

In this study, Liao et al. comprehensively investigated how cancer drivers genetically interact. They searched for modifiers of EGFR dependency by performing CRISPR, shRNA, and expression screens in a NSCLC model, and their data provide strong support for the hypothesis that many cancer drivers can substitute for each other in certain contexts.
Keywords: cancer drivers, EGFR, genetic interaction
Abstract
A large number of cancer drivers have been identified through tumor sequencing efforts, but how they interact and the degree to which they can substitute for each other have not been systematically explored. To comprehensively investigate how cancer drivers genetically interact, we searched for modifiers of epidermal growth factor receptor (EGFR) dependency by performing CRISPR, shRNA, and expression screens in a non-small cell lung cancer (NSCLC) model. We elucidated a broad spectrum of tumor suppressor genes (TSGs) and oncogenes (OGs) that can genetically modify proliferation and survival of cancer cells when EGFR signaling is altered. These include genes already known to mediate EGFR inhibitor resistance as well as many TSGs not previously connected to EGFR and whose biological functions in tumorigenesis are not well understood. We show that mutation of PBRM1, a subunit of the SWI/SNF complex, attenuates the effects of EGFR inhibition in part by sustaining AKT signaling. We also show that mutation of Capicua (CIC), a transcriptional repressor, suppresses the effects of EGFR inhibition by partially restoring the EGFR-promoted gene expression program, including the sustained expression of Ets transcription factors such as ETV1. Together, our data provide strong support for the hypothesis that many cancer drivers can substitute for each other in certain contexts and broaden our understanding of EGFR regulation.
Cancer is driven by a number of distinct genetic alterations, including gain or loss of chromosomes and chromosomal segments, translocations, and point mutations that result in inactivation of tumor suppressor genes (TSGs) or activation of oncogenes (OGs). Attempts to identify these cancer drivers based on patterns of mutagenesis in tumors have uncovered a bewilderingly large number of genes that bear the signature of genetic selection in tumors. Rather than a defined number of cancer drivers, there exists a continuum of genes that appear with increasingly lower frequency and potency in a pan-cancer analysis (Davoli et al. 2013). This has also been referred to as mountains and hills and the “long tail” of cancer drivers (Wood et al. 2007; Leiserson et al. 2015; Cho et al. 2016).
Unraveling how the genes in this large network contribute to tumorigenesis poses a significant challenge for geneticists. While it is clear that certain basic conditions related to the classic hallmarks of cancer (e.g., immortalization and deregulated proliferation) must be satisfied during tumor development (Hanahan and Weinberg 2011), it is currently unclear how many cancer genes operate to achieve these conditions. From a theoretical perspective, it makes sense that many cancer drivers may perform similar functions and be partially interchangeable during tumor evolution. They may act either to genetically modify a shared central oncogenesis pathway or in parallel pathways that provide the cell with equivalent functions to drive tumorigenesis. Thus, it is likely that many genes on these TSG and OG lists will genetically interact to modify common conditions of oncogenesis.
One of the most extensively studied oncogenic pathways is the receptor tyrosine kinase (RTK)–RAS–phosphoinositol-3-kinase (PI3K) pathway. This pathway is activated in the majority of solid tumors and has been examined extensively both biochemically and genetically. Among RTKs, perhaps the most studied is the epidermal growth factor receptor (EGFR). EGFR activates cellular signaling pathways such as PI3K/AKT, RAS/RAF/MEK/ERK, and JAK/STAT, leading to increased cell proliferation and survival (Chong and Janne 2013). Activating EGFR mutations occur in ∼10%–30% of tumors of patients with non-small cell lung cancer (NSCLC), a leading cause of cancer-related deaths (Stewart et al. 2015). These mutations confer sensitivity to EGFR inhibitors (EGFRis) such as gefitinib and a variety of later-generation inhibitors (Lynch et al. 2004; Paez et al. 2004; Wang et al. 2016). Although EGFR mutant NSCLCs typically respond dramatically to EGFRis, these responses are not universal, as the overall response rate is ∼71%. Even among the initial responders, most inevitably develop acquired resistance to EGFRi therapies within a year of treatment (Mok et al. 2009; Rosell et al. 2009; Thress et al. 2015). The resistance mechanism is unknown in up to 30% of patients (Majem and Remon 2013).
Given its central role in driving oncogenesis, the existing knowledge of the pathway, and the many tools available, the EGFR pathway is well suited for examining genetic interactions with other known and putative cancer drivers. This is supported by existing evidence of genetic interactions of EGFR with other drivers of tumorigenesis (Sharifnia et al. 2014). For example, patients bearing EGFR mutations are known to evolve resistance to EGFRi therapies by virtue of mutations in other cancer drivers. In addition to mutations in EGFR itself, low expression of NF1 (de Bruin et al. 2014) or PTEN (Sos et al. 2009; Yamamoto et al. 2010), amplification of the MET RTK (Engelman et al. 2007), amplification of the HER2 (ERBB2) RTK (Takezawa et al. 2012), and activation of KRAS, PIK3CA, BRAF, and MAPK1 (ERK) (Sartore-Bianchi et al. 2009; Diaz et al. 2012; Ercan et al. 2012; Misale et al. 2012; Ohashi et al. 2012) can confer EGFRi resistance. Thus, it is likely that additional drivers will also genetically interact with the EGFR pathway.
To test the hypothesis that cancer drivers can genetically interact and substitute for one another to drive proliferation and survival, we investigated TSG and OG drivers for their ability when mutated to partially replace EGFR in EGFR-dependent tumor cells by performing CRISPR, shRNA, and OG expression screens in parallel in a NSCLC model. We took advantage of an algorithm called TUSON (Tumor Suppressor and Oncogene) Explorer to identify TSGs and OGs (Davoli et al. 2013). This method quantifies the likelihood that a gene is a cancer driver based on the distortion of its mutational signature from the pattern expected for a “neutral” gene. For example, TSGs will have higher ratio of loss of function (LOF) to benign mutations than neutral genes (Fig. 1A). Here, we show that this genetic approach successfully recovered previously validated TSGs and OGs that interact genetically with the EGFR pathway. We also identified novel TSGs that have not been linked previously to EGFRi resistance. We further characterized the mechanisms underlying gefitinib resistance mediated by several novel TSGs. Among these, we showed that mutation of PBRM1, a subunit of the SWI/SNF complex, attenuated the effect of gefitinib in part by sustaining AKT pathway function during EGFR inhibition. We also showed that mutation of CIC, a transcriptional repressor, partially restored the EGFR gene expression program upon EGFR inhibition in NSCLC cells in part through sustained activation of ETV1, resulting in gefitinib resistance. These findings provide new biochemical insight into EGFR signaling and support the general notion that cancer drivers are part of a robust joint network that can compensate for the loss of any one member.
Figure 1.

Genetic screens identify modifiers of the cellular response to reduced EGFR signaling. (A) Schematic of the pipeline used by TUSON Explorer to predict TSGs and OGs. Adapted from Dewar et al. (2013) with permission from Elsevier. (B) Outline of the genetic screening strategy. (C) Enrichment score (Z-score determined by MAGeCK [model-based analysis of genome-wide CRISPR–Cas9 knockout]) of gefitinib treatment plotted against vehicle (DMSO) treatment for genes in the CRISPR, shRNA, and ORF screens. The top 15 genes of each screen (false discovery rate [FDR] < 0.05) are highlighted. One outlier in the shRNA screen, AAMP (enrichment score for gefitinib >100, FDR = 0.96) was excluded in the plot of shRNAs. (*) Mutant form; (#) mutant form 2. (D) The number of overlapping genes observed between the top hits of the CRISPR and shRNA screens compared with the number expected for a random overlap. (E) Distribution of the pooled LOF mutation fraction based on a permutation of a group of 30 genes from the TSG library (combined FDR > 0.1 in the screens; blue) or whole genome (excluding genes in the TSG library; gray) in EGFR mutant or EGFR wild-type tumors. The dot represents the behavior of the top 30 hits from our screen mapped on the distribution of the TSGs. The P-value was calculated based on the relative distribution of the TSG library.
