

Abstract
Synapsins are epilepsy susceptibility genes that encode phosphoproteins reversibly associated with synaptic vesicles. Synapsin II (SynII) gene deletion produces a deficit in inhibitory synaptic transmission, and this defect is thought to cause epileptic activity. We systematically investigated how SynII affects synchronous and asynchronous release components of inhibitory transmission in the CA1 region of the mouse hippocampus. We found that the asynchronous GABAergic release component is diminished in SynII-deleted (SynII(−)) slices. To investigate this defect at different interneuron subtypes, we selectively blocked either N-type or P/Q-type Ca2+ channels. SynII deletion suppressed the asynchronous release component at synapses dependent on N-type Ca2+ channels but not at synapses dependent on P/Q-type Ca2+ channels. We then performed paired double-patch recordings from inhibitory basket interneurons connected to pyramidal neurons and used cluster analysis to classify interneurons according to their spiking and synaptic parameters. We identified two cell subtypes, presumably parvalbumin (PV) and cholecystokinin (CCK) expressing basket interneurons. To validate our interneuron classification, we took advantage of transgenic animals with fluorescently labeled PV interneurons and confirmed that their spiking and synaptic parameters matched the parameters of presumed PV cells identified by the cluster analysis. The analysis of the release time course at the two interneuron subtypes demonstrated that the asynchronous release component was selectively reduced at SynII(−) CCK interneurons. In contrast, the transmission was desynchronized at SynII(−) PV interneurons. Together, our results demonstrate that SynII regulates the time course of GABAergic release, and that this SynII function is dependent on the interneuron subtype.
SIGNIFICANCE STATEMENT Deletion of the neuronal protein synapsin II (SynII) leads to the development of epilepsy, probably due to impairments in inhibitory synaptic transmission. We systematically investigated SynII function at different subtypes of inhibitory neurons in the hippocampus. We discovered that SynII affects the time course of GABA release, and that this effect is interneuron subtype specific. Within one of the subtypes, SynII deficiency synchronizes the release and suppresses the asynchronous release component, while at the other subtype SynII deficiency suppresses the synchronous release component. These results reveal a new SynII function in the regulation of the time course of GABA release and demonstrate that this function is dependent on the interneuron subtype.
Keywords: CCK, epilepsy, GABA, hippocampus, inhibitory, PV
Introduction
Synapsins I and II (SynI and SynII) are neuronal phosphoproteins that reversibly associate with synaptic vesicles and tether them to the cytomatrix to maintain a reserve pool of vesicles (Greengard et al., 1993; Shupliakov et al., 2011). Converging basic and clinical work has convincingly implicated the synapsins in the pathophysiology of epilepsy. At the clinical level, genetic epidemiology studies have identified a human nonsense mutation in the SynI gene associated with syndromic epilepsy (Garcia et al., 2004; Giannandrea et al., 2013) and also a contribution of SynII to a sporadic epilepsy predisposition (Cavalleri et al., 2007; Lakhan et al., 2010; Prasad et al., 2014). At the basic science level, experiments in genetically modified animals have shown that deletion of either the SynI or SynII genes results in a strong epileptic phenotype (Li et al., 1995; Rosahl et al., 1995; Etholm and Heggelund, 2009). Thus, understanding how deletion of the synapsins affects neuronal networks in the brain has a potential to illuminate key aspects of the pathophysiology of some forms of epilepsy.
There is now a broad consensus regarding the function of the synapsins at the molecular level. During synaptic activity, the rise in intraterminal calcium triggers the dissociation of synapsins from vesicles, allowing vesicles to replenish the releasable pool (Hilfiker et al., 1999; Orenbuch et al., 2012). Additionally, synapsins also appear to participate in the docking and priming of synaptic vesicles at active zones (Humeau et al., 2001; Cousin et al., 2003; Coleman et al., 2008; Messa et al., 2010), and regulate the latest stages of exocytosis by synchronizing release events (Hilfiker et al., 1998; Fassio et al., 2006; Coleman and Bykhovskaia, 2009). However, how the loss of SynI or SynII leads to the epileptic phenotype is not well understood.
It has been recognized that deletion of SynI and/or SynII differentially affects excitatory and inhibitory synaptic transmission. Deletion of the synapsins at GABAergic synapses produces a reduction in basal inhibitory neurotransmission, although no obvious changes in basal synaptic transmission are seen at glutamatergic synapses (Gitler et al., 2004). These observations led to the idea that a deletion of SynI/II produces an imbalance in excitatory and inhibitory transmission (Gitler et al., 2004; Chiappalone et al., 2009; Farisello et al., 2013) that could explain the epileptic phenotype (Fassio et al., 2011). More recently, Medrihan et al. (2013) demonstrated that deletion of SynII can also suppress the asynchronous component of inhibitory synaptic transmission. This finding led to the suggestion that SynII may regulate neuronal excitability by maintaining asynchronous GABAergic release and thus promoting tonic inhibition (Medrihan et al., 2015).
GABAergic interneurons are highly heterogeneous and make subtype specific synapses that vary in terms of their anatomy, physiology, and molecular composition (Klausberger and Somogyi, 2008). Several interneuron subtypes have been implicated in the regulation of seizure activity, including parvalbumin (PV) (Sessolo et al., 2015; Yekhlef et al., 2015; Yi et al., 2015) and cholecystokinin (CCK) expressing interneurons (Wyeth et al., 2010; Sun et al., 2014). At the present time, it is unclear whether the synapsin action is uniform across inhibitory interneurons or whether synapsins regulate inhibitory transmission selectively at specific interneuron subtypes. In the present study, we investigated how SynII deletion affects synchronous and asynchronous GABAergic synaptic transmission in the hippocampus and tested whether the SynII action is interneuron specific.
Materials and Methods
Animals.
Mice heterozygotes for the Syn2 targeted mutation were purchased from The Jackson Laboratory (strain B6;129S-Syn2tm1Sud/J). Homozygotes lines Syn II(−) and wild-type (WT) were derived from breeding heterozygotes. Genotypes of all the breeders were independently confirmed by commercial genotyping service (Transnetix). Mice with labeled PV interneurons were derived from breeding the strains B6;129P2-Pvalbtm1(cre)Arbr/J and B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (The Jackson Laboratory). All the animal colonies were kept at standard conditions at room temperature and on a 12 h dark/light cycle. The husbandry conditions were identical for all the strains under the study. All the experiments were performed on 15- to 20-d-old animals of either sex in accordance with the guidelines of the Animal Care and Use Committee of the Universidad Central del Caribe, the Wayne State University School of Medicine, and the National Institutes of Health of the United States Public Health Service.
Slice preparation.
Mice were anesthetized by isoflurane and decapitated as soon as reflexive reactions disappeared. Brains were removed rapidly and bathed in an ice-cold solution containing the following (in mm): 65.0 NaCl, 2.0 KCl, 5.0 MgSO4, 1.1 KH2PO4, 24.0 NaHCO3, 10 dextrose, and 100 sucrose, and bubbled with a mixture of 95% O2 and 5% CO2. Horizontal hippocampal slices (300 μm thickness) were cut using a vibratome (Leica VT 1200 S). The slices were washed two times in the ACSF solution (previously bubbled with the same gas mixture) containing the following (in mm): 120 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 KH2PO4, 2.5 CaCl2, 24.0 NaHCO3, 10 dextrose, 50 sucrose, with the osmolality adjusted to 295 mOsm with sucrose, and maintained in the ACSF solution at 33°C continuously bubbled with the same gas mixture for at least 1 h before the experiment.
Electrophysiology.
Slices were held submerged in the recording chamber continuously superfused with the ACSF solution at the room temperature (22°C). Recordings were acquired using the multiclamp 700B amplifier and pCLAMP 10.0 software (Molecular Devices), filtered at 2 kHz and digitized at 10 kHz. Evoked synaptic responses were recorded using pipettes filled with a high-chloride cesium-based solution containing the following (in mm): 153.3 CsCl, 1.0 MgCl2, 10.0 HEPES, 5 EGTA, 0.1 NaGTP, 5.0 QX-314-bromide, and 3.0 MgATP. A stimulation electrode filled with the ACSF solution was placed at the border of the stratum oriens and stratum pyramidale, and stimuli of a 100 μs duration were delivered at a constant voltage. Inhibitory synaptic events were recorded using AP5 (25 μm) and CNQX (50 μm) applied to the bath to block NMDA and AMPA currents, respectively.
