

Abstract
Cynomolgus macaques (Macaca fascicularis) were introduced to the island of Mauritius by humans around the 16th century. The unique demographic history of the Mauritian cynomolgus macaques provides the opportunity to not only examine the genetic background of well-established nonhuman primates for biomedical research but also understand the effect of an extreme population bottleneck on the pattern of polymorphisms in genomes. We sequenced the whole genomes of six Mauritian cynomolgus macaques and obtained an average of 20-fold coverage of the genome sequences for each individual. The overall level of nucleotide diversity was 23% smaller than that of the Malaysian cynomolgus macaques, and a reduction of low-frequency polymorphisms was observed. In addition, we also confirmed that the Mauritian cynomolgus macaques were genetically closer to a representative of the Malaysian population than to a representative of the Indochinese population. Excess of nonsynonymous polymorphisms in low frequency, which has been observed in many other species, was not very strong in the Mauritian samples, and the proportion of heterozygous nonsynonymous polymorphisms relative to synonymous polymorphisms is higher within individuals in Mauritian than Malaysian cynomolgus macaques. Those patterns indicate that the extreme population bottleneck made purifying selection overwhelmed by the power of genetic drift in the population. Finally, we estimated the number of founding individuals by using the genome-wide site frequency spectrum of the six samples. Assuming a simple demographic scenario with a single bottleneck followed by exponential growth, the estimated number of founders (∼20 individuals) is largely consistent with previous estimates.
Keywords: Mauritian cynomolgus macaque, genome sequence, population bottleneck
Introduction
Nonhuman primates, in particular macaque monkeys, are important biological resources because of their genetic similarity with humans (approximately 94% in nucleotide sequence identity), which is much higher than that of the nonprimate mammal animal models (Gibbs et al. 2007; Shively and Clarkson 2009). However, the difficulty of obtaining genetically homogenous individuals in primates hampers their use in several fields of experimental medicine. Therefore, it is important to elucidate the genetic background of macaques to be able to use these animals for future biomedical research.
The cynomolgus macaque (Macaca fascicularis) is one of the most widely used experimental animals in biomedical research, and has been used to study the effect of various medications as well as vaccines against infectious diseases. This species lives in widely distributed range in Southeast Asia, including areas of Indochina, Malaysia, Indonesia, the Philippines, and also the island of Mauritius, where the animals were only recently introduced by humans (Fooden 1976). Cynomolgus macaques are evolutionarily closely related to rhesus macaques (Macaca mulatta), another species which has been extensively studied. Polymorphisms shared between the cynomolgus and rhesus macaques suggest historical gene introgression between the two species, particularly in populations living near the boundary between their geographical distribution areas in the North of the Indochinese peninsula (Bonhomme et al. 2009; Stevison and Kohn 2009; Higashino et al. 2012). The average genetic divergence between the cynomolgus and rhesus macaques is 0.4–0.5% per site in the nuclear genome (Osada et al. 2010), which is considerably close to the average genetic diversity within each species. After the Indian government banned the export of the rhesus macaque to foreign countries in 1978, the importance of the cynomolgus macaques as an alternative resources for biomedical research has been increasingly appreciated (Wade 1978; Pavlin et al. 2009).
Previous studies have shown that cynomolgus macaques are genetically highly heterogeneous (Osada et al. 2010; Yan et al. 2011), and that this genetic heterogeneity could contribute to varied responses to drugs and pathogens (Menninger et al. 2002; Drevon-Gaillot et al. 2006) and influence various biological parameters (Aarnink, Garchon, et al. 2011; Aarnink et al. 2013). Studies using mitochondrial and nuclear genome data have revealed that cynomolgus macaque populations are divided into four major genetic groups (Smith et al. 2007; Blancher et al. 2008; Osada et al. 2010): The Indonesian-Malaysian, Indochinese, Philippine, and Mauritian populations. These four populations show different levels of genetic diversity and have different demographic histories. The Indonesian-Malaysian population is thought to be the ancestral population of cynomolgus macaques, and show the highest level of nucleotide diversity (π), estimated to be 3.0 − 3.2 × 10−3 per site (Osada et al. 2010; Higashino et al. 2012; Fan et al. 2014), which is approximately three times higher than that in the entire human population (Prado-Martinez et al. 2013). The macaque population in the Philippines shows slightly reduced genetic diversity, probably because of a recent population size contraction (Osada et al. 2013). Phylogenetic trees of the mitochondrial DNA suggest that the Philippine population was derived from the Indonesian-Malaysian population (Smith et al. 2007; Tosi and Coke 2007; Blancher et al. 2008; Kanthaswamy et al. 2008; Stevison and Kohn 2008). The Indochinese cynomolgus population is thought to have experienced a nonnegligible amount of gene introgression from the rhesus macaques (Kanthaswamy et al. 2008; Bonhomme et al. 2009; Stevison and Kohn 2009), although the historical effect of this interspecies gene flow has not been solely restricted to the Indochinese population (Osada et al. 2010).
Among the four major population groups, the Mauritian population has a particularly interesting demographic history; a small number of individuals were brought to the Mauritian island in the 16th century, where they settled to give rise to a quickly expanding population (Sussman and Tattersall 1986). Consistent with this historical record, the Mauritian population is characterized by a limited number of major histocompatibility complex (MHC) alleles (Leuchte et al. 2004; Krebs et al. 2005; Aarnink, Apoil, et al. 2011; Blancher et al. 2012), a small number of mitochondrial haplotypes (Smith et al. 2007; Tosi and Coke 2007; Blancher et al. 2008), and small numbers of microsatellite alleles at various loci (Bonhomme et al. 2008; Kawamoto et al. 2008). Because of their large population on the island (they are an invasive species in the Mauritian island) and the relatively simple configuration of their MHC alleles, Mauritian cynomolgus macaques have been used in several biomedical studies and their genome was sequenced as the first cynomolgus macaque genome (Ebeling et al. 2011). Although Mauritian cynomolgus macaques are thought to have a highly homogenous genetic background, recent studies using single nucleotide variant (SNV) markers unexpectedly identified genetic structures, indicating that there may be two or three subpopulations within the Mauritian cynomolgus macaques (Ogawa and Vallender 2014). However, these genetic structures do not correspond to their geographic distribution (Satkoski Trask et al. 2013). Through the studies of their MHC locus, it has been demonstrated that their repertoire of MHC haplotypes has been reduced by the founder effect; however, the impact of this population bottleneck on the other nuclear genes has not been well studied at the SNV level.
