

Abstract
Phenylacetaldehyde dehydrogenase catalyzes the NAD+-dependent oxidation of phenylactealdehyde to phenylacetic acid in the styrene catabolic and detoxification pathway of Pseudomonas putida (S12). Here we report the structure and mechanistic properties of the N-termininally histidine-tagged enzyme, NPADH. The 2.83Å X-ray crystal structure is similar in fold to sheep liver cytosolic aldehyde dehydrogenase (ALDH1), but has unique set of intersubunit interactions and active site tunnel for substrate entrance. In solution, NPADH occurs as 227 kDa homotetramer. It follows a sequential reaction mechanism in which NAD+ serves as both the leading substrate and homotropic allosteric activator. In the absence of styrene monooxygenase reductase, which regenerates NAD+ from NADH in the first step of styrene catabolism, NPADH is inhibited by a ternary complex involving NADH, product, and phenylacetaldehyde, substrate. Each oligomerization domain of NPADH contains a six-residue insertion that extends this loop over the substrate entrance tunnel of a neighboring subunit, thereby obstructing the active site of the adjacent subunit. This feature could be an important factor in the homotropic activation and product inhibition mechanisms. Compared to ALDH1, the substrate channel of NPADH is narrower and lined with more aromatic residues, suggesting a means for enhancing substrate specificity.
Keywords: styrene monooxygnease, phenylacetaldehyde dehydrogenase, structure, kinetics
Graphical Abstract
Introduction
Styrene is an essential component in the industrial synthesis of plastics, resins, rubbers, and a myriad other polymers. In 2010, approximately 3.75 billion kilograms of styrene were produced in the United States alone [1]. Despite its prevalent industrial utilization, styrene is a classified mutagen, a suspected carcinogen, and considered to be “immediately dangerous to life or health” by the National Institute for Occupational Safety and Health [2]. Moreover, styrene oxide, the immediate degradation product of styrene in the human liver, is a known carcinogen and considered a greater threat than styrene [3]. Together, styrene and highly reactive styrene oxide have been implicated as reproductive toxicants, neurotoxicants, and/or carcinogens [4]. The harmful properties and the emissions and effluents that inevitably accompany large scale production make the efficient removal of styrene from the environment not just desirable, but imperative to ensure public safety. The commercial value of styrene as a synthetic building block juxtaposed with issues of human and environmental toxicology have spurred structural and mechanistic studies of the enzymes involved in microbial styrene metabolism. In addition to the value of the intact styrene degradation pathway for environmental remediation efforts, its component enzymes offer unique biotechnological potential for the synthesis of chiral oxides, aldehydes, acids, and their derivatives.
Pseudomonas bacteria, are equipped with a genomically-encoded styrene catabolon and represent one of the most versatile organisms engaged in the oxidative metabolism of styrene and related aromatic and aliphatic hydrocarbons [5, 6]. In our work we have focused on enzymes from the styrene metabolic pathway of P. putida (S12), an unusually solvent tolerant strain. At the entry point of the styrene pathway, the NADH and FAD–dependent two-component styrene monooxygenase (SMO) transfers an atom from molecular oxygen to the vinyl side chain of styrene to synthesize styrene oxide [7]. This is followed by the transformation of styrene oxide to phenylacetaldehyde (PAL) by the membrane protein, styrene oxide isomerase (SOI) and the NAD+-dependent oxidation of PAL to phenylacetic acid (PAA) by phenylacetaldehyde dehydrogenase (PADH) [8]. Phenylacetate enters the central metabolic pathways upon its transformation into acetyl-CoA and succinyl-CoA through a series of reactions shared in common with phenylalanine catabolism (Figure 1) [9].
Figure 1.
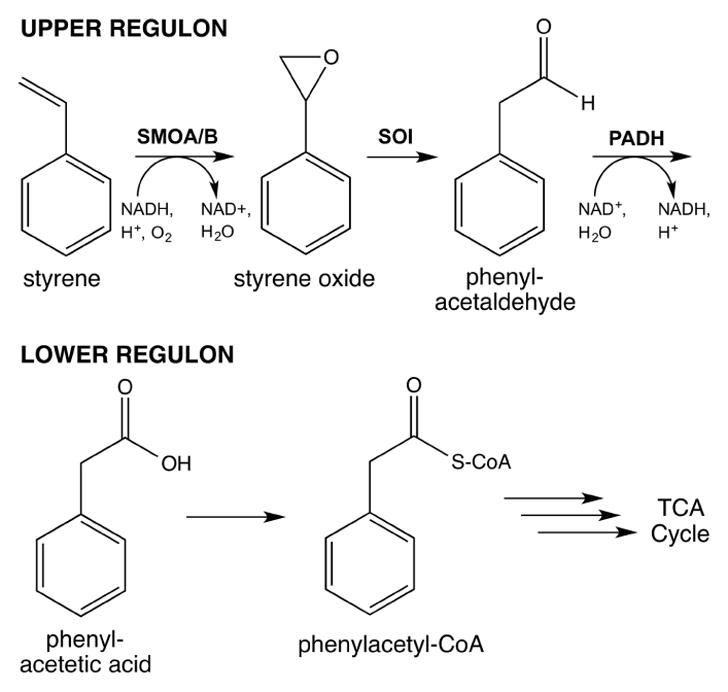
The Styrene Catabolic Pathway of P. putida.
As part of our continuing work to understand microbial styrene catabolism, we investigated the structure and mechanism of the phenylacetaldehyde dehydrogenase. In general, aldehyde dehydrogenases (ALDHs) are a structurally and mechanistically conserved family of enzymes that catalyze the oxidation of aldehydes to carboxylic acids [10, 11] and are distributed ubiquitously across all life kingdoms where they have integral biosynthetic functions ranging from the synthesis of retinoic acid from vitamin A in developmental biology [12] to the production of the lignin, a structural support polymer of plant materials [13]. ALDHs. Pyridine nucleotide-dependent ALDHs, categorized as either Class I, II or III depending on their primary sequence, share similar active site structures but differ in pyridine nucleotide and aldehyde substrate specificity, metal ion-dependence, and quaternary structure assembly [14]. Class-I and II ALDHs assemble as homotetramers and their catalytic activity is dependent on metal ions [14, 15]. Class-III enzymes assemble as homodimers and are catalytically insensitive to divalent metal ions [16].
Microbial aldehyde dehydrogenases, which are structurally and mechanistically similar to the human enzymes, are often integrated into pathways for the oxidative transformation of hydrocarbons, alcohols, amines, and amino acids into biosynthetic carbon and energy [17, 18]. In the commercial syntheisis of organic acid derivatives these enzymes represent valuable biocatalysts. In the present work we characterize the structure and mechanism of the N-terminally histidine-tagged phenylacetaldehyde dehydrogenase from the styrene catabolic pathway (NPADH) and evaluate the ability of styrene monooxygenase reductase to relieve NADH (product) inhibition and increase the catalytic efficiency of NPADH.
Materials and Methods
Cloning and Expression System
QIAGEN Genomic-tips (100G) were used to isolate genomic DNA extracted from liquid cultures of Pseudomonas putida (S12) per the manufacturers directions. Genomic DNA was concentrated and desalted by isopropanol precipitation. Primers were designed based on the reported DNA sequence of the StyD from Pseudomonas sp. (Y2) [19]. 5′-phosphorylated primers (forward: 5′-P-CCATATGAACAGTTCTCTTTCCGCA-3′ and 5′-P-reverse: AAGTTTTCATTGTTGTATCTCGCGTAA-3′) were synthesized by Invitrogen, designed for use with PfuTurbo® DNA polymerase (Tm = 72 °C) and included a 5′-NdeI restriction site. The styD gene was amplified from 100 ng of purified genomic DNA through 30-cycles of PCR in Pfu buffer containing 2.5 mM of each dNTP, and 100 ng of each primer. Next the gene was inserted into a pZeRO-2 (Invitrogen) vector that was blunt end cut with EcoRV and subsequently ligated using T4 DNA ligase. E.coli Top10 cells where then transformed with the plasmid by electroporation using a BioRad MicroPulser. Cells containing the recombinant pZeRO-2 plasmid with the styD insert were propagated, recovered by using a Qiagen Miniprep plasmid purification kit and sequence verified by Eurofins MWG Operon. The primary sequence of P. putida (S12) found to be identical to that of styD from P. florescens except for amino acid position 63 of the P. putida sequence, where an alanine is substituted for glycine.
Before transferring the gene to a pET-28b expression vector, a single Nde1 site located in the cloned gene was eliminated by using the Statagene Quick Change Mutagenesis protocol with the following primer and its complement: 5′-GCAAGGCGTCCACATGGGCCCCATGC-3′. DNA sequencing confirmed a T to C substitution and elimination of the internal Nde1 site (CATATG) in styD while preserving the histidine codon.
After removal of the Nde1 site, recombinant styD was excised from pZeRO-2 by double digestion with Nde1 and Xho1 and recovered after purification by agarose gel electrophoresis. A pET-28b plasmid from Stratagene, double digested with Nde1 and Xho1 and treated with calf intestinal phosphatase to prevent competitive recombination with the excised polycloning site, was combined with recombinant styD and ligated by reaction with T4 DNA ligase. The newly generated pET-28 (NPADH) expression vector adds a 20 amino acid His6-tag and thrombin cut site to the PADH N-terminus. Insertion of styD into the expression vector was verified by restriction endonuclease digestion followed by agarose gel electrophoresis. The sequence of the styD gene integrated in this expression vector, pET-28 (NPADH), was verified by DNA sequencing. Competent BL21(DE3) E. coli cells were transformed by heat shock, propagated and stored as stocks in 15% glycerol at −80 °C.
Expression and Purification NPADH
NPADH was expressed and purified by using nickel affinity chromatography following protocols similar to those described previously for the preparation of NSMOA [20]. Starter cultures containing 5 mL of sterile LB medium and 30 μg/mL−1 kanamycin were inoculated with BL21(DE3) E. coli cells containing pET-28(NPADH). After 16 hrs of aerobic growth at 37° C with shaking at 250 rpm, starter cultures were expanded to 6 L of the same medium divided in 6 × 2.5 L Fernbach flasks. Growth was continued until the cells reached an optical density at 600 nm of 2.5 at which point protein expression was induced by addition of 1 mM IPTG. After one hour, cell cultures were harvested using a Sorval Evolution centrifuge by spinning at 6000 rpm for 10 minutes in a GS-3 rotor. Pelleted cells were frozen and stored at −80°C.
