

Abstract
Positive strand RNA viruses are the largest group of RNA viruses on the planet and include many human, animal and plant pathogens. Cellular membranes are key players in all aspects of their life cycle, from entry and replication to assembly and exit. In particular, membranes are virally harnessed to serve as platforms for replication and as carriers to transmit viruses to other cells either as the envelope of a single virus or as the vesicle transporting a population of viruses. Studies have revealed significant convergence among different viruses to utilize membranes with specific lipid blueprints for replication and transmission. For replication many animal/human viruses utilize membranes enriched phosphatidylinositol 4-phosphate/cholesterol whereas many plant and insect-vectored animal viruses exploit ones with phosphatidylethanolamine/cholesterol. For transmission, phosphatidylserine enriched membranes are widely used as carriers for RNA and DNA viruses. Here we discuss the role of these membranes for viral pathogenesis and therapeutic development.
Keywords: virus, membrane, phosphatidylinositol 4-phosphate, phosphatidylethanolamine, phosphatidylserine, cholesterol, multivesicular body, autophagy, exosome, population
Introduction
Single positive strand or so called sense strand RNA [(+) ssRNA] viruses encompass many important pathogenic and non-pathogenic human, animal and plant viruses including poliovirus (PV), hepatitis C virus (HCV), rhinovirus, norovirus, Dengue virus, Zika virus, hoof and mouth disease, tobacco mosaic and tomato bushy stunt viruses. Perhaps more than any other family of viruses, they rely on intracellular membranes for all aspects of their lifecycle, from entry and replication to exit [1,2]. Once the sense strand RNA slips into the cytoplasm of the host cell, it is rapidly translated into structural and non-structural proteins. The non-structural proteins which make up the machinery for viral RNA replication are assembled on intracellular organelle membranes which then become platforms for viral RNA synthesis. Even though preexisting cellular organelles are harnessed, viral proteins, either alone or in conjunction with coopted host proteins, rapidly transform the protein/lipid composition and structure of these organelles to ones optimized for replication. Furthermore, after replication and assembly into new viral particles, all (+) ssRNA viruses can coopt membranes as carriers to be transported out of cells non-lytically and be transmitted to other susceptible hosts. These newly synthesized viruses can emerge as single membrane enveloped particles or as populations of viral particles, transported together to the next cell within membrane-bound vesicles [3,4]. In particular, the latter which enables trafficking of viral populations en masse from cell to cell can significantly enhance viral infectivity and replicative fitness [3,4].
Building viral replication organelles
Viral replication organelles originate from nearly every eukaryotic intracellular membrane. PV, Coxsackievirus B3 (CVB3), rhinovirus, norovirus, West Nile virus, Dengue Virus, Zika virus, HCV, hepatitis A virus (HAV) typically harness secretory pathway membranes including the endoplasmic reticulum (ER), the Golgi apparatus and the trans-Golgi network (TGN) [12,13] whereas rubella virus, brome mosaic virus (BMV) and tomato bushy stunt virus (TBSV) use the plasma membrane, the outer membrane of the mitochondria and peroxisomal membranes respectively [1,14]. Viral replication machinery is assembled on the cytoplasmic leaflet of organelle membranes prior to viral RNA synthesis. Replication on the surface of membrane platforms has several distinct advantages over replication in the cytoplasm. First, by being on a membrane, viral protein movement becomes restricted to a two dimensional plane, thus potentially saving time for proteins to find each other and form a complex, a significant advantage to getting replication going in the early stages of infection when viral proteins are a minority in the host cell [5]. Secondly, docking on membrane lipids can modulate enzymatic activity [6,7,8]. Lastly the architecture of the membranes can facilitate replication by generating pockets in which to concentrate viral machinery and protect from host innate immune defenses [2,9-11].
Electron microscopic studies of replication organelles reveal complex vesiculo-tubular structures with both positive and negative curvature membranes that resemble little the parental organelles from which they derived [9,15]. Host ESCRT proteins play critical roles in shaping replication organelle membranes during BMV and TBSV infections. For example, the combined actions of host ESCRT and BMV viral 1a proteins generate the negative curvature needed to form spherules in the outer mitochondrial membrane where subsequently BMV replication machinery is concentrated and BMV RNA is synthesized [16]. Similarly, in TBSV infections the peroxisomal membrane is remodeled into spherules by a combination of viral p33 and host ESCRT1 and ESCRTIII protein complexes [17]. Furthermore, host reticulon proteins are frequently recruited by viral machinery to stabilize the resulting positive curvature domains generated by negative curvature induction [18,19]. Notably the size of an individual membrane spherule is partly regulated by the size of the genomic RNA replicating within it. For example, spherules with a diameter of 66nm are generated when replicating full-length 4.8kB genomic TBSV RNA, whereas smaller spherules are generated with smaller replicating RNA molecules [17].
Hijacked host proteins appear to be also virally exploited to regulate access to and protect the viral replication complexes. In HCV and HAV infections, nuclear pore complex proteins have been shown to span replication organelle membrane tubule necks and impede nuclear localization signal (NLS) lacking innate immune effectors RIG-I and MDA5 from accessing replication sites [20] while letting through NLS containing viral replication proteins and accessory host nuclear factors including NF90 and NF110 [21]. This type of gating may also help regulate the levels of replication complexes at the organelles.
Phosphatidylinositol 4-phosphate (PI4P)- discovery of a panviral lipid required for replication
Replication organelles differ from their parental host organelles not only in terms of structure but also protein and lipid composition. Many different (+) ssRNA viruses appear to exploit a common replication organelle lipid blueprint. Using live cell imaging methodologies in combination with lipidomic and proteomic approaches, investigations into the lipid composition of PV, CVB3 and HCV replication organelles have revealed membranes that are highly enriched in phosphatidylinositol 4-phosphate (PI4P) lipids [22,23]. Significantly, these findings have been confirmed for the membranes of replication organelles formed during rhinovirus, echovirus, Aichi virus, enterovirus 71, and encephalomyocarditis virus (EMCV) infections among others [22-27] (Figure 1).
Figure 1. (+) ssRNA viruses exploit PE and PI4P lipid enriched organelles for replication.
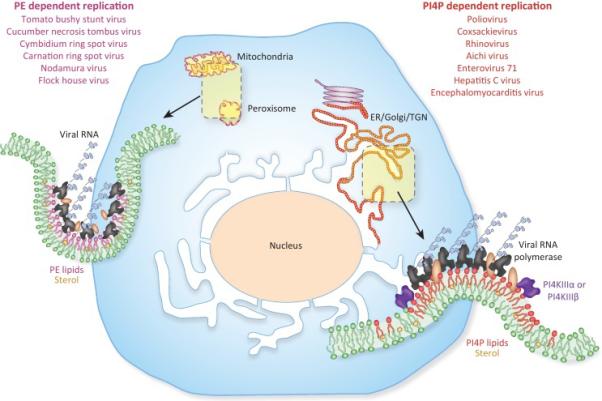
Many plant and insect vectored animal (+) ssRNA viruses utilize the surface of PE/cholesterol rich membranes for genome replication. Organelles with pre-existing pools of PE and cholesterol such as mitochondria and peroxisomes are hijacked and further enriched in these lipids. In contrast, many human and animal (+) ssRNA viruses appear to rely on PI4P and cholesterol enriched membranes for replication. Upon infection, these viruses hijack host Type III PI4 kinases and cholesterol trafficking pathways to transform the secretory pathway membranes (ER, Golgi, TGN) into replication organelles that are highly enriched in PI4P and cholesterol. The PE/cholesterol and PI4P/cholesterol enriched membranes facilitate viral RNA synthesis by helping dock and concentrate viral replication proteins; by stimulating viral enzymatic reactions; and by generating high curvature membrane pockets that can concentrate and segregate viral replication machinery from the host innate immune defenses.