The extreme population bottleneck in Mauritian cynomolgus macaques also provides evolutionary insight into how, in such circumstances, deleterious mutations can accumulate in genomes. Theoretical studies predict that the reduction of effective population size reduces the efficacy of natural selection and could result in the segregation and fixation of slightly deleterious mutations (Ohta 1973). Extreme population bottleneck reduces genetic diversity; hence, may cause a decline of the average fitness of a population. Mauritian cynomolgus macaques have well thrived in the island and rapidly expanded their population size (Sussman and Tattersall 1986). The well-documented demographic history of Mauritian cynomolgus macaque may provide a good opportunity for investigating the effect of extreme population bottleneck on the genome-wide pattern of polymorphisms.
To date, whole-genome sequences of one Malaysian (Higashino et al. 2012), one Vietnamese (Yan et al. 2011), and one Mauritian (Ebeling et al. 2011) cynomolgus macaque have been analyzed. More recently, large-scale genome sequencing of Mauritian cynomolgus macaques was performed to find genetic causes of viral susceptibility (Ericsen et al. 2014). However, analyzing randomly sampled individuals to infer the past demography of the Mauritian cynomolgus macaques has not yet been conducted. Clarifying the genetic background of Mauritian cynomolgus macaques is of great importance for both biomedical and evolutionary research. Here, we report the whole-genome sequences of six Mauritian cynomolgus macaques with an approximately 20-fold coverage of the genome.
Materials and Methods
DNA Sequencing
We extracted DNA from blood samples of wild-caught male Mauritian cynomolgus macaques, initially used for studying the sex-matched response to SIV infection (Aarnink, Dereuddre-Bosquet, et al. 2011). At sampling, there was no evidence that they were closely related with each other. Genome sequencing libraries of approximately 450 bp length were constructed for each of the six macaques. Paired-end sequences of 100 bp were determined using HiSeq2000 (Illumina Inc, San Diego, CA). The library construction, sequencing, and initial quality check were performed at Beijing Genomics Institute (Shenzhen, China).
SNV Calling
Reads were mapped on the draft genome of the rhesus macaque (rheMac2), the draft Y chromosome sequence (Hughes et al. 2012), and the mitochondrial genome (DDBJ/GenBank/EMBL accession number: AY612638) using the BWA aln/sampe algorithm with default parameter settings, except for quality trimming score of −15 (Li and Durbin 2009). Among the samples, the average mapping rate of reads was 93.3%. Potential polymerase chain reaction duplicates were marked using Picard software (http://picard.sourceforge.net, last accessed March 5, 2015). SNVs were jointly called on all six samples using the Best Practice pipeline of the Genome Analysis Toolkit software package (Mckenna et al. 2010), which includes base quality score recalibration, insertion/deletion (indel) realignment, SNV calling, and variant quality score recalibration (Van Der Auwera et al. 2002; Depristo et al. 2011). After calling the initial set of variants, further application of the following hard filters was employed: FS > 60.0, HaplotypeScore > 13.0, MQ < 40.0, MQRankSum < −12.5, QD < 2.0, ReadPosRankSum < −8.0. SNVs on fragmented scaffolds (chrUr) were not included in analysis. Heterozygosity within individuals and nucleotide diversity (π) were estimated using only high coverage sites (≥10-folds). All raw read sequences and initial sets of variants are deposited into the public database (EMBL-EBI accession number: PRJEB7871).
Principal Component Analysis and Population Tree
Principal component analysis (PCA) was conducted using the smartpca program in the EIGENSOFT software package (Patterson et al. 2006). Extraction, filtering, and processing of data were performed using custom-made perl scripts. A population tree was constructed using three additional genome sequences of macaques (Yan et al. 2011; Higashino et al. 2012). PHYLIP software (Felsenstein 1989) was used to generate the distance matrix for the macaque individuals by Nei’s genetic distance (Nei 1972). For the tree construction, the allele frequency data of SNV sites (coverage ≥ 10) found in all nine individuals were used. The phylogenetic tree was constructed using the neighbor-joining method (Saitou and Nei 1987) implemented in MEGA6 (Tamura et al. 2013).
Pairwise Sequentially Markovian Coalescent Method
The analysis was performed using Pairwise Sequentially Markovian Coalescent (PSMC) software (Li and Durbin 2011). Consensus genome sequences for PSMC input were constructed using samtools and vcf2fq utility (Li et al. 2009). The time interval parameter of 4 + 25*2 + 4 + 6 and the number of iterations of 25 were used for the parameters of PSMC.
Prediction of Disease Causing Mutations
To infer the disease causality of nonsynonymous mutations in the Mauritian cynomolgus macaques, we identified respective nonsynonymous mutations in human orthologs and predicted the functional effect using PolyPhen-2 (Adzhubei et al. 2010). Gene annotation of the rhesus macaque followed the annotation in a previous study (Higashino et al. 2012). The human–macaque ortholog information was retrieved from the Ensembl database (Flicek et al. 2014). Human and macaque protein sequences were aligned using ClustalW (Thompson et al. 1994) and only the sites that have the same amino acid residues between human and macaque reference proteins in the alignment were analyzed through the PolyPhen-2 website (http://genetics.bwh.harvard.edu/pph2/, last accessed March 5, 2015). From this analysis, we had a final functional prediction of 7,976 nonsynonymous mutations.