Frozen cell pellets (~ 27 g) were thawed in a 100 mL stainless steel beaker with 50 mL of lysis buffer comprised of 50 mM potassium phosphate, pH 7, 10 mM imidazole, 300 mM NaCl and 100 μM EDTA. The protease inhibitor PMSF was not included in this preparation as it inactivates NPADH, presumably by modifying the active site cysteine. The thawed cell suspension was cooled to 4 °C in an ice bucket and disrupted by sonication for a total time of 3 minutes at 20% power with a Branson Sonifier equipped with a 3/4–inch probe. The resulting suspension was spun in the Sorval Evolution centrifuge at 4°C for 30 minutes at 20,000 rpm in an SS-34 rotor.
Using BioRad BioLogic FPLC system, the supernatant was loaded onto a 1.5 × 20 cm Omnifit column containing 25 mL of Sigma His-Select nickel affinity resin pre-equilibrated with a low imidazole buffer (50 mM potassium phosphate, pH 7, 10 mM imidazole and 300 mM NaCl). The column was washed at 3 mL/min with 3 column volumes of low imidazole buffer after which the imidazole concentration was increased to 250 mM over a 50 mL linear gradient with NPADH eluting from the column at ~200 mM imidazole. NPADH-containing fractions were identified based on absorbance at 280 nm. SDS-PAGE was run under both reducing and non-reducing conditions to confirm the presence, purity and molecular weight of the protein. The theoretical molecular weight and ε280 of NPADH were computed by ProtParam to be 54607.3 Da and 56,120 M−1cm−1, respectively [21].
The column fractions were pooled and concentrated to ~60 μM using YM-30 Centriprep concentrators after the addition of 1 mM DTT. Concentrated protein was diluted with glycerol for long-term storage as 50% glycerol stock solutions at −80°C. NPADH used for mechanistic studies was exchanged into appropriate buffers by gel filtration through a BioGel P6 desalting column.
Absorbance and Fluorescence Spectroscopy
Routine absorbance and fluorescence kinetic and endpoint measurements were made by using Molecular Devices SpectraMax 190 and M2e microplate readers with 96-well Corning CoStar polyprolylene microplates. For studies of NPADH with phenylacetaldehyde (PAL) and other hydrophobic substrates, adsorbtion of the substrate onto the surface of the microplate becomes problematic, so in these cases a fused silica microplate was used to avoid loss of substrate.
Fluorescence-monitored titrations of NPADH with NADH and NAD+ were completed by using a Horiba Jobin-Yvon Fluorolog-3 spectrafluorimeter. Rapid kinetic studies were completed by using a previously described Applied Photophysics SX-17 Stopped Flow Spectrophotometer [22].
PAL was prepared as a stock solution at millimolar concentrations in either water or buffer. The concentration of PAL was calculated using the molar extinction coefficient of 184 M−1cm−1 at 254 nm. NAD+ and NADH concentrations were measured by using their respective molar extinction coefficients of 18,000 M−1cm−1 at 260 nm and 6,220 M−1cm−1 at 340 nm. The cytochrome c concentration was computed as previously described [22].
Kinetic Data Fitting and Analysis
Data were fit by inputting equations described in the text into the non-linear least squares curve fitting programs Kaleidagraph 4.1.2 and Graph Pad Prism 4.0b. Best fitting curves are presented.
Crystallization and Structure Determination
Purified PADH was exchanged into a 10 mM Tris, pH 7.5 buffer containing 1 mM βME using an Amicon Utra-4 centrifugal filter with a 30 kDa MW cutoff, and concentrated to ~5–7 mg/mL. The protein was crystallized in 100–200 mM trisodium citrate, pH 8.5 and 12–16% PEG 3350 by using the sitting drop vapor diffusion method at 4 °C and combining equal volumes of protein and mother liquor. Small crystals of 0.1 μm × 0.1 μm × 0.1 μm grew within a week. Crystals were flash frozen in a cryo solution containing the mother liquor and 25% glycerol before data collection at the Caltech Molecular Observatory on the Stanford Synchrotron Radiation Laboratory beam line 12–2 at 100 K. The crystals belonged to the space group P212121 and had unit dimensions a = 112.0 Å, b = 118.7 Å and c =304.7 Å (Table 1). The reflections were indexed and scaled using HKL2000 [23].
Table 1.
X-ray Data Collection and Refinement Statistics.
| Data Collection Statistics | |
|---|---|
| Beamline | SSRL BL12-2 |
| Wavelength (Å) | 0.979 |
| Temperature (K) | 100 |
| Space group | P212121 |
| Unit Cell (Å) | 112.09 × 118.69 × 304.74 |
| Resolution range (Å) | 49.99-2.83 |
| Total reflections | 93501 |
| Unique reflections | 89099 |
| Completeness (%)a | 95.7 (98.3) |
| I/σ (I)a | 5.6 (1.9) |
| Rsym (%)a,b | 11.4 (36.1) |
|
| |
| Refinement Statistics | |
|
| |
| Rcryst (%)b | 25.4 |
| Rfree (%)c | 25.8 |
| Average B-value (Å2) | 27.02 |
| r.m.s. deviation bond length (Å) | 0.013 |
| r.m.s. deviation bond angles (°) | 1.54 |
| Tetramers per ASU | 2 |
| No. Protein Atoms | 27306 |
| No. Non-Protein Atoms | 0 |
| Water Molecules | 0 |
| PDB Code | 4QYJ |
Values in parentheses are for the highest resolution shell.
Rsym = ΣiΣhkl|Ii(hkl) − <I(hkl)>|/Σhkl<I(hkl)>, where Ii(hkl) is the ith measured diffraction intensity and <I(hkl)> is the mean intensity for the Miller index (hkl).
Rcryst = Σhkl||Fo(hkl)| − |Fc(hkl)||/Σhkl|Fo(hkl)|.
Rfree = Rcryst for a test set of reflections (5% in each case).
Molecular replacement was performed with Phaser using ALDH1 (PDB accession code 1BXS) as a starting model [12, 24]. Model building and subsequent refinements were carried out in Coot and REFMAC5 using NCS and TLS parameters [25, 26]. The final R and Rfree were 25.4% and 25.8%, respectively (Table 1). The root mean square deviation (r.m.s.d.) between all backbone atoms in the two protein molecules (eight subunits) comprising the asymmetric unit ranged between 0.13–0.25 Å2. The overall stereochemistry of the model is good, with r.m.s. deviations from standard geometry values of 0.013 Å for bonds and 1.54° for angles. Final model validation was performed with PROCHECK under default settings in the Auto Deposition Input Tool (ADIT) [27]. 91.91%, 7.88%, and 0.21% of residues refined within preferred, allowed, and disallowed conformations, respectively. The only residue falling into a disallowed conformation is Pro 143, which localizes to a disordered region of the oligomerization domain on each of the eight subunits and adopts a cis conformation. Residues 1–15 were not observed due to disorder.
Results
Purification of NPADH
Over expression of NPADH from pET-28NPADH in 6 L of BL21(DE3) E.coli cell culture followed by Ni2+-affinity chromatography yielded ~55 mg protein. Assay by reducing SDS-PAGE indicated the protein was >90% pure and had a monomer molecular weight of ~55 kDa, which was in good agreement with the theoretical molecular weight (Figure S1). Similar results were observed by non-reducing SDS-PAGE, suggesting the absence of inter-subunit disulfide bonds. In analytical gel filtration studies using a 300 × 7.8 mm BioRad SEC 250–5 column, NPADH eluted as a 227 ± 21 kDa protein (Figure S2), which implies a tetrameric (α4) composition similar to that observed for related Class I and II aldehyde dehydrogenases [28].
