

Abstract
ErbB2 is a transmembrane tyrosine kinase whose surface overexpression is linked to tumorigenesis and poor prognosis in breast cancer patients. Two models have emerged that account for the high surface distribution of ErbB2. In one model, the surface pool is dynamic and governed by a balance between endocytosis and recycling, whereas in the other it is retained, static, and excluded from endocytosis. These models have contrasting implications for how ErbB2 exerts its biological function and how cancer therapies might down-regulate surface ErbB2, such as the antibody trastuzumab (Herceptin) or the Hsp90 inhibitor geldanamycin. Little is known, however, about how these treatments affect ErbB2 endocytic trafficking. To investigate this issue, we examined breast carcinoma cells by immunofluorescence and quantitative immunoelectron microscopy and developed imaging and trafficking kinetics assays using cell surface fluorescence quenching. Surprisingly, trastuzumab does not influence ErbB2 distribution but instead recycles passively with internalized ErbB2. By contrast, geldanamycin down-regulates surface ErbB2 through improved degradative sorting in endosomes exclusively rather than through increased endocytosis. These results reveal substantial dynamism in the surface ErbB2 pool and clearly demonstrate the significance of endosomal sorting in the maintenance of ErbB2 surface distribution, a critical feature of its biological function.
INTRODUCTION
ErbB2 (Her2) is a transmembrane tyrosine kinase belonging to a surface receptor family that includes epidernal growth factor receptor (EGFR; Yarden and Sliwkowski, 2001). ErbB2 does not bind ligand, but instead acts as a preferred heterodimerization partner for ligand-activated sibling members to amplify mitogenic signaling. These distinctive properties of ErbB2 are explained structurally by the “open” conformation of its extracellular domain, which mimics the activated ligand-bound state of EGFR (Garrett et al., 2003). This structure appears not to lead readily to ErbB2 self-association and ligand-independent activation, which occurs only with high surface overexpression or in the context of a transmembrane domain Val to Glu point mutation neu. Both of these conditions are oncogenic (Bargmann et al., 1986; Di Fiore et al., 1987).
In human breast cancer, the ErbB2 gene is amplified and overexpressed in ∼25% of cases and is associated with poor prognosis (Slamon et al., 1987). Several mAbs to the ErbB2 extracellular domain have been described that inhibit ErbB2-transformation in vitro and have antitumor properties in vivo (Drebin et al., 1985; Hudziak et al., 1989; Hurwitz et al., 1995; Klapper et al., 1997). Many of these mAbs are proposed to trigger surface down-regulation via bivalent antibody-driven ErbB2 transactivation and endocytic degradation. In 1998 the FDA approved a recombinant humanized form of one such antibody, trastuzumab, for the treatment of ErbB2-overexpressing breast cancer (Cobleigh et al., 1999; Slamon et al., 2001). A different class of mAbs instead disrupts ligand-activated mitogenic signaling (Klapper et al., 1997; Agus et al., 2002). A recombinant humanized version of one such antibody, pertuzumab (Omnitarg), blocks the ErbB2 dimer interface and is currently in phase II clinical trials (Franklin et al., 2004).
Another treatment for ErbB2-overexpressing cancer under clinical study is inhibition of the chaperone Hsp90 by the benzoquinoid ansamycin antibiotic geldanamycin (GA) or its derivatives (Isaacs et al., 2003). The potent antitumor activity of these compounds is due to destabilization of Hsp90 client proteins, key components of growth and survival signaling pathways. Surface ErbB2 is particularly sensitive to GA and undergoes several changes that precede its degradation, including ubiquitination, fragmentation of the cytosolic domain, and extensive internal redistribution (Mimnaugh et al., 1996; Tikhomirov and Carpenter, 2000). Hence endocytic trafficking of ErbB2 is likely to be important in the mechanism of action of multiple ErbB2-directed cancer therapies.
Despite its significance to cancer therapy, little is known about ErbB2 endocytosis. Although antibody studies suggested that ErbB2 might exhibit inducible endocytic degradation akin to that of EGF-stimulated EGFR, subsequent chimeric receptor studies showed that EGFR is unique in this regard and that all other ErbB receptors including ErbB2 have no such property (Sorkin et al., 1993; Baulida et al., 1996). Moreover, ErbB2 potentiates EGFR signaling by diverting it from EGF-induced down-regulation (Harari and Yarden, 2000, and references within). Two contrasting models of ErbB2 endocytosis have emerged to account for its high surface distribution and protective effect on EGFR down-regulation. In one model, ErbB2 efficiently recycles after basal endocytosis and diverts activated heterodimeric EGFR to a similar fate (Harari and Yarden, 2000; Hendriks et al., 2003). In a second model, ErbB2-containing dimers are retained on the cell surface and excluded from endocytosis (Wang et al., 1999; Hommelgaard et al., 2004). Though both models imply that ErbB2 endocytic trafficking is key to normal function and perhaps overexpression-induced oncogenesis, they have contrasting implications for how a therapeutic antibody or GA might down-regulate surface ErbB2. The first model suggests that modulating endosomal sorting might suffice, whereas the second model implies a block on endocytosis must be relieved. It remains to be established, however, whether antibodies or GA redistribute and degrade ErbB2 through increased endocytosis, enhanced endosomal sorting to the lysosomal pathway, or both.
To understand endocytic trafficking of ErbB2 and how cancer therapy might alter or exploit this process, we undertook detailed morphologic and kinetic studies in human breast carcinoma cells. To overcome the obstacle posed by ErbB2's predominant cell surface distribution, we developed a surface fluorescence-quenching technique that enables quantification and visualization of ErbB2 endocytic trafficking despite inefficient internalization. We find that the surface ErbB2 pool is not static but instead dynamic, undergoing basal endocytosis combined with rapid, efficient recycling. In contrast to previous observations (Hudziak et al., 1989; Kumar et al., 1991; Baselga and Albanell, 2001; Cuello et al., 2001; Citri et al., 2002), we find that trastuzumab does not down-regulate surface ErbB2 but instead efficiently recycles with the receptor after endocytosis, demonstrating that surface down-regulation is not important to its mechanism of action. We also identify the site of action of GA on ErbB2 endocytic trafficking functionally and ultrastructurally and show that GA acts exclusively through improved lysosomal sorting from the endosome without influencing its rate of endocytosis from the surface.
MATERIALS AND METHODS
Materials and Cell Lines
GA was obtained from Calbiochem (San Diego, CA). Trastuzumab, pertuzumab, mAb 7C2, and recombinant ErbB2 extracellular domain (ErbB2-ECD) were generated at Genentech and have been described previously (Fendly et al., 1990; Kelley et al., 1992). Two rabbit antibodies reactive to the cytoplasmic tail of ErbB2 used in immunoblotting and immunoelectron microscopy were obtained from Santa Cruz Biotechnology (C-18, Santa Cruz, CA) and Dako (Carpinteria, CA), respectively. mAbs recognizing CD63, Lamp1, and Lamp2 used in immunofluorescence microscopy were obtained from the Developmental Studies Hybridoma Bank at the University of Iowa, and those against CD63 used in immunoelectron microscopy, transferrin (Tf) receptor (TfR), and actin (C4) were from CLB (Amsterdam, The Netherlands), Zymed (San Francisco, CA), and MP Biomedicals (Aurora, OH), respectively. mAb to γ-adaptin (mAb 100.3) was kindly provided by E. Ungewickell (Hannover Medical School). Fluorescent reagents and rabbit polyclonal anti-Alexa-488 IgG were obtained from Molecular Probes (Eugene, OR). HRP-conjugated anti-mouse and anti-rabbit IgG were obtained from Jackson ImmunoResearch (West Grove, PA). 125I-trastuzumab was prepared by the Assay and Reagent Chemistry Group at Genentech by the lactoperoxidase method (specific activity ∼15 μCi/μg, 125I/protein molar ratio ∼1). ErbB2 antibodies were fluorescent labeled [488-] by reaction with Alexa-488 succinimidyl ester according to the manufacturer's instructions. Gold-conjugated 1-nm Nanogold (NG) trastuzumab was prepared using the monomaleimido Nanogold labeling reagent from Nanoprobes (Yaphank, NY) according to the manufacturer's instructions (labeling ratio = 0.93 NG/antibody). A silver enhancing kit, R-GENT SE-EM, was purchased from Aurion (Wageningen, The Netherlands). Most cell lines were obtained from ATCC (Rockville, MD). A stably ErbB2-transfected human breast cell line (MCF7-ErbB2 sublone 18) was described previously (Benz et al., 1993). Cell seeding was performed 18–24 h in advance and used at 50–80% confluency unless otherwise specified.