Site Frequency Spectrum
Folded site frequency spectrum (fSFS) of ith occurrence was defined as and , where Ci represents the number of variants observed for i chromosomes and n is the number of sampled chromosomes. The number of sampled chromosomes in our study is 12 (diploid chromosomes of six individuals). To correct the excess of SNVs that are heterozygous in all individuals, most of which are thought to be due to genotyping error (see also Results and Discussion section), we applied a simple correction method assuming the Hardy–Weinberg equilibrium. We assumed that the allele frequency of all SNV sites that showed heterozygosity in all six individuals was 0.5, for which the highest proportion (1/2) of heterozygotes is expected. It should be noted that this assumption is conservative. We denoted the observed number of SNV sites that showed heterozygosity in all six individuals by C6_H6, with a corresponding expected probability of 0.56. The observed number of SNV sites that have a frequency of 0.5 and are not heterozygous in all samples is . If is the true number of mutations that are heterozygous in all individuals, the following relationship should hold: . The number of was estimated by solving this equation.
Estimating the Number of Founders
The level of the past population bottleneck was inferred by fitting expected fSFS to the observed fSFS using the analytical formula obtained by Marth et al. (2004). We considered a single bottleneck event, followed by exponential growth, which has two population genetic parameters to be estimated: The time of the bottleneck (Tb) and the size of the bottleneck (Nb). The ancestral population size (Na) and the current population size (N0) were fixed for each estimation. Because the model is scalable to any population size, we estimated fSFS for when Na is 100,000, and scaled the parameters after fitting. The deviance of expected to observed fSFS was evaluated using χ2 statistics for very small intervals for each Tb and Nb. In addition, we confirmed that the analytical formula and coalescent simulations gave highly consistent expected fSFS using our population growth model (data not shown).
Results and Discussion
Identification of SNVs and Estimation of Nucleotide Diversity
We obtained 100-bp-length Illumina paired-end sequences from six unrelated Mauritian cynomolgus macaques and mapped the reads to the reference rhesus macaque genome (see Materials and Methods section). We did not map the reads to the reference genome of the Mauritian cynomolgus macaque (Ebeling et al. 2011), because the rhesus macaque reference has better gene annotation and previous studies have shown that rhesus macaque genomes are sufficiently close to cynomolgus macaque genomes for read mapping by typical short-read mappers (Yan et al. 2011; Higashino et al. 2012). The average coverage is approximately 20-fold for each individual (table 1). In total, we identified approximately 21.8 million SNVs and 1.9 million indels against the reference genome among the six macaques on the autosomes. Because all samples are males, sex chromosomes and mitochondrial genome are all haploid genomes in our samples. Therefore, we mainly focused on the pattern of SNVs on the autosomes in this study. Summary of SNVs identified on the sex chromosomes is shown in supplementary table S1, Supplementary Material online. Each individual has an average of 5.8 million heterozygous and 8.3 million homozygous SNVs, and these numbers are highly consistent among individuals. Here, homozygous variants are defined against the reference genome sequence of rhesus macaque. Estimation of genetic diversity within the Mauritian cynomolgus macaque population was 2.28 × 10−3 for nucleotide diversity (π). We retrieved the previously published Malaysian cynomolgus macaque genomes and estimated heterozygosity using the same criteria for SNV identification (π = 2.96 × 10−3). The heterozygosity of;?>the Mauritian cynomolgus macaques was 23% smaller than the Malaysian cynomolgus macaque, which is thought to have very high genetic diversity.
Table 1.
Summary of Variant Calling in Six Mauritian Cynomolgus Macaques
| Sample ID | Average Sequencing Deptha | Total SNV | Heterozygous SNV | Homozygous SNV | Heterozygosity |
|---|---|---|---|---|---|
| Tlse-8102 | 20.80 | 14,048,997 | 5,676,969 | 8,372,028 | 0.00225 |
| (MCM1) | |||||
| Tlse-8141 | 20.42 | 14,045,116 | 5,814,396 | 8,230,720 | 0.00231 |
| (MCM2) | |||||
| Tlse-8249 | 19.14 | 14,175,393 | 5,947,573 | 8,227,820 | 0.00236 |
| (MCM3) | |||||
| Tlse-9204 | 20.33 | 14,116,067 | 5,837,878 | 8,278,189 | 0.00232 |
| (MCM4) | |||||
| Tsle-9413 | 19.61 | 14,150,408 | 5,883,805 | 8,266,603 | 0.00234 |
| (MCM5) | |||||
| Tlse-9859 | 20.42 | 14,080,208 | 5,753,557 | 8,326,651 | 0.00229 |
| (MCM6) | |||||
| Malaysian cynomolgus macaque | 26.1 | 12,758,246 | 7,177,728 | 5,580,518 | 0.00296 |
aReads mapped on autosomes.
Genetic Relationship between and within Populations
In addition to the Malaysian cynomolgus macaque genome, we retrieved the two more previously published macaque genomes (Vietnamese cynomolgus macaque and Chinese rhesus macaque). Genetic relationships among the six Mauritian cynomolgus macaque individuals were examined using PCA plot (fig. 1). We confirmed that no individuals were closely overlapped in the plot. A plot including all nine macaque genomes is presented in the supplementary figure S1, Supplementary Material online. We further examined whether the Mauritian cynomolgus macaques are genetically closer to the Malaysian or to the Indochina cynomolgus macaques. Figure 2 shows the phylogenetic relationship of the four macaque populations. Consistent with the results from mitochondrial data (Smith et al. 2007), the Mauritian cynomolgus macaques are genetically closer to the Malaysian cynomolgus macaques. Because genome sequences of the Indonesian populations have not been analyzed, we were not able to determine the detailed origin of the Mauritian cynomolgus macaques.
