

ABSTRACT
Non-homologous end joining (NHEJ) is a major DNA double-strand break (DSB) repair pathway that functions in all phases of the cell cycle. NHEJ repairs genotoxic and physiological DSBs, such as those generated by ionizing radiation and during V(D)J recombination at antigen receptor loci, respectively. DNA end joining by NHEJ relies on the core factors Ku70, Ku80, XRCC4, and DNA Ligase IV. Additional proteins also play important roles in NHEJ. The XRCC4-like factor (XLF) participates in NHEJ through its interaction with XRCC4, and XLF deficiency in humans leads to immunodeficiency and increased sensitivity to ionizing radiation. However, XLF is dispensable for NHEJ-mediated DSB repair during V(D)J recombination in murine lymphocytes, where it may have redundant functions with other DSB repair factors. Paralog of XRCC4 and XLF (PAXX) is a recently identified NHEJ factor that has structural similarity to XRCC4 and XLF. Here we show that PAXX is also dispensable for NHEJ during V(D)J recombination and during the repair of genotoxic DSBs in lymphocytes. However, a combined deficiency of PAXX and XLF blocks NHEJ with a severity comparable to that observed in DNA Ligase IV-deficient cells. Similar to XLF, PAXX interacts with Ku through its C-terminal region, and mutations that disrupt Ku binding prevent PAXX from promoting NHEJ in XLF-deficient lymphocytes. Our findings suggest that the PAXX and XLF proteins may have redundant functions during NHEJ.
KEYWORDS: double-strand break repair, non-homologous end joining, PAXX, Pre-B cells, RAG, V(D)J recombination, XLF
Introduction
DNA double-strand breaks (DSBs) are generally repaired by homologous recombination (HR) or non-homologous end joining (NHEJ).1,2 HR functions to repair DSBs in the S and G2 phases of the cell cycle using the sister chromatid as a template for precise restoration of DNA sequences.2 NHEJ functions in all phases of the cell cycle and is the primary pathway of DSB repair in G1-phase cells. NHEJ rejoins broken DNA ends in a manner that is often imprecise, with the loss and gain of nucleotides at the join.1,2 The core factors that are absolutely required for NHEJ in all settings include Ku70, Ku80, DNA Ligase IV, and XRCC4.1,2 In addition, several other factors participate in NHEJ that may have redundant or more specialized functions.1-4
In developing lymphocytes, antigen receptor gene assembly occurs through the process of V(D)J recombination.5,6 The recombination activating gene (RAG) endonuclease, composed of RAG1 and RAG2 proteins, recognizes recombination signal sequences (RSs) and generates DSBs at the border of RSs and the flanking recombining V (variable), D (diversity) or J (joining) gene segments.6 RAG cleavage leads to the formation of a pair of hairpin-sealed coding ends (CEs) and a pair of blunt signal ends (SEs). The two coding ends are joined to form a coding join (CJ), and the 2 signal ends are joined to form a signal join (SJ).7 In addition to the core NHEJ proteins, this joining requires other factors including the Artemis endonuclease, which opens hairpin-sealed CEs before they can be joined.1,7 Artemis activity depends on the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs).1,7 Thus, cells deficient in either Artemis or DNA-PKcs exhibit a nearly complete block in CJ formation but no defect in SJ formation.1,7 The ataxia–telangiectasia mutated (ATM) kinase also functions in the repair of RAG DSBs, in part by promoting the stability of the broken DNA ends generated by RAG cleavage in a complex before they are joined by NHEJ.8 Although neither ATM nor DNA-PKcs appear to be essential for SJ formation, a combined deficiency of these proteins leads to a nearly complete block in this process.9,10 Thus, ATM and DNA-PKcs have overlapping activities that are critical for NHEJ-mediated DSB repair.