Global Structure of NPADH
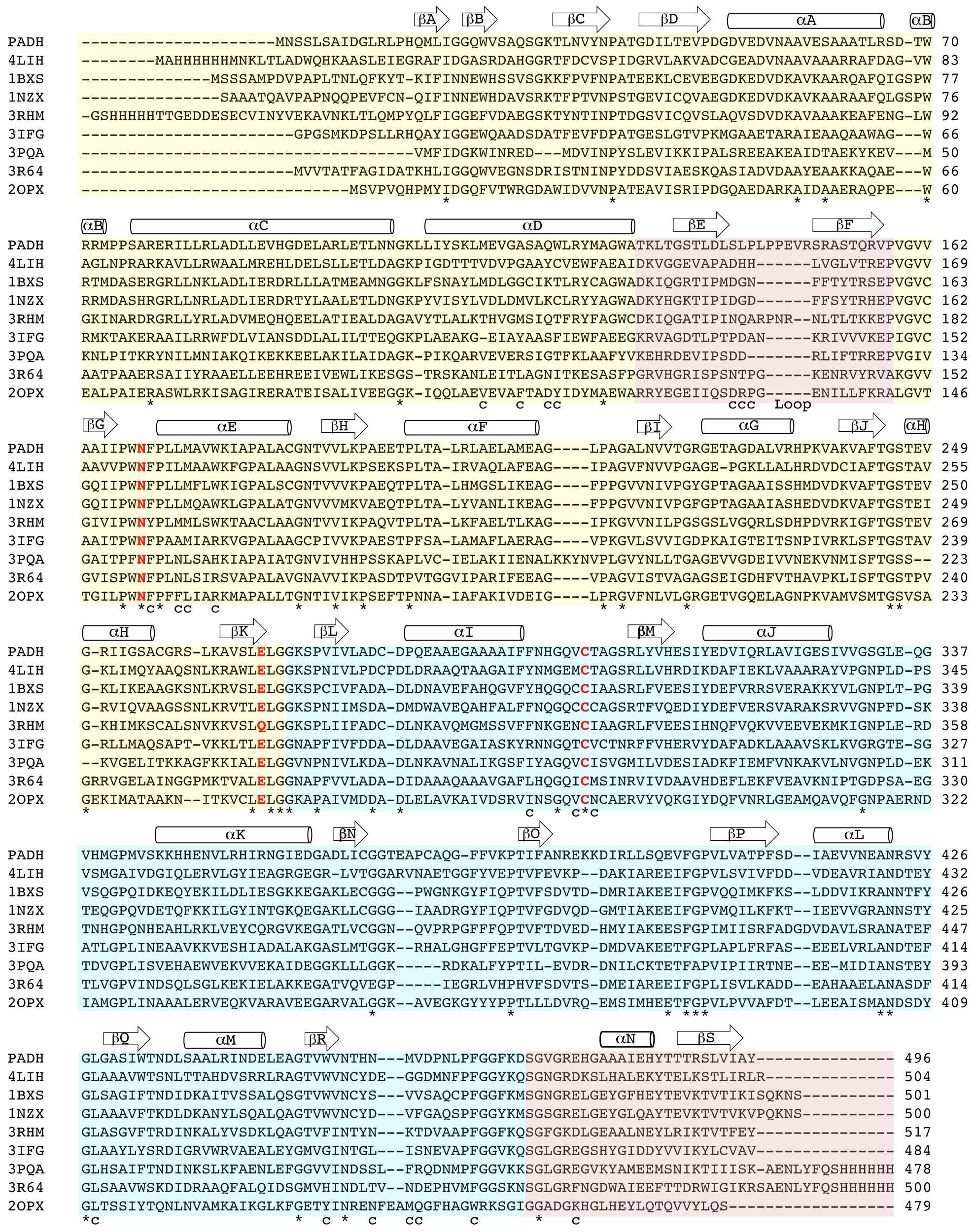
Each 496 amino acid PADH subunit consists of three domains: an N-terminal NAD+-binding domain (residues 1–130 and 159–269), a catalytic domain (residues 270–471), and an oligomerization domain (131–158 and 472–496) (Figures S3 & S4) [12]. The subunits assemble into an intricately connected homotetramer (Figure 2 and S4). The three stranded β-sheet of the oligomerization domain (βE, βF, βS) pairs with the seven-stranded β-sheet of catalytic domain (βL-βR) of a neighboring subunit, establishing “hugging” dimers within the overall tetramer (Figures 2 and S4). To complete the tetramer, βE of the oligomerization domain in one dimer pairs with βE′ the oligomerization domain of a neighboring dimer to complete a 10-stranded β-sheet that spans three subunits. Near the tip of each oligomerization domain is a channel to the active site pocket of the adjacent subunit (Figure 2A). Opposite the active site pocket, as defined by the catalytic residues Cys 301 and Glu 267, is the NAD+-binding domain. All four NAD+-binding pockets are located on the periphery of the enzyme.
Figure 2.

Global fold of PADH. A) Surface representation of the PADH dimer depicting the location of the substrate channel (*). B) PADH tetramer. Orientation and proximity of the oligomerization domain to the substrate channel for C) PADH (yellow) and BcPADH (blue) and D) ALDH1.
The closest structural homolog to NPADH is sheep liver aldehyde dehydrogenase ALDH1 (PDB ID: 1BXS), which catalyzes the conversion of retinal to retinoic acid and has 46% identity and 64% similarity to PADH [12]. Superimposing a NPADH monomer onto ALDH1 gives an r.m.s.d. between α-carbon atoms of 0.87 Å2 (Figure S5). The most significant topological difference between the structures exists in the oligomerization domain, where NPADH contains six additional amino acids (L144PPEVER149) (Figures 2C & 2D). In a homologous PADH structure from Burkholderia cenocepacia J2315 (BcPADH) (PDB ID: 4O5H), which was recently solved by the Seattle Structural Genomics Consortium and has 49% identity and 65% similarity to PADH, this loop contains the same number of amino acids as NPADH and adopts a different orientation (Figure 2C). The insertion may have catalytic implications, as these additional residues extend the oligomerization domain of each subunit over the entrance to the substrate channel of the neighboring subunit, thereby narrowing or occluding substrate access.
Substrate Channel and Active Site
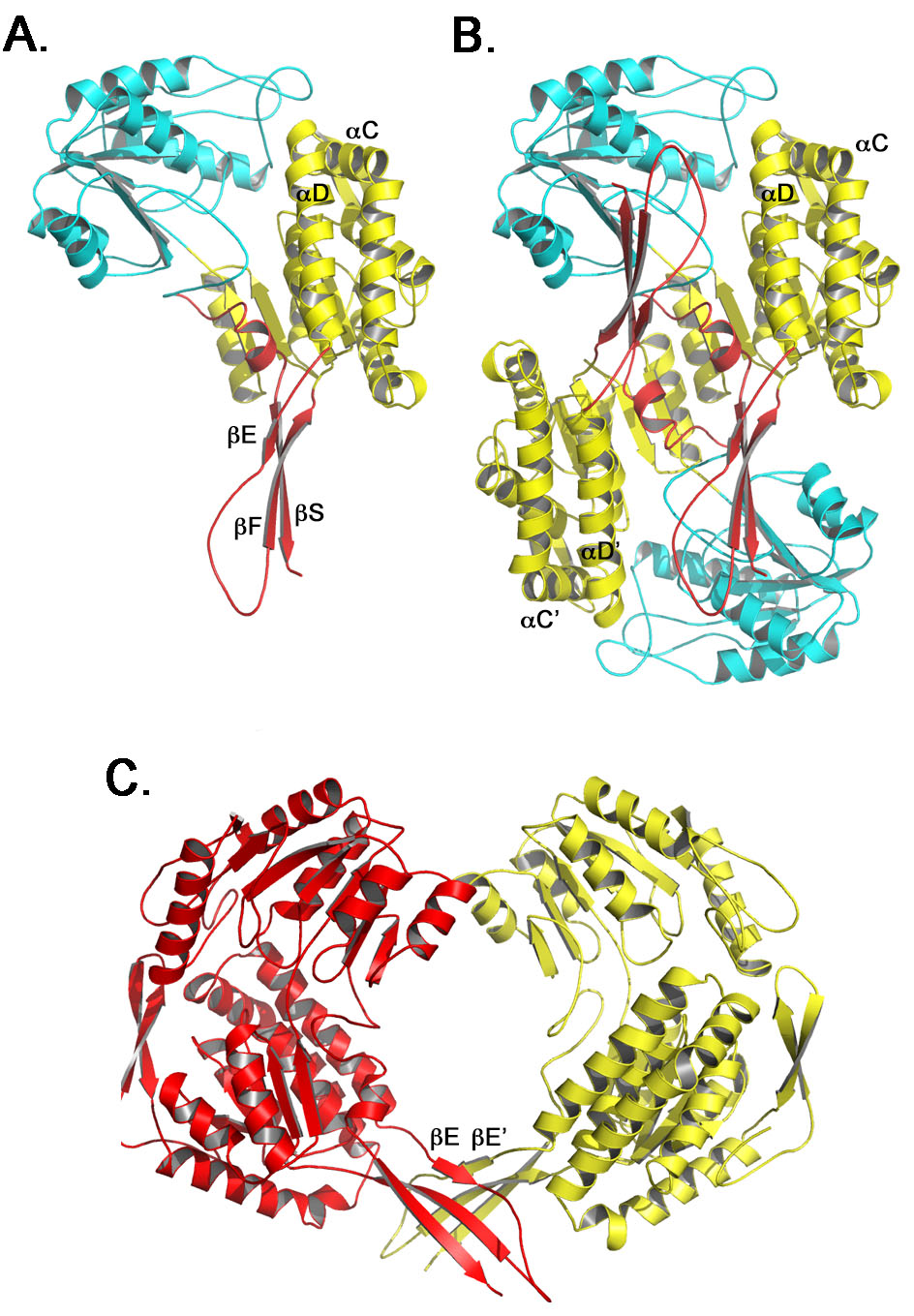
The entrance to the active site lies adjacent to the oligomerization domain and is approximately 7–8 Å by 6–7 Å (Figure 2). By comparison, the molecular dimensions of benzene are ~5.3 Å by 1.4 Å. The catalytic cysteine, Cys 301, sits at the back of the substrate channel ~15 Å below the surface. In general, mostly hydrophobic residues line the channel. These include Met 114, Ala 118, Gln 121, Trp 122, Asn 169, Phe 170, Leu 173, Met 174, and Trp 177, and Glu 267 of the NAD+-binding domain; Phe 295, Val 300, Cys 301, Thr 302, Leu 428, Trp 453, Asn 458, Met 459, Val 460, Phe 466, and Glu 477 of the catalytic domain; and Ser 141′, Leu 142′, and Pro 143′ of the oligomerization domain from the neighboring subunit (Figure 3 and S7). The aromatic amino acids lining the channel, which include Phe 170, Phe 295, Phe 466, and Trp 177, tenably assist in substrate docking and selectivity by π-stacking with the phenylacetaldehyde substrate and positioning it appropriately near the catalytic Cys 301.
Figure 3.

A) Stereoview of the PADH active site and substrate channel. NAD+ (green) was modeled in into the apo-PADH structure using the existing structure of ALDH1 with bound NAD+ (1BXS). Because E267 is disordered in PADH, E267 from BcPADH (yellow) is depicted in its place to provide a likely model of the apo-active site. B) Active site water molecule in BcPADH.
A majority of conserved residues in NPADH localize to the active site and NAD+-binding pocket [29]. At the interface between the two pockets are the catalytic Cys 301 and Glu 267 residues, which serve as the general nucleophile and general base for the reaction, respectively, as determined by previous studies of homologous ALDHs (Figures 3 & 4) [16]. The Glu 267 side chain, however, is not observed in all NPADH subunits and has high B-factors, likely due to positional disorder. For BcPADH, the Glu side chain faces away from the active site pocket and nestles into a small cavity. These differences are consistent with previous observations made in ALDH structures, implying flexibility of the proton abstracting carboxylate side chain. For example, the analogous glutamate in ALDH1 has weak electron density and higher B-factors [12], while the same residue was observed to adopt two different orientations in structures of Streptococcus mutans glyeraldehyde-3-phosphate dehydrogenase from [30]. These structures, as well as studies of several other ALDHs, have suggested that the catalytic Glu has two conformations: a “passive conformer” that tucks away to allow hydride transfer to the nicotinamide ring, and an “active conformer” that abstracts a proton from the thioester-deacylating water.