Fig. 1.—
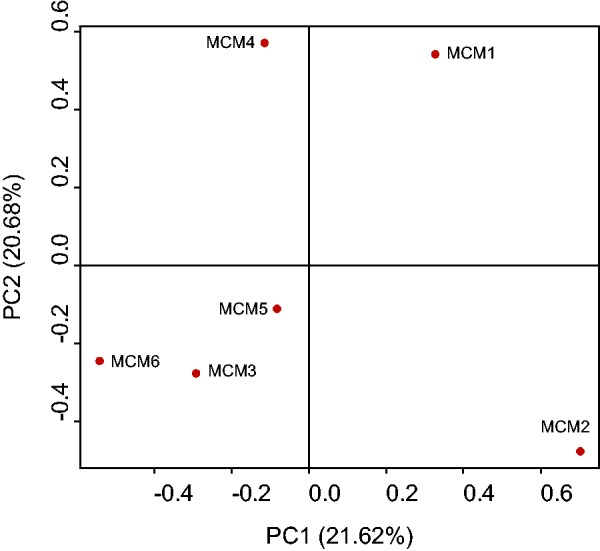
PCA plot of the six Mauritian cynomolgus macaque individuals. The individual ID is given beside each data point. The x- and y-axes represent the first and second principal components, respectively.
Fig. 2.—

Phylogenetic relationships of the four populations. MFA and MMU designate M. fascicularis and M. mulatta, respectively. The branch length represents Nei’s genetic distance. Bootstrap confidence values (percentile) are shown upon the branches.
In addition, the past demography was estimated using the PSMC method (Li and Durbin 2011). The inferred demographic histories are shown in figure 3. The six Mauritian cynomolgus macaques showed a very similar trend of past demography. This result indicates that they are not derived from genetically distinct origins, which agrees with that of the mitochondrial data (Smith et al. 2007). However, we should note that PSMC would not work for the Mauritian cynomolgus macaques to properly scale time and population size because this analysis has a limitation in inferring very recent population size changes. The actual bottleneck in the Mauritian cynomolgus macaques was very recent to be inferred by PSMC. If the genome-wide heterozygosity were dramatically changed by very recent demographic events, scaling parameters would fail. In this study, our purpose of using PSMC was to check whether the population size trajectories overlap with each other, and not for estimating demographic parameters themselves; therefore, in figure 3, we only showed parameter values scaled by N0 = 10,000, which was arbitrarily determined. The confidence intervals of population size estimation are shown in the supplementary figure S2, Supplementary Material online. In order to infer the recent demography we applied the method using information on polymorphism frequency, which is described in the later section.
Fig. 3.—

The change in population size inferred by PSMC. Six individuals, MCM1–MCM6, are labeled by red, blue, green, black, purple, and orange lines, respectively. The time from the present and effective population size are shown in the x- and y-axes, respectively. Note that the time and the size were arbitrarily scaled by baseline effective population size (N0) equal to 10,000 (see Results and Discussion).
Site Frequency Spectrum of SNVs
To elucidate a more detailed pattern of the polymorphisms in the Mauritian cynomolgus macaques, we calculated the SFS of mutations in the six samples. Because we cannot assume that the reference rhesus macaque genome has ancestral states, the spectrum is folded (see Materials and Methods section). Before evolutionary inference, we carefully examined the potential genotyping errors that could affect the pattern of SFS. We found an excess of SNVs for which all six macaques were heterozygous (H6 sites); this fraction of H6 sites was enriched with nonsynonymous mutations. Fixation of segmental duplication with subsequent mutations may cause false identification of heterozygosity at such sites; alternatively, these sites would be observed when one of the duplicated loci is not present in the reference genome sequence. If either of these cases were the cause of the H6 sites, we would expect these sites to have higher genome sequencing coverage. To evaluate this, we compared the occurrence of H6 sites with the average coverage depth among the six genomes at those sites. Repeat regions of the genome were excluded from this analysis to avoid the complex effect of repetitive sequences. The results showed that the coverage distribution for H6 sites was skewed toward higher coverage, and the coverage distribution for nonsynonymous and synonymous sites among the H6 sites was more strongly biased toward higher coverage (fig. 4). Although we cannot identify the reason for these systematic biases, this pattern of miscalling should be carefully interpreted in future whole-genome sequencing studies. To remove the potential genotyping errors for the analysis of fSFS data, we corrected the miscalling of H6 sites by assuming the Hardy–Weinberg equilibrium (see Materials and Methods).
Fig. 4.—
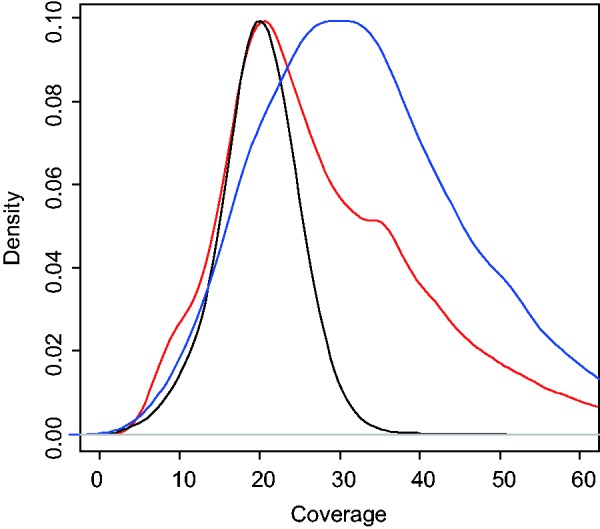
Genome sequencing coverage of SNV sites. The height of the lines shows the density estimation of read coverage. The black, red, and blue lines represent the estimated density for all SNV sites, noncoding H6 sites, and coding H6 sites, respectively. H6 sites are the sites where all six samples are heterozygous.