The XRCC4-like factor (XLF) functions during NHEJ in non-lymphoid cells, including the repair of RAG DSBs generated in plasmid substrates after transient expression of RAG.11-16 XLF binds to the Ku heterodimer and to XRCC4, 2 core NHEJ factors, and forms filaments with XRCC4 that stabilize broken DNA ends prior to joining.14,17-22 Paradoxically, in lymphoid cells there is a minimal requirement for XLF during RAG DSB repair by NHEJ.12 However, a combined deficiency of XLF and either H2AX or 53BP1 leads to a block in DSB repair in lymphoid cells even though the singular loss of any of these proteins does not lead to notable NHEJ defects.23-25 Moreover, a combined loss of XLF and either the DNA-PKcs or ATM kinase leads to a more significant block in NHEJ-mediated RAG DSB repair than a deficiency of any of these factors alone.24,26 Finally, when combined with XLF deficiency, expression of a C-terminally truncated form of RAG2 also leads to a block in NHEJ.27 Although XLF could have overlapping activities with several of these functionally diverse factors, it seems likely that many of these factors will have distinct activities, which when compromised in the setting of XLF deficiency, leads to notable defects in NHEJ-mediated DSB repair.
The paralog of XLF and XRCC4 (PAXX) was recently discovered based on its structural similarity with XLF and XRCC4 and on its association with other DNA repair factors.28-30 XLF and PAXX bind each other and both bind to Ku70/Ku80 and XRCC4-DNA Ligase IV, suggesting that these proteins may be in a single complex.28-30 Like XLF, PAXX deficiency leads to defects in NHEJ in non-lymphoid cells.28-30 Here we use an approach to assay chromosomal V(D)J recombination in murine pre-B cells after RAG expression is induced.8 We show that while deficiencies in either XLF or PAXX do not lead to defects in RAG DSB repair in pre-B cells, the combined deficiency of XLF and PAXX blocks the repair of RAG and genotoxic DSBs with a severity similar to that observed in DNA Ligase IV-deficient cells. These findings support the notion that XLF and PAXX have redundant NHEJ activities.
Results
Analysis of V(D)J recombination in abl pre-B cells: We have generated Abelson murine leukemia virus transformed pre-B cell lines, hereafter referred to as abl pre-B cells.8 Treatment of abl pre-B cells with the v-Abl kinase inhibitor, imatinib, leads to G1 cell cycle arrest, the induction of RAG, and V(D)J recombination at the immunoglobulin kappa light chain (Igk) locus and within chromosomally integrated retroviral recombination substrates.8 We generated a new retroviral recombination substrate, pMSCV-RSS-GFP-INV (hereafter pMGINV), that exhibits more stable expression than previous substrates (Fig. 1A).8 pMGINV has an IRES-Thy1.2 cDNA that serves as a marker of infected cells (Fig. 1A). It also contains an anti-sense GFP cDNA flanked by RSs that mediate rearrangement by inversion, which places the GFP cDNA in the sense orientation leading to GFP expression (Fig. 1A).
Figure 1.