Results
The central hypothesis motivating this study is that cancer mutations often impact the same pathways or control parallel pathways that can substitute for each other. To test this notion, we investigated the RTK EGFR pathway. While EGFR has been extensively studied both biochemically and genetically, it has not been systematically probed for its interactions with all known and putative cancer drivers. To explore these interactions for genes with TSG properties, we generated both a CRISPR and an shRNA library containing 10 guide RNAs (gRNAs) or 10 shRNAs per gene to a list of ∼500 genes whose LOF has been implicated in driving tumorigenesis by the TUSON Explorer algorithm (Davoli et al. 2013). Each library also contained 1000 gRNAs or 1000 shRNAs targeting the Escherichia coli genome as negative controls. To explore the genetic interactions with EGFR for genes with OG properties, we generated a barcoded ORF lentivirus library of ∼50 selected genes whose mutational signatures implicate them as potential OGs by TUSON Explorer (Fig. 1A; Davoli et al. 2013). We set out to determine which alterations could substitute for EGFR signaling using a chemical inhibitor of EGFR, gefitinib. We performed screens using a NSCLC cell line, PC9, which harbors an activating EGFR mutation and is sensitive to gefitinib. CRISPR and shRNA have different mechanisms and off targets, thus providing complementary means of assessing the functional contribution of TSGs to EGFRi resistance. Genes that retain function at low expression levels are likely to be missed in shRNA screens due to their incomplete depletion. In contrast, genes that are essential for cell viability cannot be assessed in CRISPR screens. Partial depletion by shRNA will be useful in these cases. In addition, as gene regulatory networks are highly interconnected and contain multiple feedback loops, the response to knockout and depletion can be markedly different (Shalem et al. 2015). By performing these complementary CRISPR and shRNA screens in parallel with the ORF screen, we were able to obtain a broad genetic view of the EGFR-interacting pathways.
The schematic of the CRISPR, shRNA, and ORF screens is outlined in Figure 1B and described in detail in the Materials and Methods. In each screen, cells were treated with either DMSO or 30 nM gefitinib for ∼17 d. This intermediate concentration of gefitinib was not completely lethal but caused a significant lengthening of the doubling time of PC9 cells, allowing more subtle suppressors of reduced EGFR function to be detected. We used the MAGeCK (model-based analysis of genome-wide CRISPR–Cas9 knockout) scoring algorithm (Li et al. 2014) to rank the performance of individual genes. For TSG screens, the negative controls were incorporated in the MAGeCK analysis to generate null distributions and calculate the P-value and false discovery rate (FDR) for each gene. The top 15 hits from each screen are shown in Figure 1C. For TSG screens, each screen successfully recovered previously validated TSGs that impact sensitivity to EGFR inhibition, including NF1 and PTEN, as well as novel TSGs that have not been linked previously to EGFR pathway function. The rank and FDR of each gene in the two TSG screens are summarized in Supplemental Table S1. The top 30 genes based on the combined rank of the two screens (combined FDR < 0.001) are marked in red. The rank and FDR of each gene in the OG screen are shown in the Supplemental Table S1. This screen identified a number of key regulators in the EGFR pathway previously implicated in EGFRi resistance, including EGFR itself, ERRB2, NRAS, HRAS, BRAF, and MAP2K1, as well as two canonical cell cycle regulators: CDK6 and CCND1. Since the majority of the strong interactions with EGFRi resistance was known previously, we focused the rest of our efforts on characterizing the novel TSG hits.
The top-scoring genes from the TSG screen showed significant overlap between CRISPR and shRNA screens (Fig. 1E). For example, the number of overlapping genes in the top 20 of CRISPR and top 20 of shRNA is 10 (P = 1.1 × 10−10). As noted previously (Shalem et al. 2014), CRISPR exhibited greater consistency among the gRNAs targeting the top candidate genes compared with shRNAs. For the top 20 genes, on average, 62.5% of gRNAs targeting each gene ranked among the top 5% of enriched gRNAs, whereas 40.2% of shRNAs targeting each gene ranked among the top 5% of enriched shRNAs (Supplemental Fig. S1A). Notably, the shRNA screen was able to recover genes essential for cell viability that were missed in the CRISPR screen. One example is ZC3H18 (Supplemental Fig. S1B; Blomen et al. 2015).
If the top TSG hits in this screen perform a function equivalent to that of EGFR mutants, they would be expected to be largely epistatic and would lack selective power in an EGFR mutant background. To examine this, we calculated the LOF fraction (the ratio of nonsense, frameshift, and high-impact missense mutations vs. total mutations) for a group of the top 30 TSG hits (combined FDR < 0.001) using the lung adenocarcinoma TCGA (The Cancer Genome Atlas) data set. The higher the LOF fraction, the more selective pressure is on those mutations in a given tumor set. In tumors bearing a wild-type, but not mutant, EGFR gene, the LOF fraction of the top 30 hits was much higher than the average of all the genes in the TSG library (P = 0.04). This suggests that the top TSG hits are more strongly selected in EGFR wild-type tumors. Such selective pressure suggests that they act like activating EGFR mutations and theoretically could, when mutant, impact EGFR signaling in EGFR mutant tumors when treated with an EGFRi.
We also identified three genes among our top 30 hits whose focal deletion/LOF mutations are mutually exclusive with activating EGFR mutations (Supplemental Fig. S1C). Two of these, NF1 and KEAP1, were previously known to be involved in EGFRi resistance, while the third, THRAP3, is novel. The mutual exclusivity between the inactivation of these TSGs and activating EGFR mutations observed in patients further suggests that such events operate in the same or functionally redundant pathways.
Down-regulation of PBRM1 attenuates the effect of gefitinib by sustaining AKT pathway activation during EGFR inhibition
A prominent candidate that emerged from the screens is the SWI/SNF complex. Six genes encoding subunits of SWI/SNF complexes have been ranked among the top 500 TSGs by the TUSON algorithm and were represented in our libraries. Among those, PBRM1, ARID2, and ARID1A scored in the top 30 of both CRISPR and shRNA screens (combined FDR < 0.001), while ARID1B and SMARCB1 scored only in the CRISPR screen (FDR < 0.001) (Fig. 2A). SWI/SNF complexes interact with transcription factors, coactivators, and corepressors and are capable of mobilizing nucleosomes at target promoters and enhancers to modulate gene expression. The mechanism by which mutation of each individual subunit promotes oncogenesis and the function of mutated SWI/SNF complexes in cancer are now active areas of investigation (Helming et al. 2014).
Figure 2.
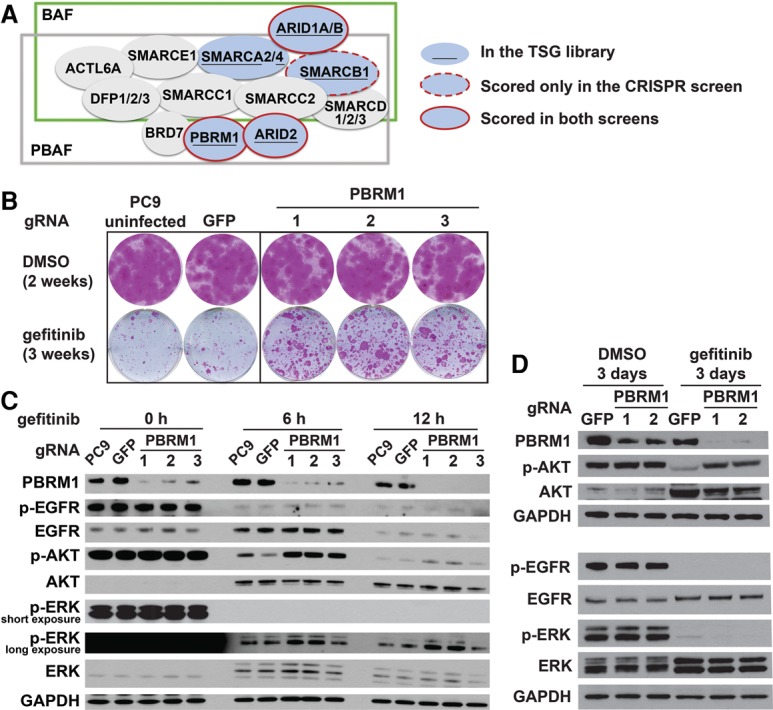
Multiple subunits of the SWI/SNF complex scored as modifiers of EGFR pathway function. (A) Schematic of the BAF and PBAF complexes showing which subunits scored in our screens as modifiers of EGFR dependency. (B) PBRM1 down-regulation enhances survival of PC9 cells in long-term colony formation assays. Cells were fixed and stained after treatment with DMSO or 30 nM gefitinib for the indicated times. Uninfected PC9 cells and GFP gRNA-expressing PC9 cells were used as controls. (C) Immunoblot analysis of PBRM1 mutant PC9 cells treated with 30 nM gefitinib for 0, 6, and 12 h. Cells were incubated with medium containing 0.5% serum 18 h before and during drug treatment. (D) Immunoblot analysis of PBRM1 mutant PC9 cells treated with 30 nM gefitinib for 3 d with the indicated antibodies.