125I-trastuzumab Uptake and Catabolism
Cells in six-well plates were incubated 60 min on ice with 0.8 nM 125I-trastuzumab and then washed extensively and transferred to 37°C for the indicated times. At each time point, the medium was collected and separated into TCA-precipitable and -nonprecipitable fractions. Cell surface radioactivity was stripped three times on ice with 2 M urea, 50 mM glycine, 150 mM NaCl, pH 2.4, for 5 min each, and supernatants were pooled. Cells were then lysed with 8 M urea, 150 mM NaCl, pH 2.4. Radioactivity of the four fractions from each time point was quantified in a gamma counter.
Indirect Immunofluorescence Confocal Microscopy
Cells on poly-l-lysine–coated multiwell glass slides were surface-labeled on ice 60 min with 1 μg/ml trastuzumab or pertuzumab in binding medium (20 mM HEPES, 3% BSA, in serum-free DME, pH 7.4). For a signal specificity control, the antibody was first preincubated 15 min at 37°C with 40 μg/ml recombinant ErbB2-ECD. Cells were washed three times and incubated at 37°C for the indicated time with or without 5 μg/ml Alexa-594–conjugated (594-) Tf and/or 1 μM GA. Cells with lysosomal protease inhibitors were preincubated 1–2 h 37°C with 100 μg/ml leupeptin and 5 nM pepstatin A. Cells were fixed in 3% PFA in PBS, treated with 50 mM NH4Cl, 0.1 M glycine in PBS, and then permeabilized (0.4% saponin, 1% BSA, and 2% normal goat serum in PBS). All subsequent primary antibody incubations, fluorescent-conjugated secondary antibody incubations, and wash steps were performed in the saponin blocking buffer. Cells were imaged with a Leica TCS SP laser scanning confocal microscope (Bannockburn, IL) equipped with a Leica DM R microscope and a HCX PL APO 63×/1.32–0.6 NA oil immersion objective lens.
Down-regulation of Surface ErbB2
Cells on six-well plates at ∼20% confluency were incubated at 37°C for the indicated intervals in growth medium with or without continuous presence of 10 μg/ml trastuzumab and/or 1 μM GA. Cells were then incubated on ice for 15 min with 0.05% trypsin, 5.3 mM EDTA in PBS, treated with an equal volume of FBS, and removed with a rubber policeman. Cells were sedimented at 800 × g and analyzed either by flow cytometry or immunoblotting. For flow cytometry, cells were incubated 1 h on ice with 488–7C2, a fluorescent labeled noncompetitive mAb to ErbB2-ECD (Fendly et al., 1990). Cells were then washed and fixed in 2% PFA, and mean fluorescence of 10,000 cells was quantified using a Beckman Coulter Epics XL-MCL single argon laser flow cytometer (Fullerton, CA). For immunoblotting, cells were either 1) lysed on ice for 15 min with 1% TX-100, 25 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, 5 mM EDTA, and a protease inhibitor tablet (Roche, Mannheim, Germany), followed by centrifugation for 10 min at 20,000 × g and addition of the supernatant to an equal volume of gel loading buffer containing 10% SDS and 10 mM DTT, or 2) lysed directly in gel loading buffer followed by microprobe sonication. Samples were boiled 5 min, resolved by SDS-PAGE (4–20%), and transferred to nitrocellulose, which was subsequently cut into horizontal strips and probed as indicated for the ErbB2 carboxy terminus, γ-adaptin, or actin. Strips were then probed with HRP-conjugated secondary antibodies, and signals were detected by enhanced chemoluminescence (Pierce, Rockford, IL).
Uptake and Endocytosis Assays Using Cell Surface Fluorescence Quenching
SKBr3 cells on six-well plates were preincubated for 60 min at 37°C with or without 1 μM GA, surface-labeled for 60 min on ice with 1 μg/ml 488-trastuzumab or 488-pertuzumab in binding medium, and then washed three times with cold binding medium and incubated at 37°C for the indicated intervals to internalize surface fluorescence. In some dual color experiments, 5 μg/ml Alexa 647–conjugated (647-) Tf was included in both steps. Cells were rapidly chilled, detached and sedimented as described above, and incubated on ice 30 min with (quenched samples) or without (nonquenched samples) 25 μg/ml or the indicated concentration of anti-Alexa 488 IgG in PBS with 3% FBS and 1 μg/ml propidium iodide. Cells were then inactivated by adding ∼2 volumes of cold 2% PFA/PBS and immediately analyzed by flow cytometry. Mean fluorescence intensity of 10,000 cells gated for viability by exclusion of propidium iodide (routinely 70–80% viable) was quantified by flow cytometry as described above or, for dual color experiments, a Beckman Coulter Cytomics FC 500 dual argon ion/red helium–neon flow cytometer. Internalized fluorescence was calculated from quenched and nonquenched sample data after correcting for incomplete surface quenching as described in Supplementary Figure 1. We determined surface antibody and Tf endocytic rate constants as described previously by generating non–steady state internalization plots (Lund et al., 1990) or steady state internal/surface plots (Wiley and Cunningham, 1982), respectively. Briefly, non–steady state internalization plots were generated by plotting internalized fluorescence vs. the time integral of the surface fluorescence S (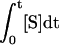 ) calculated by the trapezoidal rule: (Δt/2)([S]t0 + 2[S]t1 + 2[S]t2 +... + 2[S]tn-1 + [S]tn). Use of the time integral of the surface fluorescence rather than time removes the influence of surface antibody depletion that occurs during the course of the assay on uptake kinetics to provide a more intrinsic measure of antibody internalization. Surface antibody depletion in these assays however was very small (<10% depletion by minute 4).
) calculated by the trapezoidal rule: (Δt/2)([S]t0 + 2[S]t1 + 2[S]t2 +... + 2[S]tn-1 + [S]tn). Use of the time integral of the surface fluorescence rather than time removes the influence of surface antibody depletion that occurs during the course of the assay on uptake kinetics to provide a more intrinsic measure of antibody internalization. Surface antibody depletion in these assays however was very small (<10% depletion by minute 4).
Recycling Assay Using Cell Surface Fluorescence Quenching
SKBr3 cells preincubated at 37°C for 60 min with or without 1 μM GA were surface-labeled and incubated (pulsed) 10 min at 37°C as described for endocytosis assays above. In some single-color experiments, we instead pulsed with 5 μg/ml 488-Tf. Cells were rapidly chilled, surface-quenched on ice 30 min with 25 μg/ml anti-Alexa 488 IgG and/or 0.5 mg/ml unlabeled holotransferrin, and then warmed to 37°C (chased) in continuous presence of the quenching agents for the indicated intervals. Cells were then rapidly chilled, detached, surface-quenched, and analyzed by flow cytometry as described above. Percentage of internalized fluorescence (pulse) remaining at each chase time point was calculated as the difference between pulsed and nonpulsed cell fluorescence, normalized to this value at chase time = 0. During preparation of our manuscript, a similar surface-quenching technique was described for measuring recycling kinetics of the cation-independent mannose 6-phosphate receptor and furin (Schapiro et al., 2004; Lin et al., 2004).
Quantitative Immunoelectron Microscopy
Cells in 25-cm2 flasks were surface-labeled on ice with 1–10 μg/ml trastuzumab or 1 μg/ml NG-trastuzumab in binding medium and then incubated for the specified times at 37°C with or without 1 μM GA as described for confocal microscopy above. Cells were fixed in either 4% PFA or 2% PFA/0.2% glutaraldehyde and processed for immunogold labeling of ultrathin cryosections as described previously (Slot et al., 1991). Alternatively, NG-trastuzumab in ultrathin cryosections was silver-enhanced for 40–80 min (control sections were negative). For quantification of trastuzumab and ErbB2 immunogold labeling, at least 10 cell profiles containing a nucleus were randomly sampled on single labeled sections, and all gold particles within 25 nm of a particular membrane were scored as localized to that membrane. The frequency of ErbB2 and TfR label in coated vs. uncoated endosomal limiting membrane domains was determined by pooling data from 25 randomly sampled endosomal vacuoles. Only those endosomes that labeled positive for ErbB2 or TfR and containing at least 1 internal vesicle or a bilayered coat were counted. This procedure was repeated three times for each condition for calculation of SEM. All comparisons were tested for statistical significance by the Student's t test.