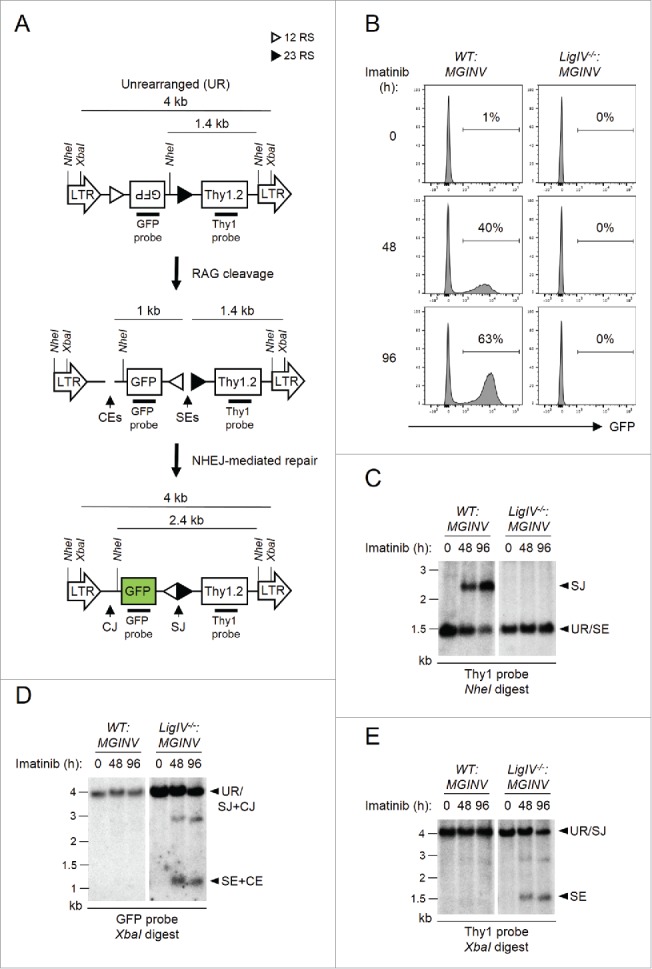
Rearrangement of pMGINV in abl pre-(B)cells. (A) Schematic of unrearranged (UR) pMGINV, its signal end (SE) and coding end (CE) intermediates following RAG cleavage, and the resulting signal join (SJ) and coding join (CJ) products. The long-terminal repeats (LTR), GFP cDNA, Thy1.2 cDNA, NheI and XbaI sites, recombination signals (RSs, represented by open and filled triangles), and Thy1 and GFP probes are shown. (B) WT:MGINV and LigIV−/−:MGINV abl pre-B cells were treated with imatinib for 0, 48, or 96 hours (h). The percentage of GFP-positive cells is indicated. (C-E) WT:MGINV and LigIV−/−:MGINV abl pre-B cells were treated with imatinib for the indicated times (h), and Southern blot analyses were carried out on NheI-digested (C) or XbaI-digested (D, E) genomic DNA isolated from these cells and hybridized with the Thy1 (C, E) or GFP (D) probe. Hybridizing bands from unrearranged pMGINV (UR), SJs, SEs, both CJs and SJs (SJ+CJ) and both SEs and CEs (SE+CE) are indicated. Molecular weight markers (kB) are also indicated.
pMGINV was introduced into both wild type (WT:MGINV) and DNA Ligase IV-deficient (LigIV−/−:MGINV) abl pre-B cells. Treatment of these cells with imatinib leads to the generation of RAG DSBs at pMGINV in both WT:MGINV and LigIV−/−:MGINV abl pre-B cells (Fig. 1B-E). In WT:MGINV abl pre-B cells, these RAG DSBs are normally repaired, forming a CJ and a SJ and leading to GFP expression (Fig. 1A and B). Southern blotting reveals robust SJ formation in WT:MGINV abl pre-B cells, as indicated by the 2.4 kb NheI Thy1 probe hybridizing fragment after RAG induction (Fig. 1A and C). Hybridization of XbaI digested WT:MGINV genomic DNA with the GFP (Fig. 1D) and Thy 1 (Fig. 1E) probes failed to reveal 1 kb and 1.4 kb fragments, respectively, indicative of unrepaired CEs and SEs (Fig. 1A, D and E). Induction of RAG in LigIV−/−:MGINV abl pre-B cells did not lead to the GFP expression due to the requirement for DNA Ligase IV to repair RAG DSBs (Fig. 1B). Indeed, Southern blot analysis of genomic DNA from LigIV−/−:MGINV abl pre-B cells after RAG induction revealed hybridizing fragments generated by unrepaired SEs and CEs (Fig. 1A, D and E) and failed to reveal hybridizing fragments indicative of SJ formation (Fig. 1A and C). Together, these data establish that the pMGINV retroviral recombination substrate can be used to assay RAG cleavage and NHEJ-mediated DSB repair during V(D)J recombination in abl pre-B cells.