The mechanistic role of PBRM1 in tumorigenesis remains elusive. PBRM1 is required for p21 expression and cell cycle arrest in breast cancer cells upon transforming growth factor-β (TGFβ) treatment (Xia et al. 2008). However, in primary mouse embryonic fibroblasts (MEFs), PBRM1 suppresses p21 expression, and deletion of PBRM1 triggers cell cycle arrest and cellular senescence (Lee et al. 2016). Direct links between EGFR signaling and PBRM1 or other SWI/SNF components have not yet been established.
To validate the effects of PBRM1 loss on the phenotype of EGFR pathway impairment, we performed colony formation assays in which cells were cultured in the presence of gefitinib for 3 wk. PBRM1 mutation using three different gRNAs substantially enhanced cell survival in these assays (Fig. 2B). PBRM1 mutation also enhanced survival of PC9 cells treated with erlotinib or third-generation EGFRi AZD9291 but had no effect on cisplatin treatment (Supplemental Fig. S2A). Given the role of PBRM1 on p21 regulation in other cellular contexts, we examined the p21 levels of PBRM1 gRNA-expressing and control gRNA-expressing PC9 cells treated with DMSO or gefitinib. PBRM1 mutation did not affect the basal level of p21 in PC9 cells or reduce the induction of p21 by gefitinib treatment (Supplemental Fig. S2B). Because two other SWI/SNF subunits, SMARCE1 and ARID1A, have been reported to negatively regulate EGFR mRNA levels (Papadakis et al. 2015), we sought to examine whether loss of PBRM1 also affects EGFR expression. PBRM1 mutation by CRISPR in PC9 cells did not affect the level of EGFR or phosphorylation of EGFR in the absence or presence of gefitinib (Fig. 2C).
We next examined the phosphorylation status of several downstream signaling proteins of EGFR. We found that treatment of PC9 cells with gefitinib quickly induced the levels of AKT while reducing its phosphorylation (p-AKT). We found that PBRM1 mutant cells showed slower rates of reduction of p-AKT than parental and control gRNA-expressing cells after 6 and 12 h of gefitinib treatment (Fig. 2C), but then the levels of p-AKT fully rebounded to untreated levels by 3 d (Fig. 2D). Gefitinib completely abolished ERK phosphorylation, and that effect did not rebound by 3 d of treatment. These results indicate that PBRM1 loss attenuates the effects of gefitinib on the downstream AKT pathway, which is likely to ameliorate the effects on EGFR pathway inhibition.
Loss of CIC confers resistance of NSCLC cell lines to EGFRis
Among the novel hits that scored in both the CRISPR and shRNA screens, CIC (combined FDR = 0) stood out because a related gene has been reported to transcriptionally repress EGFR target genes during Drosophila development. In Drosophila, LOF of the CIC ortholog Capicua bypasses the requirement for EGFR signaling in vein cell determination (Roch et al. 2002). CIC mutations increase the rate of cell proliferation without affecting cell size and bypass the requirement for EGFR signaling in imaginal discs (Tseng et al. 2007). However, the potential role of CIC loss in drug resistance has not been investigated.
To validate that CIC loss mediates gefitinib resistance, we performed a multicolor competition assay in which PC9 cells labeled with GFP were infected with a virus encoding Cas9 and a gRNA to CIC (referred to here as CIC knockdown cells) and were mixed with control Cas9–gRNA-infected cells labeled with mRuby in a 50:50 ratio. After DMSO or gefitinib treatment, the percentage of CIC knockdown cells was then quantified as the percentage of GFP cells using FACS. Knockdown of CIC protein was confirmed by immunoblotting (Fig. 3A). The percentage of CIC knockdown cells remained ∼50% under DMSO treatment, indicating that CIC does not regulate basal proliferation of PC9 cells. However, when treated with gefitinib, CIC knockdown cells were selectively enriched (Fig. 3B). CIC knockdown also enhanced survival of PC9 cells treated with erlotinib or the third-generation EGFRi AZD9291 (Supplemental Fig. S3A). We confirmed the effects of CIC knockdown in another NSCLC cell line, H1975. H1975 cells harbor an activating EGFR mutation (L858R) and the gatekeeper mutation T790M, which confers resistance to gefitinib and erlotinib but not AZD9291. CIC knockdown using two different gRNAs substantially enhanced cell survival under AZD9291 treatment in the colony formation assays (Fig. 3C,D), suggesting that the role of CIC is cell type- and EGFRi-independent.
Figure 3.
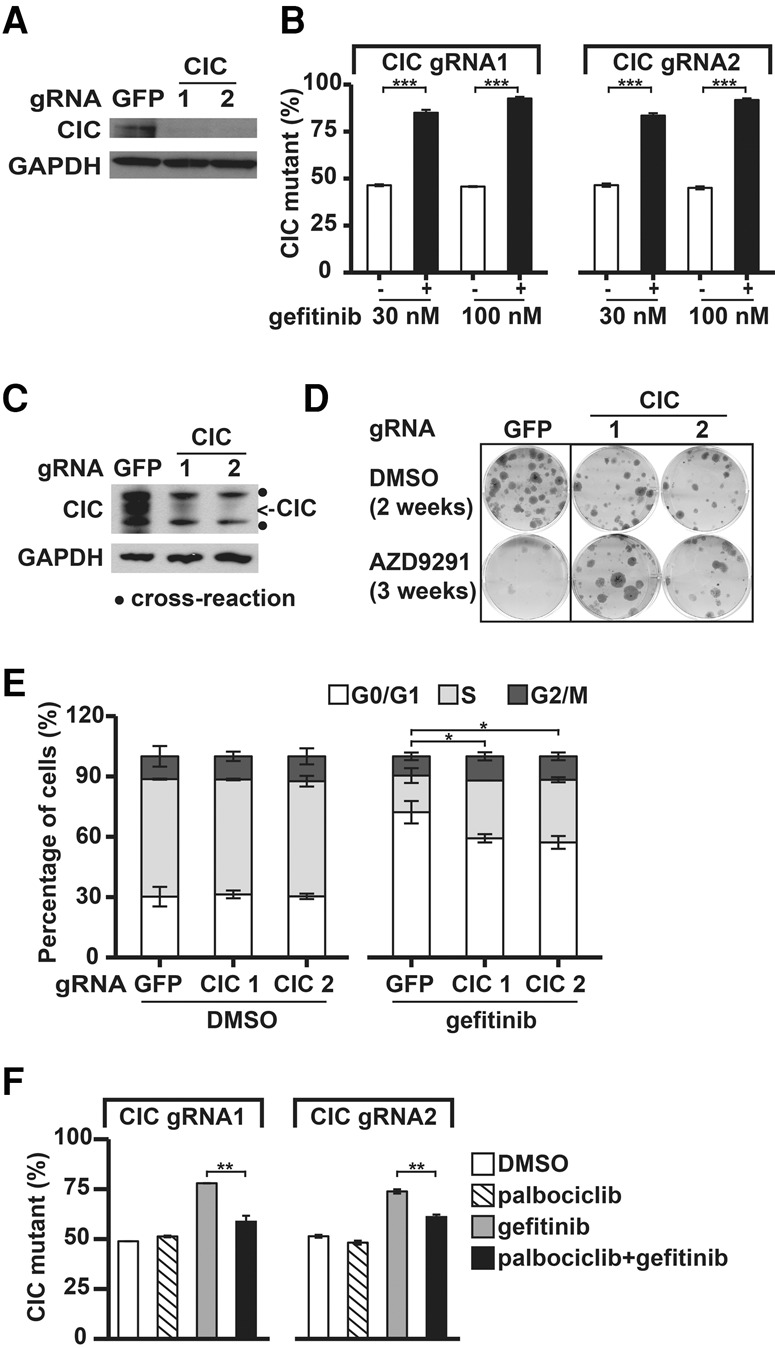
CIC LOF enhances growth of cells with impaired EGFR signaling. (A) Immunoblot analysis of the CIC protein in PC9 cells infected with lentivirus expressing Cas9 and the indicated gRNAs. (B) Viruses expressing Cas9 and CIC or GFP gRNAs were used to infect PC9 cells, infected cells were mixed in a 50:50 ratio and treated with either DMSO or 30 nM gefitinib for 10 d or DMSO or 100 nM gefitinib for 14 d, and the percentage of CIC mutants was quantified using FACS. (C) Immunoblot analysis of CIC protein in H1975 cells infected with lentivirus expressing Cas9 and the indicated gRNAs. The dots indicate a cross-reactive protein. (D) CIC mutation enhances survival of H1975 cells in long-term colony formation assays with EGFRis. Cells described in C were fixed and stained after treatment with DMSO or 100 nM AZD9291 for the indicated times. (E) Cell cycle profiles of CIC and GFP gRNA-expressing cells treated with DMSO or 30 nM gefitinib for 48 h. Incorporated BrdU and total DNA content (7AAD) were used to distinguish cells in the G0/G1, S, or G2/M phases of the cell cycle. Apoptotic (sub-G1) cells are not shown. (F) Competition assays as in B. After 10 d of treatment with either DMSO, 1 µM palbociclib, 30 nM gefitinib, or a combination of 1 µM palbociclib and 30 nM gefitinib, the percentage of CIC gRNA-expressing cells was quantified using FACS. Data are the means ± SD. n = 3 in all panels. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