IPSCs were elicited with 50 stimuli at a 0.1 Hz frequency. For recordings of miniature IPSCs (mIPSCs), TTX (1 μm) was applied for 10 min to block action potentials. To monitor the asynchronous release component produced by high-frequency stimulation, we delivered three successive trains of 15 pulses at a frequency of 50 Hz with an interval of 30 s between the trains. We evaluated the rundown between the tetani using a liner regression analysis, and the experiments with significant run down were discarded.
In the experiments with extracellular field stimulation, the stimulus intensity was adjusted to eliminate transmission failures. Holding potentials were maintained at −65 mV. The open pipette resistance was 3–4 mΩ, and recordings with the series resistance exceeding 20 mΩ were discarded. Calcium channel blockers ω-conotoxin GVIA (250 nm, Tocris Bioscience) and ω-agatoxin IVA (10 nm) were applied to the bath solution for 20 min.
For paired recordings, the presynaptic interneuron was recorded in current clamp, whereas the postsynaptic pyramidal cell was monitored in voltage clamp with a holding potential set to −65 mV. Presynaptic interneurons were patched with pipettes filled with potassium gluconate-based intracellular solution containing the following (in mm): 135 K-gluconate, 0.1 CaCl2, 2.0 MgCl2, 2.0 Na-ATP, 1.0 EGTA, 10 HEPES, and 5 mg/ml biocytin (B4261 Sigma), pH 7.35 (adjusted with KOH), 280 mOsm. The open pipette resistance in these experiments was 3–6 mΩ. To monitor synaptic activity, the presynaptic cell was stimulated with constant current depolarizing pulses (4 ms, 1000 pA amplitude). To evaluate the spiking frequency (SF), a depolarizing pulse of 250 pA amplitude and 500 ms duration was applied to the presynaptic interneuron. The coefficient of adaptation (CA) was measured as the ratio between the first and the last interspike interval.
Imaging was performed using an Olympus compound microscope with a 60× water-immersion objective and a confocal imaging system (Ultravie, PerkinElmer). Biocytin staining with a post hoc interneuron identification was performed as described by Marx et al. (2012). Biocytin was allowed to diffuse into the neurons for ∼30 min.
Data analysis.
The recordings were analyzed off-line using Clampfit (Molecular Devices) and in-house software QUANTAN (Bykhovskaia, 2008). The after tetanus charge and the frequency of asynchronous peaks were measured over a 500 ms time window after the end of a tetanus. The release time course was derived using the deconvolution analysis as described by Diamond and Jahr (1995), using the Fourier transform of averaged IPSCs divided over the Fourier transform of averaged mIPSCs, which was followed by an inverse Fourier transform. The script for the deconvolution procedure was written in MATLAB. The synchronous release component was defined as quanta released within 6 ms following a stimulus. The reconstruction of biocytin-stained interneurons was performed using the ImageJ plugin NeuroJ (Meijering et al., 2004). ANOVA followed by the Tukey's post hoc test was usedfor multiple comparisons. K-means algorithm was used for clustering. Cumulative distributions were compared using the Kolomogorov–Smirnov test.
Results
In pyramidal cells of the CA1 region, a burst of evoked IPSCs is followed by a brief period of asynchronous inhibitory synaptic activity. Previous studies in the dentate gyrus have shown that SynII deletion leads to the loss of the asynchronous component of evoked IPSCs (Medrihan et al., 2013). Therefore, we first examined whether this effect is generalizable to the CA1 region. We performed whole-cell recordings from pyramidal neurons and elicited evoked IPSCs using extracellular stimulation adjusted to eliminate transmission failures. This stimulation paradigm produced similar evoked IPSCs in the slices derived from WT and SynII(−) mice (Fig. 1A). Next, we examined synchronous and asynchronous release components produced by 50 Hz tetani (Fig. 1B).
Figure 1.
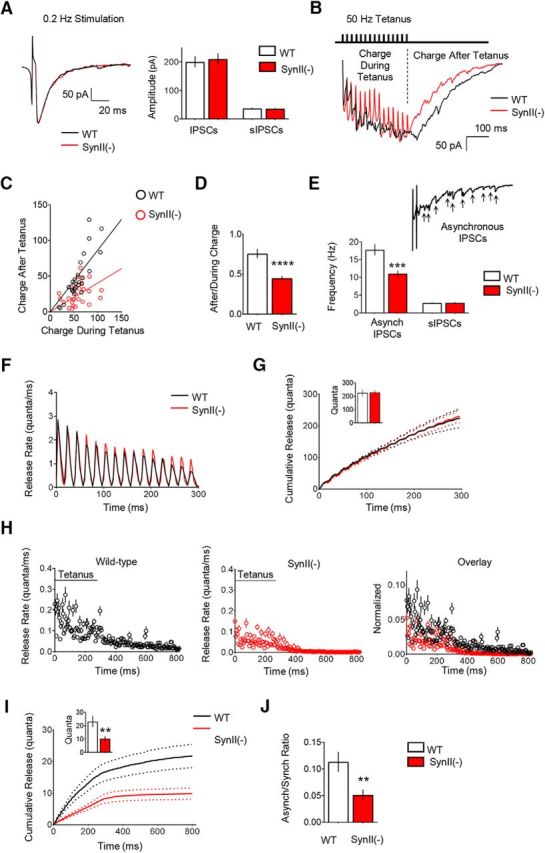
The asynchronous release component is reduced at SynII(−) inhibitory hippocampal synapses. A, Basal inhibitory transmission is similar at WT and SynII(−) slices. The stimulation intensity was adjusted to eliminate transmission failures, and this stimulation paradigm produced similar IPSC amplitudes in WT in SynII(−) lines. B, Representative postsynaptic currents elicited by 50 Hz frequency tetani at WT and SynII(−) slices illustrate the reduced after tetanus current in SynII(−) line (red). Stimulus artifacts were removed for clarity. C, The relationship between the charge transfer after a tetanus and the charge during a tetanus at individual experiments. The slope of the linear regression is significantly reduced at SynII(−) slices (p < 0.01), suggesting a selective reduction of the charge transfer after the tetanus in the Syn II(−) line. D, The After/During charge ratio is significantly reduced at SynII(−) slices (p < 0.001). E, The frequency of asynchronous current peaks is significantly reduced at SynII(−) slices (p < 0.005), whereas the sIPSC frequency is not affected. sIPSCs were collected for 5 min before the onset of the stimulation. F, The synchronous release component at WT and SynII(−) slices over the duration of the tetanus computed using deconvolution. G, The cumulative synchronous release kinetics derived by deconvolution suggest that the synchronous release component is not affected at SynII(−) slices. Dotted lines indicate confidence interval for the cumulative synchronous release. Inset, Total synchronous release over the duration of the tetanus. H, The asynchronous release component computed using deconvolution. Right, Plot (overlay) represents the data normalized by the first response in a tetanus. I, The cumulative asynchronous release component is significantly reduced at SynII(−) slices. Dotted lines indicate the confidence interval. Inset, Total asynchronous release, including that recorded during and after the tetanus (p < 0.01). J, The ratio between the cumulative asynchronous and synchronous release components is significantly reduced at SynII(−) slices (p < 0.01). Data collected from 26 slices for each genotype. **p < 0.01, ***p < 0.005, ****p < 0.001.
