat. Nanotechnol. 2011;6:615–624. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- 2.Miles BN, Ivanov AP, Wilson KA, Dogan F, Japrung D, Edel JB. Chem. Soc. Rev. 2013;42:15–28. doi: 10.1039/c2cs35286a. [DOI] [PubMed] [Google Scholar]
- 3.Kasianowicz JJ, Robertson JW, Chan ER, Reiner JE, Stanford VM. Annu. Rev. Anal. Chem. 2008;1:737–766. doi: 10.1146/annurev.anchem.1.031207.112818. [DOI] [PubMed] [Google Scholar]
- 4.Gyurcsanyi RE. TrAC, Trends Anal. Chem. 2008;27:627–639. [Google Scholar]
- 5.Friedman AK, Baker LA. In Nanoelectrochemistry. Boca Raton, FL: CRC Press; 2015. pp. 395–438. [Google Scholar]
- 6.Ying Y-L, Cao C, Long Y-T. Analyst. 2014;139:3826–3835. doi: 10.1039/c4an00706a. [DOI] [PubMed] [Google Scholar]
- 7.Deamer D. Annu. Rev. Biophys. 2010;39:79–90. doi: 10.1146/annurev.biophys.093008.131250. [DOI] [PubMed] [Google Scholar]
- 8.Dekker C. Nat. Nanotechnol. 2007;2:209–215. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Dong X, Chen P. Chem. Soc. Rev. 2012;41:2283–2307. doi: 10.1039/c1cs15270j. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Tian Y, Jiang L. Nano Today. 2016;11:61–81. [Google Scholar]
- 11.Arjmandi-Tash H, Belyaeva LA, Schneider GF. Chem. Soc. Rev. 2016;45:476–493. doi: 10.1039/c5cs00512d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Healy K, Schiedt B, Morrison AP. Nanomedicine. 2007;2:875–897. doi: 10.2217/17435889.2.6.875. [DOI] [PubMed] [Google Scholar]
- 13.Dahlin AB. Analyst. 2015;140:4748–4759. doi: 10.1039/c4an02258k. [DOI] [PubMed] [Google Scholar]
- 14.Rhee M, Burns MA. Trends Biotechnol. 2006;24:580–586. doi: 10.1016/j.tibtech.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Deamer D, Akeson M, Branton D. Nat. Biotechnol. 2016;34:518–524. doi: 10.1038/nbt.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deamer DW, Akeson M. Trends Biotechnol. 2000;18:147–151. doi: 10.1016/s0167-7799(00)01426-8. [DOI] [PubMed] [Google Scholar]
- 17.Rhee M, Burns MA. Trends Biotechnol. 2007;25:174–181. doi: 10.1016/j.tibtech.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Bayley H. Curr. Opin. Chem. Biol. 2006;10:628–637. doi: 10.1016/j.cbpa.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 19.Actis P, McDonald A, Beeler D, Vilozny B, Millhauser G, Pourmand N. RSC Adv. 2012;2:11638–11640. doi: 10.1039/C2RA21730A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayley H. Clin. Chem. 2015;61:25–31. doi: 10.1373/clinchem.2014.223016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinbock LJ, Radenovic A. Nanotechnology. 2015;26:074003. doi: 10.1088/0957-4484/26/7/074003. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths J. Anal. Chem. 2008;80:23–27. doi: 10.1021/ac802115j. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Yang Q, Wang Z. Front. Genet. 2015;5:449–449. doi: 10.3389/fgene.2014.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich SB, Krstic PS, Lindsay S, Ling XS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss JA. Nat. Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braha O, Gu L-Q, Zhou L, Lu X, Cheley S, Bayley H. Nat. Biotechnol. 2000;18:1005–1007. doi: 10.1038/79275. [DOI] [PubMed] [Google Scholar]
- 27.Gu L-Q, Braha O, Conlan S, Cheley S, Bayley H. Nature. 1999;398:686–690. doi: 10.1038/19491. [DOI] [PubMed] [Google Scholar]
- 28.Ettedgui J, Kasianowicz JJ, Balijepalli A. J. Am. Chem. Soc. 2016;138:7228–7231. doi: 10.1021/jacs.6b02917. [DOI] [PubMed] [Google Scholar]
- 29.Howorka S, Cheley S, Bayley H. Nat. Biotechnol. 2001;19:636–639. doi: 10.1038/90236. [DOI] [PubMed] [Google Scholar]
- 30.Storm AJ, Chen J, Zandbergen H, Dekker C. Phys. Rev. E. 2005;71:051903. doi: 10.1103/PhysRevE.71.051903. [DOI] [PubMed] [Google Scholar]
- 31.Butler TZ, Pavlenok M, Derrington IM, Niederweis M, Gundlach JH. Proc. Natl. Acad. Sci. U. S. A. 2008;105:20647–20652. doi: 10.1073/pnas.0807514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zahid OK, Wang F, Ruzicka JA, Taylor EW, Hall AR. Nano Lett. 2016;16:2033–2039. doi: 10.1021/acs.nanolett.6b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayub M, Stoddart D, Bayley H. ACS Nano. 2015;9:7895–7903. doi: 10.1021/nn5060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akeson M, Branton D, Kasianowicz JJ, Brandin E, Deamer DW. Biophys. J. 1999;77:3227–3233. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meller A, Nivon L, Brandin E, Golovchenko J, Branton D. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1079–1084. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deamer DW, Branton D. Acc. Chem. Res. 2002;35:817–825. doi: 10.1021/ar000138m. [DOI] [PubMed] [Google Scholar]
- 37.Harrington L, Alexander LT, Knapp S, Bayley H. Angew. Chem. 2015;127:8272–8277. doi: 10.1002/anie.201503141. [DOI] [PubMed] [Google Scholar]