To further characterize the resistance mechanism, we examined apoptosis and cell cycle distribution of CIC knockdown cells under gefitinib treatment using propidium iodide (PI) staining and BrdU labeling, respectively. Unlike loss of NF1, impairing CIC function did not protect PC9 cells from gefitinib-induced apoptosis (Supplemental Fig. S3B). However, while gefitinib induced G1/S cell cycle arrest in control cells, impairing CIC function reduced the effects of gefitinib on cell cycle arrest (Fig. 3E), suggesting that CIC loss causes gefitinib resistance in part through promoting cell cycle entry. This is consistent with our observation that CIC loss prevents the repression of cyclin D expression by gefitinib (see Fig. 4F). Cyclin D binds and activates the cyclin-dependent kinases CDK4 and CDK6 to promote cell proliferation. If this mechanism contributes to gefitinib resistance, one might expect that reducing CDK4/6 function would restore gefitinib sensitivity of CIC knockdown cells. We tested this hypothesis and found that the selective outgrowth of CIC impaired cells treated with gefitinib could be inhibited by cotreating with a CDK4/6 inhibitor, palbociclib (Fig. 3F). The effects appear to be specific to gefitinib treatment because palbociclib had little effect on these cells in the absence of gefitinib.
Figure 4.

Identification and functional evaluation of genes regulated by CIC. (A) Schematic of the RNA sequencing (RNA-seq) analysis. Cells were treated with DMSO or 30 nM gefitinib for 6 h. Differentially expressed genes (DEGs) were identified using edgeR. (B) Confirmation of CIC knockout in PC9 cells using immunoblots. (C) EGFRi resistance signature enrichment plot (using Kobayashi EGFR Signaling 24hr DN signature data sets obtained from the gene set enrichment analysis [GSEA]). The plot indicates a significant up-regulation of EGFR signatures in CIC gRNA-expressing PC9 cells compared with GFP gRNA-expressing PC9 cells during gefitinib treatment. (NES) Normalized enrichment score. (D) Protein–protein interaction (PPI) hubs among the DEGs between CIC gRNA-expressing and GFP gRNA-expressing PC9 cells during gefitinib treatment. (E) Heat map of expression levels (Z-scores) of the top up-regulated genes in CIC gRNA-expressing compared with GFP gRNA-expressing PC9 cells during gefitinib treatment. Each sample was normalized to the basal condition (GFP gRNA-infected, DMSO-treated sample 1). FDRs were calculated by comparing CIC gRNA-expressing cells with GFP gRNA-expressing cells during gefitinib treatment. The asterisk for ETV1 indicates that we were unable to calculate an FDR, as the total read counts were too low, but it ranks in the top 10 by fold change. (F) RT-qPCR analysis of mRNA expression of the indicated genes in CIC gRNA-expressing or GFP gRNA-expressing PC9 cells treated with DMSO or 30 nM gefitinib for 6 h. Data are the means ± SD. n = 3.
CIC loss mediates sustained activation of EGFR target genes during EGFR inhibition
To identify the effectors that are induced upon loss of CIC in the EGFRi resistance setting, we performed RNA sequencing (RNA-seq) using two replicates for control gRNA and three different gRNAs of CIC as three replicates. Cells were treated with DMSO or 30 nM gefitinib for 6 h (Fig. 4A). Knockdown of CIC was confirmed by immunoblotting (Fig. 4B). The full analyses of differentially expressed genes (DEGs) across different conditions are shown in Supplemental Table S2. Gene set enrichment analysis (GSEA) revealed that the EGFR target genes identified by Kobayashi et al. (2006) comprised the highest-ranking up-regulated gene set in CIC knockdown cells compared with control cells during gefitinib treatment (Fig. 4C), indicating that CIC knockdown cells were able to partially sustain EGFR signaling during gefitinib treatment. Protein–protein interaction (PPI) hub analysis indicated that MAPK1/3 (ERK1/2) and EGFR were the most significantly enriched hubs of DEGs between CIC knockdown cells and control cells during gefitinib treatment (Fig. 4D). However, CIC knockdown cells did not exhibit differential phospho-EGFR or phospho-ERK1/2 compared with control cells with or without gefitinib treatment (Supplemental Fig. S4A), suggesting that CIC regulates EGFR target genes downstream from EGFR–ERK1/2 function.
A heat map of the top 10 up-regulated genes in the CIC knockdown cells compared with control cells during gefitinib treatment is shown in Figure 4E. The top two up-regulated genes were ETV4 and ETV5. ETV1, ETV4, and ETV5 are members of the PEA3 (polyoma enhancer activator 3) subfamily of Ets transcription factors. The three PEA3 Ets transcriptional activators function as oncoproteins in several tumor types and promote cell proliferation (Oh et al. 2012). Notably, ETV1 was shown previously to restore the RAS/MAPK gene expression program upon MEK inhibition in prostate cancer cells (Hollenhorst et al. 2011). ETV1 was not included in the transcriptional analysis due to low read counts detected in RNA-seq but was significantly up-regulated in the CIC knockdown cells compared with control cells during gefitinib treatment and ranks in the top three genes when considering fold change (Fig. 4E). Using RT-qPCR, we further confirmed the mRNA levels of the three PEA3 Ets transcription factors as well as CCND1, which is of interest given that CIC knockdown bypassed cell cycle arrest induced by gefitinib. The mRNA levels of these genes were significantly reduced in response to gefitinib in control cells but remained high in CIC knockdown cells (Fig. 4F).
The CIC-regulated oncogenic Ets factor ETV1 can enhance cell growth in NSCLC cells when the EGFR pathway is impaired
Given that mRNA levels of the PEA3 Ets transcription factors were sustained in CIC knockdown cells upon gefitinib treatment, we sought to determine whether these genes contribute to the resistance caused by loss of CIC. We individually expressed ORFs of ETV1, ETV4, and ETV5 in PC9 cells and examined the effects on gefitinib resistance using the multicolor competition assay. Ectopic expression of ETV4 was toxic to PC9 cells. The percentage of GFP-labeled ETV4-expressing cells decreased to ∼0% after DMSO treatment for 10 d (Fig. 5A); thus, its role in promoting drug resistance could not be evaluated.
Figure 5.