Initially, we estimated the magnitudes of synchronous and asynchronous release components by integrating the synaptic current during and after the tetanus. Because only the asynchronous release component contributes to the charge transfer after the tetanus, whereas the tetanus charge is produced by both synchronous and asynchronous release components, the selective reduction in the charge transfer after the tetanus would indicate that the asynchronous release component is selectively reduced. We plotted the charge transfer after the tetanus as a function of the charge transfer during the tetanus for each cell (Fig. 1C). In this plot, a shallower slope of the linear regression would reflect a selective reduction of the asynchronous release component. As illustrated in Figure 1C, the linear regression analysis exhibited a reduced slope for SynII(−) slices (0.266 ± 0.05 in Syn II(−) vs 0.719 ± 0.07 in WT, ANCOVA p = 0.008). Consistently, the ratio between the charge transfer after the tetanus and the charge transfer during the tetanus (After/During charge ratio) was significantly reduced in SynII(−) slices (Fig. 1D).
As an alternative measure for the asynchronous release component, we evaluated the frequency of asynchronous IPSCs by quantifying all the current peaks following the tetanus within a 500 ms time window. We found that the frequency of asynchronous release events was significantly reduced in the SynII(−) line (Fig. 1E), even though the frequency of spontaneous IPSCs (sIPSCs) was not affected in the SynII(−) line.
Finally, we computed the release time course at WT and SynII(−) synapses using deconvolution analysis (Diamond and Jahr, 1995). The synchronous (Fig. 1F,G) and the asynchronous (Fig. 1H,I) release components were separated. The time course of the synchronous release component during the tetanus suggested that it was not altered in SynII(−) slices (Fig. 1F). This conclusion was confirmed by comparison of the cumulative synchronous release components between the genotypes (Fig. 1G). In contrast, the asynchronous release component, including that recorded during and after the tetanus, was diminished at SynII(−) slices (Fig. 1H). The analysis of the cumulative asynchronous release component demonstrated that it was significantly reduced at SynII(−) genotype (Fig. 1I). Consistently, the ratio between the asynchronous and synchronous release components was significantly reduced at SynII(−) slices (Fig. 1J).
This analysis demonstrated that the asynchronous release component was selectively reduced in SynII(−) synapses. Combined, these results identify a key role for SynII in supporting asynchronous inhibitory synaptic transmission in the CA1 region and extend previous results in the dentate gyrus (Medrihan et al., 2013, 2015).
A prominent asynchronous GABAergic release component is usually associated with CCK interneurons (Daw et al., 2009), which contribute to the regulation of epileptic activity (Sun et al., 2014). These considerations raise the possibility that the SynII-dependent asynchronous release component recorded in pyramidal cells may originate from synapses made by CCK interneurons. As a first test for this possibility, we took advantage of the finding that synaptic transmission at CCK interneurons is mediated by N-type Ca2+ channels (Hefft and Jonas, 2005). We reasoned that, if CCK interneurons were responsible for the asynchronous component that is targeted by the deletion of SynII, then the blockade of N-type calcium channels should mimic and occlude the effect of SynII deletion.
To test whether this is the case, we used N-type calcium channel blocker ω-conotoxin GVIA (Randall and Tsien, 1995). We recorded basal inhibitory transmission, as well as synchronous and asynchronous release components elicited by a tetanus (50 Hz), then applied 250 nm ω-conotoxin GVIA to the bath solution for 20 min, and repeated the initial recordings. We quantified the effect of the toxin in individual experiments by comparing the parameters derived from the current traces recorded before and after the treatment. The treatment significantly reduced the basal inhibitory transmission in both genotypes (Fig. 2A). We then evaluated how the treatment affected synchronous and asynchronous release components elicited by a tetanus at WT and SynII(−) slices (Fig. 2B). First, we calculated the frequency of asynchronous peaks (Fig. 2C) and found that it was significantly reduced by the toxin application at WT but not at SynII(−) slices. Next, we quantified the effect of the toxin on the charge transfer during the tetanus (Fig. 2D) and the charge transfer after the tetanus (Fig. 2E). We found that the charge transfer during the tetanus was similarly reduced in both lines (Fig. 2D). In contrast, the effect of the toxin on the charge transfer after the tetanus was significantly stronger in the WT line (Fig. 2E). Consistently, the After/During charge ratio was significantly reduced by the treatment at WT but not at SynII(−) slices (Fig. 2F). Surprisingly, the treatment actually increased the After/During charge ratio in the SynII(−) line (Fig. 2F). Thus, the selective blockade of N-type calcium channels using ω-conotoxin GVIA produced a preferential suppression of the asynchronous release component at WT slices and, remarkably, partly reverted the hypersynchronized phenotype observed at SynII(−) slices. These results indicate that the prominent asynchronous SynII-dependent GABAergic release component depends on N-type Ca2+ channels, and provide a tentative support for the idea that this release component may be produced by CCK cells.
Figure 2.

The blockade of N-type Ca2+ channels by ω-conotoxin GVIA reverses the inhibitory effect of SynII deletion on the asynchronous GABAergic release component. A, The blockade of N-type Ca2+ channels by ω-conotoxin GVIA (250 nm) significantly reduces the basal inhibitory transmission in both genotypes. B, Representative current traces elicited by 50 Hz tetani illustrate the reduced postsynaptic signal in both lines upon ω-conotoxin GVIA application. Notably, the effect of the treatment on the after tetanus current is stronger in WT slices. Stimulus artifacts are removed for clarity. C, The ω-conotoxin GVIA treatment significantly (p < 0.005) reduces the frequency of asynchronous peaks at WT but not at SynII(−) slices. D, The effect of ω-conotoxin GVIA on the charge transfer during the tetanus is similar at WT and SynII(−) slices. E, The effect of ω-conotoxin GVIA on the charge transfer after the tetanus is significantly stronger at WT slices (p < 0.01). F, The ω-conotoxin GVIA treatment reduces the After/During charge ratio at WT (p < 0.005) but not at SynII(−) slices. Data collected from 13 slices for each genotype. *p < 0.05, **p < 0.01, ***p < 0.005.
Because evoked GABAergic synaptic transmission depends on both N- and P/Q-type calcium channels, we next examined the effect of blocking P/Q-type calcium channels using ω-agatoxin IVA (Randall and Tsien, 1995). The application of 10 nm ω-agatoxin IVA to the bath solution for 20 min significantly reduced basal inhibitory transmission in both genotypes (Fig. 3A,B). The analysis of synchronous and asynchronous release components elicited by a tetanus (Fig. 3B) revealed that the treatment inhibited the synchronous and asynchronous release components in both genotypes to a similar extent (Fig. 3C–F). Thus, all the synaptic parameters, including the frequency of asynchronous peaks (Fig. 3C), the charge transfer during the tetanus (Fig. 3D), the charge transfer after the tetanus (Fig. 3E), and After/During charge ratio (Fig. 3F), were similarly reduced in both genotypes. These results support the idea that the interneurons dependent on P/Q-type Ca2+ channels do not make a major contribution to the asynchronous SynII-dependent GABAergic release component.
Figure 3.

The blockade of P/Q-type Ca2+ channels by ω-agatoxin suppresses both release components in WT and SynII(−) genotypes. A, The blockade of P/Q-type Ca2+ channels by ω-agatoxin IVA (10 nm) significantly reduces basal inhibitory transmission in both genotypes. B, Representative traces illustrate a uniform reduction of the postsynaptic current produced by the ω-agatoxin IVA treatment in both genotypes. C, The ω-agatoxin IVA treatment does not affect the frequency of asynchronous peaks in either of the genotypes. D, The effect of ω-agatoxin IVA on the charge transfer during the tetanus is similar at WT and SynII(−) slices. E, The effect of ω-agatoxin IVA on the charge transfer after the tetanus is similar at WT and SynII(−) slices. F, The ω-agatoxin IVA treatment does not affect the After/During charge ratio in either of the lines. Data collected from 13 slices for each genotype. *p < 0.05, **p < 0.01.