- 38.Nivala J, Mulroney L, Li G, Schreiber J, Akeson M. ACS Nano. 2014;8:12365–12375. doi: 10.1021/nn5049987. [DOI] [PubMed] [Google Scholar]
- 39.Pastoriza-Gallego M, Breton M-F, Discala F, Auvray L, Betton J-M, Pelta J. ACS Nano. 2014;8:11350–11360. doi: 10.1021/nn5042398. [DOI] [PubMed] [Google Scholar]
- 40.Van Meervelt V, Soskine M, Maglia G. ACS Nano. 2014;8:12826–12835. doi: 10.1021/nn506077e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Han Y, Zhou S, Guan X. Biosens. Bioelectron. 2014;62:158–162. doi: 10.1016/j.bios.2014.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biesemans A, Soskine M, Maglia G. Nano Lett. 2015;15:6076–6081. doi: 10.1021/acs.nanolett.5b02309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fahie M, Chisholm C, Chen M. ACS Nano. 2015;9:1089–1098. doi: 10.1021/nn506606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fahie MA, Yang B, Mullis M, Holden MA, Chen M. Anal. Chem. 2015;87:11143–11149. doi: 10.1021/acs.analchem.5b03350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kukwikila M, Howorka S. Anal. Chem. 2015;87:9149–9154. doi: 10.1021/acs.analchem.5b01764. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Montana V, Grubisic V, Stout RF, Parpura V, Gu L-Q. ACS Appl. Mater. Interfaces. 2015;7:184–192. doi: 10.1021/am5056596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soskine M, Biesemans A, Maglia G. J. Am. Chem. Soc. 2015;137:5793–5797. doi: 10.1021/jacs.5b01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Haque F, Rychahou PG, Evers BM, Guo P. ACS Nano. 2013;7:9814–9822. doi: 10.1021/nn404435v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao C, Ying Y-L, Hu Z-L, Liao D-F, Tian H, Long Y-T. Nat. Nanotechnol. 2016;11:713–718. doi: 10.1038/nnano.2016.66. [DOI] [PubMed] [Google Scholar]
- 50.Haque F, Wang S, Stites C, Chen L, Wang C, Guo P. Biomaterials. 2015;53:744–752. doi: 10.1016/j.biomaterials.2015.02.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M, Gundlach JH. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16060–16065. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuller CW, Kumar S, Porel M, Chien M, Bibillo A, Stranges PB, Dorwart M, Tao C, Li Z, Guo W, Shi S, Korenblum D, Trans A, Aguirre A, Liu E, Harada ET, Pollard J, Bhat A, Cech C, Yang A, Arnold C, Palla M, Hovis J, Chen R, Morozova I, Kalachikov S, Russo JJ, Kasianowicz JJ, Davis R, Roever S, Church GM, Ju J. Proc. Natl. Acad. Sci. U. S. A. 2016;113:5233–5238. doi: 10.1073/pnas.1601782113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J, Stein D, McMullan C, Branton D, Aziz MJ, Golovchenko JA. Nature. 2001;412:166–169. doi: 10.1038/35084037. [DOI] [PubMed] [Google Scholar]
- 54.Storm A, Chen J, Ling X, Zandbergen H, Dekker C. Nat. Mater. 2003;2:537–540. doi: 10.1038/nmat941. [DOI] [PubMed] [Google Scholar]
- 55.Li N, Yu S, Harrell CC, Martin CR. Anal. Chem. 2004;76:2025–2030. doi: 10.1021/ac035402e. [DOI] [PubMed] [Google Scholar]
- 56.Yuan J, He F, Sun D, Xia X. Chem. Mater. 2004;16:1841–1844. [Google Scholar]
- 57.Noshay A, McGrath JE. Block copolymers: overview and critical survey. New York: Elsevier; 2013. [Google Scholar]
- 58.Rodríguez-Manzo JA, Puster M, Nicolaï A, Meunier V, Drndic M. ACS Nano. 2015;9:6555–6564. doi: 10.1021/acsnano.5b02531. [DOI] [PubMed] [Google Scholar]
- 59.Wanunu M, Dadosh T, Ray V, Jin JM, McReynolds L, Drndic M. Nat. Nanotechnol. 2010;5:807–814. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 60.Venta K, Shemer G, Puster M, Rodríguez-Manzo JA, Balan A, Rosenstein JK, Shepard K, Drndic M. ACS Nano. 2013;7:4629–4636. doi: 10.1021/nn4014388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall AR. Microsc. Microanal. 2013;19:740–744. doi: 10.1017/S1431927613000500. [DOI] [PubMed] [Google Scholar]
- 62.Garaj S, Hubbard W, Reina A, Kong J, Branton D, Golovchenko JA. Nature. 2010;467:190–193. doi: 10.1038/nature09379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merchant CA, Healy K, Wanunu M, Ray V, Peterman N, Bartel J, Fischbein MD, Venta K, Luo Z, Johnson ATC, Drndic M. Nano Lett. 2010;10:2915–2921. doi: 10.1021/nl101046t. [DOI] [PubMed] [Google Scholar]
- 64.Schneider GF, Kowalczyk SW, Calado VE, Pandraud G, Zandbergen HW, Vandersypen LMK, Dekker C. Nano Lett. 2010;10:3163–3167. doi: 10.1021/nl102069z. [DOI] [PubMed] [Google Scholar]
- 65.Fischbein MD, Drndic M. Appl. Phys. Lett. 2008;93:113107. [Google Scholar]
- 66.Liu S, Lu B, Zhao Q, Li J, Gao T, Chen Y, Zhang Y, Liu Z, Fan Z, Yang F, You L, Yu D. Adv. Mater. 2013;25:4549–4554. doi: 10.1002/adma.201301336. [DOI] [PubMed] [Google Scholar]
- 67.Larkin J, Henley R, Bell DC, Cohen-Karni T, Rosenstein JK, Wanunu M. ACS Nano. 2013;7:10121–10128. doi: 10.1021/nn404326f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu K, Feng J, Kis A, Radenovic A. ACS Nano. 2014;8:2504–2511. doi: 10.1021/nn406102h. [DOI] [PubMed] [Google Scholar]