Static or Live Cell Fluorescence Imaging after Surface Quenching
SKBr3 cells on poly-l-lysine–coated glass slides or 35-mm Bioptechs TC3 environmental imaging dishes (Butler, PA) were surface-labeled with 488-trastuzumab and pulsed 5–10 min at 37°C in the absence or presence of 5 μg/ml 594-Tf or 2 μg/ml biotinylated EGF complexed to Texas Red streptavidin (TR-EGF). For static imaging, cells were rapidly chilled on ice and surface-quenched as described above. Cells were then fixed in 3% PFA/PBS, processed for indirect immunofluorescence using 594-anti-rabbit IgG, and imaged using an Applied Precision (Issaquah, WA) deconvolution microscopy system built on an Olympus IX-70 inverted microscope (Lake Success, NY) with a PL APO 60×/1.40 NA oil immersion objective lens and equipped with a Sedat Quad filter set (Chroma, Rockingham, VT) with single band excitation and emission filters, a CH350 high-resolution cooled charge coupled device camera (Applied Precision). For live cell imaging, pulse incubations were performed on a Bioptechs temperature controlled microscope stage, and chase incubations initiated without chilling by rinsing twice and adding quenching antibody in prewarmed HBSS, 20 mM HEPES, pH 7.4. Imaging was on the Deltavision system equipped with a Bioptechs objective heater using the time-lapse acquisition mode.
RESULTS
Surface Trastuzumab Accumulates Intracellularly and Is Degraded Slowly in Human Breast Cancer Cells
To examine kinetics of trastuzumab uptake and degradation in human breast carcinoma SKBr3 cells, surface ErbB2-bound 125I-trastuzumab was internalized at 37°C for various intervals and then fractionated into surface-bound, intracellular, supernatant protein-associated (dissociated), and catabolized radioactivity (Figure 1). Surface clearance of 125I-trastuzumab was very slow (T1/2 ∼11 h) and attributed to both surface dissociation (0.22% after 24 h) and degradation (0.47 after 24 h). Surface clearance not attributed to dissociation was slow and similar to catabolism kinetics (T1/2 ∼19 h). This rate does not exceed previously published turnover rates for naked ErbB2 in this cell line (Hudziak et al., 1989; Harwerth et al., 1992). Intracellular radioactivity reached a steady state within 2–4 h and then gradually decreased. Overall, this kinetic profile is similar to those of 125I-4D5 (the nonhumanized version of trastuzumab) and 125I-7C2 (a ErbB2 mAb that recognizes a distinct epitope; De Santes et al., 1992). These results indicate that surface-bound trastuzumab is not a static pool, but accumulates in cells susceptible to the growth inhibitory effects of trastuzumab, albeit with slow kinetics.
Figure 1.

Trastuzumab is slowly internalized and degraded in target breast cancer cells. SKBr3 cells were surface-labeled with 125I-trastuzumab on ice, incubated at 37°C for the indicated times, and then assayed for cell surface, internalized, and supernatant TCA-precipitable (dissociated) and TCA-nonprecipitable (catabolized) radioactivity as described in Materials and Methods.
As anticipated from the internalization kinetics above, internalized trastuzumab was detected by immunofluorescence confocal microscopy most clearly after 3 h at 37°C (Figure 2A, second panel). Detection required cell permeabilization and therefore represented accumulation in endosomes rather than on surface subdomains (unpublished data). Both surface and endosomal signals were ErbB2-specific and completely competed away by preincubating trastuzumab with an excess of recombinant ErbB2-ECD before exposure to cells (Figure 2A, third panel). If endosomal trastuzumab was attributed to ErbB2-specific rather than nonspecific antibody uptake, the signal should increase in response to GA, a drug that triggers redistribution of surface ErbB2 to endosomes and degradation (Mimnaugh et al., 1996; Tikhomirov and Carpenter, 2000). Indeed, this is what we found (Figure 2A, fourth panel).
Figure 2.

Surface-bound trastuzumab is internalized and costains with markers of recycling endosomes. (A) Surface-bound trastuzumab was internalized in BT474 cells for 0 or 3 h at 37°C with or without 1 μM GA as indicated and processed for confocal immunofluorescence microscopy. To evaluate signal specificity, trastuzumab was preincubated with excess recombinant ErbB2-ECD before exposure to cells. (B) Surface-bound trastuzumab was internalized in BT474 cells for 3 h at 37°C in the presence of lysosomal protease inhibitors with or without 594-Tf as indicated. Cells were then fixed and processed for dual-label indirect immunofluorescence confocal microscopy. (C) Experiments were performed as in B, but in the presence of 1 μM GA. Results were similar in the absence of protease inhibitors and in experiments with SKBr3 cells (unpublished data). In separate experiments, transferrin recycling after 3 h continuous incubation at 37°C was very efficient (Supplementary Figure 2). Scale bars, 20 μM.
Internalized Trastuzumab Colocalizes with Markers of the Recycling Pathway
Internalized trastuzumab colocalized with markers of recycling endosomes after 3-h uptake, including internalized 594-Tf (Figure 2B, top row) and not with markers of late endosomes and lysosomes, including CD63 (Figure 2B, middle row), Lamp2 (Figure 2B, bottom row), and Lamp1 (unpublished data). In the presence of GA, costaining with recycling endosomal markers was still significant, but costaining with late endosome/lysosome markers was also observed (Figure 2C, compare top row to bottom row). Similar results were obtained using pertuzumab (unpublished data), an antibody that recognizes a distinct epitope and has a different antitumor mechanism of action (Agus et al., 2002).
The Mechanism of Trastuzumab Action Does Not Involve Surface ErbB2 Down-regulation
The above results suggest that recycling of the trastuzumab-ErbB2 complex may account for the sustained plasma membrane levels and contradict the view that trastuzumab inhibits growth through efficient down-regulation of ErbB2. To examine this issue directly, surface ErbB2 was assayed using a fluorescent noncompetitive mAb in flow cytometric measurements (Figure 3A). Trastuzumab had no significant effect on surface ErbB2 levels in SKBr3 cells during the first day of incubation, after which a small 5–10% decline was established. These results were confirmed by immunoblotting experiments (Figure 3B). Trastuzumab also failed to rapidly down-regulate surface ErbB2 in human gastric carcinoma N87 cells (unpublished data). By contrast, GA treatment rapidly and efficiently down-regulated surface ErbB2 after a lag period of 30–60 min in both SKBr3 and human breast MCF7 cells stably transfected with ErbB2 (Figures 3, C and D, respectively). GA-induced down-regulation was only slightly faster in the presence of trastuzumab.
Figure 3.

Treatment with GA but not trastuzumab down-regulates surface ErbB2. Adherent SKBr3 cells (A and C) or MCF7-ErbB2 cells (D) were incubated at 37°C in the absence or continuous presence of 10 μg/ml trastuzumab with or without 1 μM GA treatment for the indicated time and then detached and assayed for surface ErbB2 by flow cytometry as described in Materials and Methods. (B) Adherent SKBr3 cells were incubated and detached as in A, lysed with either SDS or TX-100–containing buffer and equivalent amounts of protein immunoblotted as described in Materials and Methods.
The above results demonstrate that trastuzumab does not significantly affect surface ErbB2 level and refute the idea that surface down-regulation underlies its mechanism of action. These results also suggest that trastuzumab has little effect on ErbB2 endocytic trafficking and might therefore be a useful tool to examine this process. Indeed by immunofluorescence microscopy, we observed no significant effect of either trastuzumab or pertuzumab on ErbB2 steady state distribution after 3 h by immuno-epifluorescence (Figure 4, compare middle and bottom rows to top row), or quantitative immunoelectron microscopy (see below).
Figure 4.