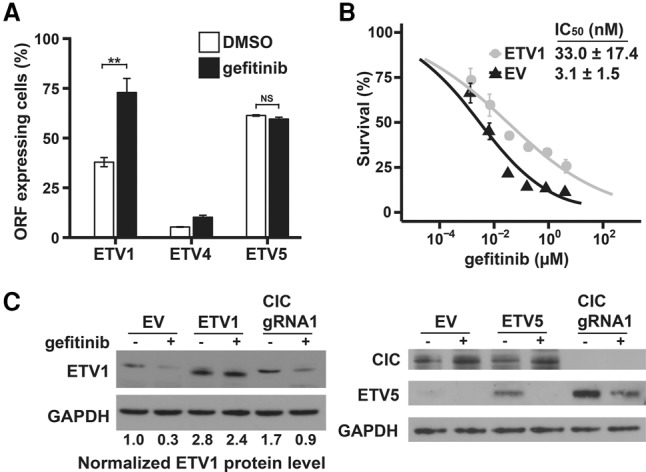
The CIC-regulated oncogenic Ets factor ETV1 can enhance cell growth when the EGFR pathway is impaired. (A) PC9 cells containing virus expressing the indicated ORFs were mixed with cells containing the empty vector (EV) in a 50:50 ratio. After 10 d of treatment with DMSO or 30 nM gefitinib, the percentage of GFP-labeled ORF-expressing cells was quantified using FACS. Data are means ± SD. n = 3. (NS) Not significant; (**) P < 0.01. (B) PC9 cells expressing ETV1 or EV were treated with the indicated concentrations of gefitinib for 72 h before cell viability was measured using a sulphorhodamine B (SRB) assay and normalized to untreated controls. Data are means ± SD. n = 3. (C) Immunoblot analysis of PC9 cells overexpressing EV or the indicated ORFs or infected with CIC gRNA and treated with 30 nM gefitinib for 12 h.
While not promoting the basal proliferation of PC9 cells, ETV1 expression conferred a significant growth advantage over the empty vector (EV) during gefitinib treatment (Fig. 5A). The IC50 of gefitinib increased 10.7-fold for ETV1-expressing cells compared with EV-expressing cells, as determined by the sulphorhodamine B (SRB) assay (Fig. 5B). Notably, consistent with the regulation of its mRNA levels, protein levels of ETV1 in the control cells were decreased upon gefitinib treatment, while in the CIC mutant cells upon gefitinib treatment, ETV1 protein levels were maintained at a level comparable with or higher than the basal level of DMSO-treated control cells (Fig. 5C; Supplemental Fig. S4B). Collectively, these results suggest that sustained activation of ETV1 and possibly other Ets factors contributes to gefitinib resistance mediated by CIC loss.
ETV5 protein was expressed at low levels in PC9 cells, and these levels were reduced upon gefitinib treatment (Fig. 5C). Basal ETV5 levels were much higher in CIC mutant cells, and, while partially reduced by gefitinib treatment, significant levels of ETV5 still remained after gefitinib treatment and could potentially contribute to gefitinib resistance. Surprisingly, while ectopic expression of ETV5 showed increased basal levels in untreated cells, addition of gefitinib reduced the protein to undetectable levels. Consistent with this, doxycycline (Dox)-induced expression of ETV5 in PC9 cells had no effect on sensitivity to gefitinib treatment (Fig. 5A). Thus, the role of ETV5 in promoting gefitinib resistance in CIC mutants remains to be established.
Discussion
We used LOF and gain-of-function genetic screens to systematically investigate genetic interactions among cancer drivers with a focus on RTK pathways. CRISPR and shRNA screening provided complementary means of assessing the functional contribution of TSGs. While these two approaches identified a significantly overlapping list of genes that influence EGFRi resistance phenotypes, they also identified unique genes. For example, the shRNA screen identified ZC3H18, which is essential for cell viability and was therefore missed in the CRISPR screen. CRISPR, on the other hand, uniquely identified NF2, which did not score in the shRNA screen. In general, gRNAs targeting the top genes behave more consistently in the CRISPR screens than shRNAs do in the shRNA screens and show stronger enrichment with more significant FDRs.
The validity of this genetic interaction screen was the identification of many genes previously known to play roles in RTK signaling, although only a few had been shown previously to impact cell growth and survival in EGFR-dependent cell lines. This is illustrated by the heat map pathway shown in Figure 6. Core members of the RTK pathway EGFR and ERBB2(HER2) scored strongly, as did HRAS and NRAS, key downstream effectors of the RTK pathway (Supplemental Table S1). Additional regulators of RAS—the RAS-GAPs NF1 and RASA1—stood out, as did SPRED1 and SPRED2, known to negatively influence RAS downstream signaling. Direct effectors of RAS such as BRAF and MAP2K1 also strongly scored, as did additional MAPK pathways that may function in various feedback mechanisms at play in RTK signaling. Genes involved in the PI3K pathway that act downstream from RTKs, such as PIK3R1, PTEN, NF2 (Pirazzoli et al. 2014), and AKT1, were also recovered in our screens. One interesting observation from the OG screen was that expression of most of the OGs that provided enrichment in the presence of gefitinib resulted in a negative enrichment score when expressed in cells bearing activated EGFR without gefitinib (Fig. 1C). A similar observation was observed previously for expression of activated KRAS in EGFR mutant tumors (Unni et al. 2015), suggesting that too much oncogenic signaling could be toxic and result in synthetic lethality. Our results suggest that this observation may not be an isolated example but instead a general rule for OGs capable of suppressing EGFR loss.
Figure 6.
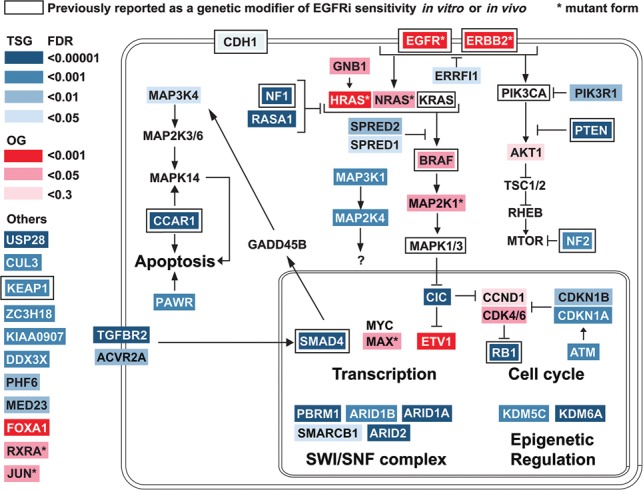
Summary and pathway schematic. Genetic interactions of TSGs and OGs with the EGFR pathway.
We observed additional pathways whose direct connection to the RTK signaling is less clear. They may represent parallel pathways that provide equivalent functions. Perturbation of the TGFβ pathway—including the two TGFβ superfamily receptors TGFBR2 and ACVR2A and their downstream effector, the transcription factor SMAD4—all showed phenotypes. SMAD4 loss has been shown previously to impact resistance to EGFRis (Cheng et al. 2015), but how it suppresses EGFRi sensitivity is not known. Other key pathways illustrated in the screen involved proliferation and survival. Genes involved in promoting apoptosis, such as CCAR and MAP3K4 (Rishi et al. 2006), were also identified. Consistent with the identification of MAP3K4, TGFβ signaling was shown to play a role in tumor inhibition by activating MAPK14 (P38 MAPK) signaling through SMAD4-dependent GADD45β expression (Takekawa et al. 2002). This could be part of the mechanism of EGFRi resistance mediated by SMAD4 loss. Among genes promoting proliferation are the central G1/S transition regulators such as the cyclin-dependent kinase CDK4/6–Cyclin D genes (Kobayashi et al. 2006) and their inhibitors, p21CDKN1A and p27CDKN1B. In addition, the downstream inactivation target of the CDK4/6 kinases, the RB1 gene (Niederst et al. 2015) that is known to restrain cyclin E and other genes necessary for promoting entry into S phase, was a strong hit in the screen. This central network may be a direct target of many TSG and OG drivers and has been linked to both RTK and TGFβ pathways in other contexts (Stalinska and Ferenc 2005; Schiewer et al. 2012; VanArsdale et al. 2015; O'Leary et al. 2016). Another key pathway known to regulate proliferation and survival identified in our screens was the MYC–MAX pathway. While MAX scored, MYC did not, but it is possible that MYC is already in excess, and its partner, MAX, is rate-limiting in this cell line. We also recovered CDH1 in our screens, down-regulation of which was shown previously to up-regulate EGFR and promote proliferation and invasiveness in NSCLC cells (Liu et al. 2013).