- 69.Wanunu M, Meller A. Nano Lett. 2007;7:1580–1585. doi: 10.1021/nl070462b. [DOI] [PubMed] [Google Scholar]
- 70.Chen P, Mitsui T, Farmer DB, Golovchenko J, Gordon RG, Branton D. Nano Lett. 2004;4:1333–1337. doi: 10.1021/nl0494001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sze JYY, Kumar S, Ivanov AP, Oh S-H, Edel JB. Analyst. 2015;140:4828–4834. doi: 10.1039/c5an01001b. [DOI] [PubMed] [Google Scholar]
- 72.Wang C, Fu Q, Wang X, Kong D, Sheng Q, Wang Y, Chen Q, Xue J. Anal. Chem. 2015;87:8227–8233. doi: 10.1021/acs.analchem.5b01501. [DOI] [PubMed] [Google Scholar]
- 73.Liu S, Dong Y, Zhao W, Xie X, Ji T, Yin X, Liu Y, Liang Z, Momotenko D, Liang D, Girault HH, Shao Y. Anal. Chem. 2012;84:5565–5573. doi: 10.1021/ac3004852. [DOI] [PubMed] [Google Scholar]
- 74.Shi W, Sa N, Thakar R, Baker LA. Analyst. 2015;140:4835–4842. doi: 10.1039/c4an01073f. [DOI] [PubMed] [Google Scholar]
- 75.Hernandez-Ainsa S, Muus C, Bell NA, Steinbock LJ, Thacker VV, Keyser UF. Analyst. 2013;138:104–106. doi: 10.1039/c2an36397f. [DOI] [PubMed] [Google Scholar]
- 76.Venkatesan BM, Polans J, Comer J, Sridhar S, Wendell D, Aksimentiev A, Bashir R. Biomed. Microdevices. 2011;13:671–682. doi: 10.1007/s10544-011-9537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kowalczyk SW, Kapinos L, Blosser TR, Magalhaes T, van Nies P, Lim RY, Dekker C. Nat. Nanotechnol. 2011;6:433–438. doi: 10.1038/nnano.2011.88. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Y, Bright LK, Shi W, Aspinwall CA, Baker LA. Langmuir. 2014;30:15351–15355. doi: 10.1021/la504097f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Macazo FC, White RJ. J. Am. Chem. Soc. 2016;138:2793–2801. doi: 10.1021/jacs.5b13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi W, Zeng Y, Zhou L, Xiao Y, Cummins TR, Baker LA. Faraday Discuss. 2016 doi: 10.1039/c6fd00133e. [DOI] [PubMed] [Google Scholar]
- 81.Hall AR, Scott A, Rotem D, Mehta KK, Bayley H, Dekker C. Nat. Nanotechnol. 2010;5:874–877. doi: 10.1038/nnano.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bell NA, Engst CR, Ablay M, Divitini G, Ducati C, Liedl T, Keyser UF. Nano Lett. 2012;12:512–517. doi: 10.1021/nl204098n. [DOI] [PubMed] [Google Scholar]
- 83.Geng J, Kim K, Zhang J, Escalada A, Tunuguntla R, Comolli LR, Allen FI, Shnyrova AV, Cho KR, Munoz D, Wang YM, Grigoropoulos CP, Ajo-Franklin CP, Frolov VA, Noy A. Nature. 2014;514:612–615. doi: 10.1038/nature13817. [DOI] [PubMed] [Google Scholar]
- 84.Gopfrich K, Li C-Y, Mames I, Bhamidimarri SP, Ricci M, Yoo J, Mames A, Ohmann A, Winterhalter M, Stulz E, Aksimentiev A, Keyser UF. Nano Lett. 2016;16:4665–4669. doi: 10.1021/acs.nanolett.6b02039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gopfrich K, Li C-Y, Ricci M, Bhamidimarri SP, Yoo J, Gyenes B, Ohmann A, Winterhalter M, Aksimentiev A, Keyser UF. ACS Nano. 2016;10:8207–8214. doi: 10.1021/acsnano.6b03759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gopfrich K, Zettl T, Meijering AEC, Hernandez-Ainsa S, Kocabey S, Liedl T, Keyser UF. Nano Lett. 2015;15:3134–3138. doi: 10.1021/acs.nanolett.5b00189. [DOI] [PubMed] [Google Scholar]
- 87.Burns JR, Seifert A, Fertig N, Howorka S. Nat. Nanotechnol. 2016;11:152–156. doi: 10.1038/nnano.2015.279. [DOI] [PubMed] [Google Scholar]
- 88.Seifert A, Gopfrich K, Burns JR, Fertig N, Keyser UF, Howorka S. ACS Nano. 2015;9:1117–1126. doi: 10.1021/nn5039433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coulter W. Means for counting particles suspended in a fluid. U.S. Patent US2656508 A. 1953 [Google Scholar]
- 90.Wei C, Bard AJ, Feldberg SW. Anal. Chem. 1997;69:4627–4633. [Google Scholar]
- 91.Umehara S, Pourmand N, Webb CD, Davis RW, Yasuda K, Karhanek M. Nano Lett. 2006;6:2486–2492. doi: 10.1021/nl061681k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.White HS, Bund A. Langmuir. 2008;24:2212–2218. doi: 10.1021/la702955k. [DOI] [PubMed] [Google Scholar]
- 93.Momotenko D, Cortes-Salazar F, Josserand J, Liu S, Shao Y, Girault HH. Phys. Chem. Chem. Phys. 2011;13:5430–5440. doi: 10.1039/c0cp02595j. [DOI] [PubMed] [Google Scholar]
- 94.Ohshiro T, Tsutsui M, Yokota K, Furuhashi M, Taniguchi M, Kawai T. Nat. Nanotechnol. 2014;9:835–840. doi: 10.1038/nnano.2014.193. [DOI] [PubMed] [Google Scholar]
- 95.Tsutsui M, Taniguchi M, Yokota K, Kawai T. Nat. Nanotechnol. 2010;5:286–290. doi: 10.1038/nnano.2010.42. [DOI] [PubMed] [Google Scholar]
- 96.Ivanov AP, Instuli E, McGilvery CM, Baldwin G, McComb DW, Albrecht T, Edel JB. Nano Lett. 2011;11:279–285. doi: 10.1021/nl103873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sawafta F, Clancy B, Carlsen AT, Huber M, Hall AR. Nanoscale. 2014;6:6991–6996. doi: 10.1039/c4nr00305e. [DOI] [PubMed] [Google Scholar]