Generation of Xlf- and Paxx-deficient abl pre-B cells
Xlf−/− abl pre-B cells were generated from Xlf−/− mice that contained a Bcl-2 transgene.24 A single copy of the pMGINV recombination substrate was introduced into these cells to generate Xlf−/−:MGINV abl pre-B cells. CRISPR/Cas9 approaches were used to generate Paxx-deficient abl pre-B cells.31 To this end, we introduced a lentiviral vector (pCW-Cas9) with a Cas9 cDNA under the control of a tetracycline-inducible promoter into WT:MGINV and Xlf−/−:MGINV abl pre-B cells.32 These cell lines exhibit robust induction of Cas9 upon doxycycline treatment (Fig. 2A). The lentiviral vector pKLV containing a guide RNA (gRNA) that targets the third exon of Paxx was introduced into WT:MGINV and Xlf−/−:MGINV abl pre-B cells that have an integrated copy of pCW-Cas9.33 After treatment with doxycycline and sub-cloning by limiting dilution, western blotting was used to identify Paxx−/−:MGINV and Xlf−/−:Paxx−/−:MGINV abl pre-B cells (Fig. 2B). Sequencing of the PAXX gene in the PAXX-deficient cells reveals deletions of various sizes (6 – 450 bp) in regions flanking the gRNA targeting sequence (Fig. S1).
Figure 2.
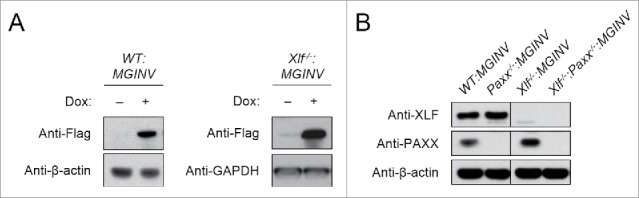
Generation of Paxx−/− and Xlf−/−:Paxx−/− abl pre-(B)cells: (A) Western blot of lysates from WT:MGINV and Xlf−/−:MGINV abl pre-B cells stably transduced with pCW-Cas9 in the absence (−) and presence (+) of doxycycline. Western blots were probed with antibodies to Flag (to detect Flag-Cas9), with β-actin or GAPDH used as protein loading controls. (B) Western blotting for XLF, PAXX, and β-actin in lysates from WT:MGINV, Xlf−/−:MGINV, Paxx−/−:MGINV and Xlf−/−:Paxx−/−:MGINV abl pre-B cells.
Deficiencies in PAXX and XLF blocks NHEJ-mediated repair of RAG DSBs
To determine whether PAXX functions in the repair of RAG DSBs, we analyzed V(D)J recombination in WT:MGINV and Paxx−/−:MGINV abl pre-B cells (Fig. 3 and Fig. S2). PAXX deficiency did not impair rearrangement of pMGINV in Paxx−/−:MGINV abl pre-B cells as compared to WT:MGINV abl pre-B cells (Fig. 3 and Fig. S2). In this regard, after induction of RAG with imatinib, the percentage of Paxx−/−:MGINV abl pre-B cells expressing GFP (indicative of RAG DSB generation and repair) was similar to that observed in WT:MGINV abl pre-B cells (Fig. 3A). In agreement, Southern blot analyses revealed robust SJ formation (Fig. 3B and Fig. S2A) and no detectable unrepaired CEs or SEs (Fig. 3C, D and Fig S2B) in Paxx−/−:MGINV abl pre-B cells. As expected, analyses of Xlf−/−:MGINV abl pre-B cells also did not reveal any defects in V(D)J recombination (Fig. 3 and Fig. S2). Thus, like XLF deficiency, PAXX deficiency in abl pre-B cells does not lead to defects in V(D)J recombination.
Figure 3.