Many TSGs and OGs not previously implicated in RTK signaling emerged from our screens, including USP28 and the CUL3–KEAP1 E3 ubiquitin ligase complex. Although damaging, mutations in KEAP1 were shown previously to mediate EGFRi resistance (Yamadori et al. 2012). Among these, the SWI/SNF complex stood out, as multiple members of that complex scored as modifiers of EGFR dependency. Two SWI/SNF subunits, SMARCE1 and ARID1A, have been reported to confer resistance to MET and ALK inhibitors by regulating EGFR expression (Papadakis et al. 2015). However, we showed that loss of PBRM1 does not affect EGFR levels. Instead, PBRM1 mutant NSCLC cells were able to restore activation of AKT in the presence of gefitinib after an initial inhibition. AKT signaling pathways are known to rebound in the continuous presence of PI3K inhibitors due to relief of various feedback mechanisms (Costa et al. 2015; Schwartz et al. 2015), but it is unknown whether the effect of EGFR inhibition works through the same mechanism as PI3K inhibitors, and, in this case, the rebound in response to gefitinib is dependent on PRBM1 mutation. Thus, how PBRM1 restrains AKT activation remains to be determined. The different mechanisms underlying the regulation of EGFR signaling by different subunits of SWI/SNF may be complex. Also, despite the role of PBRM1 in regulating p21 in other cellular contexts, knockdown of PBRM1 in PC9 cells did not affect the basal level of p21 or its level during gefitinib treatment. This may be in part because the composition of the SWI/SNF complex varies from tissue to tissue, as do its transcriptional targets (Helming et al. 2014). This level of combinatorial complexity may allow for different mechanisms depending on which subunits are expressed and in which tissues they are present.
Another tumor suppressor identified and characterized in our study is CIC. CIC has been studied primarily in Drosophila melanogaster, where it acts downstream from EGFR during fly development. Depending on the context, EGFR signaling causes the CIC protein to decrease or exit the nucleus and relocate to the cytoplasm (Jimenez et al. 2012). In some cases, neither of these events occurs, but its activity is somehow interfered with by RAS/MAPK signals (Dissanayake et al. 2011). In PC9 cells, we did not see either induction of CIC protein or increased localization to the nucleus under gefitinib treatment (data not shown), suggesting another form of inactivation in response to EGFR signaling. Regardless, these studies suggest that the relationship between CIC and EGFR found in Drosophila is conserved in humans and that CIC loss can suppress loss of EGFR signaling in both organisms.
The mechanism through which CIC loss impacts EGFR signaling and resistance to gefitinib is likely its role as a transcriptional repressor of genes that affect the ability of cells to maintain cell proliferation. Among the transcriptional targets induced in PC9 cells in the absence of CIC, we identified a D-type cyclin (CCND1) and a family of Ets-related transcription factors (ETV1, ETV4, and ETV5). Furthermore, the CDK4/6 kinase inhibitor CDKN2B is induced by gefitinib in CIC wild-type cells, but this induction was abrogated in the absence of CIC (Supplemental Table S2). A functional role for regulators of the cyclin D/CDK4/6/RB pathway in mediating part of the bypass of EGFR function in the absence of CIC was suggested by the enhanced sensitivity of CIC mutant PC9 cells to gefitinib in the presence of the CDK4/6 inhibitor palbociclib (Fig. 2D). The fact that CDK4 and CDK6 overproduction provided gefitinib resistance in the OG screen (Supplemental Table S1) further supports a role for cell cycle regulation in EGFR-driven cell proliferation.
The Ets factors, which have known oncogenic functions, are also likely to play an important role in the ability of CIC mutants to modify PC9 cells’ resistance to gefitinib. Ectopic ETV1 expression conferred gefitinib resistance to these cells. It is likely that ETV4 and ETV5 also play a role in mediating resistance to gefitinib in the absence of CIC; however, we were unable to test this because we were unable to express sufficient levels of these proteins in PC9 cells. Unexpectedly, while we were able to express detectable levels of ETV5 from an exogenous promoter, inhibition of EGFR still extinguished ETV5 protein levels, suggesting that active EGFR is promoting the expression of ETV5 both transcriptionally and post-transcriptionally. Nonetheless, the derepression of this family of oncogenic transcription factors is likely to play an important role in how CIC loss mediates resistance to EGFR inhibition in PC9 cells.
The present system-level study of the genetic interactions among cancer drivers provides strong support for the hypothesis that cancer drivers can substitute for each other in some contexts. We identified a large number of TSGs and OGs that can genetically modify the growth and survival deficiencies caused by reduced EGFR signaling. In addition to the genes already known to modify resistance to EGFR inhibition and genes directly implicated in the RTK–RAS–PI3K signaling pathway, we uncovered many new TSGs and OGs that can genetically interact with EGFR signaling. These genes may play a role in augmenting the RTK–RAS–PI3K pathway or parallel pathways that can provide cells with enhanced growth and survival functions that substitute for those lacking when EGFR signaling is impaired. Many of these genes become candidates for the 30% of tumors that acquire resistance through unknown mechanisms. This type of analysis can also be performed in cells experiencing other forms of OG addiction or TSG hypersensitivity to identify TSGs and OGs that modify their function and will provide information on the role of many relatively understudied cancer drivers.
Materials and methods
Cell culture and reagents
PC9 cells were kindly provided by J. Engelman (Massachusetts General Hospital Cancer Center and Harvard Medical School). H1975 cells were kindly provided by Kwok Kin Wong (Dana-Farber Cancer Institute and Harvard Medical School). 293T cells were purchased from American Type Culture Collection (ATCC). PC9 cells/H1975 cells and 293T cells were maintained in RPMI 1640 medium (ATCC modification) and DMEM, respectively (Thermo Fisher Scientific, Inc.), supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GE Healthcare HyClone), 100 IU/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Gefitinib, erlotinib, and AZD9291 were purchased from Selleck Chemicals. Palbociclib was a gift from K. Cichowski. The antibodies used were Tubulin (1:2500; Cell Signaling, 2128S), GAPDH (1:2500; Cell Signaling, 8884), PBRM1 (1:500; Bethyl Laboratories, A301-591A-M), p21 (1:1000; Cell Signaling, 2947S), phospho-EGFR (Tyr1068; 1:1000; Cell Signaling, 2236S), EGFR (1:1000; Cell Signaling, 2232S), phospho-ERK1/2 (T202/Y204; 1:1000; Cell Signaling, 4370P), total ERK1/2 (1:1000; Cell Signaling, 4695P), phospho-AKT (S473; 1:1000; Cell Signaling, 4060P), total AKT (1:1000; Cell Signaling, 2920S), ETV1 (1:1000; Abcam, ab184120), ETV5 (1:1000; Abcam, ab54704), and CIC (1:5000; a gift from H. Zoghbi).
TSG CRISPR library construction
A customized DNA oligonucleotide library with 10 gRNAs per gene for each of the 500 TSG candidates was synthesized on a microarray (Agilent). The gRNA oligonucleotide library was PCR-amplified by a set of specific primers and digested with BbsI. The digested gRNA was purified on a 10% TBE PAGE gel and cloned into BsmBI-digested pLentiCRISPR V2 (Addgene plasmid no. 52961).
TSG shRNA library construction
A customized DNA oligonucleotide library with 10 shRNAs per gene for each of the 500 TSG candidates was synthesized as above. The shRNA oligonucleotide library was PCR-amplified by a set of specific primers and digested with XhoI and EcoRI. The digested PCR products were purified on a 3% Nusieve gel and cloned into the XhoI/EcoRI-digested vector (pMSCV-mirE-pheS) to make pMSCV-mirE libraries. The pMSCV-mirE libraries were digested with XhoI/MluI and cloned into XhoI/MluI-cut pHAGE-Ind10-mirE. The pMSCV-mirE-pheS vector was made by two steps. First, pMSCV-PM-pheS plasmid DNA was digested with EcoRI, and a pair of complementary oligonucleotides containing the 3′mirE sequences were inserted using SLIC (Li and Elledge 2012) to generate pMSCV-mirE3′-pheS. Second, the pMSCV-mirE3′-pheS plasmid was digested with HpaI, and a pair of complementary oligonuclotides containing the 5'mirE sequence were inserted using SLIC to generate pMSCV-mirE-pheS.