- 98.Huang S, Romero-Ruiz M, Castell OK, Bayley H, Wallace MI. Nat. Nanotechnol. 2015;10:986–991. doi: 10.1038/nnano.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu S, Wall TA, Ozcelik D, Parks JW, Hawkins AR, Schmidt H. Chem. Commun. 2015;51:2084–2087. doi: 10.1039/c4cc07591a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jonsson MP, Dekker C. Nano Lett. 2013;13:1029–1033. doi: 10.1021/nl304213s. [DOI] [PubMed] [Google Scholar]
- 101.Crick CR, Albella P, Ng B, Ivanov AP, Roschuk T, Cecchini MP, Bresme F, Maier SA, Edel JB. Nano Lett. 2015;15:553–559. doi: 10.1021/nl504536j. [DOI] [PubMed] [Google Scholar]
- 102.Li Y, Nicoli F, Chen C, Lagae L, Groeseneken G, Stakenborg T, Zandbergen HW, Dekker C, Van Dorpe P, Jonsson MP. Nano Lett. 2015;15:776–782. doi: 10.1021/nl504516d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nicoli F, Verschueren D, Klein M, Dekker C, Jonsson MP. Nano Lett. 2014;14:6917–6925. doi: 10.1021/nl503034j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Belkin M, Chao S-H, Jonsson MP, Dekker C, Aksimentiev A. ACS Nano. 2015;9:10598–10611. doi: 10.1021/acsnano.5b04173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gilboa T, Meller A. Analyst. 2015;140:4733–4747. doi: 10.1039/c4an02388a. [DOI] [PubMed] [Google Scholar]
- 106.Bayley H. Curr. Opin. Biotechnol. 1999;10:94–103. doi: 10.1016/s0958-1669(99)80017-2. [DOI] [PubMed] [Google Scholar]
- 107.Ayub M, Bayley H. Curr. Opin. Chem. Biol. 2016;34:117–126. doi: 10.1016/j.cbpa.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 109.Niederweis M. Mol. Microbiol. 2003;49:1167–1177. doi: 10.1046/j.1365-2958.2003.03662.x. [DOI] [PubMed] [Google Scholar]
- 110.Faller M, Niederweis M, Schulz GE. Science. 2004;303:1189–1192. doi: 10.1126/science.1094114. [DOI] [PubMed] [Google Scholar]
- 111.Parker MW, Buckley JT, Postma JP, Tucker AD, Leonard K, Pattus F, Tsernoglou D. Nature. 1994;367:292–295. doi: 10.1038/367292a0. [DOI] [PubMed] [Google Scholar]
- 112.Degiacomi MT, Iacovache I, Pernot L, Chami M, Kudryashev M, Stahlberg H, van der Goot FG, Dal Peraro M. Nat. Chem. Biol. 2013;9:623–629. doi: 10.1038/nchembio.1312. [DOI] [PubMed] [Google Scholar]
- 113.Janda JM, Abbott SL. Clin. Infect. Dis. 1998;27:332–344. doi: 10.1086/514652. [DOI] [PubMed] [Google Scholar]
- 114.Valpuesta JM, Carrascosa JLQ. Rev. Biophys. 1994;27:107–155. doi: 10.1017/s0033583500004510. [DOI] [PubMed] [Google Scholar]
- 115.Guasch A, Pous J, Ibarra B, Gomis-Ruth FX, Valpuesta JMa, Sousa N, Carrascosa JL, Coll M. J. Mol. Biol. 2002;315:663–676. doi: 10.1006/jmbi.2001.5278. [DOI] [PubMed] [Google Scholar]
- 116.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. Nature. 2009;459:726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- 117.Libby SJ, Goebel W, Ludwig A, Buchmeier N, Bowe F, Fang FC, Guiney DG, Songer JG, Heffron F. Proc. Natl. Acad. Sci. U. S. A. 1994;91:489–493. doi: 10.1073/pnas.91.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Subbarao GV, van den Berg B. J. Mol. Biol. 2006;360:750–759. doi: 10.1016/j.jmb.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 119.Watson JD, Crick FH. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 120.Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Pavlenok M, Niederweis M, Gundlach JH. Nat. Biotechnol. 2012;30:349–353. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Craig JM, Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Nova IC, Doering K, Tickman BI, Svet MT, Gundlach JH. PLoS One. 2015;10:e0143253. doi: 10.1371/journal.pone.0143253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Laszlo AH, Derrington IM, Gundlach JH. Methods. 2016;105:75–89. doi: 10.1016/j.ymeth.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Conlan S, Zhang Y, Cheley S, Bayley H. Biochemistry. 2000;39:11845–11854. doi: 10.1021/bi001065h. [DOI] [PubMed] [Google Scholar]
- 124.Chen M, Khalid S, Sansom MS, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6272–6277. doi: 10.1073/pnas.0711561105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fahie MA, Yang B, Pham B, Chen M. ACS Sensors. 2016;1:614–622. doi: 10.1021/acssensors.6b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fahie MA, Chen M. J. Phys. Chem. B. 2015;119:10198–10206. doi: 10.1021/acs.jpcb.5b06435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Franceschini L, Brouns T, Willems K, Carlon E, Maglia G. ACS Nano. 2016;10:8394–8402. doi: 10.1021/acsnano.6b03159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maglia G, Restrepo MR, Mikhailova E, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19720–19725. doi: 10.1073/pnas.0808296105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rincon-Restrepo M, Mikhailova E, Bayley H, Maglia G. Nano Lett. 2011;11:746–750. doi: 10.1021/nl1038874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wendell D, Jing P, Geng J, Subramaniam V, Lee TJ, Montemagno C, Guo P. Nat. Nanotechnol. 