Figure 4.

The Proposed Catalytic Mechanism of PADH.
Other significant residues of catalytic interest include Asn 169, Thr 244, Lys 179, and Glu 477. Asn 169, which sits above Cys 301, may function so stabilize the oxyanion intermediate and is highly conserved among ALDHs (Figures 3 & 4) [29, 30]. Conserved Thr 244 sits opposite to Cys 301 and adjacent to Glu 267 and Lys 179. In the 2.0 Å BcPADH structure, a water molecule rests between Lys 179, Thr 244, and Glu 267, and Glu 477 (Figure 3B). After the hydride transfer step, these residues could either facilitate an increase in the pKa of Glu 267 or a decrease the pKa of the water to promote deprotonation by Glu 267 and subsequent generation of the attacking nucleophile.
NAD+-Binding Domain
The NAD+-binding domain is localized to the amino-terminal half of PADH and consists of residues 1–115 and 144–254 (Figures S3 and S5). The oligomerization domain divides the NAD+-binding domain into two distinct intervals of primary structure but does not affect the tertiary structure. Despite several attempts, crystallization of NPADH with bound cofactor was not achieved. Nevertheless, a potential NPADH NAD+-binding conformation was obtained by aligning the structures of NPADH and NAD+-bound ALDH1 (Figure S7) [12]. Modeling bound NAD+ shows that αG, αH and βJ likely cradle the nucleotide moiety in a manner analogous to ALDH1 and effectively establish the nucleotide-binding pocket. The adenine ring-binding motif of the canonical NAD+-binding Rossman fold comprises αG, βJ, αH, and βK (residues 225–267) in NPADH and is highly conserved especially the “ftGstevg” turn pattern, which is paramount in adenine and nicotinamide ring binding [29] (Figure S3 & S7). Phe 243 likely π-stacks with the NAD+ adenine ring, whereas the Gly 245 backbone hydrogen bonds with the nicotinamide amide. Conserved Phe 402 from the catalytic domain π-stacks with the nicontimamide ring.
The superposition of NAD+-bound ALDH1 onto the apo-NPADH structure reveals how αG, and to a lesser extent αF, cradle the NAD+. Comparing NPADH and ALDH1, these helices appear more open in the apo-NPADH model (Figure S5). Looking at the regions surrounding the NAD+-binding site, NAD+ binding in ALDH1 seems to induce subtle conformational changes. The cinching of αG and αH around NAD+ causes the β-sheets immediately behind the helices (βG, βH, βJ, and βK) to twist, subsequently pushing the C-terminal end of βK towards the active site. Interestingly, the putative catalytic general base Glu 267 resides at the βK C-terminus. These structural changes may suggest why NAD+-binding in ALDH1 and NPADH precedes substrate binding (vide infra) [12]. Perhaps binding NAD+ causes a conformational shift that either ushers Glu 267 towards Cys 301 and/or changes the local environment to raise the pKa of Glu 267, thereby promoting basicity.
Substrate and Product Inhibition of NPADH
When assayed at 25°C in 50 mM POPSO buffer at pH 8 in the presence of a saturating concentration of PAL (120 μM), purified NPADH was found to have a turnover number of 12.3 ± 0.2 s−1 per monomer and an apparent KM for NAD+ of 207 ± 9 μM (Figure 5). To test product inhibition, similar assays were carried out in the presence of NADH and PAA. These data were fit by using Equation 1 in which [P] represents the concentration of either product (Figure 5). The apparent product dissociation constants of NADH and PAA under these conditions were 146 ± 14 μM and 172 ± 20 mM respectively.
Figure 5.
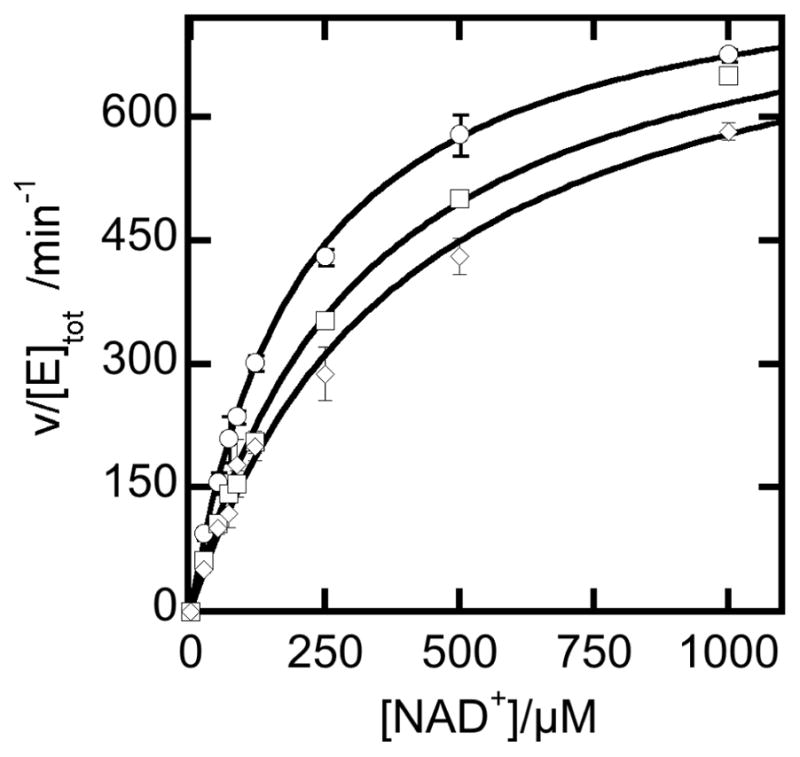
Steady state reaction of NPADH in the presence or absence of product inhibitors. Reactions were carried out at 25°C in 50 mM POPSO buffer pH 8 containing 120 μM PAL and 0.4 μM enzyme. Reactions included either no inhibitor (❍), 95.2 mM phenylacetic acid (□), or 100 μM NADH (◇).
| (1) |
It should be noted that the definitions of the apparent KM and Vmax parameters are functions of the substrate and product concentration. The exact relationship of the reaction rate constants to the apparent steady state parameters is readily determined by application of the King and Altman method [31]. Details on the origin of the equations used to fit these data are provided the supplementary material (Scheme S2 and the associated supplementary equations).
pH Dependence of Catalysis by NPADH
The catalytic activity of NPADH has an optimum operating range between pH 8 and 10 (Figure S3). This pH dependence is similar to that previously reported for related aldehyde dehydrogenases and is consistent with the role of the conserved active site cysteine as a nucleophile in catalysis. A more complete analysis of the pH dependence of steady-state reaction mechanism may help to establish more definitively the structural origins of the macroscopic pKa values the in the reaction mechanism of NPADH.
Pyridine Nucleotide Complexes of NPADH
For many aldehyde dehydrogenases, NADH binds competitively with NAD+ and forms a nonproductive “dead-end” complex during catalysis [14]. To characterize this behavior in NPADH, titrations of NPADH with NAD+ and NADH were evaluated to estimate the binding affinities of the oxidized and reduced pyridine nucleotides under equilibrium conditions. Mg2+ was included in these studies due to a previously established role in pyridine nucleotide binding [14]. The extent of NPADH saturation with NADH was evaluated based on the increase in the intrinsic fluorescence of NADH that occurs upon binding of NADH to the apo enzyme. The maximum change in fluorescence intensity was obtained by exciting at 340 nm and recording fluorescence emission at 469 nm. Data from the titration of apoNPADH with NADH were corrected for the inner filter effect on the fluorescence excitation path and are plotted in Figure 6A [32]. The initial hyperbolic increase in fluorescence is due to the saturation of NPADH active sites with NADH. The subsequent linear increase in fluorescence occurs as excess, unbound NADH accumulates in the later part of the titration. Data were fit with the quadratic expression given in Equation 3a, which represents a mathematical description of this system at equilibrium. The binding affinity of NADH for NPADH was estimated to be 44.5 ± 9.2 μM.
Figure 6.

The binding of pyridine nucleotides to NPADH as monitored by fluorescence. Assays were performed in a 20 mM POPSO, pH 7.0 buffer with 2 mM Mg2+ at 25 °C. (A) Increase in fluorescence emission monitored at 469 nm in the titration of 8.8 μM NPADH with NADH. (B) Decrease in 469 nm fluorescence emission observed in the titration 7.0 μM NPADH equilibrated with 50 μM NADH with NAD+. Samples were excited at 340 nm and all fluorescence data were corrected for the inner filter effect. Equations 3a and 3b were used to generate fits through the data points in panels A and B, respectively.
| (3a) |
The affinity of NAD+ for NPADH was determined to be 48.7 ± 0.8 μM by analysis of the fluorescence decreases when NAD+ is titrated into a solution of NPADH equilibrated with NADH (Figure 6B). Data were fit with equation 3b, which describes the hyperbolic decrease in fluorescence associated with the displacement of NADH from the active site of NPADH by the competitive binding of NAD+. The origin and definition of terms used in this equation are provided in supplementary material.
| (3b) |
In this analysis, we detect only a single class of pyridine nucleotide binding site at equilibrium. For this reason, the total enzyme concentration term included in equations 3a and 3b refers to the total NPADH monomer concentration. This observation is in agreement with the crystal structure of the human mitochondrial aldehyde dehydrogenase co-crystallized with NAD+, which shows all four active sites to be fully occupied by bound pyridine nucleotides [14].
Effects of Mg2+ and Mn2+ on NPADH kinetics
Divalent metal ions were found to have concentration-dependent activating and inhibiting effects on steady-state catalysis by NPADH. These properties were evaluated by kinetic assays in which the concentrations of NAD+ and PAL were held constant while Mg2+ and Mn2+ were varied. The reaction rates were computed from linear fits through the initial increase in absorbance at 340 nm corresponding to the formation of NADH formation and plotted as a function of increasing Mg2+ and Mn2+ concentrations (Figure 7). The rate increases initially and then decreases with metal ion concentration. The threshold values marking the interchange of activating and inhibiting effects are unique to each metal ion. In the presence of NADH, the inhibitory effects are more severe and the threshold marking the interchange metal-ion activation and inhibition shifts to lower Mg2+ and Mn2+ concentrations.
Figure 7.