CRISPR and shRNA screen
The CRISPR and shRNA screens were performed in three and two replicates, respectively. Each library contained 10 gRNAs or 10 shRNAs per gene for each of the 500 TSG candidates and 1000 gRNAs or 1000 shRNAs targeting the E. coli genome as negative controls. PC9 cells were transduced using 4 µg/mL polybrene (Sigma) at a low multiplicity of infection ([MOI] = 0.2) with an average representation of ∼1000 cells per gRNA/shRNA. Transduced cells were then selected with 1 µg/mL puromycin (Clontech) for 3 d. The shRNA library was constructed in a Dox-inducible vector. After puro selection, Dox was added 3 d prior to gefitinib treatment, allowing the targeted genes to be knocked down. For the CRISPR screen, cells were passaged for 9 d, allowing enough time for genome modification by Cas9. Cells from the initial time point were harvested, and the cells were then split into two arms and treated with either DMSO or 30 nM gefitinib for ∼17 d. Cells from the end time point (DMSO treatment and gefitinib treatment for 17 d) were then harvested. Genomic DNAs containing shRNA or gRNA were amplified by PCR and subsequently subjected to next-generation sequencing. The MAGeCK scoring algorithm (Li et al. 2014) was used to rank the performance of individual genes based on enrichment, comparing the gefitinib treatment group with the DMSO treatment group. The negative controls were incorporated in the MAGeCk analysis to generate null distributions and calculate the P-value and FDR for each gene. Combined FDRs of the two screens were generated using combined P-values generated by Fisher's method and were used as the FDRs for TSGs in Figure 6.
OG screen
The OG screen was performed in three replicates. A lentiviral library containing a partial list of barcoded OGs (K Mengwasser, T Davoli, Y Leng, Q Xu, E Wooten, and S Elledge, in prep.) identified by TUSON under the control of a Dox-inducible TRE promoter and containing the rtTA gene was introduced into PC9 cells. Transduced cells were then selected with 1 µg/mL puromycin (Clontech) for 3 d and grown for 7 d. Dox (1 µg/mL) was added, and cells were grown for 3 d, allowing the genes to be expressed. Next, either DMSO or 30 nM gefitinib was added, and cells were grown for ∼17 d in medium containing Dox. DNA was harvested, processed, and analyzed as described above.
Analysis of the somatic mutation data set and focal deletion data set
The data sets of somatic mutations and focal deletions of lung adenocarcinoma patients were from TCGA (http://cancergenome.nih.gov). We classified patients harboring the following activating mutations in EGFR as EGFR mutant patients: p.L858R, p.TSPKANKE751del, p.KANKEI754del, p.LREAT747del, p.ELR746del, p.ELREA746del, p.G719A, p.G719C, p.G719S, p.V765A, p.T783A, and p.L861Q. After excluding patients with mutations >1000, we ended up with 50 EGFR mutants and 443 EGFR wild-type patients.
We used the PolyPhen2 Hum-Var prediction model (Adzhubei et al. 2010) to weigh the functional impact of each missense mutation. Based on the possible or probable damage in the Hum-Var prediction by PolyPhen2, we defined missense mutations with a Hum-Var score >0.447 as damaging. We defined LOF mutations as nonsense, frameshift, and “damaging missense” (corresponding to possibly or probably damaging in the Hum-Var prediction with a score >0.447). We defined a gene as focally deleted if its log2 copy number change was lower than −0.2.
Because of the low number of EGFR mutant tumors in our data set, most genes had few mutations and could not be analyzed individually. Therefore, we pooled 30 genes as a group to reduce noise and gain statistical power. We then calculated the average percentage of LOF mutations (LTop30) for our top 30 TSG hits in EGFR mutant and wild-type patients as follows:
We then performed 10,000 permutations using a random group of 30 genes to get the distribution of LTSG (using genes in the TSG library with rank position >65 in the screens, FDR > 0.1) or Lgenome (using all genes in the genome, excluding the ones in the TSG library). The P-value of LTop30 was calculated based on the distribution of LTSG.
The analysis of mutual exclusivity of each gene with activating EGFR was performed using the Fisher exact test (one-tailed test) based on 447 patients who had both mutation and focal deletion data.
Multicolor competition assay
GFP-labeled PC9 cells were transduced with the indicated gRNA/ORFs, and mRuby-labeled PC9 cells were transduced with GFP gRNA/EV. After puromycin selection and 3 d of 1 µg/mL Dox treatment for ORF-expressing cells, GFP-labeled PC9 cells were mixed with mRuby-labeled PC9 cells in a 50:50 ratio in six-well plates. After treatment of DMSO or the indicated dose of drug, the drug and medium were replaced every 2 d, and the percentage of GFP-labeled gRNA/ORF-expressing cells was quantified using FACS (BD, LSR II).
Colony formation assay
Single-cell suspensions were seeded into six-well plates (200 cells per well) and incubated overnight before continuous treatment of DMSO for 2 wk or 30 nM gefitinib for 3 wk, with drug and medium replaced every 2 d. At the end of treatment, cells were fixed with 10% trichloroacetic acid, washed, and stained with SRB as described previously (Vichai and Kirtikara 2006).
Cell viability assay
The effects of gefitinib on cell proliferation were determined by SRB as described previously (Vichai and Kirtikara 2006).
Apoptosis and cell cycle analysis
Cells seeded in six-well plates (5 × 104 cells per well) were treated with DMSO or 30 nM gefitinib for 48 h. For apoptosis analysis, cells were then fixed with 70% (v/v) cold ethanol overnight at −20°C and stained with PI solution (20 µg/mL PI [Sigma-Aldrich], 0.1% Triton X-100, 8 µg/mL RNase). For cell cycle analysis, cells were pulsed with BrdU (10 µM final concentration) for 1 h prior to fixation. BrdU labeling and staining were performed using the APC BrdU flow kit (BD Biosciences) according to the manufacturer's instructions. Samples were analyzed by flow cytometry (BD, LSR II).
Quantitative RT-qPCR
Total RNA was isolated using the RNAeasy minikit (Qiagen), and cDNA was synthesized using SuperScript IV (Invitrogen) according to the manufacturer's instructions. Quantitative RT-qPCR was performed in triplicate using the TaqMan gene expression master mix (Invitrogen) with TaqMan gene expression assay (Life Technologies) on an Applied Biosystems Fast 7500 machine using GAPDH as the endogenous normalization control. The IDs for the TaqMan assays used were as follows: GAPDH (Hs99999905_m1), ETV1 (Hs00951951_m1), ETV4 (Hs003833 61_g1), ETV5 (Hs00927557_m1), and CCND1 (Hs00765553_m1).
Gene expression profiling
PC9 cells were transduced with pLentiCRISPR V2 with gRNA targeting GFP or CIC. Infected cells were selected using 1 µg/mL puromycin (Clontech) for 3 d. One week after transduction, cells were treated with DMSO or 30 nM gefitinib for 6 h. Total RNA was isolated using the RNAeasy minikit (Qiagen). RNA-seq libraries were generated using NEBNext ultra RNA library preparation kit (New England Biolabs). RNA-seq was performed using two replicates for control GFP gRNA and three different gRNAs of CIC as three replicates. Fifty-base-pair single-end sequencing was performed using an Illumina HiSeq 2500. Reads were aligned to the hg19 genome using HiSat2 (Pertea et al. 2016), transcripts and frequencies were assessed from the aligned data by subread (Liao et al. 2013), and edgeR (Robinson et al. 2010) was used to identify DEGs and generate the CPM expression values used for GSEA (http://www.broadinstitute.org/gsea) (Subramanian et al. 2005). PPI hubs were identified using Enrichr (Kuleshov et al. 2016). Z-scores were used to generate a heat map of relative expression levels. Each sample was normalized to the basal condition (GFP-gRNA-infected, DMSO-treated sample 1) by subtracting the Z-score of the basal condition from the Z-score of each sample for each gene as Znomalized to basal = Zsample − ZGFP gRNA, DMSO sample 1.
Plasmids, cloning and viral transduction
gRNAs were cloned into pLentiCRISPR V2 (Addgene, plasmid no. 52961) as described previously (Sanjana et al. 2014).