2009;4:765–772. doi: 10.1038/nnano.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Geng J, Wang S, Fang H, Guo P. ACS Nano. 2013;7:3315–3323. doi: 10.1021/nn400020z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Soskine M, Biesemans A, Moeyaert B, Cheley S, Bayley H, Maglia G. Nano Lett. 2012;12:4895–4900. doi: 10.1021/nl3024438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Franceschini L, Soskine M, Biesemans A, Maglia G. Nat. Commun. 2013;4:2415–2422. doi: 10.1038/ncomms3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sauer-Budge AF, Nyamwanda JA, Lubensky DK, Branton D. Phys. Rev. Lett. 2003;90:238101. doi: 10.1103/PhysRevLett.90.238101. [DOI] [PubMed] [Google Scholar]
- 135.Angevine CE, Seashols-Williams SJ, Reiner JE. Anal. Chem. 2016;88:2645–2651. doi: 10.1021/acs.analchem.5b03631. [DOI] [PubMed] [Google Scholar]
- 136.Johnson RP, Fleming AM, Burrows CJ, White HS. J. Phys. Chem. Lett. 2014;5:3781–3786. doi: 10.1021/jz502030e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Johnson RP, Fleming AM, Jin Q, Burrows CJ, White HS. Biophys. J. 2014;107:924–931. doi: 10.1016/j.bpj.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jin Q, Fleming AM, Johnson RP, Ding Y, Burrows CJ, White HS. J. Am. Chem. Soc. 2013;135:19347–19353. doi: 10.1021/ja410615d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ding Y, Fleming AM, White HS, Burrows C. J. ACS Nano. 2015;9:11325–11332. doi: 10.1021/acsnano.5b05055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johnson RP, Fleming AM, Beuth LR, Burrows CJ, White HS. J. Am. Chem. Soc. 2016;138:594–603. doi: 10.1021/jacs.5b10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Johnson RP, Perera RT, Fleming AM, Burrows CJ, White HS. Faraday Discuss. 2016 doi: 10.1039/c6fd00058d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou S, Wang L, Chen X, Guan X. ACS Sensors. 2016;1:607–613. doi: 10.1021/acssensors.6b00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nivala J, Marks DB, Akeson M. Nat. Biotechnol. 2013;31:247–250. doi: 10.1038/nbt.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pastoriza-Gallego M, Rabah L, Gibrat G, Thiebot B, van der Goot FG, Auvray L, Betton J-M, Pelta J. J. Am. Chem. Soc. 2011;133:2923–2931. doi: 10.1021/ja1073245. [DOI] [PubMed] [Google Scholar]
- 145.Cressiot B, Braselmann E, Oukhaled A, Elcock AH, Pelta J, Clark PL. ACS Nano. 2015;9:9050–9061. doi: 10.1021/acsnano.5b03053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ji Z, Wang S, Zhao Z, Zhou Z, Haque F, Guo P. Small. 2016;12:4572–4578. doi: 10.1002/smll.201601157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Soskine M, Biesemans A, De Maeyer M, Maglia G. J. Am. Chem. Soc. 2013;135:13456–13463. doi: 10.1021/ja4053398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Campos EJ, McVey CE, Astier Y. Anal. Chem. 2016;88:6214–6222. doi: 10.1021/acs.analchem.5b03558. [DOI] [PubMed] [Google Scholar]
- 149.Li T, Liu L, Li Y, Xie J, Wu HC. Angew. Chem., Int. Ed. 2015;54:7568–7571. doi: 10.1002/anie.201502047. [DOI] [PubMed] [Google Scholar]
- 150.Krasilnikov OV, Rodrigues CG, Bezrukov SM. Phys. Rev. Lett. 2006;97:018301. doi: 10.1103/PhysRevLett.97.018301. [DOI] [PubMed] [Google Scholar]
- 151.Baaken G, Halimeh I, Bacri L, Pelta J, Oukhaled A, Behrends JC. ACS Nano. 2015;9:6443–6449. doi: 10.1021/acsnano.5b02096. [DOI] [PubMed] [Google Scholar]
- 152.Lee J, Bayley H. Proc. Natl. Acad. Sci. U. S. A. 2015;112:13768–13773. doi: 10.1073/pnas.1510565112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pulcu GS, Mikhailova E, Choi L-S, Bayley H. Nat. Nanotechnol. 2015;10:76–83. doi: 10.1038/nnano.2014.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Derrington IM, Craig JM, Stava E, Laszlo AH, Ross BC, Brinkerhoff H, Nova IC, Doering K, Tickman BI, Ronaghi M, Mandell JG, Gunderson KL, Gundlach JH. Nat. Biotechnol. 2015;33:1073–1075. doi: 10.1038/nbt.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC, Craig JM, Langford KW, Samson JM, Daza R, Doering K, Shendure J, Gundlach JH. Nat. Biotechnol. 2014;32:829–833. doi: 10.1038/nbt.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Bartlett JJ, Pavlenok M, Gundlach JH. Proc. Natl. Acad. Sci. U. S. A. 2013;110:18904–18909. doi: 10.1073/pnas.1310240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kumar S, Tao C, Chien M, Hellner B, Balijepalli A, Robertson JWF, Li Z, Russo JJ, Reiner JE, Kasianowicz JJ, Ju J. Sci. Rep. 2012;2:684. doi: 10.1038/srep00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang B, Galusha J, Shiozawa PG, Wang G, Bergren AJ, Jones RM, White RJ, Ervin EN, Cauley CC, White HS. Anal. Chem. 2007;79:4778–4787. doi: 10.1021/ac070609j. [DOI] [PubMed] [Google Scholar]