Activating and inhibitory effects of divalent metal ions on steady-state catalysis by NPADH. A) The rate of NADH production by 0.1 μM NPADH reacting with 10 μM PAL and 50 μM NAD+ in 50 mM POPSO buffer at pH 7 in the presence (□) or absence (■) of 50 μM NADH as a function of (A) Mg2+ and (B) Mn2+. Best fits through the data according to equation 4 are represented by the solid curves passing through the plotted data.
In evaluating various reaction schemes that could explain the observed trends, the metal ion activating and inhibitory effects were best described by a model in which metal ions bind reversibly at two unique sites. A kinetic model expanding the sequential reaction mechanism of NPADH to include the activating and inhibitory effects of metal ions (Mx) is included in supplementary material (Scheme S4) together with the derivation and definition of the constant terms of equation 4 used to fit the data plotted in Figure 8.
Figure 8.
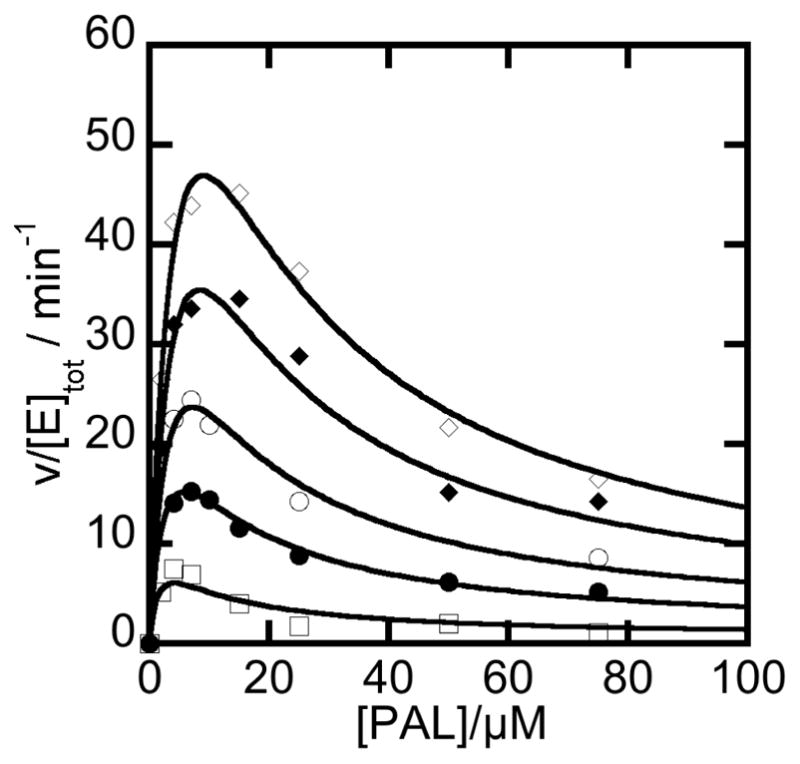
Global fitting of the steady state NPADH reaction as a function NAD+ and PAL concentration. NAD+ concentrations (increasing from the lowest curve to the highest curve) were 50, 100, 150, 250, and 500 μM. Initial rates from the reaction of 1 μM NPADH with various concentrations of PAL and NAD+ in a 50 mM POPSO, 2 mM Mg2+ buffer at pH 7 were globally fit according to the model shown in (Scheme S4) by using equation 5b. The fit converged with a global R2 value of 0.99 and absolute sum of squares of 59.5.
| (4) |
Nonlinear least squares fits of this model converge to give excellent agreement with the data (R2 values > 0.99). The best-fitting numerator and denominator parameters are given in Table S1. The close agreement between data and model dependent fit provides support for the proposed inhibitory role of the second metal ion binding site in the catalytic mechanism of NPADH..
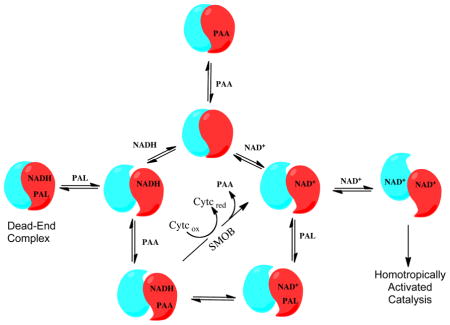
Homotropic Allosteric Activation of NPADH by NAD+
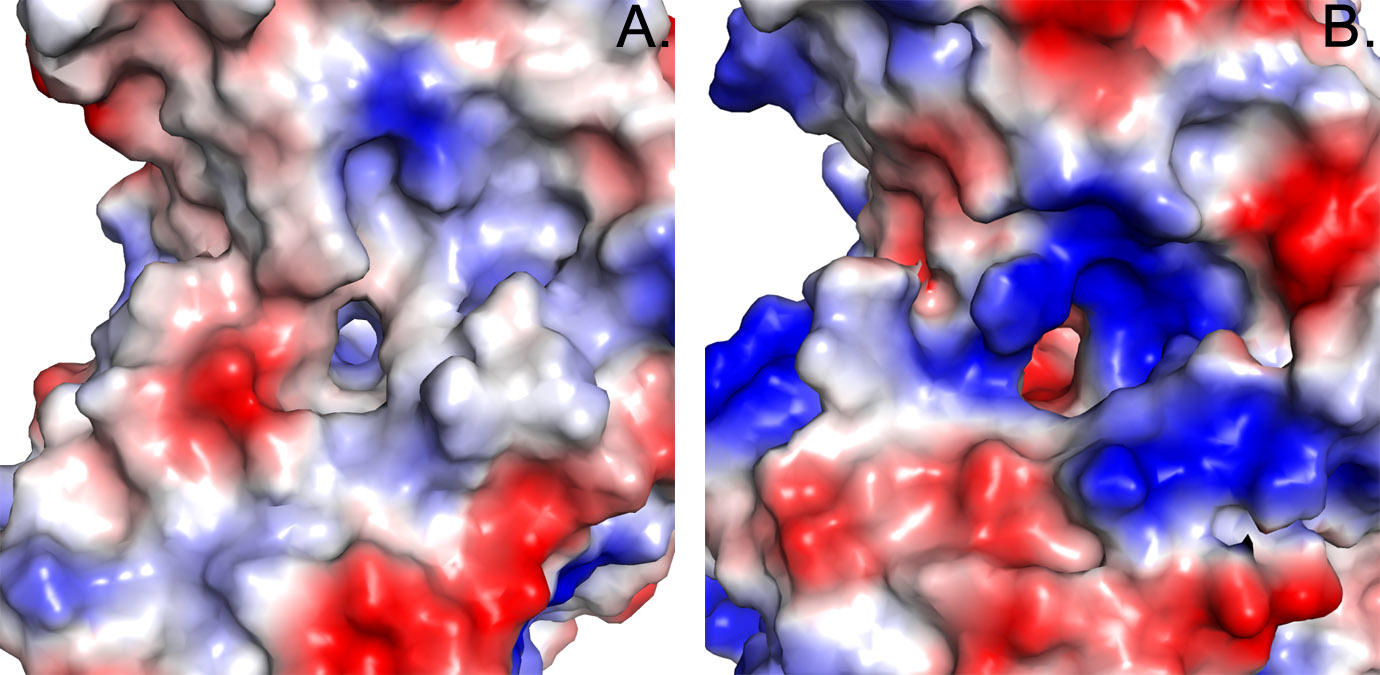
Half-sites reactivity in which only two monomers of each tetramer engage catalytically to transform aldehyde to acid product, was previously reported based on single turnover studies of tetrameric aldehyde dehydrogenases [33]. The pair of non-catalytically active sites of each protomer are thought to take on a regulatory role that serves to tune the rate of catalysis. In view of the previous findings, the steady-state kinetic data from the reaction of NPADH with NAD+ and PAL were evaluated over a broad range of substrate concentrations and different kinetic models used to fit the data were compared. The dependence of the NPADH steady-state reaction rate of on substrate concentration is presented in Figure 8. A hyperbolic increase in reaction rate is observed as a function of increasing NAD+ concentration over the full range of aldehyde concentrations. An example of this trend is more easily visualized in the cross section of data plotted as a function of [NAD+] at 120 μM PAL (Figure 5). The data trend is more complicated as the aldehyde concentration is increased at constant [NAD+]. The reaction rate initially increases as a function of added aldehyde. At higher concentrations, the reaction rate drops with increases in [PAL]. As noted above, this type of substrate inhibition is thought to occur through the formation of a ternary “dead-end” complex of NPADH with substrate, PAL, and product, NADH.
A steady-state rate expression describing a sequential mechanism of catalysis and the substrate inhibition (Scheme 1) was derived by the King and Altman method and is given in equation 5a. A and B correspond to NAD+ and PAL.
Scheme 1.

Summary of Steady-State Reactions Catalyzed by NPADH
| (5a) |
Although this equation provides an excellent fit through the PAL concentration-dependence data at any fixed concentration of NAD+, the best fit at each NAD+ concentration returns a unique best-fitting set of kinetic parameters. When the three-dimensional data set is fit globally with the imposed constraint that kinetic constants in the numerator and denominator are preserved in all data sets, the fit converges, but the observed NAD+ dependence of the reaction is very poorly represented (global R2 = 0.92). The best fit according to this scheme fails to model the dynamic increase in reaction rate with increasing [NAD+] at all of the aldehyde concentrations evaluated. The aldehyde concentration dependence of the reaction rate is very poorly modeled at low concentrations of NAD+. At high aldehyde concentrations, the fit converges on a narrow range of limiting velocities. This projection is incongruent with the data, which retains a strong NAD+ concentration dependence at even at high aldehyde concentrations. The results from fitting with this model are presented in Figure S10 with the best-fitting constant and coefficient terms presented in Table S2.