Steady state ErbB2 distribution is not significantly affected by trastuzumab or pertuzumab. SKBr3 cells were incubated with 647-Tf for 3.5 h at 37°C in the absence (top row) or presence of 10 μg/ml 488-trastuzumab (middle row) or 488-pertuzumab (bottom row). Cells were then fixed and processed for indirect immuno-epifluorescence microscopy using an antibody recognizing the cytoplasmic tail of ErbB2 (second panels). Scale bars, 20 μM.
Surface Fluorescence Quenching Reveals Trastuzumab Uptake and Efficient Endocytic Recycling
The above data suggest that trastuzumab recycles after endocytosis. If recycling is efficient, the rate of trastuzumab endocytosis may be substantially faster than implied by the slow uptake observed in Figures 1 and 2. We therefore developed pulse-chase methods to measure trastuzumab endocytosis and recycling directly. To this end, a variety of acid-stripping conditions were explored, none of which removed surface trastuzumab without deleterious effects on cells (unpublished data). As an alternative, we used fluorescent-conjugated antibodies, 488-trastuzumab and 488-pertuzumab, and removed surface fluorescence with a quenching antibody that recognizes the Alexa-488 moiety (Zheng and Zagotta, 2000). Surface quenching quantified by flow cytometry was 80–90% efficient for 488-trastuzumab (Figure 5A) and 90–95% for 488-pertuzumab (unpublished data). Internalization kinetics of surface-bound 488-trastuzumab or 488-pertuzumab were measured in SKBr3 cells by pulsing at 37°C for various intervals, surface quenching on ice, and then quantifying by flow cytometry. Figure 5B shows that the resulting uptake curves for 488-trastuzumab and 488-pertuzumab are similar and biphasic, with initial rapid uptake followed by deceleration to a slower phase after 4–8 min. Experiments with the corresponding 488-Fab fragments were complicated by weaker construct fluorescence and more labile cell surface binding and required longer incubations for reliable uptake data. These experiments showed similar though somewhat diminished internalization at 120 min compared with the IgG forms (Figure 5C). Uptake was the same regardless of whether or not soluble IgG or Fab was continually present during the pulse, as fluid-phase uptake measured in the presence of excess recombinant ErbB2-ECD or unlabeled IgG or Fab was undetectable (unpublished data).
Figure 5.

SKBr3 cell surface fluorescence quenching facilitates ErbB2 antibody uptake measurements and imaging. (A) Cells were surface-labeled on ice with 488-trastuzumab, washed, and then detached on ice (no 37°C incubation) as described in Materials and Methods. Cells were then sedimented and resuspended with the indicated concentrations of anti-Alexa-488 IgG on ice. Mean fluorescence intensity was measured by flow cytometry as described in Materials and Methods. Results were normalized to that of the control (nonquenched) sample. (B) Cells were surface-labeled with 488-trastuzumab or 488-pertuzumab, washed, and incubated at 37°C for the indicated intervals to internalize the surface-bound fluorescence. Cells were then rapidly chilled, detached, sedimented, and surface-quenched on ice with 25 μg/ml anti-Alexa 488 IgG, and % internalization calculated from flow cytometric data as described in Materials and Methods. Inset: rapid phase (0–5 min) portion of the data. (C) Cells were incubated continuously for 120 min with soluble 488-trastuzumab, 488-pertuzumab, or their corresponding 488-Fab fragments and then processed as in B to calculate % internalized. (D) Cells with surface-bound 488-trastuzumab were pulsed at 37°C for 5 min, rapidly chilled, surface-quenched, PFA-fixed, and then processed for immunofluorescence microscopy to detect surface quenching antibody using 647-anti-rabbit IgG as described in Materials and Methods. For comparison, the left panel inset shows 488-trastuzumab after a 15-min pulse but no surface quenching (20-fold lower exposure). Scale bars, 20 μM.
The substantial deceleration observed after a few minutes of uptake is consistent with the proposal that internalized trastuzumab efficiently recycles after endocytosis. Two other potential explanations we considered were 1) commencement of degradation and 2) deceleration of the endocytic rate due to saturation of the endocytic machinery. We eliminated degradation because it is not significant until several hours of incubation (see Figure 1), and inclusion of effective concentrations of lysosomal protease inhibitors did not alter the uptake profile (unpublished data). To determine if efficient recycling rather than reduced endocytosis accounted for the biphasic uptake profile, we measured recycling directly in pulse-chase experiments. After a 10-min internalization pulse with 488-trastuzumab, cells were surface-quenched on ice, returned to 37°C (chased) in continuous presence of quenching antibody, and then detached and analyzed by flow cytometry. Accordingly as internalized conjugate recycled to the surface during the chase, it was quenched resulting in decreased cell fluorescence over time (Figure 6A, •). Recycling was rapid and efficient, with ∼50% recycled within 5 min and 85% within 30 min. By comparison, Tf recycling was somewhat slower, with 50% recycled within ∼10 min (Figure 6, B and C, •). 488-Tf recycling kinetics measured by the quenching technique was similar to that derived from the classical method of chasing with excess unlabeled Tf, validating the surface-quenching technique (Figure 6B). Overall these results directly demonstrate rapid and efficient recycling of internalized trastuzumab. Similar results were observed with 488-pertuzumab and corresponding Fab fragments (unpublished data).
Figure 6.

Internalized trastuzumab efficiently recycles in the absence but not the presence of GA. (A) Adherent untreated (•) or GA-pretreated (▪) SKBr3 cells with surface-bound 488-trastuzumab were pulsed or not for 10 min at 37°C and then chased for the indicated intervals, processed for flow cytometry, and % pulse remaining calculated as described in Materials and Methods. Each data point is a mean of two similar experiments except for 30-min points, which are means of four separate experiments (±SEM). Similar results were obtained with 488-pertuzumab and with the corresponding 488-Fab fragments (unpublished data). (B) Recycling assays were performed as in A, but instead pulsing with 488-Tf and chasing with continuous presence of quenching antibody (•), excess unlabeled Tf (▴), or no additive (○). For comparison, a trastuzumab recycling assay was run in parallel (plain line). (C) 488- or 647-Tf recycling assays were performed on untreated (•) or GA pretreated (▪) SKBr3 cells using unlabeled Tf during the chase. 647-Tf assays were performed simultaneously in the same samples as A. Each data point is a mean of 1–3 similar experiments.
Surface quenching also facilitated pulse-chase imaging experiments. After only a 5-min pulse, we observed 488-trastuzumab in endosomes by video-enhanced epifluorescence microscopy upon surface quenching (Figure 5D, left panel). The quenching antibody itself was used as a cell surface marker and was absent from these puncta, confirming their identity as endosomes (Figure 5D, compare middle with left and right panels). To visualize 488-trastuzumab endocytic trafficking dynamics, cells were imaged live by dual color time-lapse epifluorescence microscopy (Supplementary Videos 1–4, Supplementary Figures and Tables). 594-Tf and Texas Red EGF (TR-EGF) were used as dynamic markers for recycling and lysosome-bound pathways, respectively. We reasoned that receptor dissociation of TR-EGF in the context of high ErbB2 expression would render it a lysosome-bound fluid phase marker (Harari and Yarden, 2000; Hendriks et al., 2003). Indeed 488-trastuzumab trafficking after a 10-min pulse was highly dynamic and similar to that of 594-Tf both spatially and temporally and distinct from that of TR-EGF. In large endosomes, outer membrane regions were relatively enriched with 488-trastuzumab and 594-Tf (Supplementary Videos 1 and 2), whereas inner regions were relatively enriched with TR-EGF (Supplementary Video 3) or another fluid phase marker, 594-dextran (unpublished data). Although the cells with internalized TR-EGF became relatively redder (i.e., less green) over time, cells with internalized 594-Tf did not, a finding consistent with the idea that 488-trastuzumab and 594-Tf recycle, whereas TR-EGF is retained. In all, these time-lapse images support to the idea that trastuzumab recycles and demonstrate the dynamic nature of ErbB2 endocytic trafficking.