Studies of PV and CVB3 infections, revealed PI4P to be generated in situ at replication organelles, through viral recruitment and stimulation of preexisting pools of host Type III phosphatidylinositol-4-kinase β (PI4KIIIβ) enzymes [22,28]. This resulted in nearly 6-fold increase in whole cell PI4P levels within a matter of hours post infection. PI4KIIIβ was also the source of PI4P lipids in human rhinovirus [7,26] and Aichi virus infections [24], whereas HCV and EMCV infections largely relied upon another member of the PI4K family, PI4KIIIα, to generate the PI4P lipids at their replication organelles [23,27-31].
It is notable that all of these viruses have evolved to harness type III PI4 kinases rather than type II PI4 kinases, which are also present in mammalian cells. Although both type II and type III enzymes catalyze PI4P production from phosphatidylinositol, type II enzymes are smaller and differ from type III enzymes in their catalytic domains [28]. Potentially the larger type III kinases afford more interaction sites for regulation by viral proteins. Type III kinases also have effectors, such as Rab11 (see below) that enable viruses to recruit other molecules, including other lipids such as cholesterol to replication organelle membranes. In addition, while there is some overlap in subcellular distribution, type II enzymes mainly localize to and regulate host endocytic pathways [28] and thus may provide a much smaller membrane surface area for establishing viral replication platforms then the secretory pathway membranes, not to mention maintaining endocytic pathways may be vital for the viral lifecycle, giving viruses access to extracellular nutrients and other factors.
Biogenesis of PI4P lipid enriched replication platforms
In all eukaryotic cells, secretory pathway membranes are highly dynamic as demonstrated by the Golgi apparatus: at steady state it is maintained through continual membrane flux, anterograde and retrograde, between itself and the ER. Arf1, a small ras family GTPase, plays a major role in the maintenance of organelles within the secretory pathway. Upon activation (by swapping GDP for GTP), it recruits to membranes of the ER, Golgi and TGN a variety of effectors including lipid modifying enzymes PI4KIIIβ and phospholipase D, membrane shaping coat proteins and their adaptors including coatomer, clathrin and GGA proteins, cytoskeletal proteins and regulators etc. The combined activities of these effectors create specialized membrane domains, sort, move and target cargo containing membranes, and in essence helping generate and maintain the secretory pathway [32-34]. While the spatio-temporal regulation of effector selection and recruitment is still not well understood [32-34], PV and CVB3 3A proteins were found to modulate this process [22,35]. Early in infection, when viral 3A levels are low, PV and CVB3 use preexisting Golgi and TGN membranes as sites of viral RNA synthesis presumably because their preexisting PI4P lipids levels are relatively high compared to other cellular organelles [22,36]. But increasing 3A production (due to positive feedback between viral RNA synthesis and viral RNA translation) results in a selective enhancement of PI4KIIIβ enzymes to Arf1 containing membrane sites which include the ER-Golgi intermediate compartment, the Golgi apparatus and the TGN, all the while suppressing the recruitment of Arf1 dependent membrane coat proteins and many other effectors [22]. Consequently, the dynamics of membrane trafficking among the secretory organelles is profoundly disrupted and the Golgi and TGN are eventually disassembled with the Golgi membranes being resorbed back into the ER. Without coat proteins and other Arf1 effectors to sort and traffic out secretory cargo and rebuild the secretory organelles, the membranes emerging from ER exit sites become instead organelles to replicate viral RNA, highly enriched in host PI4KIIIβ enzymes and PI4P lipids as well as viral replication proteins including the 3A protein and the viral RNA polymerase [22]. Remarkably ectopic expression of viral 3A proteins alone is sufficient to bring about this dramatic reorganization of the secretory pathway membranes [22] and is mediated by interactions among 3A, Arf1, the guanosine exchange factor GBF1 of Arf1 [22,35], the Golgi adaptor protein acyl-coenzyme A binding domain containing protein 3 (ACBD3) [37-39] and PI4KIIIβ [40]. In HCV infections the secretory pathway is largely unaffected and viral NS5A proteins recruit host PI4KIIIα enzymes directly to ER domains that lie outside of ER exit sites from where membranes are trafficked out to secretory organelles. Although Arf1 and GBF1 have also been implicated in HCV replication and nucleocapsid assembly their precise roles are currently unclear.
An unexplored area of investigation is what if any the impact of disrupting secretory pathway dynamics and/or high levels of PI4P lipids are on cellular physiology. In chronic or persistent viral infections, such as those observed with CVB3 or HCV, replication at some level is maintained over many years with higher levels of PI4P lipids being reported in infected tissues in vivo [23]. While in vivo secretory activity is largely unchanged for HCV, the degree of in vivo secretory perturbation is unknown for PV, CVB3 and rhinovirus infections and has been speculated that if present may be beneficial to viruses by inhibiting the cell surface presentation of MHC class I molecules to the immune system [41]. Alternatively, non-lytic and chronically infecting viruses may be able to maintain secretory activity in vivo and still obtain sufficient quantities of PI4P lipids to sustain replication by potentially setting up a negative feedback loop among PI4K enzyme recruitment/activation, secretory suppression and viral RNA synthesis/viral protein translation pathways, where the latter will become attenuated when PI4P levels get too high and/or secretion is blocked.
Type III PI4 kinases as panviral therapeutic targets
The exploitation of host Type III PI4 kinases by multiple different viruses, makes them attractive panviral therapeutic candidates [29] and targeting a host protein rather than a viral protein, decreases the likelihood of developing resistance quickly. Indeed, in vitro studies, acutely inhibiting PI4KIIIβ or PI4KIIIα kinases with small molecules such as PIK93, depleting them from the host cell, overexpressing kinase dead mutants or ectopically expressing phosphatases to lower PI4P levels, have all been shown to be successful in blocking viral RNA synthesis in many different types of infections [22,23,31,42,43], while not significantly affecting cell viability. The latter is likely due to compensation of host PI4P needs by redundant enzymatic activities provided by other, such as type II, PI4 kinase family members [28]. In contrast to in vitro studies, results from in vivo studies have been harder to interpret. In some studies, animals treated with PIK93 or closely related small molecules, while protected from viral replication, have manifested gut epithelial toxicity and lymphoid cell proliferation while other studies have found little to no adverse effects [42,44]. Some of the toxicities observed appear to be due to cross reactivity of PIK93 or its structural analogs with Class III PI3 kinases and Class 1B PI3Kγ, whose catalytic sites are closely related to type III PI4 kinase catalytic sites [45]. Given the potential significance of developing a single panviral therapeutic that can be effective against many types of viral infections, it will be important to continue to develop and test new type III PI4K inhibitors such as the recently developed class of PI4KIIIβ inhibitors that have significantly less cross reactivity with other lipid kinases [45].