A high quality global fit through the data was possible by expanding the catalytic model to include a homotropic allosteric activation effect associated with the binding of NAD+. One of the simplest versions of this model was evaluated. In this case, NPADH is considered to be a dimer of α2 protomers and the allosteric influence of NAD+-binding to NPADH extends only through the interacting monomer interfaces of the protomer subunits. Possible inter-protomer interactions are neglected. The reaction scheme describing this model (Scheme S5) was used to derive a steady-state rate equation by the King and Altman method (equation 5b) in which A and B represent NAD+ and PAL, respectively.
| (5b) |
This expansion of the original kinetic model to include homotropic allosteric activation by NAD+ results in an improved fit with a global R2 value of 0.99 (Figure 8). Statistical comparison of the globally fit models by the F-test with α = 0.05 resulted in a p < 0.0001 ruling in favor of the homotropoic allosteric model [34]. Because the fitting expression is parameter rich, the best-fitting values of the constant and coefficient terms can not be estimated with high precision. Kinetic parameters resulting in the best fit through the data shown in Figure 8 are provided in Table S4...
Pyridine Nucleotide Exchange between NPADH and SMOB
Our kinetic analysis indicates that NADH binds tightly to NPADH as a competitive inhibitor and in an unproductive “dead-end” complex with NPADH and phenylacetaldehyde (Scheme 1). Assuming that the reported cytosolic concentration of NADH in actively growing E.coli (~83 μM) is similar to that found in Pseudomonas,[35] we can expect PADH to be significantly inhibited under these conditions and inhibit the styrene catabolic pathway. This inhibition may be offset by the activity of styrene monooxygenase reductase (SMOB), which efficiently binds and oxidizes NADH to NAD+ in first step of styrene metabolism [22]. To evaluate this possibility, a rate limiting concentration of NPADH and catalytic excess of SMOB were combined in a reaction initiated by the addition of PAL and NAD+. The reaction also included 30 μM FAD as coenzyme for SMOB and 40 μM horse heart cytochrome c as a reporter molecule (Figure 10). In this catalytic system NADH produced by the transformation of PAL to PAA by NPADH is used as a substrate by SMOB in the reduction of FAD. Rapid intermolecular electron-transfer then occurs from the reduced FAD to the pool of oxidized cytochrome c [22]. Because this steady-state transformation is ultimately limited by the rate of NADH synthesis by NPADH, the observed rate of cytochrome c reduction provides information about NPADH catalysis as influenced by the NADH-scavenging activity of SMOB. The initial rate data recorded in this study are presented in Figure 10. The computed rate of NADH production, instead of the experimentally observed rate of cytochrome c reduction, is presented to allow direct comparison of the data sets presented. With SMOB included as an NADH scavenger, the reaction is no longer significantly impacted by substrate inhibition. This observation supports the hypothesized role of SMOB in preventing “dead-end” inhibition of NPADH.
The best fit through the available data gives an apparent Km of 31.1 ± 5.1 μM for PAL compared with as very similar value of 22.0 ± 2.6 μM for the same reaction in the absence of SMOB. The apparent Vmax values differ more significantly. In the presence of SMOB, the apparent Vmax is 445.6 ± 31.0 μM min.−1, which is 3-times lower than the value obtained in the absence of SMOB (Vmax(app) =1229.2 ± 103.5 μM min−1).
Discussion
Aldehyde dehydrogenases are an essential element in diverse pathways spanning biosynthesis, catabolism, and cellular detoxification [36]. Though theme and variation exist in quarternary structure, pyridine nucleotide preference, and substrate specificity, the core structural and mechanistic features of the ALDHs have remained remarkably conserved over millions of years [17, 18].
As a member of the extended family of aldehyde dehydrogenases, NPADH is most closely associated with the Class I enzymes based on its primary amino acid sequence and homotetrameric structure. The enzyme follows a sequential steady-state mechanism with NAD+ as the leading substrate, and is regulated by an apparent pKa consistent with the role of cysteine as a catalytic nucleophile [37–39]. The Class II, human mitochondrial aldehyde dehydrogenase has an alternate nitroreductase activity that is thought to involve two cysteines adjacent to the catalytic cysteine [40]. These are absent from the active site of NPADH. Moreover, we have not observed evidence for covalent bonding of the active site cysteine with the nicotinamide ring as has been reported for related enzymes [41, 42]. Outside of these differences, many catalytic and structural features of the human mitochondrial enzyme are shared in common with NPADH and have provided great insight in our understanding of the bacterial enzyme.
Substrate and Product Inhibition
Our steady state kinetic data indicate the phenylacetic acid product binds weakly to NPADH and likely contributes the unidirectionality of aldehyde oxidation. At very high concentrations, PAA interferes with NAD+ binding to NPADH, but this is not thought to be physiologically significant as down stream catabolic enzymes would funnel PAA into central metabolic pathways. Thus, there is no evolutionary pressure for this catabolic pathway to develop reversibility.
In isolation, NPADH is very sensitive to aldehyde substrate and NADH product inhibition. This effect is difficult to reconcile if one considers the function of NPADH as an isolated enzyme. In context of the functional catalytic pathway, this may represent a control mechanism that is coupled to activity of styrene monooxygenase and other NADH-consuming oxidative enzymes in the cell. As oxidative metabolism decreases due to decreasing levels of styrene and/or molecular oxygen, NADH levels will rise and down-regulate the activity of NPADH.
Upon partial reassembly of the catalytic pathway, the coupled activities of NPADH and styrene monooxygenase reductase very efficiently recycle NADH to NAD+ (Scheme 1). The apparent KM of NPADH for phenylacetaldehyde is slightly changed in the SMOB-coupled reaction, however, the dead-end inhibition of NPADH is relieved and the apparent Vmax is decreased by a factor of three. The decrease in Vmax could be the result of a rate limiting NPADH-SMOB interaction involved in the pyridine nucleotide-exchange reaction. Additional studies are needed to resolve the extent to which direct coenzyme channeling between NPADH and SMOB occurs styrene metabolism.
Allosteric Activation & Subunit Cooperativity
The reaction mechanisms of ALDHs have been shown previously to be sensitive to both homotropic and heterotrophic allosteric regulation. The human mitochondrial aldehyde dehydrogenase is heterotropically regulated by low molecular weight effectors molecules [43, 44] and the oriental variant of this enzyme binds NAD+ with positive cooperativity [45]. Isozymes of more distantly related glyceraldehyde 3-phosphate dehydrogenase have been previously shown to be either allosterically activated or inhibited by pyridine nucleotides [46].
In the present work we evaluated the binding of pyridine nucleotides to NPADH under equilibrium conditions and found our data to be well represented by a model of identical, non-interacting sites. Under conditions of steady-state turnover, NAD+ functions as a homotropic allosteric activator of catalysis. Collectively this result implies that NAD+ binding at one NPADH subunit enhances catalysis by the neighboring subunit of each protomer without a significantly changing in pyridine nucleotide binding affinity.
In the mechanism of half sites reactivity, our data implies that NAD+ is binding with essentially the same affinity when interacting as a substrate or homotropic allosteric effector. The inter-subunit communication responsible for the half-sites reactivity observed for the aldehyde dehydrogenases may differentially affect the catalytically relevant conformations of pyridine nucleotides without significantly influencing the pyridine nucleotide binding equilibria [33]. Possible structural changes accounting for this activation are discussed below.
Structural Elements Controlling Substrate Specificity
ALDHs have been shown to act on such structurally diverse substrates as retinal, 10′-formyltetrahydrofolate, γ-glutamyl-γ-aminobutyraldehyde, lactaldehyde, methylmalonate semialdehyde, glyceraldehyde-3-phosphate, benzaldehyde, 1-pyrroline-5-carboxylate, and succinate-semialdehyde, to name a few. A detailed understanding of the substrate channel becomes essential if one were to engineer specific pathways for either the biosynthesis or bioremediation of alternative substrates, or modulate the flux of metabolites through the SMO upper catabolon [47]. Comparisons between NPADH and ALDH family members highlight several differences in the substrate channel that could account for the smaller substrate preference of NPADH. The smaller dimensions of the channel opening, which are 7–8 Å by 6–7 Å for NPADH compared to 17–19 Å by 7–8 Å for ALDH1, appear to be the most significant. The reduced aperture of the channel entrance likely functions as a selectivity filter that screens for smaller aldehyde substrates. Comparisons to ALDH1 suggest the reduced size of the NPADH substrate channel can be attributed to a few critical amino acid substitutions, alternate rotamer conformations, and shifts in α-carbon backbone positioning. Channel volume reducing ALDH1 to NPADH substitutions (PADH numbering) include G118A, T122W, C300V, S458N, and V459M, of which W122 has the most significant effect (Figure 3). The majority of these substitutions are located at the periphery of the entrance tunnel, distal to the catalytic residues.
Additional factors that may contribute to substrate selectivity are the length and positioning of the oligomerization domain loop and the electrostatics at the entrance to the substrate channel. In NPADH, the oligomerization domain is longer and can occlude the substrate channel. Sequence and structural comparisons of NPADH to ALDH1 and some additional ALDH family members indicate a shorter loop in the oligomerization domain and unimpeded access to the entrance to the substrate (Figure 2) [12, 48–50] [30, 51] [52]. Among the different ALDHs, the electostactic surfaces at the channel entrance vary significantly. For NPADH, the surface of the of the substrate channel entrance is neither strongly positive nor negative (Figure S9). By contrast, ALDHs acting on negatively charged substrates, like succinic semialdehyde or glyceraldehye-3-phosphate, tend of have basic residues lining the channel opening [52] [50].
Comparing PADH to ALDHs with a similar substrate preferences can provide additional structural insight into selectivity, however, at this time the benzaldeyde dehydrogenase structure (3R64) solved by structural genomics has significant portions of the substrate channel (residues 456–471, PADH numbering) missing, presumably due to disorder. By contrast, BcPADH preserves nearly all of the channel residues found in Pseudomonas PADH, making it difficult to assess which amino acids and channel features are the most significant for controlling substrate specificity.
The Oligomerization Domain and Subunit Interactions
The oligomerization domains of all four subunits form the core of PADH and are responsible not only for the effective dimerization observed within the homotetramer, but also for the association of these dimers to form the tetramer. Comparing PADH to BcPADH reveals the oligomerization loop in these proteins are of similar length and sequence, but adopt distinctly different conformations on the surface whereby the loop orientation in BcPADH more readily occludes the channel entrance (Figure 2C). Although further inspection of the BcPADH structure does not reveal why this loop adopts a different orientation, the higher flexibility in this region as well as the observed closing and/or narrowing of the entrance to the substrate channel may support the notion of a cooperative catalytic mechanism present in PADH. The oligomerzation domain is connected directly to catalytically pertinent structural elements, specifically αC, a constituent of a three-helix bundle lining the substrate entrance tunnel, and βG, which forms the base of the NAD+-binding pocket. The C-terminal end of βG contains several residues predicted to interact with NAD+, which is then followed in sequence by αD, another member of the three-helix bundle comprising the substrate channel. αD contains Phe 170 and Trp 177, both of which could contribute to substrate binding (Figure 3).