GA Triggers ErbB2 Down-regulation via Reduced Recycling Rather than Increased Endocytosis
Down-regulation of surface ErbB2 in response to GA requires a change in the balance between ErbB2 endocytosis and recycling, because the biosynthetic contribution is very small for proteins with a long half-life such as ErbB2. The surface down-regulation profile (see Figure 3B) indicates that this imbalance develops fully after ∼60-min exposure to GA irrespective of the presence or absence of trastuzumab. To determine if GA down-regulates ErbB2 by increasing endocytosis, decreasing recycling, or both, the kinetics of these processes were determined in cells pretreated with GA for 60 min using our fluorescent antibody endocytosis and recycling assays described above. We reasoned that because antibody treatment has no significant effect on steady-state distribution of ErbB2 and very little effect on the down-regulation kinetics of geldanamycin, endocytosis, and recycling kinetics of antibody in these experiments would essentially reflect that of naked Her2. Recycling assays revealed that GA reduced recycling efficiency, resulting in >3-fold increase in the amount of conjugate retained (Figure 6A). Similar results were observed with 488-pertuzumab (unpublished data). By contrast, the effect of GA on Tf recycling was much less pronounced (Figure 6C). To determine the effect of GA on the endocytic rate constant (Ke), the initial linear uptake phase (0–4 min) was measured, displayed as internalization plots, and slopes were determined. No significant influence of GA on endocytic rate of 488-trastuzumab, 488-pertuzumab, or 647-Tf was observed (Figure 7, A–C). Our observed endocytic rates expressed as the fraction of the surface pool internalized per minute (0.01–0.02 min–1) are comparable to those reported previously for ErbB2 using a Fab-based assay (Hendriks et al., 2003) and ∼60-fold slower than 647-Tf. Overall, these results demonstrate that GA exerts its down-regulating effect on ErbB2 at the level of endosomal sorting rather than endocytosis.
Figure 7.

GA does not influence endocytosis rates during surface ErbB2 down-regulation. (A) Adherent untreated (•) or GA-pretreated (▪) SKBr3 cells were surface-labeled with 488-trastuzumab and pulsed at 37°C for 0–4 min, then detached, and surface-quenched, and internalization was calculated from flow cytometric data and plotted against the time integral of the surface fluorescence as described in Materials and Methods. Data points are means of three separate experiments. (B) Endocytic rate constants (Ke) were determined from the slopes of each individual experiment in A or similar experiments using 488-pertuzumab, and means of triplicate experiments were plotted (± SEM). Statistical p values were calculated using the two-tailed t test. (C) 647-Tf endocytosis was measured simultaneously in samples from A. Each data point was derived from means of three separate experiments and plotted as described in Materials and Methods.
Electron Microscopy Reveals Trastuzumab and ErbB2 in the Recycling Pathway and GA-induced Sorting to the Lysosomal Pathway
If GA affects endosomal sorting rather than endocytosis, why does internalized trastuzumab in GA-treated cells appear to costain better by immunofluorescence microscopy with 594-Tf than with late endosome/lysosome markers (see Figure 2C)? To address this question and characterize trastuzumab and ErbB2 localization more precisely, we performed 1) quantitative immunoelectron microscopy using antibodies to detect either trastuzumab or the cytoplasmic tail of ErbB2 or to improve sensitivity in single labeling experiments, and 2) silver enhancement of 1 nm NG-trastuzumab. Surface-bound trastuzumab, like ErbB2 (see below) and in line with previous observations (De Potter and Quatacker, 1993; Hommelgaard et al., 2004), was enriched on microvillus-like protrusions, was found occasionally within clathrin-coated pits, and was absent from noncoated plasma membrane invaginations (Figure 8, A and B, Supplementary Table 1). Immunogold label for internalized trastuzumab in BT474 cells increased slowly over time at 37°C (Table 1); uptake at 5 min and 3 h corresponded to that in SKBr3 cells determined by flow cytometry (compare to Figure 5B). Internalized trastuzumab was found mainly on small (≤60 nm) tubules and vesicles adjacent to endosomal vacuoles and dispersed in the cytoplasm (Figure 8, C–E, and Table 1). The large majority of these vacuoles were early endosomes, as indicated by their vacuolar morphology, association with TfR-positive recycling tubulovesicular membranes, and distinction from CD63-rich lysosomes (Figures 8, D and E). Though scarcity of gold particles precluded quantitative colocalization, trastuzumab-positive tubulovesicular membranes were morphologically identical to and often admixed with TfR-positive membranes, suggesting that internalized trastuzumab was indeed localized to the recycling pathway.
Figure 8.

Surface and internal distribution of trastuzumab. SKBr3 (A–D) or BT474 (E) cells with surface-bound trastuzumab (A, 10 nm gold; D and E, 15 nm gold) or NG-trastuzumab (B and C) were pulsed for the indicated time at 37°C and then processed for electron microscopy as described in Materials and Methods. (A and B) Surface trastuzumab was found frequently on microvillus-like cell protrusions (asterisks) and occasionally within clathrin-coated pits (arrow). (C) Internalized NG-trastuzumab was found mainly on small tubules and vesicles (diameter ≤ 60 nm, arrows). (D) Trastuzumab (arrows) occurs on a recycling tubule extending from an early endosome, adjacent to similar tubules marked by TfR (10 nm gold). (E) Trastuzumab-positive (arrows) early endosomes are distinguished from lysosomes (L) by morphology and CD63 (10 nm gold) labeling. EE, early endosome. Scale bars, 200 nm.
Table 1.
Normalized trastuzumab distribution with or without GA
| Endosomes and lysosomes
|
||||
|---|---|---|---|---|
| Intracellular (% of total) | Tubulovesicular, <60 nm (% of intracellular) | Internal mb (% of intracellular) | Limiting mb (% of intracellular) | |
| 0 min | 1.4 ± 0.3 | ND | ND | ND |
| 5 min | 6.2 ± 1.0 | ND | ND | ND |
| 3 h | 17.0 ± 2.5 | 69.2 ± 3.5 | 15.7 ± 2.7 | 11.8 ± 2.2 |
| 3 h +GA | 39.3 ± 5.2a | 20.1 ± 3.8a | 73.1 ± 3.6a | 6.3 ± 1.9 |
BT474 cells with surface-bound trastuzumab were incubated ± GA for the indicated time at 37°C and then fixed and processed for quantitative detection of trastuzumab by immunogold electron microscopy as described in Materials and Methods. Normalization to the number of total (left column) or intracellular gold particles (right columns) was performed individually for each cell profile prior to mean and SE calculations. Not included is a small population of endocytic 60-150-nm vesicles comprising <4% of the intracellular label. Non-normalized data are presented in Supplementary Table 1. Qualitatively similar results were observed for SKBr3 cells (unpublished data). mb, membrane; GA, geldanamycin; ND, not determined.
p < 0.001 in comparison with corresponding 3-h result.
By contrast, incubation with GA for 3 h triggered extensive internalization of surface-bound trastuzumab into multivesicular endosomal bodies (MVBs; Figure 9 and Table 1). Most striking was the accumulation within internal vesicles of these endosomes. In distinction from lysosomes, trastuzumab-positive endosomes had immature characteristics, such as weak CD63 labeling, association with peripheral TfR-positive tubules, and frequent presence of limiting membrane bilayered coats (Figures 9, A and B, and 10D). This finding explains why internalized trastuzumab in GA-treated cells costains by immunofluorescence microscopy better with Tf than late endosome/lysosome markers despite its entry into the lysosomal degradative pathway and highlights the importance of ultrastructural localization.
Figure 9.

GA-induced redistribution of internalized trastuzumab. BT474 cells with surface-bound trastuzumab (trast, 15 nm gold) were incubated with GA for 3 h at 37°C and processed for double immunogold labeling. (A) Internalized trastuzumab is predominantly localized to internal vesicles of MVBs that are distinct from lysosomes (L) by morphology and CD63 (10 nm gold) labeling level. (B) Trastuzumab-positive MVBs have immature features: association with recycling tubules positive for TfR (10 nm gold, arrows) and frequent presence of bilayered coats. Inset: a recycling tubule (arrow) extends from a trastuzumab-positive endosome toward a group of TfR-containing recycling vesicles. Arrowheads, bilayered coats. Scale bars, 200 nm.
Figure 10.

ErbB2 has a distribution similar to that of internalized trastuzumab and is sorted GA to bilayered coats on endosomes in SKBr3 (A, C, and D) and BT474 (B) cells. (A) Surface ErbB2 is concentrated on microvillus-like protrusions (asterisks) and infrequently observed in clathrin-coated pits (arrow). (B) Internal nonbiosynthetic ErbB2 is localized mainly on small tubulovesicular membranes (arrows). (C) GA induces redistribution of ErbB2 on endosomal limiting membranes to bilayered coats (arrowheads) in trastuzumab-treated cells. (D) Sorting of ErbB2 to bilayered coats (arrowheads) occurs after only 45 min of GA treatment. EE, early endosome. Scale bars, 200 nm.