- 159.Losic D, Santos A. Nanoporous alumina: fabrication, structure, properties and applications. Vol. 219. New York: Springer; 2015. [Google Scholar]
- 160.Morris CA, Friedman AK, Baker LA. Analyst. 2010;135:2190–2202. doi: 10.1039/c0an00156b. [DOI] [PubMed] [Google Scholar]
- 161.Takami T, Park BH, Kawai T. Nano convergence. 2014;1:1–12. doi: 10.1186/s40580-014-0017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haywood DG, Saha-Shah A, Baker LA, Jacobson SC. Anal. Chem. 2015;87:172–187. doi: 10.1021/ac504180h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kwok H, Briggs K, Tabard-Cossa V. PLoS One. 2014;9:e92880. doi: 10.1371/journal.pone.0092880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Briggs K, Kwok H, Tabard-Cossa V. Small. 2014;10:2077–2086. doi: 10.1002/smll.201303602. [DOI] [PubMed] [Google Scholar]
- 165.Kowalczyk SW, Grosberg AY, Rabin Y, Dekker C. Nanotechnology. 2011;22:315101. doi: 10.1088/0957-4484/22/31/315101. [DOI] [PubMed] [Google Scholar]
- 166.Kuan AT, Lu B, Xie P, Szalay T, Golovchenko JA. Appl. Phys. Lett. 2015;106:203109. doi: 10.1063/1.4921620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Feng J, Liu K, Graf M, Lihter M, Bulushev RD, Dumcenco D, Alexander DTL, Krasnozhon D, Vuletic T, Kis A, Radenovic A. Nano Lett. 2015;15:3431–3438. doi: 10.1021/acs.nanolett.5b00768. [DOI] [PubMed] [Google Scholar]
- 168.Crick CR, Sze JY, Rosillo-Lopez M, Salzmann CG, Edel JB. ACS Appl. Mater. Interfaces. 2015;7:18188–18194. doi: 10.1021/acsami.5b06212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schneider GF, Xu Q, Hage S, Luik S, Spoor JNH, Malladi S, Zandbergen H, Dekker C. Nat. Commun. 2013;4:2619. doi: 10.1038/ncomms3619. [DOI] [PubMed] [Google Scholar]
- 170.Banerjee S, Wilson J, Shim J, Shankla M, Corbin EA, Aksimentiev A, Bashir R. Adv. Funct. Mater. 2015;25:936–946. doi: 10.1002/adfm.201403719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Postma HWC. Nano Lett. 2010;10:420–425. doi: 10.1021/nl9029237. [DOI] [PubMed] [Google Scholar]
- 172.Traversi F, Raillon C, Benameur SM, Liu K, Khlybov S, Tosun M, Krasnozhon D, Kis A, Radenovic A. Nat. Nanotechnol. 2013;8:939–945. doi: 10.1038/nnano.2013.240. [DOI] [PubMed] [Google Scholar]
- 173.Feng J, Liu K, Bulushev RD, Khlybov S, Dumcenco D, Kis A, Radenovic A. Nat. Nanotechnol. 2015;10:1070–1076. doi: 10.1038/nnano.2015.219. [DOI] [PubMed] [Google Scholar]
- 174.Heerema SJ, Dekker C. Nat. Nanotechnol. 2016;11:127–136. doi: 10.1038/nnano.2015.307. [DOI] [PubMed] [Google Scholar]
- 175.Ferrari AC, Bonaccorso F, Fal’ko V, Novoselov KS, Roche S, Boggild P, Borini S, Koppens FHL, Palermo V, Pugno N, Garrido JA, Sordan R, Bianco A, Ballerini L, Prato M, Lidorikis E, Kivioja J, Marinelli C, Ryhanen T, Morpurgo A, Coleman JN, Nicolosi V, Colombo L, Fert A, Garcia-Hernandez M, Bachtold A, Schneider GF, Guinea F, Dekker C, Barbone M, Sun Z, Galiotis C, Grigorenko AN, Konstantatos G, Kis A, Katsnelson M, Vandersypen L, Loiseau A, Morandi V, Neumaier D, Treossi E, Pellegrini V, Polini M, Tredicucci A, Williams GM, Hee Hong B, Ahn J-H, Min Kim J, Zirath H, van Wees BJ, van der Zant H, Occhipinti L, Di Matteo A, Kinloch IA, Seyller T, Quesnel E, Feng X, Teo K, Rupesinghe N, Hakonen P, Neil SRT, Tannock Q, Lofwander T, Kinaret J. Nanoscale. 2015;7:4598–4810. doi: 10.1039/c4nr01600a. [DOI] [PubMed] [Google Scholar]
- 176.Bell NA, Muthukumar M, Keyser UF. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2016;93:022401. doi: 10.1103/PhysRevE.93.022401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Verschueren DV, Jonsson M. P., Dekker, C. Nanotechnology. 2015;26:234004. doi: 10.1088/0957-4484/26/23/234004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Galla L, Meyer AJ, Spiering A, Sischka A, Mayer M, Hall AR, Reimann P, Anselmetti D. Nano Lett. 2014;14:4176–4182. doi: 10.1021/nl501909t. [DOI] [PubMed] [Google Scholar]
- 179.Sischka A, Galla L, Meyer AJ, Spiering A, Knust S, Mayer M, Hall AR, Beyer A, Reimann P, Golzhauser A, Anselmetti D. Analyst. 2015;140:4843–4847. doi: 10.1039/c4an02319f. [DOI] [PubMed] [Google Scholar]
- 180.Keyser UF, Koeleman BN, van Dorp S, Krapf D, Smeets RMM, Lemay SG, Dekker NH, Dekker C. Nat. Phys. 2006;2:473–477. [Google Scholar]
- 181.Carlsen AT, Zahid OK, Ruzicka J, Taylor EW, Hall AR. ACS Nano. 2014;8:4754–4760. doi: 10.1021/nn501694n. [DOI] [PubMed] [Google Scholar]
- 182.Kowalczyk SW, Dekker C. Nano Lett. 2012;12:4159–4163. doi: 10.1021/nl301719a. [DOI] [PubMed] [Google Scholar]
- 183.Smeets RMM, Keyser UF, Krapf D, Wu M-Y, Dekker NH, Dekker C. Nano Lett. 2006;6:89–95. doi: 10.1021/nl052107w. [DOI] [PubMed] [Google Scholar]