The amino acid network within the oligomerization domain may provide a basis for inter-subunit communication in the homotropic allosteric activation of NPADH. In our studies, we detect no linkage of pyridine nucleotide binding. The transmission of NAD+ binding energy between subunits required for the observed NAD+-dependent homotropic activation NPADH must occur later in catalysis, downstream of the initial pyridine nucleotide-binding reaction.
It remains to be established whether NPADH is regulated by further allosteric interactions. In high throughput screening studies, a small molecule, Alda-1 (N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide), was recently discovered to function as an allosteric activator of the human mitochondrial aldehyde dehydrogenase [43]. In comparing the structure of NPADH with the human mitochondrial enzyme co-crystalized with Alda-1, several of the side chains defining the Alda-1 binding pocket are conserved or occupy very similar positions as those in the Pseudomonas enzyme. In particular, Met 114, Leu 173, Phe 295, and Cys 301 in the structure of NPADH occur at positions corresponding to the Alda-1 interacting side chains Met 122, Leu 173, Phe 296, and Cys 302 in the mitochondrial enzyme. We therefore expect that Alda-1 or related small molecules may function as heterotrophic allosteric activators of NPADH.
For some ALDH family members, such as those belonging to a group of hyperthermophilic glyceraldehyde 3-phophate dehydrogenases, the base of the oligomerization domain supports an allosteric binding site for glucose-1-phosphate, fructose-6-phosphate, ADP, and AMP [50]. The residues comprising the binding site for these effectors are not strictly conserved in NPADH. Moreover, there is no obvious clustering of residues in the analogous area on the NPADH surface that would suggest a likely small-molecule binding site.
Structural changes coupled to the binding of NAD+
The superposition of NAD+-bound NAD+ ALDH1 onto the apo-NPADH structure reveals αG, and to a lesser extent αF, cradle NAD+. Comparing NPADH and ALDH1, these helices appear more open in the apo-NPADH model (Figure S7) Looking at the regions surrounding the NAD+-binding site, NAD+ binding in ALDH1 seems to induce subtle conformational changes. The cinching of αG and αH around NAD+ causes the β-sheets immediately behind the helices (βG, βH, βJ, and βK) to twist, subsequently pushing the C-terminal end of βK towards the active site. Interestingly, the putative catalytic general base Glu 267 resides at the βK C-terminus. These structural changes may suggest why NAD+-binding in ALDH1 and NPADH precedes substrate binding [12]. It is also possible that binding NAD+ causes a conformational shift that either ushers Glu 267 towards Cys 301 or changes the local environment to raise the pKa of Glu 267 and thereby promote basicity.
Role of Metal Ions in the Catalytic Mechanism of NPADH
The catalytic activity of Class I and Class II aldehdyde dehydrogenases is dependent on the presence of divalent metal ions, which are thought to bind and orient pyridine nucleotides in the active site in different conformations [14, 48, 53]. One conformation of NAD+ activates hydride transfer from the thiohemiacetal intermediate to NAD+ and subsequent hydrolysis of the thioester intermediate while a second conformation inhibits the dissociation of NADH from the enzyme as a product complex [14, 48, 53]. This idea is supported by crystallographic data showing alternate orientations of pyridine nucleotide-binding in the active site solved in the presence and absence of divalent metal ions and by kinetic studies demonstrating interchanges in the rate limiting step as a function of metal concentration [12, 14, 48].
We observe similar metal ion-dependent activation and inhibition of NPADH activity and have evaluated several models for their ability to describe this behavior. The closest agreement between the kinetic data and model dependent fit tend to support both an activating and inhibitory metal binding site in the catalytic mechanism of NPADH. The activating divalent metal binding site may be best described as the direct interaction of the metal ion with the pyrophosphate linkage joining the nicotinamide mononucleotide and adenosine mononucleotide components of the pyridine nucleotide structure, such as in human mitochondrial aldehyde dehydrogenase [54] (Figure S7). A second mononuclear metal binding site, occupied by Mg2+ was detected in this same structure [54]. The Mg2+ in this site assumes a roughly octahedral geometry and is coordinated by the backbone carbonyl oxygens of Val 40, Asp 109, Glu 196, and Val 345, as well as a monodentate interaction with a carboxylate oxygen of Asp 109. The sixth ligand is a crystallographically resolved water molecule. A backbone alignment suggests the corresponding potential coordinate ligands in the structure of NPADH are Tyr 45, Asn 103, Gln 196, Val 344 (Figure S7). To explain the inhibitory effect of the second metal binding site, its relative proximity to the pyridine nucleotide-binding site and positive charge may interfere with the binding of positively charged NAD+ and possibly affect the orientation of the nicotinamide ring in a way that increases the activation energy and decreases the rate constant for hydride-transfer. The neutral nicotinamide ring of NADH, however, would be relatively unaffected by occupancy of the mononuclear metal site and therefore stabilize the NADH–bound form.
Divalent metal ions magnesium, manganese, and calcium, have been observed to impact the catalytic activity of aldehyde dehydrogenases [14, 55–57]. More recently reported crystal structures of the human mitochondrial aldehyde dehydrogenase with allosteric activator, Alda-1 [43], or alternate substrate, nitroglycerin bound [40], include a sodium ion occupying the mononuclear metal site in place of the magnesium observed in the NAD+-bound structure. Further work is needed to better resolve the monouclear metal ion-binding site and its role in catalysis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9.
Effect of SMOB on NADH inhibition. Reactions were carried out with 1 μM NPADH, 1 mM NAD+ and increasing amounts of PAL in 50 mM POPSO, 2 mM Mg2+ buffer at pH 7 in the presence (●) or absence (❍) of 1 μM SMOB, 30 μM FAD, and 40 μM cytochrome c.
Highlights for Review.
NAD+ serves as both the leading substrate and a homotrophic allosteric activator.
PADH is inhibited by a dead end catalytic complex of substrate and product NADH.
SMO offsets inhibition by use of NADH in the first step of styrene catabolism.
A variable length intersubunit loop in ALDHs possibly effects homotropic activation.
Aldehyde specificity appears fine tuned by channel size, loop length and flexibility.
Acknowledgments
This work was supported by NIHSC1 GM081140 to Dr. George Gassner and a Camille and Henry Dreyfus startup award to Dr. Matthew Sazinsky. Shahram Emami was Supported by a Beckman Fellowship. The authors thank Auric Kantz for helping in the preparation of protein for these studies, Diana Quinonez for assistance with the analytical gel filtration studies, and Andrew Skinner for helping us to complete some of the steady-state assays. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Abbreviations
- ALDH
aldehyde dehydrogenase
- ALDH1
sheep liver cytosolic aldehyde dehydrogenase
- ALDH2
bovine mitochondrial aldehyde dehydrogenase
- BME
β-mercaptoethanol
- CoA
coenzyme A
- P-SEA
protein secondary element assignment
- (N)PADH
(N-terminally histidine-tagged)phenylacetaldehyde dehydrogenase
- rmsd
root mean square deviation
- SMO
styrene monoxygenase
- SMOA
styrene monooxygenase reductase component
- SMOB
styrene monoxygenase oxygenase component
- SOI
styrene oxide isomerase
- NADH
nicotinamide adenine dinucleotide, reduced form
- NAD+
nicotinamide adenine dinucleotide, oxidized form
- PAL
phenylacetaldehyde
- PAA
phenylacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Styrene Uses and Market Data. 2010 http://www.icis.com/v2/chemicals/9076472/styrene/uses.html%3E.