Immunolabeling for ErbB2 produced similar results in untreated as in trastuzumab-treated cells, i.e., with 70–80% of the label at the plasma membrane and the intracellular label in comparable amounts in endocytic and biosynthetic compartments (Table 2, Supplementary Table 2). As with trastuzumab, surface ErbB2 was prominent on microvillus-like protrusions and occasionally observed within clathrin-coated pits (Figure 10A, Supplementary Table 2). In particular, internal ErbB2 was mainly associated with small tubulovesicular membranes, irrespective of trastuzumab treatment (Figure 10B, Table 2). Importantly and in agreement with the results above, trastuzumab did not significantly down-regulate or significantly shift ErbB2 distribution (Table 2 and Supplementary Table 2). A small increase in ErbB2 internalization after 3 h with trastuzumab was noted but was not statistically significant (Table 2, p > 0.05). By contrast GA with or without trastuzumab induced a significant shift of ErbB2 from small tubulovesicular structures to internal membranes of MVBs as observed for internalized trastuzumab (compare Table 2 with Table 1).
Table 2.
Normalized ErbB2 distribution with or without trastuzumab or GA
| Endosomes and lysosomes
|
|||||
|---|---|---|---|---|---|
| Endocytic (% of NB) | Tubulovesicular (% of endocytic) | Internal mb (% of endocytic) | Limiting mb (% of endocytic) | Coated limiting endosomal mb (% of limiting mb) | |
| Untreated | 10.2 ± 1.9 | 86.1 ± 3.7 | 6.1 ± 1.8 | 5.4 ± 2.2 | 0 ± 0 |
| +T, 0 min | 11.6 ± 1.6 | 79.8 ± 5.8 | 7.4 ± 2.8 | 12.2 ± 5.6 | 0 ± 0 |
| +T, 5 min | 13.0 ± 1.9 | 78.7 ± 4.8 | 9.0 ± 3.7 | 3.4 ± 1.7 | 0 ± 0 |
| +T, 3 h | 16.6 ± 2.6 | 70.5 ± 3.2 | 15.1 ± 3.0 | 10.3 ± 2.6 | 4.2 ± 3.7 |
| +GA, T, 3 h | 41.2 ± 3.7a | 33.0 ± 5.7a | 54.5 ± 5.8a | 11.9 ± 2.6 | 37.9 ± 9.1a |
| +GA, 3 h | 37.2 ± 3.9a | 45.5 ± 5.8a | 41.4 ± 7.9a | 11.5 ± 3.5 | 14.3 ± 12.0 |
SKBr3 cells ± surface-bound trastuzumab (T) were incubated at 37°C ± GA (G) as indicated and then fixed and processed for quantitative detection of ErbB2 by immunogold electron microscopy as described in Materials and Methods. Normalization to the number of total non-biosynthetic (NB) (left column), endocytic (middle three columns), or endosomal limiting membrane localized gold particles was performed for each cell profile individually prior to mean and SE calculations. Not included is a small population of endocytic and undefined 60-150-nm vesicles comprising <5% of the intracellular label. Non-normalized data are presented in Supplementary Table 2. Qualitatively similar results were observed for BT474 cells (unpublished data). NB, non-biosynthetic; mb, membrane; T, trastuzumab; GA, geldanamycin.
p < 0.01, in comparison with corresponding +T, 3-h result.
These findings demonstrate that GA with or without trastuzumab redistributed surface ErbB2 to internal vesicles of endosomes, an event indicating entry into the lysosomebound degradative pathway (Longva et al., 2002). To determine if this redistribution was attributed to altered endosomal sorting as suggested from the recycling experiments above (see Figure 6A), we quantified the percentage of ErbB2 gold particles on the limiting membrane of endosomal vacuoles associated with a cytoplasmic bilayered membrane coat. Partitioning into this characteristic coat has been implicated as an early event in the endosomal sorting of membrane proteins away from recycling and into the degradative pathway (Sachse et al., 2002). Indeed, GA treatment for 3 h substantially increased the percentage of limiting membrane ErbB2 associated with bilayered coats (Figure 10C, Table 2). Most importantly, sorting to coated membrane regions was evident after only 45 min with GA (Figure 10D, Table 3), a time when surface ErbB2 down-regulation commences but GA-induced internal accumulation is minimal (Figure 3B and Table 3). Relative to untreated cells, the percentage of ErbB2 associated with coated membranes was increased >3-fold after 45 min with GA (Table 3). By contrast, immunolabeling for TfR revealed no such GA-induced sorting. These results together with the kinetics data above (see Figures 6 and 7) strongly support the contention that GA exerts its down-regulating effect through improved endosomal sorting rather than increased endocytosis.
Table 3.
ErbB2 redistribution on MVB-limiting membranes after 45 min with GA
| ErbB2 endocytic (% of NB) | ErbB2 endo/lyso coated limiting mb (% of limiting mb) | TfR endo/lyso coated limiting mb (% of limiting mb) | |
|---|---|---|---|
| Control | 13.6 ± 1.2 | 11.9 ± 3.8 | 8.3 ± 0.6 |
| GA | 22.1 ± 3.9 | 38.9 ± 3.5a | 10.9 ± 2.7 |
SKBr3 cells were incubated ± GA for 45 min at 37°C and then fixed and processed for quantitative detection of ErbB2 or TfR by immunogold electron microscopy as described in Materials and Methods. Normalization to the total number of gold particles on the endosomal limiting membrane was performed per sample of 25 endosomal vacuoles prior to mean and SE calculations (n = 3). NB, non-biosynthetic; endo/lyso, endosomes and lysosomes; mb, membrane.
Statistical significance relative to control (p < 0.01).
DISCUSSION
In this study we used surface fluorescence quenching techniques combined with immunoelectron microscopy to characterize quantitatively and spatially endocytosis and recycling of ErbB2-antibody complexes. We believe trafficking of these complexes largely reflects that of naked ErbB2 for several reasons. First, these antibodies do not stimulate (Sliwkowski et al., 1999; Agus et al., 2002), down-regulate (Figure 3), or significantly redistribute (Figure 4, Table 2, and Supplementary Table 2) surface ErbB2. Second, these antibodies have almost no influence on GA-dependent ErbB2 down-regulation kinetics (Figure 3, B and C). If antibody binding were to instead mobilize a significant static pool of surface ErbB2, we should expect large differences in the down-regulation kinetics in light of our finding that GA acts at the level of endosomal sorting rather than endocytosis (see below). Third, the two antibodies used in this study show similar trafficking behavior (Figures 5, B and C, and 7B, and unpublished data) yet bind to distinct epitopes (Fendly et al., 1990) and have different mechanisms of action (Sliwkowski et al., 1999; Agus et al., 2002). Fourth, uptake and recycling kinetics for these antibodies were similar to that of their corresponding Fab fragments, suggesting that the influence of antibody bivalency in our system is not large (Figure 5C and unpublished data). Overall our results contradict the notion that surface ErbB2 is static and instead support the idea of a dynamic equilibrium between ErbB2 endocytosis and recycling. These findings, however, do not exclude the possibility that plasma membrane retention limits to some degree ErbB2 endocytosis. Indeed in agreement with recent observations of others (Hommelgaard et al., 2004), we found that plasma membrane ErbB2 was only occasionally (0.1–0.4%) localized to clathrin-coated pits (Supplementary Table 2). We also found that ErbB2 endocytosis is ∼60-fold slower than that of TfR (Figure 7), roughly corresponding to the differential occurrence of these receptors in coated pits as estimated by these authors. Hence our results suggest that if ErbB2 is indeed retained on the cell surface, such retention does not result in a static surface pool, but rather one with considerable residual dynamism.