- 184.Wanunu M, Morrison W, Rabin Y, Grosberg AY, Meller A. Nat. Nanotechnol. 2010;5:160–165. doi: 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wanunu M, Sutin J, McNally B, Chow A, Meller A. Biophys. J. 2008;95:4716–4725. doi: 10.1529/biophysj.108.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Carson S, Wilson J, Aksimentiev A, Wanunu M. Biophys. J. 2014;107:2381–2393. doi: 10.1016/j.bpj.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Plesa C, van Loo N, Ketterer P, Dietz H, Dekker C. Nano Lett. 2015;15:732–737. doi: 10.1021/nl504375c. [DOI] [PubMed] [Google Scholar]
- 188.Carson S, Wick ST, Carr PA, Wanunu M, Aguilar CA. ACS Nano. 2015;9:12417–12424. doi: 10.1021/acsnano.5b05782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Plesa C, Verschueren D, Pud S, van der Torre J, Ruitenberg JW, Witteveen MJ, Jonsson MP, Grosberg AY, Rabin Y, Dekker C. Nat. Nanotechnol. 2016 doi: 10.1038/nnano.2016.153. [DOI] [PubMed] [Google Scholar]
- 190.Plesa C, van Loo N, Dekker C. Nanoscale. 2015;7:13605–13609. doi: 10.1039/c5nr02793d. [DOI] [PubMed] [Google Scholar]
- 191.Carlsen AT, Zahid OK, Ruzicka JA, Taylor EW, Hall AR. Nano Lett. 2014;14:5488–5492. doi: 10.1021/nl501340d. [DOI] [PubMed] [Google Scholar]
- 192.Plesa C, Kowalczyk SW, Zinsmeester R, Grosberg AY, Rabin Y, Dekker C. Nano Lett. 2013;13:658–663. doi: 10.1021/nl3042678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pedone D, Firnkes M, Rant U. Anal. Chem. 2009;81:9689–9694. doi: 10.1021/ac901877z. [DOI] [PubMed] [Google Scholar]
- 194.Li W, Bell NAW, Hernandez-Ainsa S, Thacker VV, Thackray AM, Bujdoso R, Keyser UF. ACS Nano. 2013;7:4129–4134. doi: 10.1021/nn4004567. [DOI] [PubMed] [Google Scholar]
- 195.Larkin J, Henley RY, Muthukumar M, Rosenstein JK, Wanunu M. Biophys. J. 2014;106:696–704. doi: 10.1016/j.bpj.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nir I, Huttner D, Meller A. Biophys. J. 2015;108:2340–2349. doi: 10.1016/j.bpj.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yusko EC, Johnson JM, Majd S, Prangkio P, Rollings RC, Li J, Yang J, Mayer M. Nat. Nanotechnol. 2011;6:253–260. doi: 10.1038/nnano.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wei R, Gatterdam V, Wieneke R, Tampe R, Rant U. Nat. Nanotechnol. 2012;7:257–263. doi: 10.1038/nnano.2012.24. [DOI] [PubMed] [Google Scholar]
- 199.Plesa C, Ruitenberg JW, Witteveen MJ, Dekker C. Nano Lett. 2015;15:3153–3158. doi: 10.1021/acs.nanolett.5b00249. [DOI] [PubMed] [Google Scholar]
- 200.Yu J-S, Lim M-C, Huynh DTN, Kim H-J, Kim H-M, Kim Y-R, Kim K-B. ACS Nano. 2015;9:5289–5298. doi: 10.1021/acsnano.5b00784. [DOI] [PubMed] [Google Scholar]
- 201.Squires A, Atas E, Meller A. Sci. Rep. 2015;5:11643. doi: 10.1038/srep11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bell NAW, Keyser UF. J. Am. Chem. Soc. 2015;137:2035–2041. doi: 10.1021/ja512521w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kong J, Bell NAW, Keyser UF. Nano Lett. 2016;16:3557–3562. doi: 10.1021/acs.nanolett.6b00627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bell NAW, Keyser UF. Nat. Nanotechnol. 2016;11:645–651. doi: 10.1038/nnano.2016.50. [DOI] [PubMed] [Google Scholar]
- 205.Liu X, Skanata MM, Stein D. Nat. Commun. 2015;6:6222. doi: 10.1038/ncomms7222. [DOI] [PubMed] [Google Scholar]
- 206.Marshall MM, Ruzicka J, Zahid OK, Henrich VC, Taylor EW, Hall AR. Langmuir. 2015;31:4582–4588. doi: 10.1021/acs.langmuir.5b00457. [DOI] [PubMed] [Google Scholar]
- 207.Japrung D, Bahrami A, Nadzeyka A, Peto L, Bauerdick S, Edel JB, Albrecht T. J. Phys. Chem. B. 2014;118:11605–11612. doi: 10.1021/jp506832u. [DOI] [PubMed] [Google Scholar]
- 208.Ivanov AP, Instuli E, McGilvery CM, Baldwin G, McComb DW, Albrecht T, Edel JB. Nano Lett. 2011;11:279–285. doi: 10.1021/nl103873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hansma P, Drake B, Marti O, Gould S, Prater C. Science. 1989;243:641–643. doi: 10.1126/science.2464851. [DOI] [PubMed] [Google Scholar]
- 210.Bard AJ, Fan FRF, Kwak J, Lev O. Anal. Chem. 1989;61:132–138. [Google Scholar]
- 211.Chen C-C, Zhou Y, Baker LA. Annu. Rev. Anal. Chem. 2012;5:207–228. doi: 10.1146/annurev-anchem-062011-143203. [DOI] [PubMed] [Google Scholar]
- 212.Amemiya S, Bard AJ, Fan F-RF, Mirkin MV, Unwin PR. Annu. Rev. Anal. Chem. 2008;1:95–131. doi: 10.1146/annurev.anchem.1.031207.112938. [DOI] [PubMed] [Google Scholar]
- 213.Bard AJ, Mirkin MV. Scanning electrochemical microscopy. Boca Raton, FL: CRC Press; 2012. [Google Scholar]
- 214.Seifert J, Rheinlaender J, Novak P, Korchev YE, Schaffer TE. Langmuir. 2015;31:6807–6813. doi: 10.1021/acs.langmuir.5b01124. [DOI] [PubMed] [Google Scholar]
- 215.Nashimoto Y, Takahashi Y, Ida H, Matsumae Y, Ino K, Shiku H, Matsue T. Anal. Chem. 2015;87:2542–2545. doi: 10.1021/ac5046388. [DOI] [PubMed] [Google Scholar]
- 216.Takahashi Y, Shevchuk AI, Novak P, Murakami Y, Shiku H, Korchev YE, Matsue T. J. Am. Chem. Soc. 2010;132:10118–10126. doi: 10.1021/ja1029478. [DOI] [PubMed] [Google Scholar]