To test this idea more directly, we conducted paired double-patch recordings from basket interneurons located in the stratum pyramidale, or in the region at the border of the stratum pyramidale and stratum oriens, connected to CA1 pyramidal neurons (Fig. 4A,B). Because interneurons in this area are heterogeneous, we first thought to distinguish between different classes. Consistent with previous work, we could distinguish two broad types of interneurons in this area based upon IPSC kinetics (Fig. 4C). One of the types was characterized by slower IPSC kinetics and likely corresponded to dendritic targeting interneurons, including the somatostatin-expressing interneurons (Savanthrapadian et al., 2014). Within this class, the basal GABAergic transmission did not differ between WT and SynII(−) genotypes (IPSC peak amplitude 36.9 ± 7.7 pA, n = 8 for WT vs 43.5 ± 4.4 pA, n = 7 for SynII(−); p = 0.48, data not shown); therefore, these interneurons were not studied further. The second class of neurons was characterized by fast IPSC kinetics and likely corresponded to perisomatic innervating interneurons (Maccaferri et al., 2000; Bartos et al., 2002). These interneurons form a heterogeneous class that includes PV- and CCK-expressing cells. We next focused on this cell group.
Figure 4.

Paired recordings from inhibitory basket cells connected to CA1 neurons at the stratum pyramidale reveal two classes of interneurons with fast and slow IPSC kinetics. A, Paired recordings from GABAergic basket cells connected to pyramidal neurons. B, A GABAergic basket cell (red) synaptically connected to a pyramidal neuron (blue). Biotin staining shows axon arbors predominantly located in stratum pyramidale (SP). SO, Stratum oriens; s.rad, stratum radiatum; SLM, stratum lacunosum-moleculare. C, All the examined connected pairs fall into two clusters according to the rise and decay times of the recorded IPSCs. Examples of IPSCs with fast and slow kinetics recorded from different interneuron types are shown on the left.
Persisomatic innervating PV and CCK interneurons can be distinguished by their SF, with CCK cells being regular spiking (RS) and PV cells being fast spiking (FS). The other parameters include the CA, the coefficient of variation (CV) of synaptic currents, the paired pulse ratio (PPR), and the failure rate (FR) of synaptic transmission (Neu et al., 2007; Daw et al., 2009; Bartos and Elgueta, 2012; Hu et al., 2014). A recent study demonstrated that cluster analysis can be used to classify these interneuron subtypes according to their intrinsic synaptic and spiking properties (Kohus et al., 2016). We conducted cluster analysis based upon five variables, including spiking (SF and CA) and synaptic (CV, FR, and PPR) parameters (Fig. 5; Table 1). The cluster analysis was performed using the K-means algorithm, which relies on the Euclidean distance between the clusters. As illustrated in Figure 5, the population of interneurons sampled in our experiments sorted into two distinct clusters that we have termed FS and RS. For each parameter, significant differences were observed between the two clusters (Table 1, p < 0.05 between interneuron subtypes for all the parameters). Compared with the RS cells, the FS interneurons exhibited an increase in SF (Fig. 5A), an increase in CA (Fig. 5B), a decrease in CV, a lack of transmission failures (FR = 0, Fig. 5C), and a decrease in PPR (Fig. 5D). We did not detect any statistically significant difference in any of the five parameters between WT and SynII(−) genotypes (Table 1, p > 0.05 between the genotypes for all the parameters), although the RS SynII(−) cells showed a slightly decreased FR (Fig. 4C) and PPR (Fig. 4D), raising the possibility that some of the parameters of synaptic transmission within this interneuron subtype may be affected by SynII deletion. As illustrated in the 3D plot (Fig. 5E) depicting the three parameters (SF, CA, and PPR), the entire population of the sampled interneurons subdivided into two distinct clusters. This analysis indicated that the physiological properties of the cells in the FS cluster matched those previously reported for PV+ basket cells (Bartos and Elgueta, 2012; Hu et al., 2014), whereas the properties of interneurons in the RS cluster matched those of the CCK basket cells (Neu et al., 2007; Daw et al., 2009). Thus, these results support the idea that the perisomatic innervating interneurons sampled in the current study represent two neuronal subtypes that we can tentatively identify as PV- and CCK-expressing interneurons.
Figure 5.

Perisomatic interneurons with fast IPSC kinetics constitute two interneuron subtypes, FS and RS. All the connected pairs are classified according to five parameters: SF (A), CA (B), CV (C), FR (C), and PPR (D). *Significant difference (p < 0.05) between the interneuron subtypes (two-way ANOVA). A, Spiking frequencies differ between neuron subtypes but not between the genotypes. B, RS interneurons have a lower CA, which is manifested as a stronger decay in SF. C, RS interneurons have a higher variability in IPSC amplitudes, CV, and a higher failure rate. D, RS interneurons have a higher PPR. E, A 3D plot showing that all the interneuron subtypes fell into two distinct clusters according to three physiological parameters: SF, CA, and PPR. WT and SynII(−) genotypes were pooled together because all the plotted parameters were similar for WT and SynII(−) lines.
Table 1.
Two-way ANOVA of electrophysiologic parameters measured by paired recordings
| Parameter | Variable | p value |
|---|---|---|
| PPR | Neuron type | 0.0004 |
| Genotype | 0.0512 | |
| SF | Neuron type | <0.0001 |
| Genotype | 0.9375 | |
| FR (%) | Neuron type | 0.0482 |
| Genotype | 0.3183 | |
| CV | Neuron type | 0.0014 |
| Genotype | 0.956 | |
| CA | Neuron type | <0.0001 |
| Genotype | 0.7881 |
To directly address this interpretation, we took advantage of Cre-Lox strategy to derive animals with fluorescently labeled PV interneurons. We used hippocampal slices from these animals to selectively stimulate PV basket interneurons situated in striatum pyramidale of the CA1 region of the hippocampus (Fig. 6A) and record from connected pyramidal neurons (Fig. 6B). We found that the magnitudes of synaptic responses (Fig. 6C), as well as synaptic and spiking parameters obtained for PV expressing cells (PV+, Fig. 6D–G) were similar to those obtained for FS interneurons (presumed PV). As illustrated in the 3D plot depicting the three parameters (SF, CA, and PPR, Fig. 6G), FS and PV+ cells form a uniform cluster. These experiments support our interneuron classification, which was based initially on electrophysiological properties (Fig. 5), suggesting the two identified classes of interneurons correspond to PV basket cells (FS) and CCK basket cell (RS).
Figure 6.

Spiking and synaptic parameters of FS basket interneurons match the parameters obtained for PV+ basket interneurons. A, The layer of labeled PV cells in the CA region of the hippocampus. B, Paired recordings from PV+ interneurons connected to pyramidal neurons. Top to bottom, DIC image showing the recording configuration, an epifluorescence image of the same field of view showing the presynaptic PV+ cell, and an overlay. C, IPSC amplitudes are similar for PV+ and FS cells. D, Spiking frequencies are similar for PV+ and FS cells. E, CAs are similar for PV+ and FS cells. F, Coefficients of variation are similar for PV+ and FS cells. G, PPRs are similar for PV+ and FS cells. H, The 3D plot depicting the three parameters, CF, CA, and PPR, shows that PV+ and FS interneurons fall within the same cluster. Data collected from nine PV+ cells.
To test the idea that the effect of SynII deletion on asynchronous synaptic transmission may be interneuron subtype specific, we analyzed the synchronous and asynchronous release components obtained in paired recordings upon a high-frequency stimulation tetanus (50 Hz). As illustrated in Figure 7, SynII deletion reduced the asynchronous release component at RS (presumed CCK) interneurons. This was evident from a reduced number of asynchronous current peaks (Fig. 7B), a left shift in the cumulative distribution of the after tetanus charge transfer (Fig. 7C, middle), and a reduced After/During charge ratio (Fig. 7C, right) at SynII(−) neurons. The deconvolution analysis (Fig. 7D–G) demonstrated that the cumulative asynchronous release component was significantly reduced in SynII(−) neurons (Fig. 7E). Interestingly, we have also detected a significant increase in the synchronous release component at SynII(−) neurons (Fig. 7G). Consistently, the ratio between the cumulative asynchronous and synchronous release components was reduced at SynII(−) neurons (Fig. 7H). These results suggest that SynII desynchronizes the release process at CCK interneurons and maintains a prominent asynchronous release component.
Figure 7.