The consequence of ErbB2 dynamism is that rapid surface down-regulation can be accomplished by diverting to degradative trafficking without altering basal endocytosis. Such appears to be the case for GA. Although GA down-regulates ErbB2 within a few hours (Figure 3, B and C), it has no discernible effect on endocytosis rate per se (Figures 7, A and B), while significantly reducing recycling efficiency (Figure 6A). More explicitly, because the basal endocytic rate is ∼2% per minute and rapid recycling is ∼50% efficient in the presence of GA, altered sorting alone could account for a down-regulation rate as high as ∼1% per minute. This estimate indeed corresponds to the GA-induced down-regulation kinetics we observed experimentally (Figures 3, C and D). Previously, it was shown that GA-induced ErbB2 degradation coincides with its ubiquitination, proteolytic fragmentation of the cytoplasmic domain, and internal redistribution of the surface pool (Mimnaugh et al., 1996; Tikhomirov and Carpenter, 2000). Although it might be presumed that ubiquitination induced by GA serves as an endocytosis and/or lysosomal sorting signal, the precise mechanism by which GA regulates ErbB2 endocytic trafficking is hitherto unknown. Moreover, the model in which ErbB2 is retained in a static plasma membrane pool implies that GA induces ErbB2 endocytosis. Our results however, demonstrate that GA does not influence ErbB2 endocytosis, but rather diverts basally endocytosed ErbB2 from a recycling to a degradative pathway. Because high surface distribution appears to be critical for the ability of ErbB2 to interfere with ligand-induced EGFR down-regulation and for ErbB2-associated tumorigenesis, this finding underscores the significance of endosomal recycling in ErbB2 biological function.
The abrupt deceleration in uptake kinetics at ∼5 min observed in Figure 5B can be accounted for by commencement of rapid and efficient recycling, a phenomenon rendered less efficient by GA treatment. What explains the slow phase rise in uptake that occurs subsequently in Figure 5B? Notably, our surface-quenching assays are best suited for short-term assays; prolonged incubation at 37°C would increase the influence of artifacts on the results such as intracellular antibody dissociation from ErbB2 and increased construct fluorescence upon degradation (Supplementary Figure 3B). Degradative routing alone, however, provides an incomplete explanation because it implies a trastuzumab turnover rate that is much faster than we observe (T1/2 ∼11 h, Figure 1). Importantly, both flow cytometric data in Figure 5B and quantitative immunoelectron microscopy data in Table 1 indicate that only ∼6% of the antibody had been internalized within 5 min. By comparison, ErbB2 label quantification reveals that at steady state, the endocytic pool comprises as much as 10–17% of total nonbiosynthetic ErbB2 (Table 2). In light of the predominantly tubulovesicular distribution pattern we see for internalized trastuzumab at 3 h (Table 1), a time when internalized antibody distribution more closely reflects that of steady-state ErbB2 (compare Table 1 with Table 2), we think a likely explanation for the slow phase rise in internalized antibody is the gradual filling of a kinetically distinct, slowly recycling internal pool. Indeed the notion of two kinetically distinct recycling routes back to the cell surface is well established (Maxfield and McGraw, 2004). The reason for the transient plateau in uptake observed between 5 and 16 min is less clear, but might possibly be attributed to recovery from prior low-temperature incubations.
A surprising result is that trastuzumab does not down-regulate surface ErbB2 in SKBr3 cells (Figures 3 and 4, Table 2), a cell line sensitive to the growth-inhibitory effects of this antibody in vitro. Though the mechanism of action of trastuzumab in vitro is not completely understood and might involve a combination of mechanisms, surface down-regulation of ErbB2 is widely believed to be important (Sliwkowski et al., 1999; Mendelsohn and Baselga, 2000; Baselga and Albanell, 2001; Yarden, 2001). This notion in part stems from a substantial body of work demonstrating the connection between down-regulation and antitumor activity for bivalent antibodies that either have agonistic properties or recognize the activated rat or human oncogenic neu mutant version of the receptor (Drebin et al., 1985; Harwerth et al., 1992; Hurwitz et al., 1995; Klapper et al., 1997). Indeed trastuzumab was initially thought to be agonistic as well (Scott et al., 1991), though this property was subsequently shown to be largely attributed to an in vitro artifact of nonionic detergent lysis (Sliwkowski et al., 1999). In agreement, molecular modeling of the trastuzumab-ErbB2 complex reveals that despite its bivalency, trastuzumab is very unlikely to drive dimerization interface interactions to form ErbB2 homodimers, as one might expect for an antibody with ligand-like properties (Supplementary Figure 4). Nonetheless, several studies have reported data showing apparent ErbB2 down-regulation in SKBr3 and other cell lines after treatment with trastuzumab (Hudziak et al., 1989; Kumar et al., 1991; Baselga and Albanell, 2001; Cuello et al., 2001; Citri et al., 2002). The kinetics of the effect in SKBr3 cells varies greatly between studies, with the fastest showing >50% down-regulation within 2 h (Citri et al., 2002) and the slowest showing slow down-regulation commencing after 24 h and reaching a maximum at 72–96 h (Cuello et al., 2001). Although the latter finding suggests down-regulation might be indirect and a consequence rather than a cause of growth inhibition, the former finding suggests a direct effect on the ErbB2 surface pool.
Our results contradict both of these views and instead show that the down-regulating effect of trastuzumab is not significant. The reasons for these discrepancies are unclear. Notably, our data derives from four independent techniques, three of which (immunofluorescence microscopy, flow cytometry, and quantitative immunoelectron microscopy) are unaffected by changes in sample protein resulting from growth inhibition. If trastuzumab does not down-regulate ErbB2, how does it inhibit ErbB2-dependent tumor cell growth? In vivo, studies in mice suggest that the host immune response might contribute substantially to the antitumor effect (Clynes et al., 2000). This however does not explain the ability of trastuzumab to inhibit cell growth in vitro. Trastuzumab does not disrupt association with other ErbB family members, as does pertuzumab (Agus et al., 2002). Other mechanisms have been proposed, including inhibition of metalloprotease-mediated cleavage of the ErbB2-ECD, resulting in a truncated constitutively active receptor (Molina et al., 2001; See Note Added in Proof).
In summary, our results reveal that surface ErbB2 in target breast cancer cells is dynamic rather than static. Trastuzumab does not down-regulate surface ErbB2 but instead passively recycles after ErbB2 endocytosis. GA by contrast rapidly down-regulates surface ErbB2 through improved degradative sorting following basal endocytosis.
Supplementary Material
Acknowledgments
We thank Susan Palmieri for her helpful advice with immunofluorescence microscopy, Peter Schow and Katharine Grimmer for advice with flow cytometry, and René Scriwanek and Marc van Peski for their excellent photographical work.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04–07–0591. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04–07–0591.
Abbreviations used: 488-, Alexa-488–conjugated; 547-, Alexa-594–conjugated; 647-, Alexa-647–conjugated; ErbB2-ECD, recombinant ErbB2 extracellular domain; GA, geldanamycin; MVB, multivesicular endosomal bodies; NG, 1-nm Nanogold; Tf, transferrin; TfR, transferrin receptor; TR-EGF, Texas Red–conjugated EGF.