In figure 5A, fSFS for nonsynonymous, synonymous, and noncoding sites is shown. Notably, nonsynonymous and synonymous sites are defined among the six Mauritian cynomolgus macaque alleles. Compared with a neutral expectation with a constant population size, Mauritian cynomolgus macaques harbor significantly fewer low-frequency polymorphisms, particularly singletons (P < 10−15; χ2 test). A reduction of population size is expected to affect low-frequency polymorphisms more than common-frequency polymorphisms (Luikart et al. 1998). Because π is more sensitive to the difference in common-frequency polymorphisms, we expected that π would not be greatly affected by the very recent population bottleneck (Tajima 1989).
Fig. 5.—
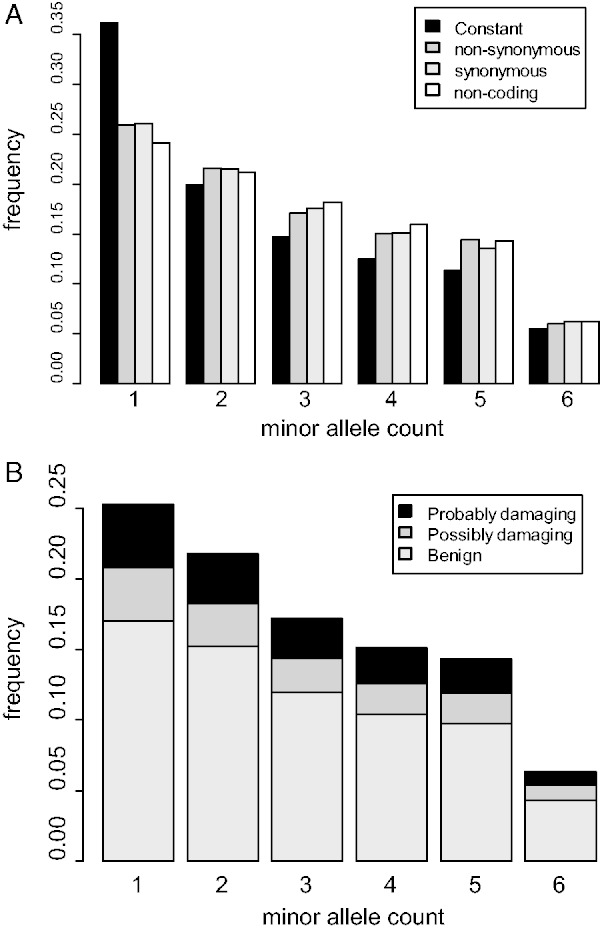
fSFS for nonsynonymous (dark gray), synonymous (light gray), and noncoding (white) sites among the six Mauritian cynomolgus macaque individuals. The expected frequency with constant population size is shown by the black bar (A). Prediction of disease-causing mutations was performed using PolyPhen-2. Probably damaging, possibly damaging, and benign nonsynonymous mutations are shown in the black, dark gray, and light gray bars, respectively (B).
Interestingly, the patterns of fSFS for noncoding, synonymous, and nonsynonymous mutations are not strongly different within the Mauritian cynomolgus macaque population. In particular, most of the large-scale population genetic studies in humans (e.g., Fujimoto et al. 2010) have found an excess of low frequency nonsynonymous mutations; however, this was not observed in the Mauritian macaque, although the difference between synonymous and nonsynonymous mutations was statistically significant (P < 10−15; χ2 test). In addition, nonsynonymous and synonymous mutations showed similar level of singletons (P = 0.67; χ2 test). Because recent population bottleneck mostly affects the pattern of rare polymorphisms, mutations segregating within the macaque population at low frequencies were rapidly lost during the bottleneck period, and the time elapsed since the bottleneck has been short to allow for the appearance of new mutations.
We examined the phenotypic effect of mutations using predictions of disease causality in human genes. Predictions of the functional effect on nonsynonymous SNVs were performed using PolyPhen-2 (Adzhubei et al. 2010), which predicts the potential impact of an amino acid substitution based on protein structure and evolutionary conservation. We excluded all H6 mutations from the disease-causing mutation analyses because most of them are likely genotyping errors. The fraction of potentially damaging mutations for each fSFS category is shown in figure 5B. The excess of damaging mutations in the singleton class was not statistically significant (P = 0.17; χ2 test), which is considerably different from the pattern in humans (Andrés et al. 2009).
On an average, Mauritian cynomolgus macaques have 10,565 nonsynonymous and 13,533 synonymous heterozygous SNVs. The ratio of nonsynonymous to synonymous polymorphisms was 0.78, which is significantly higher than the ratio observed in the Malaysian cynomolgus macaque individual (0.68; Higashino et al. 2012; P < 10−15; χ2 test). The higher ratio in the Mauritian cynomolgus macaques indicates that more deleterious mutations are segregating with greater frequency in the population. In addition, we found that 12,467 nonsynonymous and 17,749 synonymous changes are fixed among the Mauritian cynomolgus macaque samples compared with the reference rhesus macaque genome.
Considering that the proportion of heterozygosity at nonsynonymous SNVs/synonymous SNVs is higher in Mauritian individuals than in the Malaysian individual, we concluded that the pattern of polymorphisms in the Mauritian cynomolgus macaques has been predominantly shaped by a strong genetic drift and has overwhelmed by the power of purifying selection during the population bottleneck.
However, the data also showed that, at the same time, low-frequency nonsynonymous polymorphisms have been effectively removed from the population by genetic drift. Therefore, there have been both gain and loss of deleterious mutations in the population. The observation is consistent with recent theoretical and experimental studies in humans, which found that the genetic load of the population is not strongly affected by recent demographic changes (Lohmueller 2014; Simons et al. 2014; Do et al. 2015).