The sequences targeted by the gRNAs used were as follows (if the first base of the targeted sequence is not G, it was changed to G during cloning for expression under U6 promoter): GFP, GGGCGAGGAGCTGTTCACCG; PBRM1_1, CGAGACTATAAGGATGAACA; PBRM1_2, GCAATGGTCTTGAGATCTAT; PBRM1_3, TCATTAGGGCACCAAAGCGA; CIC_1, GCAACCTGCCAGCCACCCAG; CIC_2, GGGGTACACAGCCTGGACGG; CIC_3, GGGGCGGCAGTGGGTAAAGG; and NF1, GAGAGAAAATAAAACCCCAG.cDNA for ETV4 was from PlasmID. cDNAs for ETV1 and ETV5 were from the human ORFeome library version 8.1. Stop codons were added using QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). cDNAs were subcloned into pHAGE-TREx-BC-Dest vector via LR recombinase reaction (Invitrogen).
To produce lentiviruses, 293T cells were transfected with vector DNA, pRev, pTat, pHIV Gag/pol, and pVSVG. Viruses were harvested 48 h after transfection and filtered (45-µm pore size). TransIT-293 (Mirus) was used to transfect 293T cells.
Cells were transduced with 4 µg/mL polybrene, and infected cells were selected using 1 µg/mL puromycin (Clontech) for 3 d. pLentiCRISPR-V2-infected cells were subjected to multicolor competition assay, cell viability assay, colony formation assay, or immunoblot analysis 1 wk after infection to allow for genome modification by Cas9. Cells expressing ORFs were treated with 1 µg/mL Dox for 3 d prior to subsequent experiments for ORF expression and maintained in medium containing 1 µg/mL Dox during subsequent experiments.
Statistical analysis
Data are presented as the mean ± SD, and significance was analyzed using the Student's t-test. Differences were considered significant when P < 0.05.
Supplementary Material
Acknowledgments
We thank K. Naxerova, P. Bruno, C. Wang, E. Watson, T. Martin, and the other members of the Elledge laboratory for critical comments on the manuscript and advice. We also thank H. Zoghbi, M. Brown, K. Cichowski, and A. Toker for advice and reagents. This work was supported with funding from the Ludwig Foundation. S.J.E. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.291948.116.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. 2015. Gene essentiality and synthetic lethality in haploid human cells. Science 350: 1092–1096. [DOI] [PubMed] [Google Scholar]
- Cheng H, Fertig EJ, Ozawa H, Hatakeyama H, Howard JD, Perez J, Considine M, Thakar M, Ranaweera R, Krigsfeld G, et al. 2015. Decreased SMAD4 expression is associated with induction of epithelial-to-mesenchymal transition and cetuximab resistance in head and neck squamous cell carcinoma. Cancer Biol Ther 16: 1252–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho A, Shim JE, Kim E, Supek F, Lehner B, Lee I. 2016. MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biol 17: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Janne PA. 2013. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 19: 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, et al. 2015. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell 27: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. 2013. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155: 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, Gettinger S, Walther Z, Wurtz A, Heynen GJ, et al. 2014. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov 4: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486: 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake K, Toth R, Blakey J, Olsson O, Campbell DG, Prescott AR, MacKintosh C. 2011. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem J 433: 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043. [DOI] [PubMed] [Google Scholar]
- Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, et al. 2012. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov 2: 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Helming KC, Wang X, Roberts CW. 2014. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 26: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. 2011. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev 25: 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez G, Shvartsman SY, Paroush Z. 2012. The Capicua repressor—a general sensor of RTK signaling in development and disease. J Cell Sci 125: 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Shimamura T, Monti S, Steidl U, Hetherington CJ, Lowell AM, Golub T, Meyerson M, Tenen DG, Shapiro GI, et al. 2006. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res 66: 11389–11398. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Dai F, Zhuang L, Xiao ZD, Kim J, Zhang Y, Ma L, You MJ, Wang Z, Gan B. 2016. BAF180 regulates cellular senescence and hematopoietic stem cell homeostasis through p21. Oncotarget 7: 19134–19146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. 2015. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 47: 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MZ, Elledge SJ. 2012. SLIC: a method for sequence- and ligation-independent cloning. Methods Mol Biol 852: 51–59. [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2013. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41: e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Su L, Liu X. 2013. Loss of CDH1 up-regulates epidermal growth factor receptor via phosphorylation of YBX1 in non-small cell lung cancer cells. FEBS Lett 587: 3995–4000. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139. [DOI] [PubMed] [Google Scholar]
- Majem M, Remon J. 2013. Tumor heterogeneity: evolution through space and time in EGFR mutant non small cell lung cancer patients. Transl Lung Cancer Res 2: 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486: 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. 2009. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361: 947–957. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. 2015. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6: 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Shin S, Janknecht R. 2012. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta 1826: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, et al. 2012. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci 109: E2127–E2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary B, Finn RS, Turner NC. 2016. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13: 417–430. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304: 1497–1500. [DOI] [PubMed] [Google Scholar]
- Papadakis AI, Sun C, Knijnenburg TA, Xue Y, Grernrum W, Holzel M, Nijkamp W, Wessels LF, Beijersbergen RL, Bernards R, et al. 2015. SMARCE1 suppresses EGFR expression and controls responses to MET and ALK inhibitors in lung cancer. Cell Res 25: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11: 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirazzoli V, Nebhan C, Song X, Wurtz A, Walther Z, Cai G, Zhao Z, Jia P, de Stanchina E, Shapiro EM, et al. 2014. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell Rep 7: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishi AK, Zhang L, Yu Y, Jiang Y, Nautiyal J, Wali A, Fontana JA, Levi E, Majumdar AP. 2006. Cell cycle- and apoptosis-regulatory protein-1 is involved in apoptosis signaling by epidermal growth factor receptor. J Biol Chem 281: 13188–13198. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch F, Jimenez G, Casanova J. 2002. EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 129: 993–1002. [DOI] [PubMed] [Google Scholar]
- Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, et al. 2009. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361: 958–967. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. 2009. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 69: 1851–1857. [DOI] [PubMed] [Google Scholar]
- Schiewer MJ, Augello MA, Knudsen KE. 2012. The AR dependent cell cycle: mechanisms and cancer relevance. Mol Cell Endocrinol 352: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, et al. 2015. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 27: 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F. 2015. High-throughput functional genomics using CRISPR–Cas9. Nat Rev Genet 16: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifnia T, Rusu V, Piccioni F, Bagul M, Imielinski M, Cherniack AD, Pedamallu CS, Wong B, Wilson FH, Garraway LA, et al. 2014. Genetic modifiers of EGFR dependence in non-small cell lung cancer. Proc Natl Acad Sci 111: 18661–18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, et al. 2009. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 69: 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalinska L, Ferenc T. 2005. [The role of TGF-β in cell cycle regulation]. Postepy Hig Med Dosw (Online) 59: 441–449. [PubMed] [Google Scholar]
- Stewart EL, Tan SZ, Liu G, Tsao MS. 2015. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res 4: 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M, Tatebayashi K, Itoh F, Adachi M, Imai K, Saito H. 2002. Smad-dependent GADD45β expression mediates delayed activation of p38 MAP kinase by TGF-β. EMBO J 21: 6473–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, et al. 2012. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2: 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. 2015. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21: 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng AS, Tapon N, Kanda H, Cigizoglu S, Edelmann L, Pellock B, White K, Hariharan IK. 2007. Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr Biol 17: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. 2015. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung carcinoma. Elife 4: e06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanArsdale T, Boshoff C, Arndt KT, Abraham RT. 2015. Molecular pathways: targeting the cyclin D–CDK4/6 axis for cancer treatment. Clin Cancer Res 21: 2905–2910. [DOI] [PubMed] [Google Scholar]
- Vichai V, Kirtikara K. 2006. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 1: 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wang X, Goldstein D, Crowe PJ, Yang JL. 2016. Next-generation EGFR/HER tyrosine kinase inhibitors for the treatment of patients with non-small-cell lung cancer harboring EGFR mutations: a review of the evidence. Onco Targets Ther 9: 5461–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. 2007. The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113. [DOI] [PubMed] [Google Scholar]
- Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T, Memeo L, Hibshoosh H, Parsons R. 2008. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res 68: 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamadori T, Ishii Y, Homma S, Morishima Y, Kurishima K, Itoh K, Yamamoto M, Minami Y, Noguchi M, Hizawa N. 2012. Molecular mechanisms for the regulation of Nrf2-mediated cell proliferation in non-small-cell lung cancers. Oncogene 31: 4768–4777. [DOI] [PubMed] [Google Scholar]
- Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, et al. 2010. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res 70: 8715–8725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.