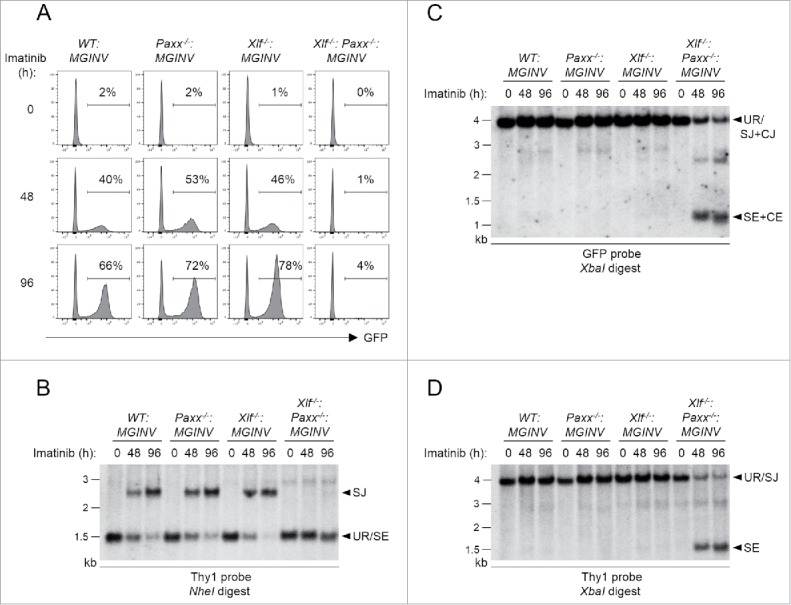
RAG DSB repair in PAXX- and XLF-deficient abl pre-(B)cells: (A) WT:MGINV, Xlf−/−:MGINV, Paxx−/−:MGINV and Xlf−/−:Paxx−/−:MGINV abl pre-B cells were treated with imatinib for 0, 48, or 96 hours (h). The percentage of GFP-positive cells is indicated. (B-D) Southern blot analyses of NheI-digested (B) or XbaI-digested (C, D) genomic DNA isolated from imatinib-treated abl pre-B cells in (A) and hybridized with the Thy1 (B, D) or GFP (C) probe. Hybridizing bands for different pMGINV rearrangements are indicated as described in Fig. 1A. Molecular weight markers are indicated (kb).
We next examined abl pre-B cells deficient in both XLF and PAXX (Xlf−/−:Paxx−/−:MGINV) (Fig. 3 and Fig. S2). In striking contrast to Paxx−/−:MGINV and Xlf−/−:MGINV abl pre-B cells, Xlf−/−:Paxx−/−:MGINV abl pre-B cells exhibit a dramatic defect in V(D)J recombination, with very few cells expressing GFP after RAG induction with imatinib (Fig. 3A). This is due to a block in the repair of RAG DSBs, as evidenced by Southern blot analyses that reveal very little SJ formation and an accumulation of unrepaired CEs and SEs similar in magnitude to that observed in LigIV−/−:MGINV abl pre-B cells (Fig. 3B-D, Fig. 1D, E and Fig. S2). We conclude that the combined deficiency in XLF and PAXX leads to a defect in NHEJ of similar severity to that observed in DNA Ligase IV-deficient abl pre-B cells.