- 217.Comstock DJ, Elam JW, Pellin MJ, Hersam MC. Anal. Chem. 2010;82:1270–1276. doi: 10.1021/ac902224q. [DOI] [PubMed] [Google Scholar]
- 218.O’Connell MA, Wain A. J. Anal. Chem. 2014;86:12100–12107. doi: 10.1021/ac502946q. [DOI] [PubMed] [Google Scholar]
- 219.O’Connell MA, Lewis JR, Wain A. J. Chem. Commun. 2015;51:10314–10317. doi: 10.1039/c5cc01640a. [DOI] [PubMed] [Google Scholar]
- 220.Sen M, Takahashi Y, Matsumae Y, Horiguchi Y, Kumatani A, Ino K, Shiku H, Matsue T. Anal. Chem. 2015;87:3484–3489. doi: 10.1021/acs.analchem.5b00027. [DOI] [PubMed] [Google Scholar]
- 221.Shi W, Baker LA. RSC Adv. 2015;5:99284–99290. [Google Scholar]
- 222.Momotenko D, McKelvey K, Kang M, Meloni GN, Unwin PR. Anal. Chem. 2016;88:2838–2846. doi: 10.1021/acs.analchem.5b04566. [DOI] [PubMed] [Google Scholar]
- 223.Chen C-C, Zhou Y, Morris CA, Hou J, Baker LA. Anal. Chem. 2013;85:3621–3628. doi: 10.1021/ac303441n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhou Y, Chen C-C, Weber AE, Zhou L, Baker LA, Hou J. Tissue barriers. 2013;1:e25585. doi: 10.4161/tisb.25585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhou L, Gong Y, Sunq A, Hou J, Baker LA. Anal. Chem. 2016;88:9630–9637. doi: 10.1021/acs.analchem.6b02392. [DOI] [PubMed] [Google Scholar]
- 226.Sa N, Baker LA. J. Am. Chem. Soc. 2011;133:10398–10401. doi: 10.1021/ja203883q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sa N, Lan W-J, Shi W, Baker LA. ACS Nano. 2013;7:11272–11282. doi: 10.1021/nn4050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Plett T, Shi W, Zeng Y, Mann W, Vlassiouk I, Baker LA, Siwy ZS. Nanoscale. 2015;7:19080–19091. doi: 10.1039/c5nr06340j. [DOI] [PubMed] [Google Scholar]
- 229.McKelvey K, Perry D, Byers JC, Colburn AW, Unwin PR. Anal. Chem. 2014;86:3639–3646. doi: 10.1021/ac5003118. [DOI] [PubMed] [Google Scholar]
- 230.Perry D, Al Botros R, Momotenko D, Kinnear SL, Unwin PR. ACS Nano. 2015;9:7266–7276. doi: 10.1021/acsnano.5b02095. [DOI] [PubMed] [Google Scholar]
- 231.Perry D, Paulose Nadappuram B, Momotenko D, Voyias PD, Page A, Tripathi G, Frenguelli BG, Unwin PR. J. Am. Chem. Soc. 2016;138:3152–3160. doi: 10.1021/jacs.5b13153. [DOI] [PubMed] [Google Scholar]
- 232.Wang Y, Shan X, Wang S, Tao N, Blanchard P-Y, Hu K, Mirkin MV. Anal. Chem. 2016;88:1547–1552. doi: 10.1021/acs.analchem.5b04382. [DOI] [PubMed] [Google Scholar]
- 233.Babakinejad B, Jonsson P, Lopez Cordoba A, Actis P, Novak P, Takahashi Y, Shevchuk A, Anand U, Anand P, Drews A, Ferrer-Montiel A, Klenerman D, Korchev YE. Anal. Chem. 2013;85:9333–9342. doi: 10.1021/ac4021769. [DOI] [PubMed] [Google Scholar]
- 234.Ivanov AP, Actis P, Jonsson P, Klenerman D, Korchev Y, Edel JB. ACS Nano. 2015;9:3587–3595. doi: 10.1021/acsnano.5b00911. [DOI] [PubMed] [Google Scholar]
- 235.Wang Y, Cai H, Mirkin MV. Chem Electro Chem. 2015;2:343–347. [Google Scholar]
- 236.Nashimoto Y, Takahashi Y, Zhou Y, Ito H, Ida H, Ino K, Matsue T, Shiku H. ACS Nano. 2016;10:6915–6922. doi: 10.1021/acsnano.6b02753. [DOI] [PubMed] [Google Scholar]
- 237.Saha-Shah A, Green CM, Abraham DH, Baker LA. Analyst. 2016;141:1958–1965. doi: 10.1039/c6an00028b. [DOI] [PubMed] [Google Scholar]
- 238.Saha-Shah A, Weber AE, Karty JA, Ray SJ, Hieftje GM, Baker LA. Chem. Sci. 2015;6:3334–3341. doi: 10.1039/c5sc00668f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Momotenko D, Page A, Adobes-Vidal M, Unwin PR. ACS Nano. 2016;10:8871–8878. doi: 10.1021/acsnano.6b04761. [DOI] [PubMed] [Google Scholar]
- 240.Li J, Gershow M, Stein D, Brandin E, Golovchenko JA. Nat. Mater. 2003;2:611–615. doi: 10.1038/nmat965. [DOI] [PubMed] [Google Scholar]
- 241.Kowalczyk SW, Wells DB, Aksimentiev A, Dekker C. Nano Lett. 2012;12:1038–1044. doi: 10.1021/nl204273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Carson S, Wanunu M. Nanotechnology. 2015;26:074004. doi: 10.1088/0957-4484/26/7/074004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Pud S, Verschueren D, Vukovic N, Plesa C, Jonsson MP, Dekker C. Nano Lett. 2015;15:7112–7117. doi: 10.1021/acs.nanolett.5b03239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Langer J, Novikov SM, Liz-Marzan LM. Nanotechnology. 2015;26:322001. doi: 10.1088/0957-4484/26/32/322001. [DOI] [PubMed] [Google Scholar]
- 245.Nam S, Choi I, Fu C-c, Kim K, Hong S, Choi Y, Zettl A, Lee LP. Nano Lett. 2014;14:5584–5589. doi: 10.1021/nl503159d. [DOI] [PubMed] [Google Scholar]
- 246.Li Y, Nicoli F, Chen C, Lagae L, Groeseneken G, Stakenborg T, Zandbergen HW, Dekker C, Van Dorpe P, Jonsson MP. Nano Lett. 2015;15:776–782. doi: 10.1021/nl504516d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rothemund PWK. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 248.Hernandez-Ainsa S, Keyser UF. Nanoscale. 2014;6:14121–14132. doi: 10.1039/c4nr04094e. [DOI] [PubMed] [Google Scholar]
- 249.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]