- 2.C. Center of Disease Control and Prevention. Immediately Dangerous to Life and Health Concentrations (ILDH): Styrene, NIOSH Publications and Products. 2011. [Google Scholar]
- 3.Panke S, Witholt A, Schmid A, Wubbolts MG. Towards a Biocatalyst for (S)-Styrene Oxide Production: Characterization of the Styrene Degradation Pathway of Pseudomonas sp. Strain VLB120. Appl Environ Microbiol. 1998;64:2032–2043. doi: 10.1128/aem.64.6.2032-2043.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Przybulewska K, Wieczorek A, Nowak I. Isolation of microorganisms capable of styrene degradation. Polish J Environ Stud. 2006:777–783. [PubMed] [Google Scholar]
- 5.Xue J, Yu Y, Bai Y, Wang L, Wu Y. Marine Oil-Degrading Microorganisms and Biodegradation Process of Petroleum Hydrocarbon in Marine Environments: A Review. Curr Microbiol. 2015;71(2):220–8. doi: 10.1007/s00284-015-0825-7. [DOI] [PubMed] [Google Scholar]
- 6.Kuepper J, Ruijssenaars HJ, Blank LM, de Winde JH, Wierckx N. Complete genome sequence of solvent-tolerant Pseudomonas putida S12 including megaplasmid pTTS12. J Biotechnol. 2015;200:17–8. doi: 10.1016/j.jbiotec.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 7.Montersino S, Tischler D, Gassner GT, van Berkel WJH. Catalytic and Structural Features of Flavoprotein Hydroxylases and Epoxidases. Adv Synth Catal. 2011;353:2301–2319. [Google Scholar]
- 8.Hartmans S, van der Werf MJ, de Bont JAM. Bacterial Degradation of Styrene Involving a Novel Flavin Adenine Dinucleotide-Dependent Styrene Monooxygenase. Appl Environ Micro. 1990;56(5):1347–1351. doi: 10.1128/aem.56.5.1347-1351.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teufel R, Mascaraque V, Ismail W, Voss M, Perera J, Eisenreich W, Haehnel W, Fuchs G. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(32):14390–5. doi: 10.1073/pnas.1005399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hempel J, Perozich J, Chapman T, Rose J, Boesch JS, Liu ZJ, Lindahl R, Wang BC. Aldehyde dehydrogenase catalytic mechanism. A proposal. Advances in experimental medicine and biology. 1999;463:53–9. doi: 10.1007/978-1-4615-4735-8_7. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida A, Rzhetsky A, Hsu LC, Chang C. Human aldehyde dehydrogenase gene family. European journal of biochemistry/FEBS. 1998;251(3):549–57. doi: 10.1046/j.1432-1327.1998.2510549.x. [DOI] [PubMed] [Google Scholar]
- 12.Moore SA, Baker HM, Blythe TJ, Kitson KE, Kitson TM, Baker EN. Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure. 1998;6(12):1541–51. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 13.Nair RB, Bastress KL, Ruegger MO, Denault JW, Chapple C. The Arabidopsis thaliana reduced epidermal fluorescence1 gene encodes an aldehyde dehydrogenase involved in ferulic acid and sinapic acid biosynthesis. Plant Cell. 2004;16(2):544–54. doi: 10.1105/tpc.017509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho KK, Allali-Hassani A, Hurley TD, Weiner H. Differential effects of Mg2+ ions on the individual kinetic steps of human cytosolic and mitochondrial aldehyde dehydrogenases. Biochemistry. 2005;44(22):8022–9. doi: 10.1021/bi050038u. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Brown CS, Weiner H. Mechanism of the magnesium ion activation of the catalytic activity of horse liver aldehyde dehydrogenase. Advances in experimental medicine and biology. 1980;132:181–8. doi: 10.1007/978-1-4757-1419-7_19. [DOI] [PubMed] [Google Scholar]
- 16.Liu ZJ, Sun YJ, Rose J, Chung YJ, Hsiao CD, Chang WR, Kuo I, Perozich J, Lindahl R, Hempel J, Wang BC. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nature structural biology. 1997;4(4):317–26. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 17.Perozich J, Nicholas H, Wang BC, Lindahl R, Hempel J. Relationships within the aldehyde dehydrogenase extended family. Protein science : a publication of the Protein Society. 1999;8(1):137–46. doi: 10.1110/ps.8.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson B, Brocker C, Thompson DC, Black W, Vasiliou K, Nebert DW, Vasiliou V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum Genomics. 2011;5(4):283–303. doi: 10.1186/1479-7364-5-4-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Velasco A, Alonso S, Garcia JL, Perera J, Diaz E. Genetic and functional analysis of the styrene catabolic cluster of Pseudomonas sp. strain Y2. Journal of bacteriology. 1998;180(5):1063–71. doi: 10.1128/jb.180.5.1063-1071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantz A, Chin F, Nallamothu N, Nguyen T, Gassner GT. Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch Biochem Biophys. 2005;442(1):102–16. doi: 10.1016/j.abb.2005.07.020. [DOI] [PubMed] [Google Scholar]; Arch Biochem Biophys. 2005;442:102–116. doi: 10.1016/j.abb.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 21.HCG, Gasteiger EA, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 22.Morrison E, Kantz A, Gassner GT, Sazinsky MH. Structure and mechanism of styrene monooxygenase reductase: new insight into the FAD-transfer reaction. Biochemistry. 2013;52(35):6063–75. doi: 10.1021/bi400763h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 25.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Cryst D. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: Model building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, Thorton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 28.Rodriguez-Zavala JS, Allali-Hassani A, Weiner H. Characterization of E. coli tetrameric aldehyde dehydrogenases with atypical properties compared to other aldehyde dehydrogenases. Protein science : a publication of the Protein Society. 2006;15(6):1387–96. doi: 10.1110/ps.052039606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hempel J, Nicholas H, Lindahl R. Aldehyde dehydrogenases: Widespread structural and functional diversity within a shared framework. Protein Science. 1993;2:1890–1900. doi: 10.1002/pro.5560021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cobessi D, Tete-Favier F, Marchal S, Branlant G, Aubry A. Structural and biochemical investigations of the catalytic mechanism of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. Journal of molecular biology. 2000;300(1):141–52. doi: 10.1006/jmbi.2000.3824. [DOI] [PubMed] [Google Scholar]
- 31.King EL, Altman C. A Systematic Method of Deriving Rate Laws for Enzyme Catalyzed Reactions. J Phys Chem. 1956;60:1375–1378. [Google Scholar]
- 32.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; New York, NY: 2006. [Google Scholar]
- 33.Weiner H, Wei B, Zhou J. Subunit communication in tetrameric class 2 human liver aldehyde dehydrogenase as the basis for half-of-the-site reactivity and the dominance of the oriental subunit in a heterotetramer. Chemico-Biological Interactions. 2001:47–56. doi: 10.1016/s0009-2797(00)00221-0. [DOI] [PubMed]
- 34.Motulsky HaC, Arthur . Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting. Oxford University Press Inc; 198 Madison Ave., New York, New York 100016: 2004. [Google Scholar]
- 35.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nature chemical biology. 2009;5(8):593–9. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4(6):697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Weiner H. Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry. 1995;34(1):237–43. doi: 10.1021/bi00001a028. [DOI] [PubMed] [Google Scholar]
- 38.Farres J, Wang TT, Cunningham SJ, Weiner H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry. 1995;34(8):2592–8. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez-Segura L, Velasco-Garcia R, Munoz-Clares RA. Modulation of the reactivity of the essential cysteine residue of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa. Biochem J. 2002;361(Pt 3):577–85. doi: 10.1042/0264-6021:3610577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lang BS, Gorren AC, Oberdorfer G, Wenzl MV, Furdui CM, Poole LB, Mayer B, Gruber K. Vascular bioactivation of nitroglycerin by aldehyde dehydrogenase-2: reaction intermediates revealed by crystallography and mass spectrometry. J Biol Chem. 2012;287(45):38124–34. doi: 10.1074/jbc.M112.371716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsybovsky Y, Donato H, Krupenko NI, Davies C, Krupenko SA. Crystal structures of the carboxyl terminal domain of rat 10-formyltetrahydrofolate dehydrogenase: implications for the catalytic mechanism of aldehyde dehydrogenases. Biochemistry. 2007;46(11):2917–29. doi: 10.1021/bi0619573. [DOI] [PubMed] [Google Scholar]
- 42.Wymore T, Deerfield DW, 2nd, Hempel J. Mechanistic implications of the cysteine-nicotinamide adduct in aldehyde dehydrogenase based on quantum mechanical/molecular mechanical simulations. Biochemistry. 2007;46(33):9495–506. doi: 10.1021/bi700555g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17(2):159–64. doi: 10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheikh S, Weiner H. Allosteric inhibition of human liver aldehyde dehydrogenase by the isoflavone prunetin. Biochem Pharmacol. 1997;53(4):471–8. doi: 10.1016/s0006-2952(96)00837-4. [DOI] [PubMed] [Google Scholar]
- 45.Wei B, Ni L, Hurley TD, Weiner H. Cooperativity in nicotinamide adenine dinucleotide binding induced by mutations of arginine 475 located at the subunit interface in the human liver mitochondrial class 2 aldehyde dehydrogenase. Biochemistry. 2000;39(18):5295–302. doi: 10.1021/bi000028n. [DOI] [PubMed] [Google Scholar]
- 46.Kuravsky ML, Barinova KV, Asryants RA, Schmalhausen EV, Muronetz VI. Structural basis for the NAD binding cooperativity and catalytic characteristics of sperm-specific glyceraldehyde-3-phosphate dehydrogenase. Biochimie. 2015;115:28–34. doi: 10.1016/j.biochi.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 47.Luengo JM, Garcia JL, Olivera ER. The phenylacetyl-CoA catabolon: a complex catabolic unit with broad biotechnological applications. Molecular microbiology. 2001;39(6):1434–42. doi: 10.1046/j.1365-2958.2001.02344.x. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Miller SJ, Hurley TD. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry. 2003;42(23):7100–9. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 49.Tsybovsky Y, Krupenko SA. Conserved catalytic residues of the ALDH1L1 aldehyde dehydrogenase domain control binding and discharging of the coenzyme. J Biol Chem. 2011;286(26):23357–67. doi: 10.1074/jbc.M111.221069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lorentzen E, Hensel R, Knura T, Ahmed H, Pohl E. Structural Basis of allosteric regulation and substrate specificity of the non-phosphorylating glyceraldehyde 3-Phosphate dehydrogenase from Thermoproteus tenax. Journal of molecular biology. 2004;341(3):815–28. doi: 10.1016/j.jmb.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 51.Langendorf CG, Key TL, Fenalti G, Kan WT, Buckle AM, Caradoc-Davies T, Tuck KL, Law RH, Whisstock JC. The X-ray crystal structure of Escherichia coli succinic semialdehyde dehydrogenase; structural insights into NADP+/enzyme interactions. PloS one. 2010;5(2):e9280. doi: 10.1371/journal.pone.0009280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan Z, Yin B, Wei D, Yuan YR. Structural basis for cofactor and substrate selection by cyanobacterium succinic semialdehyde dehydrogenase. Journal of structural biology. 2013;182(2):125–35. doi: 10.1016/j.jsb.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Dickinson FM, Hart GJ. Effects of Mg2+, Ca2+ and Mn2+ on sheep liver cytoplasmic aldehyde dehydrogenase. Biochem J. 1982;205(2):443–8. doi: 10.1042/bj2050443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ni L, Zhou J, Hurley TD, Weiner H. Human liver mitochondrial aldehyde dehydrogenase: three-dimensional structure and the restoration of solubility and activity of chimeric forms. Protein science : a publication of the Protein Society. 1999;8(12):2784–90. doi: 10.1110/ps.8.12.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi K, Weiner H. Magnesium stimulation of catalytic activity of horse liver aldehyde dehydrogenase. Changes in molecular weight and catalytic sites. J Biol Chem. 1980;255(17):8206–9. [PubMed] [Google Scholar]
- 56.Vallari RC, Pietruszko R. Interaction of Mg2+ with human liver aldehyde dehydrogenase. I. Species difference in the mitochondrial isozyme. J Biol Chem. 1984;259(8):4922–6. [PubMed] [Google Scholar]
- 57.Vallari RC, Pietruszko R. Interaction of Mg2+ with human liver aldehyde dehydrogenase. II. Mechanism and site of interaction. J Biol Chem. 1984;259(8):4927–33. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.