Other panviral lipids regulating viral replication
Phosphatidylethanolamine
Many plant as well as some insect-vectored animal (+) ssRNA viruses appear to be dependent on phosphatidylethanolamine (PE) enriched membranes for replication and depletion of PE significantly inhibits viral RNA synthesis [6,8, 46,47]. The plant viruses TBSV, Cymbidium ring spot virus and Cucumber Necrosis Tombus virus all generate their PE enriched replication organelles from primarily peroxisome membranes while Carnation Ring Spot virus and the insect-vectored animal viruses Nodamura and flock house exploit the mitochondrial outer membrane for this purpose [6, 47]. In uninfected cells, both peroxisomes and mitochondria already are already high in PE but PE levels are further enhanced upon infection, largely as a consequence of redistribution from other subcellular organelles. Although the mechanism of this redistribution is unclear, a single TBSV protein, p33, is sufficient to accomplish it [46], akin to the roles played by 3A proteins in PV and CVB3 infections [22] and NS5A proteins in HCV infections [23].
PE is one of the most abundant lipid species in bacterial membranes. To date only very few bacterial (+) ssRNA viruses have been discovered, mostly members of the Leviviridae family. While the Leviviridae Qβ replicase can replicate RNA in solution in vitro it will be important to determine whether in vivo its activity is facilitated by membranes (i.e., bacterial cytoplasmic membrane) and particularly PE lipids. Although the pool of investigated plant and insect-vectored animal (+) ssRNA viruses is small, findings point to a preference for PE enriched membranes as replication platforms (and hence use of mitochondrial and peroxisomal membranes) (Figure 1). It remains to be determined if PE preference is more widespread among these viruses and whether it suggests an evolutionary proximity between these viruses and current or ancestral bacterial (+) ssRNA viruses.
Cholesterol
Frequently cholesterol is also found enriched along with PI4P or PE at replication organelles [7,48-50]. Cholesterol is well known to regulate protein and lipid packing and recent investigations have revealed that it is required for the formation of large (i.e., more than a few nanometers) and stable phosphoinositide or PE enriched lipid domains [51]. Unlike phospholipids, cholesterol does not have a large head group protruding from the membrane and is hypothesized to act as a spacer to reduce the intermolecular electrostatic repulsive forces among phosphoinositide lipids [51,52] as well as ordering acyl chains of PE lipids and thereby stabilizing the negative curvature membrane domains induced by it [53] (Figure 1). Consistent with this idea, depleting cholesterol from PV, CVB3 or rhinovirus replication organelles results in smaller PI4P enriched replication organelles and significantly reduced viral RNA synthesis [7].
In PV, CVB3, rhinovirus and echovirus infected cells, >90% of the plasma membrane cholesterol pools are redistributed to the replication organelles within 2 hours of infection [7]. These cholesterol pools are internalized and transferred to Rab11 positive recycling endosomes, which then traffic to replication organelles to deliver the cholesterol cargo [7,50]. Rab11 is itself an effector of PI4KIIIβ in all eukaryotic cells [54] and physical complex formation between Rab11 and PI4KIIIβ is found to be highly enhanced (> 4 fold) over uninfected cells during infection with PV or CVB3, even though the levels of either Rab11 or PI4KIIIβ are unchanged [7]. Thus by increasing the levels of PI4KIIIβ at replication organelles and exploiting the existing affinity between PI4KIIIβ and Rab11, these viruses appear to target cholesterol filled recycling endosomes to ER-based replication organelles [7,54]. These findings raise the possibility of the development of novel therapeutics that disrupt the interaction interface between PI4KIIIβ and Rab11, rather than targeting catalytic domains and as such they may exhibit less toxicity in vivo.
Non-vesicular cholesterol transfer proteins, Osh and oxysterol binding protein (OSBP), also play roles in enriching for cholesterol at viral replication organelles [55]. Osh proteins have been shown to transfer cholesterol to TBSV replication sites and stimulate replication [47,56] and OSBP depletion leads to some inhibition of HCV, PV, CVB3 and rhinovirus replication [25,57]. OSBP proteins have been previously demonstrated in uninfected cells to swap PI4P for cholesterol, in order to transport cholesterol against its concentration gradient [58]. Similarly, in PI4P-dependent viral infections, OSBP proteins may also facilitate the transfer of cholesterol against its concentration gradient, from cholesterol rich Rab11 endosomes to replication organelles, in exchange for PI4P from the latter. Since PI4P is produced in large quantities continually throughout infection [22], its levels at the replication sites would likely not be significantly depleted. Further support for feedback between PI4P and cholesterol levels during infection comes from inhibition of PI4KIIIα recruitment in HCV infected cells, which leads to a decrease in both PI4P and cholesterol pools at the replication organelles [48].
Lipid regulation of viral RNA synthesis
Although much remains to be known about how PI4P, PE, and cholesterol facilitate viral replication, recent findings have begun to shed some light. Though positive strand RNA replication takes place on membranes, many viral replication components including the RNA dependent RNA polymerases (RdRp) of PV, CVB3, rhinovirus, flock house virus and TBSV are soluble proteins. Thus it has been suggested that specific lipids may facilitate membrane recruitment of these proteins [22]. Investigations with PV and CVB3 RNA polymerases have revealed a highly selective binding site for PI4P lipids [22] and studies with TBSV replication proteins have also shown that the viral p33 and p92 RNA polymerase binding to PE helps them form a membrane associated complex [46]. In addition, the clustered PI4P or PE lipid enriched domains may facilitate concentration and even polymerization of viral replication proteins that in turn can enhance viral RNA synthesis. Indeed, electron microscopic studies of isolated replication organelles from PV infected cells have revealed two-dimensional membrane associated viral RNA polymerase arrays [59].
Secondly PI4P and PE has been shown to modulate viral enzymatic activities. The autocatalytic cleavage of the RNA priming PV 3CDpol proteins, which are the precursors of the polymerase (3Dpol) and protease (3C), is significantly attenuated when it is bound to PI4P lipids [7]. As RNA priming is a pre-requirement for RNA elongation, docking 3CDpol proteins on PI4P lipids may be a mechanism by which these viruses regulate the rate of proteolytic cleavage (in cis or trans) and thus ensure that there will always be sufficient levels of primed viral RNA for elongation. Similarly, binding to PE lipids has been shown to directly stimulate the enzymatic activity of TBSV RNA polymerase’ and facilitate their association with viral RNA [8] whereas binding to phosphatidylglycerol molecules can inhibit TBSV polymerases enzymatic activities [47].