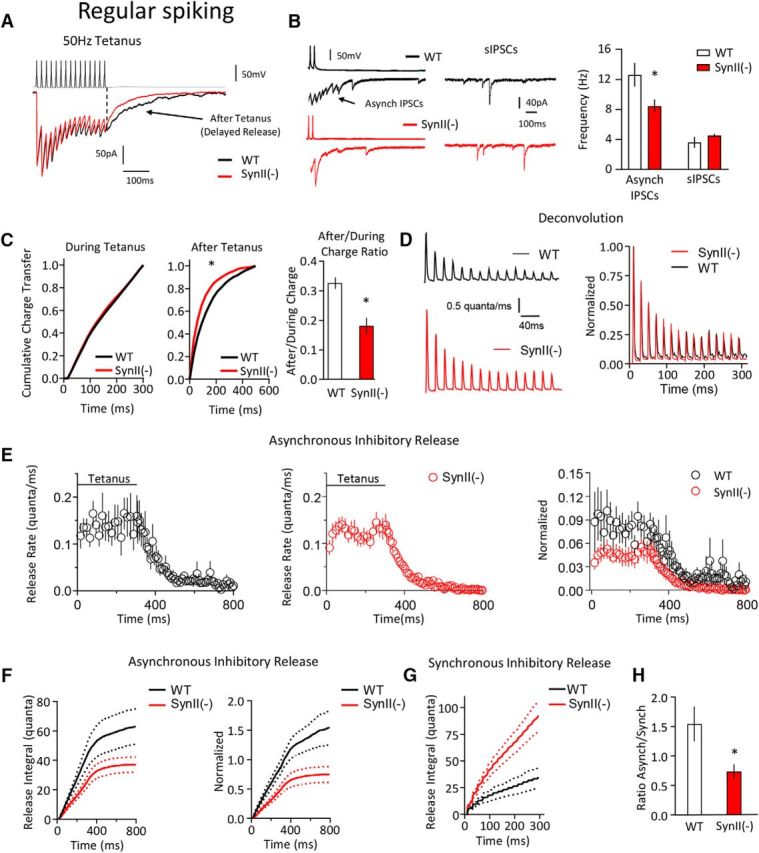
SynII deletion suppresses the asynchronous release component at regular-spiking (presumed CCK) interneurons. A, Representative traces illustrating that the after tetanus current is decreased at SynII(−) neurons. B, Asynchronous IPSC frequency is significantly (p < 0.05) reduced at SynII(−) neurons, whereas the sIPSC frequency is not affected. Left, Representative traces show asynchronous and spontaneous IPSCs. C, Cumulative probability plots show that SynII has no effect on the charge transfer during the tetanus (left), whereas the charge transfer after the tetanus is significantly reduced at SynII(−) neurons (middle, the distribution is shifted to the left, p < 0.05, Kolomogorov–Smirnov test). The After/During charge ratio is significantly decreased at SynII(−) neurons (right, p < 0.05). D, The synchronous release component at WT and SynII(−) neurons computed using the deconvolution. Right, Plot represents data normalized by the first response in a tetanus. E, The asynchronous release component computed using deconvolution. Right, Plot represents data normalized by the first response in a tetanus. F, The cumulative asynchronous release component is significantly decreased at SynII neurons (p < 0.05). Dotted lines indicate the confidence interval. G, The cumulative synchronous release component is significantly increased at SynII neurons (p < 0.05). Dotted lines indicate the confidence interval. H, The ratio between the cumulative asynchronous and synchronous release components is reduced at SynII(−) neurons (p < 0.05). Data collected from six pairs for each genotype. *p < 0.05.
In contrast, SynII deletion produced a desynchronization of the release process at FS (presumed PV) interneurons (Fig. 8), including an increase in the asynchronous release component and a decrease in the synchronous release component. This was manifested as an increase in the frequency of asynchronous current peaks at SynII(−) slices (Fig. 8B), a right shift in the cumulative distribution of the after tetanus charge transfer (Fig. 8C), and an increase in the After/During charge ratio (Fig. 8C, right). The deconvolution analysis revealed a decrease in the synchronous release component (Fig. 8D,G) and an increase in the asynchronous release component (Fig. 8E,F) in SynII(−) genotype, producing an overall desynchronization of the release time course (Fig. 8H) in SynII(−) PV neurons.
Figure 8.
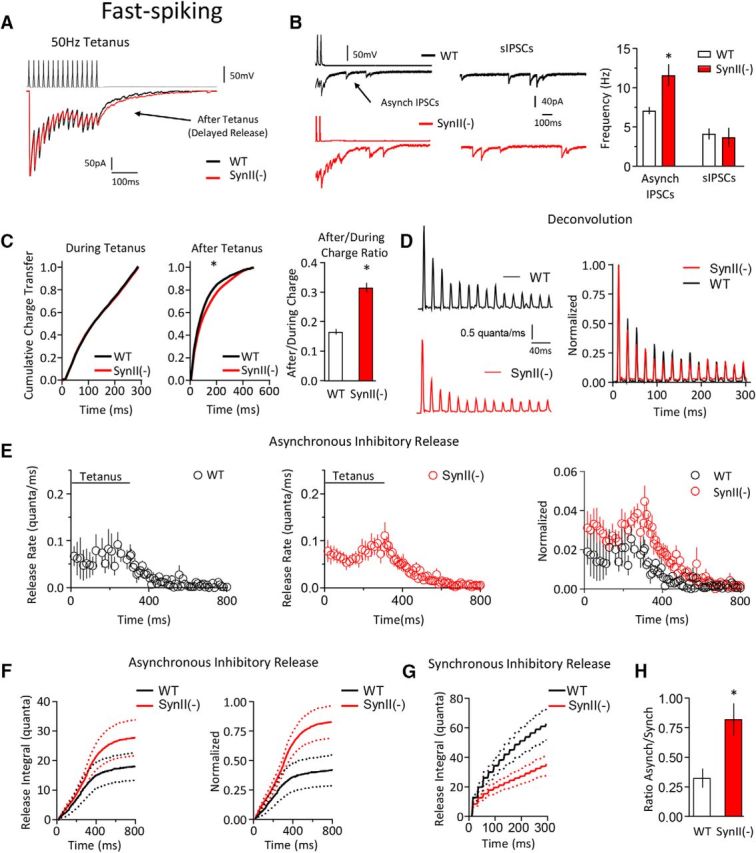
SynII deletion suppresses the synchronous release component and enhances the asynchronous release component at FS (PV) interneurons. A, Representative traces illustrate a mild selective increase in the after tetanus current in the SynII(−) line. B, The frequency of asynchronous IPSCs is significantly (p < 0.05) increased at SynII(−) neurons, whereas the sIPSC frequency is not affected. Left traces, Examples of asynchronous and spontaneous IPSCs. C, The charge transfer after the tetanus is selectively increased at SynII(−) neurons. This is evident from the right shift in the cumulative distribution representing the after tetanus charge (p < 0.05, Kolomogorov–Smirnov test) and a significant increase in the After/During charge ratio at SynII(−) neurons (p < 0.05). D, The synchronous release component at WT and SynII(−) KO interneurons computed using deconvolution. E, The asynchronous release component computed using deconvolution. Right plot, Data normalized by the first response in a tetanus. F, The cumulative asynchronous release component is significantly increased at SynII neurons (p < 0.05). Dotted lines indicate the confidence interval. G, The cumulative synchronous release component is significantly decreased at SynII neurons (p < 0.05). Dotted lines indicate the confidence interval. H, The ratio between the total asynchronous and synchronous release components is significantly increased at SynII(−) neurons (p < 0.05). Data collected from six pairs for each genotype. *p < 0.05.