Note Added in Proof. While this manuscript was under review, an article by Nagata et al. (2004. Cancer Cell 6, 117–127) was published suggesting that rapid activation of the lipid phosphatase PTEN may underlie the mechanism of action of trastuzumab. Their findings are consistent with our conclusions and provide an alternative explanation for the mechanism of action of trastuuzumab.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Agus, D.B. et al. (2002). Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2, 127–137. [DOI] [PubMed] [Google Scholar]
- Bargmann, C.I., Hung, M.C., and Weinberg, R.A. (1986). Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45, 649–657. [DOI] [PubMed] [Google Scholar]
- Baselga, J., and Albanell, J. (2001). Mechanism of action of anti-HER2 monoclonal antibodies. Ann. Oncol. 12(Suppl 1), S35–S41. [DOI] [PubMed] [Google Scholar]
- Baulida, J., Kraus, M.H., Alimandi, M., Di Fiore, P.P., and Carpenter, G. (1996). All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 271, 5251–5257. [DOI] [PubMed] [Google Scholar]
- Benz, C.C., Scott, G.K., Sarup, J.C., Johnson, R.M., Tripathy, D., Coronado, E., Shepard, H.M., and Osborne, C.K. (1993). Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res. Treat. 24, 85–95. [DOI] [PubMed] [Google Scholar]
- Citri, A. et al. (2002). Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 21, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes, R.A., Towers, T.L., Presta, L.G., and Ravetch, J.V. (2000). Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 6, 443–446. [DOI] [PubMed] [Google Scholar]
- Cobleigh, M.A. et al. (1999). Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 17, 2639–2648. [DOI] [PubMed] [Google Scholar]
- Cuello, M., Ettenberg, S.A., Clark, A.S., Keane, M.M., Posner, R.H., Nau, M.M., Dennis, P.A., and Lipkowitz, S. (2001). Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 61, 4892–4900. [PubMed] [Google Scholar]
- De Potter, C.R., and Quatacker, J. (1993). The p185erbB2 protein is localized on cell organelles involved in cell motility. Clin. Exp. Metastasis 11, 453–461. [DOI] [PubMed] [Google Scholar]
- De Santes, K., Slamon, D., Anderson, S.K., Shepard, M., Fendly, B., Maneval, D., and Press, O. (1992). Radiolabeled antibody targeting of the HER-2/neu oncoprotein. Cancer Res. 52, 1916–1923. [PubMed] [Google Scholar]
- Di Fiore, P.P., Pierce, J.H., Kraus, M.H., Segatto, O., King, C.R., and Aaronson, S.A. (1987). erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 237, 178–182. [DOI] [PubMed] [Google Scholar]
- Drebin, J.A., Link, V.C., Stern, D.F., Weinberg, R.A., and Greene, M.I. (1985). Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell 41, 697–706. [DOI] [PubMed] [Google Scholar]
- Fendly, B.M., Winget, M., Hudziak, R.M., Lipari, M.T., Napier, M.A., and Ullrich, A. (1990). Characterization of murine monoclonal antibodies reactive to either the human epidermal growth factor receptor or HER2/neu gene product. Cancer Res. 50, 1550–1558. [PubMed] [Google Scholar]
- Franklin, M.C., Carey, K.D., Vajdos, F.F., Leahy, D.J., De Vos, A.M., and Sliwkowski, M.X. (2004). Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328. [DOI] [PubMed] [Google Scholar]
- Garrett, T.P. et al. (2003). The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 11, 495–505. [DOI] [PubMed] [Google Scholar]
- Harari, D., and Yarden, Y. (2000). Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 19, 6102–6114. [DOI] [PubMed] [Google Scholar]
- Harwerth, I.M., Wels, W., Marte, B.M., and Hynes, N.E. (1992). Monoclonal antibodies against the extracellular domain of the erbB-2 receptor function as partial ligand agonists. J. Biol. Chem. 267, 15160–15167. [PubMed] [Google Scholar]
- Hendriks, B.S., Opresko, L.K., Wiley, H.S., and Lauffenburger, D. (2003). Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: quantitative analysis of HER2 overexpression effects. Cancer Res. 63, 1130–1137. [PubMed] [Google Scholar]
- Hommelgaard, A.M., Lerdrup, M., and Van Deurs, B. (2004). Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol. Biol. Cell 15, 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudziak, R.M., Lewis, G.D., Winget, M., Fendly, B.M., Shepard, H.M., and Ullrich, A. (1989). p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 9, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz, E., Stancovski, I., Sela, M., and Yarden, Y. (1995). Suppression and promotion of tumor growth by monoclonal antibodies to ErbB-2 differentially correlate with cellular uptake. Proc. Natl. Acad. Sci. USA 92, 3353–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs, J.S., Xu, W., and Neckers, L. (2003). Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 3, 213–217. [DOI] [PubMed] [Google Scholar]
- Kelley, R.F., O'Connell, M.P., Carter, P., Presta, L., Eigenbrot, C., Covarrubias, M., Snedecor, B., Bourell, J.H., and Vetterlein, D. (1992). Antigen binding thermodynamics and antiproliferative effects of chimeric and humanized anti-p185HER2 antibody Fab fragments. Biochemistry 31, 5434–5441. [DOI] [PubMed] [Google Scholar]
- Klapper, L.N., Vaisman, N., Hurwitz, E., Pinkas-Kramarski, R., Yarden, Y., and Sela, M. (1997). A subclass of tumor-inhibitory monoclonal antibodies to ErbB-2/HER2 blocks crosstalk with growth factor receptors. Oncogene 14, 2099–2109. [DOI] [PubMed] [Google Scholar]
- Kumar, R., Shepard, H.M., and Mendelsohn, J. (1991). Regulation of phosphorylation of the c-erbB-2/HER2 gene product by a monoclonal antibody and serum growth factor(s) in human mammary carcinoma cells. Mol. Cell. Biol. 11, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.X., Mallet, W.G., Huang, A.Y., and Maxfield. F.R. (2004). Endocytosed cation-independent mannose 6-phosphate receptor traffics via the endocytic recycling compartment en route to the trans-Golgi network and a subpopulation of late endosomes. Mol. Biol. Cell 15, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longva, K.E., Blystad, F.D., Stang, E., Larsen, A.M., Johannessen, L.E., and Madshus, I.H. (2002). Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 156, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, K.A., Opresko, L.K., Starbuck, C., Walsh, B.J., and Wiley, H.S. (1990). Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J. Biol. Chem. 265, 15713–15723. [PubMed] [Google Scholar]
- Maxfield, F.R., and McGraw, T.E. (2004). Endocytic recycling. Nat. Rev. Mol. Cell. Biol. 5, 121–132. [DOI] [PubMed] [Google Scholar]
- Mendelsohn, J., and Baselga, J. (2000). The EGF receptor family as targets for cancer therapy. Oncogene 19, 6550–6565. [DOI] [PubMed] [Google Scholar]
- Mimnaugh, E.G., Chavany, C., and Neckers, L. (1996). Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J. Biol. Chem. 271, 22796–22801. [DOI] [PubMed] [Google Scholar]
- Molina, M.A., Codony-Servat, J., Albanell, J., Rojo, F., Arribas, J., and Baselga, J. (2001). Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 61, 4744–4749. [PubMed] [Google Scholar]
- Sachse, M., Urbe, S., Oorschot, V., Strous, G.J., and Klumperman, J. (2002). Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell. 13, 1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro, F.B., Soe, T.T., Mallet, W.G., and Maxfield, F.R. (2004). Role of cytoplasmic domain serines in intracellular trafficking of furin. Mol. Biol. Cell. 15, 2884–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, G.K., Dodson, J.M., Montgomery, P.A., Johnson, R.M., Sarup, J.C., Wong, W.L., Ullrich, A., Shepard, H.M., and Benz, C.C. (1991). p185HER2 signal transduction in breast cancer cells. J. Biol. Chem. 266, 14300–14305. [PubMed] [Google Scholar]
- Slamon, D.J., Clark, G.M., Wong, S.G., Levin, W.J., Ullrich, A., and McGuire, W.L. (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- Slamon, D.J. et al. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Sliwkowski, M.X., Lofgren, J.A., Lewis, G.D., Hotaling, T.E., Fendly, B.M., and Fox, J.A. (1999). Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin. Oncol. 26, 60–70. [PubMed] [Google Scholar]
- Slot, J.W., Geuze, H.J., Gigengack, S., Lienhard, G.E., and James, D.E. (1991). Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin, A., Di Fiore, P.P., and Carpenter, G. (1993). The carboxyl terminus of epidermal growth factor receptor/erbB-2 chimerae is internalization impaired. Oncogene 8, 3021–3028. [PubMed] [Google Scholar]
- Tikhomirov, O., and Carpenter, G. (2000). Geldanamycin induces ErbB-2 degradation by proteolytic fragmentation. J. Biol. Chem. 275, 26625–26631. [DOI] [PubMed] [Google Scholar]
- Wang, Z., Zhang, L., Yeung, T.K., and Chen, X. (1999). Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell. 10, 1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, H.S., and Cunningham, D.D. (1982). The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J. Biol. Chem. 257, 4222–4229. [PubMed] [Google Scholar]
- Yarden, Y. (2001). Biology of HER2 and its importance in breast cancer. Oncology 61(Suppl 2), 1–13. [DOI] [PubMed] [Google Scholar]
- Yarden, Y., and Sliwkowski, M.X. (2001). Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2, 127–137. [DOI] [PubMed] [Google Scholar]
- Zheng, J., and Zagotta, W.N. (2000). Gating rearrangements in cyclic nucleotidegated channels revealed by patch-clamp fluorometry. Neuron 28, 369–374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.