While ESCRT and reticulon proteins play important roles in sculpting replication organelles, lipids themselves either alone or through interactions with host and viral proteins can also induce particular organelle morphologies that are potentially conducive to replication. For example, in PI4P lipid enriched membrane monolayers, the large headgroup to acyl chain ratio of PI4P lipids bends the monolayer away from the headgroups to form positive curvature membrane domains, whereas the conical PE lipids with their small headgroups bend the membrane monolayer towards each other to form negative membrane curvature domains (Figure 1) [60]. Along with host PI4P binding FAPP2 proteins, which are found at replication organelles, the PI4P enriched positive curvature tubular domains may be further pronounced and stabilized [61] to generate complex three dimensional membrane bound spaces that both concentrate as well as spatially segregate viral replication machinery. Indeed, depleting either PI4P lipids or FAPP2 proteins from PV infected cells dramatically alters the morphology of replication organelles and inhibits viral replication [23,31,62].
Widespread role for membranes in facilitating non-lytic viral exit and transmission
Convention has long dictated that enveloped viruses (i.e. those with a membrane around their nucleocapsids) escape the cell by budding out from the surface or by budding into an exocytic compartment and that cell lysis is not a pre-requisite for their exit and/or transmission to the next cell, although lysis may occur eventually due to stress from the viral infection or being cleared by the immune system. Instead, for so-called non-enveloped viruses such as PV, CVB3, rhinovirus, norovirus, rotavirus, HAV (among many others), cell lysis has always been considered to be a prerequisite for exit and transmission. However, recent discovery of an enveloped form of HAV, in cell culture and in sera from infected patients, has blurred this separation between enveloped and non-enveloped viruses [63]. HAV after replication and assembly on endoplasmic reticulum derived membranes has been shown to be internalized in multivesicular bodies (MVB) and released to the extracellular environment, in small vesicles, so-called exosomes (Figure 2) [63]. This MVB to plasma membrane pathway exists in uninfected cells as well [64] and is an important source of extracellular vesicles for cellular communication at a distance [64,65]. However, in contrast to the conventional MVB to lysosome maturation pathway, this plasma membrane targeted secretory MVB pathway is poorly understood and much of what is known is focused on the processes regulating the biogenesis of the exosomes at the MVBs (e.g. ESCRT, tetraspannins proteins) rather than their release, with the exception of a potential role for Rab27 GTPases [64]. Soon after the discovery of an exosomal HAV, other non-enveloped viruses, including Hepatitis E virus (HEV), PV, CVB3 and rhinovirus [3,66-68], were also found to be released from cells in extracellular vesicles and all prior to cell lysis [3,67]. The discovery of non-enveloped viral particles released from cells non-lytically and transmitted in membrane-bound vesicles, much like bona fide enveloped viral particles, suggest that membranes have a universally central role in viral exit and transmission.
Figure 2. RNA viruses exit cells in PS lipid enriched membranes as single or populations of viral particles.
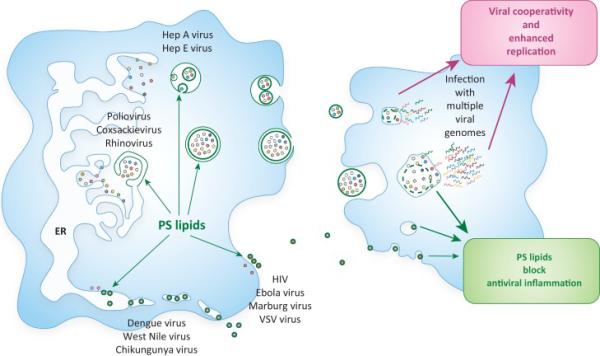
Both enveloped and non-enveloped viruses exploit PS-enriched membranes to be released from cells either as single particles (Dengue, West Nile, Ebola, VSV, HIV) or as populations (PV, CVB3, rhinovirus, HAV, HEV). PS lipids are obtained by budding into the ER; by budding off from the plasma membrane; or by being captured in secretory autophagosomes and multivesicular bodies (MVB) and subsequently released to the outside in extracellular vesicles. The PS-enriched membranes can potentially suppress a widespread, adaptive immune response against the virus. In addition, enhanced infection efficiency is observed by viruses traveling as populations within vesicles (e.g. PV, CVB3, rhinovirus [3]). This is potentially due to a vesicle being able to simultaneously deliver multiple viral genomes into the host cell, allowing subsequent cooperative interactions to take place among viral quasispecies that benefit replication.
While HEV much like HAV appears to harness secretory MVB's [63,66], PV, Coxsackievirus and rhinovirus all exploit the host secretory autophagy pathway to exit cells (Figure 2) [3,67-69]. Autophagy is primarily a degradative pathway, triggered in times of cellular stress, where cytoplasmic cargo are selectively captured within double-membraned autophagosomes, which can originate from a variety of cellular sources including the endoplasmic reticulum [70,71], mitochondria [72] and even the plasma membrane [73]. Autophagy is a highly regulated process with cargo receptors for specific types of cargo [71]. In canonical autophagy once autophagosomes have formed, they typically fuse with lysosomes and their contents are degraded into protein, lipid and carbohydrate building blocks for the cell [70,71]. However, in PV and CVB3 infected cells, the newly assembled viral particles at the PI4P/cholesterol rich replication organelles are captured within ER-derived autophagosomes [3] and rather than fusing with lysosomes they instead are trafficked to and fuse with the plasma membrane [3]. PV carrying autophagosomes appear to lack at least one of the SNARE machinery, syntaxin 17, required for lysosomal fusion [3]. Fusion with the plasma membrane results in releasing a vesicle (the inner membrane bound compartment of the former autophagosome) filled with infectious virions to the extracellular space. The secretory autophagy pathway is also exploited in uninfected cells to secrete IL1β [74,75], synuclein [76], amyloid β protein [77], bone morphogens and mineralizing agents [78] and even whole intact organelles, the latter in reticulocytes undergoing maturation into red blood cells [79]. Nevertheless, almost nothing is known regarding what molecular machinery regulates whether an autophagosome becomes degradative or secretory and how plasma membrane targeting and fusion is accomplished [74]. It will be important to determine also if both secretory and degradative pathways co-exist in PV and CVB3 infected cells and if any molecular cues exist on viral proteins that allow selective recognition by one pathway over another.
Once the vesicles carrying viral cargo are released, the subsequent infection is virus-receptor dependent and not simply mediated by membrane-membrane fusion between the vesicle and host cell as at least for HAV, PV, CVB3 and rhinovirus pre-incubating the susceptible cells with virus receptor blocking antibodies can inhibit infection [3,63]. This suggests that the vesicle membranes are ruptured, during or after uptake into the new host cell, as prior to that the viruses are also resistant to neutralizing antibodies [63,80], the latter a potentially significant immune evasion mechanism for this mode of transmission by non-enveloped viruses. Nevertheless, this raises the important question of how this membrane is eventually ruptured. Recent evidence points to endosomal lipases and cholesterol transfer proteins such as the Niemann-Pick transporter playing a role in the process [80]. In addition, the viral cargo itself could facilitate this rupture as many non-enveloped viral capsid proteins including the adenovirus protein VI, rotavirus VP5 and the VP4 proteins from rhinovirus and HAV, under acidified conditions (such as those that would be encountered within endosomes), undergo conformational changes that are sufficient to penetrate and disrupt membranes [81-84].