These results demonstrate that SynII regulates the synchrony of GABA release elicited by a tetanus and that this regulation is interneuron type specific, suggesting that SynII may regulate the time course of GABAergic transmission. To test this idea, we investigated the time course of basal GABAergic synaptic transmission at PV and CCK interneurons. We compared synaptic responses triggered by a single action potential in connected pairs between perisomatic interneurons and pyramidal cells in both genotypes. For the RS (presumed CCK) interneurons, the deletion of SynII produced an increase in the IPSC amplitude (Fig. 9A,G) and an acceleration of the IPSC decay (Fig. 9A,B) which produced a leftward shift in the cumulative distribution of the charge transfer (Fig. 9C, p = 0.008, Kolomogorov–Smirnov test). In contrast, the total IPSC charge was not significantly increased (Fig. 9H, p = 0.399). To test the idea that these changes may stem from hypersynchronization of release following deletion of SynII, we examined the ratio of the IPSC charge to the IPSC amplitude in individual sweeps. This ratio can serve as a measure of the release synchrony, with higher ratios corresponding to more asynchronous responses. As illustrated in Figure 8I, we found that the charge to amplitude ratio at RS SynII(−) interneurons is significantly decreased, suggesting that basal release becomes more synchronized after Syn II(−) deletion at RS (CCK) interneuron synapses.
Figure 9.

SynII deletion desynchronizes basal transmission at regular spiking (CCK) interneurons and synchronizes transmission at FS (PV) interneurons. A, Representative traces illustrate higher IPSC amplitudes and faster kinetics at RS SynII(−) neurons. B, The average IPSC traces normalized by the peak amplitudes demonstrate a narrower time course at the currents recorded from FS SynII(−) pairs. C, The normalized cumulative charge plot shows a significantly faster IPSC kinetics at the SynII(−) line (p < 0.05, Kolomogorov–Smirnov test). D–F, At FS interneurons, SynII deletion produces a slower IPSC kinetics. G, SynII deletion increases the IPSC amplitude at RS interneurons and decreases the IPSC amplitude at FS interneurons (p < 0.05, one-way ANOVA followed by the Tukey's test). H, SynII(−) deletion does not affect the IPSC charge at either of the interneuron subtypes, suggesting that the release magnitude is not affected. I, The ratio between the IPSC charge and amplitude, as a measure of IPSC kinetics. SynII(−) deletion significantly reduces the ratio at RS interneurons and significantly increases the ratio at FS interneurons (p < 0.05, one-way ANOVA followed by the Tukey's test), suggesting that SynII regulates the release kinetics at FS and RS interneurons in opposite ways. *p < 0.05.
In contrast to the results outlined above, a similar analysis performed for the FS (PV) interneurons indicated that SynII deletion produced a reduction in the IPSC amplitude (Fig. 9D) and a broadening of its time course (Fig. 9E), which resulted in a rightward shift in the cumulative distribution of charge (p = 0.001, Kolomogorov–Smirnov test, Fig. 9F). Consistently, SynII deletion produced a significantly increased IPSC charge to amplitude ratio (Fig. 9I). These results suggest that deletion of SynII results in a desynchronization of basal release at FS (PV) interneuron synapses. Together, these results identify an interneuron subtype dependent effect of SynII on the timing of GABA release in the CA1 region.
Together, these results indicate that SynII regulates the time course of GABAergic transmission in a neuron type-specific manner. The analysis of the time course of GABA release elicited either by a tetanus (Figs. 7, 8) or by a single action potential (Fig. 9) suggested that SynII synchronizes release events at RS (presumed CCK) interneurons and desynchronizes the release events at FS (PV) interneurons. In line with these findings, the results presented in Figures 2 and 3 demonstrate that the asynchronous release component is either decreased or increased at SynII(−) slices, depending on the Ca2+ channel specificity. SynII deletion desynchronized the release in the slices treated with the N-type Ca2+ channel blocker (Fig. 2) and synchronized the release in the slices treated with the P/Q-type Ca2+ channels blocker (Fig. 3). These findings indicate that SynII may desynchronize GABA release in the interneurons dependent on N-type Ca2+ channels (CCK) and synchronize the release in the interneurons dependent on P/Q-type Ca2+ channels (PV).
Discussion
We investigated the role of SynII in the regulation of inhibitory transmission at hippocampal slices and found that SynII controls the release time course, as well as the asynchronous release component. Importantly, we found that this SynII function is interneuron specific. Thus, SynII deficiency synchronizes the basal release and diminishes the asynchronous release component at RS (presumed CCK) basket interneurons dependent on N-type Ca2+ channels. In contrast, SynII deficiency desynchronized release at FS (presumed PV) basket interneurons dependent on P/Q-type Ca2+ channels.
It is generally agreed that synapsins regulate inhibitory transmission and that this synapsin function is likely to be critical for the regulation of neuronal excitability. However, the specific impact of synapsins on GABAergic transmission is still debated. Thus, several studies have reported that synapsins maintain synchronous GABAergic transmission (Gitler et al., 2004; Chiappalone et al., 2009; Fassio et al., 2011). However, it has also been reported (Medrihan et al., 2013) that SynII desynchronizes release, maintaining the asynchronous release component and probably inhibiting the synchronous release component. Finally, a recent study (Song and Augustine, 2016) suggested that SynII accelerates the release process, whereas SynII deficiency prolongs the release kinetics.
In addressing this apparent controversy, it is important to consider that inhibitory GABAergic synaptic transmission in hippocampus and cortex involves multiple classes of interneurons that make synapses of varying molecular composition and distinct physiological properties (Klausberger and Somogyi, 2008). This raises the possibility that synapsins may differentially regulate transmission in different classes of interneurons. Indeed, consideration of the results from multiple previous studies supports the idea that the effect of synapsin on neuronal secretion depends on synapse type, presumably being sensitive to the neuron-specific molecular machinery. Thus, synapsin deletion had no effect on basal glutamatergic transmission (Gitler et al., 2004), slightly reduced cholinergic transmission in a Ca2+-dependent manner (Samigullin et al., 2004; Coleman et al., 2008), and had a severe effect on inhibitory transmission (Gitler et al., 2004; Medrihan et al., 2013). This nonuniformity should be taken into consideration when addressing the synapsin function.
To test whether the regulation of inhibitory transmission by SynII may be interneuron specific, we performed double-patch paired recordings from hippocampal perisomatic basket cells connected to CA1 pyramidal neurons. We detected two distinct classes of inhibitory interneurons, FS and RS. According to their spiking and synaptic parameters, these two classes of cells matched PV- and CCK-expressing basket interneurons, respectively. The match between the PV and FS interneurons was also supported by the experiments performed at transgenic animals with fluorescently labeled PV cells.
Strikingly, we found that SynII deletion affected these two interneuron subtypes in opposite directions. At RS (presumed CCK) basket interneurons, SynII deletion drastically diminished the asynchronous release component and synchronized basal transmission. At FS (PV) interneurons, the effect of SynII deletion was opposite, as it produced an increase in the asynchronous release component and desynchronization of basal transmission. This result suggests that SynII affects the release kinetics, and depending on the neuron subtype it may accelerate or decelerate the release process. Such a function was demonstrated earlier for SynI, which was shown to accelerate the release process at several preparations (Hilfiker et al., 1998; Fassio et al., 2006; Coleman and Bykhovskaia, 2009). The results of the present study show that SynII gene is similarly critical for the regulation of the release kinetics of inhibitory transmission.
The specific molecular mechanisms that define this SynII function and its neuron specificity remain to be unraveled. Because expression levels of synapsins vary across neuronal subtypes (Fassio, Raimondi et al., 2011), the different expression levels can influence the way synapsins control synaptic transmission. However, a detailed analysis of the synapsin expression (Song and Augustine, 2016) demonstrated that all the synapsin I and synapsin II isoforms are expressed broadly in GABAergic synapses of the hippocampus. Furthermore, this study demonstrated that each of SynII isoforms can rescue the impairments in inhibitory transmission produced by SynII deletion, including the release magnitude and IPSC kinetics. These finding suggest that the differential expression of SynII isoforms is unlikely to explain the opposing effects of SynII deletion at different interneuron subtypes.