Estimation of Demography
To investigate whether the observed fSFS agrees with the extreme population bottleneck from the known historical record, we estimated the level of population bottleneck by fitting expected fSFS to observed fSFS. To this end, we assume a simple demographic scenario, where a small number of individuals were introduced to the island from the ancestral population with a constant population size, followed by a quickly increased population size with exponential growth (fig. 6). Four parameters: Ancestral effective population size (Na), current effective population size (N0), the effective population size at bottleneck (Nb), and timing of bottleneck (Tb), are involved in this model. Our approach estimated Nb for a given Na, N0, and Tb by fitting observed fSFS to expected fSFS with grid sampling of Nb and Tb (Marth et al. 2004). Because the number of analyzed sites is so large (approximately 16 million), the confidence intervals of the estimate became small. Therefore, although we need be careful in the interpreting these results, here we present only the maximum-likelihood point estimates of Nb for a given Na, N0, and Tb.
Fig. 6.—
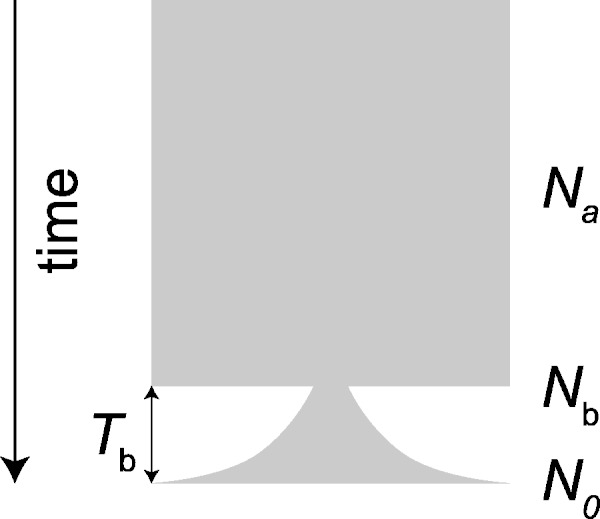
Proposed demographic model for estimating the number of founding individuals. The width of the shaded area represents effective population sizes. We assumed that the bottleneck occurred at time Tb and the number of Nb individuals were randomly selected at the bottleneck from an ancestral population with constant population size of Na. After the bottleneck the size of population recovered to N0 with exponential growth.
According to the historical record, introduction of macaques to the Mauritian islands was around 400–500 years ago. The estimation of a generation time for macaques is uncertain, ranging from 5 to 12 years (Gage 1998). Any bias in generation time estimation would affect the accuracy of the estimation of Nb. Therefore, we used the two long-term demographic studies of Japanese macaques (Macaca fuscata) to calculate the average time of reproduction of females, which yielded a generation time of 9.6–11.4 years (Koyama et al. 1992; Fujimoto et al. 2010). In this study, we applied the estimation of 10-year generation time, which means the population bottleneck occurred 40–50 generations ago. In the following estimation, we assumed Tb = 40. Assuming Na = 30,000 and N0 = 30,000, the estimated number of individuals during the bottleneck is 16. This estimated number of founders is robust against the assumption of Na and N0. For example, Nb = 15 assuming Na = 50,000 and N0 = 25,000, Nb = 14. In table 2, the estimated numbers of founders with different values of Na and N0 are shown. The range of the estimated number of founders does not contradict the previous microsatellite (Bonhomme et al. 2008) and mitochondrial data (Smith et al. 2007).
Table 2.
Estimated Number of Founders with Different Na and N0
| Na = 50,000 | Na = 30,000 | |
|---|---|---|
| N0 = Na | 15 | 16 |
| N0 = Na/2 | 17 | 18 |
| N0 = Na/5 | 20 | 21 |
In addition to the exponential growth model, we examined logistic growth models. These models showed a better fit than the previous study using microsatellite data (Bonhomme et al. 2008). In general, the logistic growth models yielded a smaller number of founding individuals than the exponential growth model. This is because the logistic model has more rapid growth in the early phase, which makes the bottleneck less effective. Using similar parameter settings as the study of Bonhomme et al. (2008), a generation time of 5 years and a growth rate of 0.3, we obtained a slightly smaller number of founding individuals (2–8 founders) than the estimated number by Bonhomme et al. (12 founders). However, in this study, we did not thoroughly apply different growth models because our data have a limited sample size and may not have enough power to infer a very recent demographic history.
The estimated number of founders assuming exponential growth may be overestimated because reports of large-scale haplotyping in the MHC region have identified only seven founding MHC haplotypes in the Mauritian cynomolgus macaque population (Wiseman et al. 2007; Mee et al. 2009; Budde et al. 2010; Blancher et al. 2012; Aarnink et al. 2014) and eight haplotypes in the killer cell immunoglobulin-like receptor (KIR) region (Bimber et al. 2008). In this scenario, the lower limit of the founding individuals is 4, which is closer to our estimated number assuming logistic growth. However, the probability of allele loss is highly dependent on the initial pattern of population growth; this is difficult to accurately estimate, and the process could be highly stochastic with a small number of founders.
Natural selection could have preserved the number of alleles at the MHC locus; in particular, a recent study found that there is MHC class I semi-incompatibility between mother and offspring in cynomolgus macaques; thus, natural selection would act against the loss of MHC alleles in this population (Aarnink et al. 2014). It is of interest to further investigate the effect of natural selection on the genetic diversity at the MHC locus in future studies using whole-genome sequences of more individuals from the populations from which the Mauritian macaques originated.
Conclusions
In this article, we report the genome sequences of six Mauritian cynomolgus macaques. The pattern of polymorphisms in these animals shows a reduced level of genetic diversity, particularly in low-frequency polymorphisms. This pattern agrees well with the historical record of an extreme population bottleneck during the founding of this population. The low efficacy of purifying selection on their genomes may provide the further insight into the specific phenotypic characteristics in Mauritian cynomolgus macaques. The smaller genetic diversity in this population is of great importance for better reproducibility of drug testing and viral infection experiments. In addition, the whole-genome sequences of the Mauritian cynomolgus macaques provide further insights into the genetic basis of variation among macaques for drug and viral response in future biomedical research.