Phosphatidylserine lipids play a panviral role in facilitating viral infectivity
Anionic PS lipids are a major component of the host lipid repertoire. After de novo synthesis from PC and/or PE at the ER, they can remain in the ER, be transferred to the mitochondria [85,86], or to the inner leaflet of the plasma membrane, the latter in exchange for PI4P lipids [87-89]. From the plasma membrane they can then be trafficked to endosomes, MVBs and lysosomes. PS lipids are present in the membrane of many bona fide enveloped (+) and (−) ssRNA viruses including Dengue, West Nile and Chikungunya, Ebola, Marburg and Vesiculo Stomatitis virus (VSV); retroviruses including human immune deficiency virus (HIV); and even DNA viruses such as Vaccinia virus (Figure 2) [90]. They are also enriched in the membranes carrying non-enveloped viral particles including the secretory autophagosomes carrying PV, CVB3 and rhinovirus populations as well as the extracellular vesicles derived from them [3] and the exosomes carrying HAV and HEV (Figure 2) [85]. While enveloped viruses pick up PS lipids by budding from the plasma membrane or into the ER lumen [90]; the PS lipids of secretory autophagosomes potentially originate from increased PS synthesis at the ER/replication organelles; from increased trafficking of PS from the plasma membrane to the ER/replication organelles, possibly with the Rab11 carriers containing sterols [7]; and/or by decreased transfer of PS from the ER to the plasma membrane as a consequence of high PI4P levels in the former [22].
Presence of PS lipids in the membranes of many non-enveloped and enveloped viruses, suggest there may be a significant role for them in viral pathogenesis. Indeed, PS lipids are well known potent anti-inflammatory agents that modulate the immune responses of the host [91]. As part of normal cell turn over in the body, apoptotic cells are recognized and phagocytosed by both professional phagocytes (e.g. macrophages, monocytes) and non-professional phagocytes (e.g. epithelial cells, fibroblasts). Apoptotic cells are sensed by phagocytes because of their exposure of PS lipids from the inner leaflet to the outer leaflet of the plasma membrane. These exposed PS lipids are recognized by PS receptors on phagocytes, either through direct binding such as with the TIM1 receptor or by indirect binding via bridging molecules to AXL receptors [91-93]. This results in phagocytic uptake of PS-enriched membranes and in professional phagocytic cells like macrophages leads to trigger of an anti-inflammatory response [92,93]. Growing evidence suggests that PS-enriched enveloped and non-enveloped viruses exploit this pathway, so-called apoptotic mimicry [94] to be internalized [3, 90]. Indeed, blocking TIM, AXL and other PS receptors or masking PS with PS-binding proteins such as Annexin V significantly inhibits binding, endocytosis and infection by Chikungunya, Dengue, West Nile Virus, Ebola, Marburg, Arenaviruses, PV, CVB3 and rhinovirus [3,90]. But in addition to facilitating internalization, the panviral exploitation of PS lipids may be to suppress inflammation and a wider immune response to the virus. In vivo studies, in the context of the immune system will be needed to test this hypothesis. In addition to internalization and anti-inflammation, PS lipids and perhaps other molecular cues on the vesicles, may also play a role in determining viral tropism in the host.
The presence of PS lipids across the membranes of so many different viruses also provides an opportunity for the development of panviral therapeutics targeting these lipids. Bavituximab, a PS-targeting moclonal antibody has shown success in blocking infectivity of PS-enriched enveloped virions such as Ebola and Lassa fever viruses in in vitro assays and in small animal studies (soares 2008; Dowall and Hewson 2015). Further studies, including those with non-enveloped virions will be needed to evaluate its full clinical potential.
En masse viral transmission and increased infection efficiency due to extracellular membrane vesicles
Finally, the exploitation of membranes for exit by non-enveloped viruses has also led to reexamining the long held belief that viruses only travel between and infect cells as single infectious units. Remarkably, each secretory autophagosome derived vesicle was found to carry a population of infectious PV, CVB3 or rhinovirus particles at once [3,4,65]. Given a single PV particle is ~ 30nm in size, an autophagosome of ~500nm diameter can in theory pack many hundreds of PV particles. Even smaller vesicles, such as exosomes which are typically ~100nm in diameter, have the capacity to traffic multiple HAV or HEV viral particles [63]. While this novel type of viral transmission was first reported with (+) ssRNA viruses [3], recently a giant DNA virus, Marseille virus, has been discovered to also traffic between cells within micrometer vesicles, with each vesicle carrying hundreds of viral particles at once [95].
One of the implications of this novel type of viral transmission is that it enables single cells to be infected simultaneously with multiple viral particles (Figure 2). Significantly this type of viral transmission appears to yield greater infection efficiency and higher replication rates in a given population of cells compared to infection with similar numbers of free independent viral particles [3,4,65,95]. For RNA viruses in general, collective travel and infection may be an important mechanism to overcome the drawbacks of errors during replication, due to many viral RNA polymerases lacking proofreading, that result in heterogeneous populations of viral quasispecies [96]. Even small genetic differences among quasispecies can manifest themselves, in the next infection cycle, in differing rates of RNA translation, RNA synthesis, particle assembly, resistance to innate immune defenses, etc. [97,98]. While these mutations might determine the replicative fate of viral particle infecting a cell alone, delivered in a vesicle as part of a population, cooperative interactions among quasispecies including sharing genetic templates, replication machinery, structural proteins and innate immune defense modulators, could yield greater overall survival and long term fitness for these genomes [98] (Figure 2). Future studies will determine which types of viral cooperative interactions can enhance replicative fitness, what the relative contributions of cooperativity versus competition are and whether vesicular travel of viral populations, compared to free viral particles, leads to greater genetic diversity over time. Finally, there may be an opportunity for developing new forms of therapeutics that decrease viral replicative fitness and emergence of drug resistance by either blocking the cellular membrane trafficking pathways that enable populations of viruses to be captured and released (i.e., secretory autophagy, secretory MVBs) or by disrupting the integrity of the vesicle membranes after release from cells.
Concluding Remarks
Recent investigations have revealed the novel ways by which membranes are central to the viral lifecycle: from facilitating viral genome synthesis to enabling viral populations to be transported out of cells and enhancing viral infectivity. Future studies will determine the extent of dependence on specific lipids among viruses, including PI4P, PE, PS and others for replication and transmission; evaluate the long term impact of manipulating lipid metabolism on cell physiology; and identify the machinery regulating secretory autophagy and secretory MVB pathways (see Outstanding Questions). Progress in these areas will not only lead to novel antiviral therapeutics but also tools for manipulating these pathways in uninfected cells, in particular for those that regulate the production of extracellular vesicles, which are critical for cell to cell communication and tissue remodeling. Finally, with the revolutionary new tools in microscopy, lipidomics, proteomics and genetics, this is a very exciting time to be studying viruses, which so often ends up being the study of cellular life.