We hypothesize that the interneuron specificity of SynII function may result from the differential reliance on N- and P/Q-type presynaptic Ca2+ channels in CCK and PV cells. In support of this hypothesis, it has been demonstrated (Medrihan et al., 2013) that SynII interacts with P/Q -type channels but not with N-type channels. Thus, it is possible that SynII may position synaptic vesicles in the vicinity of P/Q-type channels, thus bringing vesicles within a Ca2+ nanodomain (Bucurenciu et al., 2008; Eggermann et al., 2011) and synchronizing the release process. In support of this hypothesis, we found that SynII deletion prolongs the release kinetics and decreases IPSC amplitudes in PV interneurons, which rely on P/Q-type Ca2+ channels, but not in CCK interneurons dependent on N-type Ca2+ channels (Hefft and Jonas, 2005; Goswami et al., 2012). Consistently, SynII deletion desynchronized release upon the blockade of N-type Ca2+ channels but not upon the blockade of P/Q-type Ca2+ channels. It should be noted that the latter finding does not agree with an earlier study (Medrihan et al., 2013), which reported a similar effect for N-type and P/Q-type Ca2+ channel blockers on the asynchronous release component. In agreement with Medrihan et al. (2013), we demonstrated that the blockade of N-type Ca2+ channels reduced the asynchronous release component at WT but not at SynII(−) slices. However, in contrast to the study cited above, we found that the blockade of P/Q-type Ca2+ channels similarly affected the synchronous and asynchronous release components in both genotypes.
Our study also demonstrated that SynII deletion synchronized the release kinetics and suppressed the asynchronous release component at CCK interneurons, which depend on N-type Ca2+ channels. The mechanisms underlying this phenomenon remain to be unraveled. However, it is possible that SynII contributes to maintaining the asynchronous release component at CCK cells by promoting Ca2+-dependent docking of vesicles to the synaptic membrane, as it was observed at the neuromuscular synapse (Coleman et al., 2008). This hypothesis remains to be tested.
It is still unclear how specifically the deficit in inhibitory transmission produces seizure activity in synapsin-deficient brain. The epilepsy in synapsin knock-out animals is age-dependent, and a number of studies suggest that seizures arise as a result of developmental compensations in neuronal networks (Fassio et al., 2011; Ketzef et al., 2011; Ketzef and Gitler, 2014). However, there is strong evidence that an excitation/inhibition imbalance in presymptomatic animals contributes to the initial epilepsy predisposition (Boido et al., 2010; Fassio et al., 2011; Farisello et al., 2013; Feliciano et al., 2013). In the present study, we demonstrated that SynII deletion reduced IPSC amplitudes and prolonged the release kinetics in PV interneurons, which have a key role in the regulation of seizure propagation (Krook-Magnuson et al., 2013; Sessolo et al., 2015). Furthermore, extensive evidence links epilepsy with asynchronous activity of GABAergic synapses through tonic inhibition (Jiang et al., 2012; Pandit et al., 2013; Medrihan et al., 2015). We demonstrated here that SynII maintains the asynchronous release component at CCK interneurons, which are thought to contribute to vulnerability of neuronal networks to seizure activity (Wyeth et al., 2010; Lee and Soltesz, 2011).
Footnotes
This study was supported by National Science Foundation Grant IOS 1145010 to M.B., National Institutes of Health Grants MH43987 and MH100850 to R.A., and National Institutes of Health Grant F31NS079096 to P.F.
The authors declare no competing financial interests.
References
- Bartos M, Elgueta C (2012) Functional characteristics of parvalbumin- and cholecystokinin-expressing basket cells. J Physiol 590:669–681. 10.1113/jphysiol.2011.226175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Frotscher M, Meyer A, Monyer H, Geiger JR, Jonas P (2002) Fast synaptic inhibition promotes synchronized gamma oscillations in hippocampal interneuron networks. Proc Natl Acad Sci U S A 99:13222–13227. 10.1073/pnas.192233099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boido D, Farisello P, Cesca F, Ferrea E, Valtorta F, Benfenati F, Baldelli P (2010) Cortico-hippocampal hyperexcitability in synapsin I/II/III knockout mice: age-dependency and response to the antiepileptic drug levetiracetam. Neuroscience 171:268–283. 10.1016/j.neuroscience.2010.08.046 [DOI] [PubMed] [Google Scholar]
- Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P (2008) Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 57:536–545. 10.1016/j.neuron.2007.12.026 [DOI] [PubMed] [Google Scholar]
- Bykhovskaia M. (2008) Making quantal analysis more convenient, fast, and accurate: user-friendly software QUANTAN. J Neurosci Methods 168:500–513. 10.1016/j.jneumeth.2007.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalleri GL, Weale ME, Shianna KV, Singh R, Lynch JM, Grinton B, Szoeke C, Murphy K, Kinirons P, O'Rourke D, Ge D, Depondt C, Claeys KG, Pandolfo M, Gumbs C, Walley N, McNamara J, Mulley JC, Linney KN, Sheffield LJ, et al. (2007) Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol 6:970–980. 10.1016/S1474-4422(07)70247-8 [DOI] [PubMed] [Google Scholar]
- Chiappalone M, Casagrande S, Tedesco M, Valtorta F, Baldelli P, Martinoia S, Benfenati F (2009) Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I-deficient cortical networks. Cereb Cortex 19:1422–1439. 10.1093/cercor/bhn182 [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bykhovskaia M (2009) Synapsin I accelerates the kinetics of neurotransmitter release in mouse motor terminals. Synapse 63:531–533. 10.1002/syn.20635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman WL, Bill CA, Simsek-Duran F, Lonart G, Samigullin D, Bykhovskaia M (2008) Synapsin II and calcium regulate vesicle docking and the cross-talk between vesicle pools at the mouse motor terminals. J Physiol 586:4649–4673. 10.1113/jphysiol.2008.154666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Malladi CS, Tan TC, Raymond CR, Smillie KJ, Robinson PJ (2003) Synapsin I-associated phosphatidylinositol 3-kinase mediates synaptic vesicle delivery to the readily releasable pool. J Biol Chem 278:29065–29071. 10.1074/jbc.M302386200 [DOI] [PubMed] [Google Scholar]
- Daw MI, Tricoire L, Erdelyi F, Szabo G, McBain CJ (2009) Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J Neurosci 29:11112–11122. 10.1523/JNEUROSCI.5760-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1995) Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15:1097–1107. 10.1016/0896-6273(95)90098-5 [DOI] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P (2011) Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci 13:7–21. 10.1038/nrn3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etholm L, Heggelund P (2009) Seizure elements and seizure element transitions during tonic-clonic seizure activity in the synapsin I/II double knockout mouse: a neuroethological description. Epilepsy Behav 14:582–590. 10.1016/j.yebeh.2009.02.021 [DOI] [PubMed] [Google Scholar]
- Farisello P, Boido D, Nieus T, Medrihan L, Cesca F, Valtorta F, Baldelli P, Benfenati F (2013) Synaptic and extrasynaptic origin of the excitation/inhibition imbalance in the hippocampus of synapsin I/II/III knockout mice. Cereb Cortex 23:581–593. 10.1093/cercor/bhs041 [DOI] [PubMed] [Google Scholar]
- Fassio A, Merlo D, Mapelli J, Menegon A, Corradi A, Mete M, Zappettini S, Bonanno G, Valtorta F, D'Angelo E, Benfenati F (2006) The synapsin domain E accelerates the exoendocytotic cycle of synaptic vesicles in cerebellar Purkinje cells. J Cell Sci 119:4257–4268. 10.1242/jcs.03194 [DOI] [PubMed] [Google Scholar]
- Fassio A, Raimondi A, Lignani G, Benfenati F, Baldelli P (2011) Synapsins: from synapse to network hyperexcitability and epilepsy. Semin Cell Dev Biol 22:408–415. 10.1016/j.semcdb.2011.07.005 [DOI] [PubMed] [Google Scholar]