Supplementary Material
Supplementary table S1 and figures S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the funds from University Toulouse III (EA3034, Université Paul Sabatier) and French Ministry of Research and by the Grant-in-Aid for Scientific Research (A), Grant number 26251040, to N.S. and N.O. They thank Dr Tomas Marques-Bonet for the assistance of data analysis and the two anonymous reviewers for helpful comments on the manuscript.
Literature Cited
- Aarnink A, Apoil PA, Takahashi I, Osada N, Blancher A. Characterization of MHC class I transcripts of a Malaysian cynomolgus macaque by high-throughput pyrosequencing and EST libraries. Immunogenetics. 2011;63:703–713. doi: 10.1007/s00251-011-0550-8. [DOI] [PubMed] [Google Scholar]
- Aarnink A, Dereuddre-Bosquet N, et al. Influence of the MHC genotype on the progression of experimental SIV infection in the Mauritian cynomolgus macaque. Immunogenetics. 2011;63:267–274. doi: 10.1007/s00251-010-0504-6. [DOI] [PubMed] [Google Scholar]
- Aarnink A, et al. Comparative analysis in cynomolgus macaque identifies a novel human MHC locus controlling platelet blood counts independently of BAK1. J Thromb Haemost. 2013;11:384–386. doi: 10.1111/jth.12092. [DOI] [PubMed] [Google Scholar]
- Aarnink A, et al. Deleterious impact of feto-maternal MHC compatibility on the success of pregnancy in a macaque model. Immunogenetics. 2014;66:105–113. doi: 10.1007/s00251-013-0752-3. [DOI] [PubMed] [Google Scholar]
- Aarnink A, Garchon HJ, et al. Impact of MHC class II polymorphism on blood counts of CD4+ T lymphocytes in macaque. Immunogenetics. 2011;63:95–102. doi: 10.1007/s00251-010-0492-6. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés AM, et al. Targets of balancing selection in the human genome. Mol Biol Evol. 2009;26:2755–2764. doi: 10.1093/molbev/msp190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimber BN, Moreland AJ, Wiseman RW, Hughes AL, O’Connor DH. Complete characterization of killer Ig-like receptor (KIR) haplotypes in Mauritian cynomolgus macaques: novel insights into nonhuman primate KIR gene content and organization. J Immunol. 2008;181:6301–6308. doi: 10.4049/jimmunol.181.9.6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancher A, Aarnink A, Savy N, Takahata N. Use of cumulative Poisson probability distribution as an estimator of the recombination rate in an expanding population: example of the Macaca fascicularis major histocompatibility complex. G3. 2012;2:123–130. doi: 10.1534/g3.111.001248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancher A, et al. Mitochondrial DNA sequence phylogeny of 4 populations of the widely distributed cynomolgus macaque (Macaca fascicularis fascicularis) J Hered. 2008;99:254–264. doi: 10.1093/jhered/esn003. [DOI] [PubMed] [Google Scholar]
- Bonhomme M, Blancher A, Cuartero S, Chikhi L, Crouau-Roy B. Origin and number of founders in an introduced insular primate: estimation from nuclear genetic data. Mol Ecol. 2008;17:1009–1019. doi: 10.1111/j.1365-294X.2007.03645.x. [DOI] [PubMed] [Google Scholar]
- Bonhomme M, Cuartero S, Blancher A, Crouau-Roy B. Assessing natural introgression in 2 biomedical model species, the rhesus macaque (Macaca mulatta) and the long-tailed macaque (Macaca fascicularis) J Hered. 2009;100:158–169. doi: 10.1093/jhered/esn093. [DOI] [PubMed] [Google Scholar]
- Budde M, et al. Characterization of Mauritian cynomolgus macaque major histocompatibility complex class I haplotypes by high-resolution pyrosequencing. Immunogenetics. 2010;62:773–780. doi: 10.1007/s00251-010-0481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, et al. No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat Genet. 2015;47:126–131. doi: 10.1038/ng.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevon-Gaillot E, Perron-Lepage M-F, Clément C, Burnett R. A review of background findings in cynomolgus monkeys (Macaca fascicularis) from three different geographical origins. Exp Toxicol Pathol. 2006;58:77–88. doi: 10.1016/j.etp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Ebeling M, et al. Genome-based analysis of the nonhuman primate Macaca fascicularis as a model for drug safety assessment. Genome Res. 2011;21:1746–1756. doi: 10.1101/gr.123117.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsen A, et al. Whole genome sequencing of SIV-infected macaques identifies candidate loci that may contribute to host control of virus replication. Genome Biol. 2014;15:478. doi: 10.1186/s13059-014-0478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, et al. Whole-genome sequencing of Tibetan macaque (Macaca thibetana) provides new insight into the macaque evolutionary history. Mol Biol Evol. 2014;31:1475–1489. doi: 10.1093/molbev/msu104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylip (version 3.2): phylogeny inference package. Cladistics. 1989;5:164–166. [Google Scholar]
- Flicek P, et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooden J. Provisional classifications and key to living species of macaques (primates: Macaca) Folia Primatol (Basel). 1976;25:225–236. doi: 10.1159/000155715. [DOI] [PubMed] [Google Scholar]
- Fujimoto A, et al. Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nat Genet. 2010;42:931–936. doi: 10.1038/ng.691. [DOI] [PubMed] [Google Scholar]
- Gage TB. The comparative demography of primates: with some comments on the evolution of life histories. Annu Rev Anthropol. 1998;27:197–221. doi: 10.1146/annurev.anthro.27.1.197. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Higashino A, et al. Whole-genome sequencing and analysis of the Malaysian cynomolgus macaque (Macaca fascicularis) genome. Genome Biol. 2012;13:R58. doi: 10.1186/gb-2012-13-7-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature. 2012;483:82–86. doi: 10.1038/nature10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthaswamy S, et al. Hybridization and stratification of nuclear genetic variation in Macaca mulatta and M. fascicularis. Int J Primatol. 2008;29:1295–1311. doi: 10.1007/s10764-008-9295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto Y, et al. Genetic diversity of longtail macaques (Macaca fascicularis) on the island of Mauritius: an assessment of nuclear and mitochondrial DNA polymorphisms. J Med Primatol. 2008;37:45–54. doi: 10.1111/j.1600-0684.2007.00225.x. [DOI] [PubMed] [Google Scholar]