Outstanding Questions.
- How widespread is PI4P/cholesterol and PE/cholesterol dependence among viruses?
- How does PI4P or PE docking regulate viral enzymatic activities?
- What is the long term impact of viral manipulation of lipid metabolism on cells?
-Do PS lipids in the membranes surrounding exiting viruses - either as a single particle or as a population of particles- impact the in vivo host immune response?
- What are the cellular and viral components regulating secretory autophagy and secretory MVB pathways?
- How are viral particles recognized by these pathways?
-How do the extracellular vesicles carrying viral particles transfer the viral genomes to the cells?
- What types of viral cooperative interactions enhance replicative fitness?
-What are the relative contributions of cooperativity versus competition?
-Does vesicular transmission of viral populations, compared to free viral particles, lead to greater genetic diversity?
Trends Box.
RNA viruses utilize membranes with specific lipid blueprints for replication and transmission.
Phosphatidylinositol 4-phosphate, phosphatidylethanolamine and cholesterol are critical lipids exploited by many RNA viruses to facilitate replication.
Membranes are critical for both enveloped and non-enveloped RNA viruses for exit and transmission.
Membrane vesicles can transport multiple viral particles together from one cell to another–a novel type of transmission which enhances infection efficiency.
Phosphatidylserine, a potent anti-inflammatory molecule, is frequently enriched in the membranes exploited for exit and transmission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol. 2008;6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Boon JA, Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol. 2010;64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 3.Chen YH, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, Brantner CA, Stempinski ES, Connelly PS, Ma HC, Jiang P, Wimmer E, Altan-Bonnet G, Altan-Bonnet N. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–30. doi: 10.1016/j.cell.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altan-Bonnet N, Chen YH. Intercellular Transmission of Viral Populations with Vesicles. J Virol. 2015;89:12242–12244. doi: 10.1128/JVI.01452-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCloskey MA, Poo MM. Rates of membrane-associated reactions: reduction of dimensionality revisited. J Cell Biol. 1986;102:88–96. doi: 10.1083/jcb.102.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu SX, Ahlquist P, Kaesberg P. Active complete in vitro replication of nodavirus RNA requires glycerophospholipid. Proceeding of the National Academy of Sciences. 1992;89:11136–11140. doi: 10.1073/pnas.89.23.11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, Dixon JL, Altan-Bonnet N. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe. 2013;14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pogany J, Nagy PD. Activation of Tomato bushy stunt virus RdRp by cellular Hsp70 is enhanced by phospholipids in vitro. J Virol. 2015:JVI.03711–14. doi: 10.1128/JVI.03711-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNAin intracellular membrane vesicles. J Virol. 2010;84:8470–8483. doi: 10.1128/JVI.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belov GA. Modulation of lipid synthesis and trafficking pathways by picornaviruses. Curr Opin Virol. 2014;9:19–23. doi: 10.1016/j.coviro.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Romero-Brey I, Bartenschlager R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses. 2016;8(6):E160. doi: 10.3390/v8060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz A, Ahlquist P. Role of host reticulon proteins in rearranging membranes for positive-strand RNA virus replication. Curr Opin Microbiol. 2012;15:519–524. doi: 10.1016/j.mib.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Short JR, Speir JA, Gopal R, Pankratz LM, Lanman J, Schneemann A. Role of Mitochondrial Membrane Spherules in Flock House Virus Replication. J Virol. 2016;90:3676–3683. doi: 10.1128/JVI.03080-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz A, Zhang J, Ollwerther A, Wang X, Ahlquist P. Host ESCRT Proteins Are Required for Bromovirus RNA Replication Compartment Assembly and Function. PLoS Pathog. 2015;11:e1004742. doi: 10.1371/journal.ppat.1004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovalev N, de Castro Martín IF, Pogany J, Barajas D, Pathak K, Risco C, Nagy PD. Role of Viral RNA and Co-opted Cellular ESCRT-I and ESCRT-III Factors in Formation of Tombusvirus Spherules Harboring the Tombusvirus Replicase. J Virol. 2016;90:3611–3626. doi: 10.1128/JVI.02775-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu MJ, Ke PY, Hsu JT, Yeh CT, Horng JT. Reticulon 3 interacts with NS4B of the hepatitis C virus and negatively regulates viral replication by disrupting NS4B self-interaction. Cell Microbiol. 2014;16:1603–1618. doi: 10.1111/cmi.12318. [DOI] [PubMed] [Google Scholar]
- 19.Diaz A, Wang X, Ahlquist P. Membrane-shaping host reticulon proteins play crucial roles in viral RNA replication compartment formation and function. Proc Natl Acad Sci U S A. 2010;107:16291–16296. doi: 10.1073/pnas.1011105107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neufeldt CJ, Joyce MA, Van Buuren N, Levin A, Kirkegaard K, Gale M, Jr, Tyrrell DL, Wozniak RW. The Hepatitis C Virus-Induced Membranous Web and Associated Nuclear Transport Machinery Limit Access of Pattern Recognition Receptors to Viral Replication Sites. PLoS Pathog. 2016;12:e1005428. doi: 10.1371/journal.ppat.1005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. Nuclear factors are involved in hepatitis C virus RNA replication. RNA. 2007;13:1675–1692. doi: 10.1261/rna.594207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Bühler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa-Sasaki K, Sasaki J, Taniguchi K. A complex comprising phosphatidylinositol 4-kinase IIIβ, ACBD3, and Aichi virus proteins enhances phosphatidylinositol 4-phosphate synthesis and is critical for formation of the viral replication complex. J Virol. 2014;88:6586–6598. doi: 10.1128/JVI.00208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roulin PS, Lötzerich M, Torta F, Tanner LB, van Kuppeveld FJ, Wenk MR, Greber UF. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe. 2014;16:677–690. doi: 10.1016/j.chom.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Spickler C, Lippens J, Laberge MK, Desmeules S, Bellavance É , Garneau M, Guo T, Hucke O, Leyssen P, Neyts J, Vaillancourt FH, Décor A, O'Meara J, Franti M, Gauthier A. Phosphatidylinositol 4-kinase III beta is essential for replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob Agents Chemother. 2013;57:3358–3368. doi: 10.1128/AAC.00303-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorobantu CM, Albulescu L, Harak C, Feng Q, van Kampen M, Strating JR, Gorbalenya AE, Lohmann V, van der Schaar HM, van Kuppeveld FJ. Modulation of the Host Lipid Landscape to Promote RNA Virus Replication: The Picornavirus Encephalomyocarditis Virus Converges on the Pathway Used by Hepatitis C Virus. PLoS Pathog. 2015;11:e1005185. doi: 10.1371/journal.ppat.1005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altan-Bonnet N, Balla T. Phosphatidylinositol 4-kinases: hostages harnessed to build panviral replication platforms. Trends Biochem Sci. 2012;37:293–302. doi: 10.1016/j.tibs.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, Leighton-Davies J, Wilson CJ, Myer V, Cornellataracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, Bouwmeester T, Mathy JE, Lin K, Compton T, Labow M, Wiedmann B, Gaither LA. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol. 2009;83:10058–10074. doi: 10.1128/JVI.02418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trotard M, Lepère-Douard C, Régeard M, Piquet-Pellorce C, Lavillette D, Cosset FL, Gripon P, Le Seyec J. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 2009;23:3780–3789. doi: 10.1096/fj.09-131920. [DOI] [PubMed] [Google Scholar]