- Feliciano P, Andrade R, Bykhovskaia M (2013) Synapsin II and Rab3a cooperate in the regulation of epileptic and synaptic activity in the CA1 region of the hippocampus. J Neurosci 33:18319–18330. 10.1523/JNEUROSCI.5293-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, Curtis A, Goodship JA (2004) Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet 41:183–186. 10.1136/jmg.2003.013680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannandrea M, Guarnieri FC, Gehring NH, Monzani E, Benfenati F, Kulozik AE, Valtorta F (2013) Nonsense-mediated mRNA decay and loss-of-function of the protein underlie the X-linked epilepsy associated with the W356× mutation in synapsin I. PLoS One 8:e67724. 10.1371/journal.pone.0067724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ (2004) Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci 24:11368–11380. 10.1523/JNEUROSCI.3795-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami SP, Bucurenciu I, Jonas P (2012) Miniature IPSCs in hippocampal granule cells are triggered by voltage-gated Ca2+ channels via microdomain coupling. J Neurosci 32:14294–14304. 10.1523/JNEUROSCI.6104-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F (1993) Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259:780–785. 10.1126/science.8430330 [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P (2005) Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8:1319–1328. 10.1038/nn1542 [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ (1998) Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci 1:29–35. 10.1038/229 [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P (1999) Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354:269–279. 10.1098/rstb.1999.0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P (2014) Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 345:1255263. 10.1126/science.1255263 [DOI] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B (2001) Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J Neurosci 21:4195–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zhu J, Liu Y, Yang M, Tian C, Jiang S, Wang Y, Guo H, Wang K, Shu Y (2012) Enhancement of asynchronous release from fast-spiking interneuron in human and rat epileptic neocortex. PLoS Biol 10:e1001324. 10.1371/journal.pbio.1001324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketzef M, Gitler D (2014) Epileptic synapsin triple knockout mice exhibit progressive long-term aberrant plasticity in the entorhinal cortex. Cereb Cortex 24:996–1008. 10.1093/cercor/bhs384 [DOI] [PubMed] [Google Scholar]
- Ketzef M, Kahn J, Weissberg I, Becker AJ, Friedman A, Gitler D (2011) Compensatory network alterations upon onset of epilepsy in synapsin triple knock-out mice. Neuroscience 189:108–122. 10.1016/j.neuroscience.2011.05.030 [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P (2008) Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57. 10.1126/science.1149381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohus Z, Káli S, Rovira-Esteban L, Schlingloff D, Papp O, Freund TF, Hájos N, Gulyás AI (2016) Properties and dynamics of inhibitory synaptic communication within the CA3 microcircuits of pyramidal cells and interneurons expressing parvalbumin or cholecystokinin. J Physiol 594:3745–3774. 10.1113/JP272231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I (2013) On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 4:1376. 10.1038/ncomms2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhan R, Kalita J, Misra UK, Kumari R, Mittal B (2010) Association of intronic polymorphism rs3773364 A>G in synapsin-2 gene with idiopathic epilepsy. Synapse 64:403–408. 10.1002/syn.20740 [DOI] [PubMed] [Google Scholar]
- Lee SY, Soltesz I (2011) Cholecystokinin: a multi-functional molecular switch of neuronal circuits. Dev Neurobiol 71:83–91. 10.1002/dneu.20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P (1995) Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A 92:9235–9239. 10.1073/pnas.92.20.9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Roberts JD, Szucs P, Cottingham CA, Somogyi P (2000) Cell surface domain specific postsynaptic currents evoked by identified GABAergic neurones in rat hippocampus in vitro. J Physiol 524:91–116. 10.1111/j.1469-7793.2000.t01-3-00091.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx M, Günter RH, Hucko W, Radnikow G, Feldmeyer D (2012) Improved biocytin labeling and neuronal 3D reconstruction. Nat Protoc 7:394–407. 10.1038/nprot.2011.449 [DOI] [PubMed] [Google Scholar]
- Medrihan L, Cesca F, Raimondi A, Lignani G, Baldelli P, Benfenati F (2013) Synapsin II desynchronizes neurotransmitter release at inhibitory synapses by interacting with presynaptic calcium channels. Nat Commun 4:1512. 10.1038/ncomms2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Ferrea E, Greco B, Baldelli P, Benfenati F (2015) Asynchronous GABA release is a key determinant of tonic inhibition and controls neuronal excitability: a study in the synapsin II−/− mouse. Cereb Cortex 25:3356–3368. 10.1093/cercor/bhu141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M (2004) Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A 58:167–176. 10.1002/cyto.a.20022 [DOI] [PubMed] [Google Scholar]
- Messa M, Congia S, Defranchi E, Valtorta F, Fassio A, Onofri F, Benfenati F (2010) Tyrosine phosphorylation of synapsin I by Src regulates synaptic-vesicle trafficking. J Cell Sci 123:2256–2265. 10.1242/jcs.068445 [DOI] [PubMed] [Google Scholar]
- Neu A, Földy C, Soltesz I (2007) Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol 578:233–247. 10.1113/jphysiol.2006.115691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenbuch A, Shalev L, Marra V, Sinai I, Lavy Y, Kahn J, Burden JJ, Staras K, Gitler D (2012) Synapsin selectively controls the mobility of resting pool vesicles at hippocampal terminals. J Neurosci 32:3969–3980. 10.1523/JNEUROSCI.5058-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Jeong JA, Jo JY, Cho HS, Kim DW, Kim JM, Ryu PD, Lee SY, Kim HW, Jeon BH, Park JB (2013) Dual mechanisms diminishing tonic GABAA inhibition of dentate gyrus granule cells in Noda epileptic rats. J Neurophysiol 110:95–102. 10.1152/jn.00727.2012 [DOI] [PubMed] [Google Scholar]
- Prasad DK, Shaheen U, Satyanarayana U, Prabha TS, Jyothy A, Munshi A (2014) Association of GABRA6 1519 T>C (rs3219151) and Synapsin II (rs37733634) gene polymorphisms with the development of idiopathic generalized epilepsy. Epilepsy Res 108:1267–1273. 10.1016/j.eplepsyres.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW (1995) Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci 15:2995–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC (1995) Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375:488–493. 10.1038/375488a0 [DOI] [PubMed] [Google Scholar]
- Samigullin D, Bill CA, Coleman WL, Bykhovskaia M (2004) Regulation of transmitter release by synapsin II in mouse motor terminals. J Physiol 561:149–158. 10.1113/jphysiol.2004.073494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savanthrapadian S, Meyer T, Elgueta C, Booker SA, Vida I, Bartos M (2014) Synaptic properties of SOM- and CCK-expressing cells in dentate gyrus interneuron networks. J Neurosci 34:8197–8209. 10.1523/JNEUROSCI.5433-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T, Carmignoto G (2015) Parvalbumin-positive inhibitory interneurons oppose propagation but favor generation of focal epileptiform activity. J Neurosci 35:9544–9557. 10.1523/JNEUROSCI.5117-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Haucke V, Pechstein A (2011) How synapsin I may cluster synaptic vesicles. Semin Cell Dev Biol 22:393–399. 10.1016/j.semcdb.2011.07.006 [DOI] [PubMed] [Google Scholar]
- Song SH, Augustine GJ (2016) Synapsin isoforms regulating GABA release from hippocampal interneurons. J Neurosci 36:6742–6757. 10.1523/JNEUROSCI.0011-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547. 10.1146/annurev.neuro.26.041002.131412 [DOI] [PubMed] [Google Scholar]
- Sun C, Sun J, Erisir A, Kapur J (2014) Loss of cholecystokinin-containing terminals in temporal lobe epilepsy. Neurobiol Dis 62:44–55. 10.1016/j.nbd.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyeth MS, Zhang N, Mody I, Houser CR (2010) Selective reduction of cholecystokinin-positive basket cell innervation in a model of temporal lobe epilepsy. J Neurosci 30:8993–9006. 10.1523/JNEUROSCI.1183-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S (2015) Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol 113:1616–1630. 10.1152/jn.00841.2014 [DOI] [PubMed] [Google Scholar]
- Yi F, DeCan E, Stoll K, Marceau E, Deisseroth K, Lawrence JJ (2015) Muscarinic excitation of parvalbumin-positive interneurons contributes to the severity of pilocarpine-induced seizures. Epilepsia 56:297–309. 10.1111/epi.12883 [DOI] [PMC free article] [PubMed] [Google Scholar]