- Koyama N, Takahata Y, Huffman M, Norikoshi K, Suzuki H. Reproductive parameters of female Japanese macaques: thirty years data from the Arashiyama troops, Japan. Primates. 1992;33:33–47. [Google Scholar]
- Krebs KC, Jin Z, Rudersdorf R, Hughes AL, O’Connor DH. Unusually high frequency MHC Class I alleles in Mauritian origin cynomolgus macaques. J Immunol. 2005;175:5230–5239. doi: 10.4049/jimmunol.175.8.5230. [DOI] [PubMed] [Google Scholar]
- Leuchte N, et al. MhcDRB-sequences from cynomolgus macaques (Macaca fascicularis) of different origin. Tissue Antigens. 2004;63:529–537. doi: 10.1111/j.0001-2815.2004.0222.x. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE. The impact of population demography and selection on the genetic architecture of complex traits. PLoS Genet. 2014;10:e1004379. doi: 10.1371/journal.pgen.1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart G, Allendorf F, Cornuet J-M, Sherwin W. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered. 1998;89:238–247. doi: 10.1093/jhered/89.3.238. [DOI] [PubMed] [Google Scholar]
- Marth GT, Czabarka E, Murvai J, Sherry ST. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee ET, et al. MHC haplotype frequencies in a UK breeding colony of Mauritian cynomolgus macaques mirror those found in a distinct population from the same geographic origin. J Med Primatol. 2009;38:1–14. doi: 10.1111/j.1600-0684.2008.00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menninger K, et al. The origin of cynomolgus monkey affects the outcome of kidney allografts under neoral immunosuppression. Transplant Proc. 2002;34:2887–2888. doi: 10.1016/s0041-1345(02)03547-9. [DOI] [PubMed] [Google Scholar]
- Nei M. Genetic distance between populations. Am Nat. 1972;106:283–292. [Google Scholar]
- Ogawa L, Vallender E. Genetic substructure in cynomolgus macaques (Macaca fascicularis) on the island of Mauritius. BMC Genomics. 2014;15:748. doi: 10.1186/1471-2164-15-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Osada N, et al. Ancient genome-wide admixture extends beyond the current hybrid zone between Macaca fascicularis and M. mulatta. Mol Ecol. 2010;19:2884–2895. doi: 10.1111/j.1365-294X.2010.04687.x. [DOI] [PubMed] [Google Scholar]
- Osada N, et al. Finding the factors of reduced genetic diversity on X chromosomes of Macaca fascicularis: male-driven evolution, demography, and natural selection. Genetics. 2013;195:1027–1035. doi: 10.1534/genetics.113.156703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlin BI, Schloegel LM, Daszak P. Risk of importing zoonotic diseases through wildlife trade, United States. Emerg Infect Dis. 2009;15:1721–1726. doi: 10.3201/eid1511.090467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Martinez J, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–475. doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Satkoski Trask J, George D, Houghton P, Kanthaswamy S, Smith DG. Population and landscape genetics of an introduced species (M. fascicularis) on the island of Mauritius. PLoS One. 2013;8:e53001. doi: 10.1371/journal.pone.0053001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shively CA, Clarkson TB. The unique value of primate models in translational research. Am J Primatol. 2009;71:715–721. doi: 10.1002/ajp.20720. [DOI] [PubMed] [Google Scholar]
- Simons YB, Turchin MC, Pritchard JK, Sella G. The deleterious mutation load is insensitive to recent population history. Nat Genet. 2014;46:220–224. doi: 10.1038/ng.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DG, Mcdonough JW, George DA. Mitochondrial DNA variation within and among regional populations of longtail macaques (Macaca fascicularis) in relation to other species of the fascicularis group of macaques. Am J Primatol. 2007;69:182–198. doi: 10.1002/ajp.20337. [DOI] [PubMed] [Google Scholar]
- Stevison LS, Kohn MH. Determining genetic background in captive stocks of cynomolgus macaques (Macaca fascicularis) J Med Primatol. 2008;37:311–317. doi: 10.1111/j.1600-0684.2008.00292.x. [DOI] [PubMed] [Google Scholar]
- Stevison LS, Kohn MH. Divergence population genetic analysis of hybridization between rhesus and cynomolgus macaques. Mol Ecol. 2009;18:2457–2475. doi: 10.1111/j.1365-294X.2009.04212.x. [DOI] [PubMed] [Google Scholar]
- Sussman RW, Tattersall I. Distribution, abundance, and putative ecological strategy of Macaca fascicularis on the Island of Mauritius, Southwestern Indian Ocean. Folia Primatol (Basel). 1986;46:28–43. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi AJ, Coke CS. Comparative phylogenetics offer new insights into the biogeographic history of Macaca fascicularis and the origin of the Mauritian macaques. Mol Phylogenet Evol. 2007;42:498–504. doi: 10.1016/j.ympev.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Van Der Auwera GA, et al. Current Protocols in Bioinformatics. p. 11.10.1-11.10.33. Hoboken (NJ): John Wiley & Sons, Inc; 2002. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade N. India bans monkey export: U.S. may have breached accord. Science. 1978;199:280–281. doi: 10.1126/science.199.4326.280. [DOI] [PubMed] [Google Scholar]
- Wiseman RW, et al. Simian immunodeficiency virus SIVmac239 infection of major histocompatibility complex-identical cynomolgus macaques from Mauritius. J Virol. 2007;81:349–361. doi: 10.1128/JVI.01841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, et al. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol. 2011;29:1019–1023. doi: 10.1038/nbt.1992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.