- 31.Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J Virol. 2011;85:8870–8883. doi: 10.1128/JVI.00059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, Luini A, Corda D, De Matteis MA. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. 1999;1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- 33.Haynes LP, Sherwood MW, Dolman NJ, Burgoyne RD. Specificity, promiscuity and localization of ARF protein interactions with NCS-1 and phosphatidylinositol-4 kinase-III beta. Traffic. 2007;8:1080–1092. doi: 10.1111/j.1600-0854.2007.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cherfils J. Arf GTPases and their effectors: assembling multivalent membrane-binding platforms. Curr Opin Struct Biol. 2014;29:67–76. doi: 10.1016/j.sbi.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Wessels E, Duijsings D, Niu TK, Neumann S, Oorschot VM, de Lange F, Lanke KH, Klumperman J, Henke A, Jackson CL, Melchers WJ, van Kuppeveld FJ. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev Cell. 2006;11:191–201. doi: 10.1016/j.devcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Van der Schaar HM, Melia CE, van Bruggen JA, Strating JR, van Geenen ME, Koster AJ, Bárcena M, van Kuppeveld FJ. Illuminating the Sites of Enterovirus Replication in Living Cells by Using a Split-GFP-Tagged Viral Protein. mSphere. 2016;1:e00104–16. doi: 10.1128/mSphere.00104-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J Virol. 2012;86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasaki J, Ishikawa K, Arita M, Taniguchi K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2013;31:754–766. doi: 10.1038/emboj.2011.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klima M, Tóth DJ, Hexnerova R, Baumlova A, Chalupska D, Tykvart J, Rezabkova L, Sengupta N, Man P, Dubankova A, Humpolickova J, Nencka R, Veverka V, Balla T, Boura E. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci Rep. 2016;6:23641. doi: 10.1038/srep23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorobantu CM, Ford-Siltz LA, Sittig SP, Lanke KH, Belov GA, van Kuppeveld FJ, van der Schaar HM. GBF1- and ACBD3-independent recruitment of PI4KIIIβ to replication sites by rhinovirus 3A proteins. J Virol. 2015;89:1913–1918. doi: 10.1128/JVI.02830-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deitz SB, Dodd DA, Cooper S, Parham P, Kirkegaard K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci U S A. 2000;97:13790–13795. doi: 10.1073/pnas.250483097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Schaar HM, Leyssen P, Thibaut HJ, de Palma A, van der Linden L, Lanke KH, Lacroix C, Verbeken E, Conrath K, Macleod AM, Mitchell DR, Palmer NJ, van de Poël H, Andrews M, Neyts J, van Kuppeveld FJ. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIβ. Antimicrob Agents Chemother. 2013;57:4971–4981. doi: 10.1128/AAC.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacLeod AM, Mitchell DR, Palmer NJ, Van de Poël H, Conrath K, Andrews M, Leyssen P, Neyts J. Identification of a series of compounds with potent antiviral activity for the treatment of enterovirus infections. ACS Med Chem Lett. 2013;4:585–589. doi: 10.1021/ml400095m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamarche MJ, Borawski J, Bose A, Capacci-Daniel C, Colvin R, Dennehy M, Ding J, Dobler M, Drumm J, Gaither LA, Gao J, Jiang X, Lin K, McKeever U, Puyang X, Raman P, Thohan S, Tommasi R, Wagner K, Xiong X, Zabawa T, Zhu S, Wiedmann B. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob Agents Chemother. 2012;56:5149–5156. doi: 10.1128/AAC.00946-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rutaganira FU, Fowler ML, McPhail JA, Gelman MA, Nguyen K, Xiong A, Dornan GL, Tavshanjian B, Glenn JS, Shokat KM, Burke JE. Design and Structural Characterization of Potent and Selective Inhibitors of Phosphatidylinositol 4 Kinase IIIβ. J Med Chem. 2016;59:1830–1839. doi: 10.1021/acs.jmedchem.5b01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu K, Nagy PD. RNA virus replication depends on enrichment of phosphatidylethanolamine at replication sites in subcellular membranes. Proc Natl Acad Sci U S A. 2015;112:E1782–E1791. doi: 10.1073/pnas.1418971112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagy PD, Pogany J, Xu K. Cell-Free and Cell-Based Approaches to Explore the Roles of Host Membranes and Lipids in the Formation of Viral Replication Compartment Induced by Tombusviruses. Viruses. 2016;8:68. doi: 10.3390/v8030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reghellin V, Donnici L, Fenu S, Berno V, Calabrese V, Pagani M, Abrignani S, Peri F, De Francesco R, Neddermann P. NS5A inhibitors impair NS5A-phosphatidylinositol 4-kinase IIIα complex formation and cause a decrease of phosphatidylinositol 4-phosphate and cholesterol levels in hepatitis C virus-associated membranes. Antimicrob Agents Chemother. 2014;58:7128–7140. doi: 10.1128/AAC.03293-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma M, Sasvari Z, Nagy PD. Inhibition of sterol biosynthesis reduces tombusvirus replication in yeast and plants. J Virol. 2010;84:2270–2281. doi: 10.1128/JVI.02003-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Albulescu L, Wubbolts R, van Kuppeveld FJ, Strating JR. Cholesterol shuttling is important for RNA replication of coxsackievirus B3 and encephalomyocarditis virus. Cell Microbiol. 2015 doi: 10.1111/cmi.12425. doi: 10.1111/cmi.12425. [DOI] [PubMed] [Google Scholar]
- 51.Jiang Z, Redfern RE, Isler Y, Ross AH, Gericke A. Cholesterol stabilizes fluid phosphoinositide domains. Chem Phys Lipids. 2014;182:52–61. doi: 10.1016/j.chemphyslip.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kooijman EE, King KE, Gangoda M, Gericke A. Ionization properties of phosphatidylinositol polyphosphates in mixed model membranes. Biochemistry. 2009;48(40):9360–9371. doi: 10.1021/bi9008616. [DOI] [PubMed] [Google Scholar]
- 53.Giang H, Schick M. How cholesterol could be drawn to the cytoplasmic leaf of the plasma membrane by phosphatidylethanolamine. Biophys J. 2014;107:2337–2344. doi: 10.1016/j.bpj.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burke JE, Inglis AJ, Perisic O, Masson GR, McLaughlin SH, Rutaganira F, Shokat KM, Williams RL. Structures of PI4KIIIβ complexes show simultaneous recruitment of Rab11 and its effectors. Science. 2014;344:1035–1038. doi: 10.1126/science.1253397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Schaar HM, Dorobantu CM, Albulescu L, Strating JR, van Kuppeveld FJ. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trends Microbiol. 2016;24:535–546. doi: 10.1016/j.tim.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barajas D, Xu K, Sharma M, Wu CY, Nagy PD. Tombusviruses upregulate phospholipid biosynthesis via interaction between p33 replication protein and yeast lipid sensor proteins during virus replication in yeast. Virology. 2014;471-473:72–80. doi: 10.1016/j.virol.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology. 2014;146:1373–1385. doi: 10.1053/j.gastro.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 59.Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science. 2002;296:2218–2222. doi: 10.1126/science.1070585. [DOI] [PubMed] [Google Scholar]
- 60.McMahon HT, Boucrot E. Membrane curvature at a glance. J Cell Science. 2015;128:1065–1070. doi: 10.1242/jcs.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lenoir M, Coskun U, Grzybek M, Cao X, Buschhorn SB, James J, Simons K, Overduin M. Structural basis of wedging the Golgi membrane by FAPP pleckstrin homology domains. EMBO Rep. 2010 Apr. 2010;11(4):279–284. doi: 10.1038/embor.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan I, Katikaneni DS, Han Q, Sanchez-Felipe L, Hanada K, Ambrose RL, Mackenzie JM, Konan KV. Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J Virol. 2014;88(21):12276–12295. doi: 10.1128/JVI.00970-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367–371. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 65.Altan-Bonnet N. Extracellular vesicles are the Trojan horses of viral infection. Curr Opin Microbiol. 2016;32:77–81. doi: 10.1016/j.mib.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagashima S, Jirintai S, Takahashi M, Kobayashi T, Tanggis, Nishizawa T, Kouki T, Yashiro T, Okamoto H. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J Gen Virol. 2014;95:2166–2175. doi: 10.1099/vir.0.066910-0. [DOI] [PubMed] [Google Scholar]
- 67.Bird SW, Maynard ND, Covert MW, Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci U S A. 2014;111:3081–3086. doi: 10.1073/pnas.1401437111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, Nguyen DP, Sayen MR, Hilton BJ, Doran KS, Segall AM, Wolkowicz R, Cornell CT, Whitton JL, Gottlieb RA, Feuer R. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015:25354–25363. doi: 10.1016/j.tcb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ktistakis NT, Tooze SA. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016:S0962–8924(16)00045-3. doi: 10.1016/j.tcb.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 72.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J, Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK. Lippincott-Schwartz J. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. doi: 10.1016/j.ceb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife. 2015;4:e11205. doi: 10.7554/eLife.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ejlerskov P, Rasmussen I, Nielsen TT, Bergström AL, Tohyama Y, Jensen PH, Vilhardt F. Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. doi: 10.1074/jbc.M112.401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–69. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 78.Rosenthal AK, Gohr CM, Mitton-Fitzgerald E, Grewal R, Ninomiya J, Coyne CB, Jackson WT. Autophagy modulates articular cartilage vesicle formation in primary articular chondrocytes. J Biol Chem. 2015;290:13028–13038. doi: 10.1074/jbc.M114.630558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mankelow TJ, Griffiths RE, Trompeter S, Flatt JF, Cogan NM, Massey EJ, Anstee DJ. Autophagic vesicles on mature human reticulocytes explain phosphatidylserine-positive red cells in sickle cell disease. Blood. 2015;126:1831–1834. doi: 10.1182/blood-2015-04-637702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yin X, Ambardekar C, Lu Y, Feng Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J Virol. 2016;90:4232–4242. doi: 10.1128/JVI.02804-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Denisova E, Dowling W, LaMonica R, Shaw R, Scarlata S, Ruggeri F, Mackow ER. Rotavirus capsid protein VP5* permeabilizes membranes. J Virol. 1999;73:3147–3153. doi: 10.1128/jvi.73.4.3147-3153.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panjwani A, Strauss M, Gold S, Wenham H, Jackson T, Chou JJ, Rowlands DJ, Stonehouse NJ, Hogle JM, Tuthill TJ. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog. 2014;10:e1004294. doi: 10.1371/journal.ppat.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shukla A, Padhi AK, Gomes J, Banerjee M. The VP4 peptide of hepatitis A virus ruptures membranes through formation of discrete pores. J Virol. 2014;88:12409–12421. doi: 10.1128/JVI.01896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wiethoff CM, Nemerow GR. Adenovirus membrane penetration: Tickling the tail of a sleeping dragon. Virology. 479. 2015;480:591–9. doi: 10.1016/j.virol.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, Grinstein S, Parton RG. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol. 2011;194:257–275. doi: 10.1083/jcb.201012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 87.Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P. INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science. 2015;349:428–432. doi: 10.1126/science.aab1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moser von Filseck J, Čopič A, Delfosse V, Vanni S, Jackson CL, Bourguet W, Drin G. INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science. 2015;349:432–436. doi: 10.1126/science.aab1346. [DOI] [PubMed] [Google Scholar]
- 89.Sohn M, Ivanova P, Brown HA, Toth DJ, Varnai P, Kim YJ, Balla T. Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions. Proc Natl Acad Sci U S A. 2016;113:4314–4319. doi: 10.1073/pnas.1525719113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amara A, Mercer J. Viral apoptotic mimicry. Nat Rev Microbiol. 2015;13:461–469. doi: 10.1038/nrmicro3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, Freimark B, Empig C, Mercer J, Schroit AJ, Schett G, Herrmann M. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23:962–978. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Henson PM, Bratton DL. Antiinflammatory effects of apoptotic cells. J Clin Invest. 2013;123:2773–2774. doi: 10.1172/JCI69344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. 2015;33:355–391. doi: 10.1146/annurev-immunol-032414-112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 95.Arantes TS, Rodrigues RA, Silva LK, Oliveira GP, de Souza HL, B Khalil JY, Oliveira DB, Torres AA, da Silva LL, Colson P, Kroon EG, da Fonseca FG, Bonjardim CA, La Scola B, Abrahão JS. The large marseillevirus explores different entry pathways by forming giant infectious vesicles. J Virol. 2016:JVI.00177–1. doi: 10.1128/JVI.00177-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Andino R, Domingo E. Viral quasispecies. Virology. 479. 2015;480:46–51. doi: 10.1016/j.virol.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bordería AV, Isakov O, Moratorio G, Henningsson R, Agüera-González S, Organtini L, Gnädig NF, Blanc H, Alcover A, Hafenstein S, Fontes M, Shomron N, Vignuzzi M. Group Selection and Contribution of Minority Variants during Virus Adaptation Determines Virus Fitness and Phenotype. PLoS Pathog. 2015;11:e1004838. doi: 10.1371/journal.ppat.1004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Combe M, Garijo R, Geller R, Cuevas JM, Sanjuán R. Single-Cell Analysis of RNA Virus Infection Identifies Multiple Genetically Diverse Viral Genomes within Single Infectious Units. Cell Host Microbe. 2015;18:424–432. doi: 10.1016/j.chom.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]