Defective genotoxic DSB repair in Paxx−/−:Xlf−/− abl pre-B cells
In non-lymphoid cells, PAXX and XLF are both required for the repair of DSBs generated by genotoxic agents.11-16, 28-30 To assess the requirement for PAXX and XLF in repairing genotoxic DSBs in lymphocytes, we monitored nuclear foci of phosphorylated H2AX (γH2AX) in G1-arrested WT:MGINV, Xlf−/−:MGINV, Paxx−/−:MGINV, Xlf−/−:Paxx−/−:MGINV and LigIV−/−:MGINV abl pre-B cells after treatment with bleocin, a radiomimetic agent (Fig. 4A and B). γH2AX forms when DNA damage response kinases, such as ATM and DNA-PKcs, phosphorylate H2AX in chromatin flanking DNA DSBs, and γH2AX is lost after DSB repair. After two hours of bleocin treatment, 35% to 45% of all cell lines examined had greater than 5 γH2AX foci (Fig. 4A and B). When cells were removed from bleocin treatment, most of the γH2AX foci resolved by 24 hours in WT:MGINV, Xlf−/−:MGINV and Paxx−/−:MGINV abl pre-B cells (Fig. 4A and B). However, as is observed in LigIV−/−:MGINV abl pre-B cells, which have a complete block in NHEJ, the percentage of Xlf−/−:Paxx−/−:MGINV cells with more than 5 γH2AX foci remains unchanged, indicative of persistent unrepaired DSBs (Fig. 4A and B). Similarly, Xlf−/−:Paxx−/−:MGINV cells were also unable to resolve γH2AX foci induced by etoposide, which induces DSBs through a mechanism distinct of bleocin (Fig. 4C and D). Thus, while there are no detectable defects in the repair of genotoxic DSBs in abl pre-B cells with isolated deficiencies in either XLF or PAXX, combined deficiency of XLF and PAXX in abl pre-B cells leads to a defect in the repair of genotoxic DSBs as severe as that observed in DNA Ligase IV-deficient abl pre-B cells.
Figure 4.
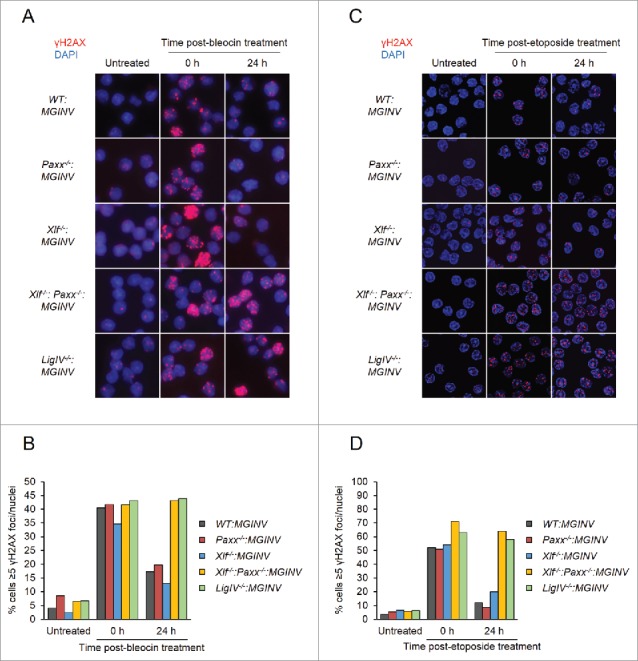
Genotoxic DSB repair in PAXX- and XLF-deficient abl pre-(B)cells: (A, C) Representative micrographs of γH2AX foci in WT:MGINV, Paxx−/−:MGINV, Xlf−/−:MGINV, Xlf−/−: Paxx−/−:MGINV, and LigIV−/−:MGINV abl pre-B cells that were untreated or treated with bleocin (A) or etoposide (C) and then allowed to recover in fresh media for 0 or 24 hours. (B, D) Quantitation of cells in (A) or (C) with ≥5 γH2AX foci per nucleus. At least 100 nuclei were scored for each condition. Data shown is representative of 3 replicate experiments.
The PAXX C-terminal domain is required for RAG DSB repair
PAXX is composed of a N-terminal globular head domain, a central coiled-coil region and a low complexity C-terminal domain (Fig. 5A). This structural arrangement is similar to that of XRCC4 and XLF.14,28-30,34 XLF and XRCC4 associate with one another through their N-terminal domains, and their C-terminal domains are known to interact with many DSB repair proteins.14,17,35-39 While there is no known function for the PAXX N-terminal head domain, mutations of this domain in human PAXX have been shown to effect its function in DSB repair in non-lymphoid cells.29 The C-terminal region of PAXX binds to Ku and mutations that disrupt this binding also effect the function of human PAXX in NHEJ.28,29 To determine whether these 2 domains are also critical for the repair of RAG DSBs in lymphocytes, we ectopically expressed wild type or mutant PAXX proteins in Xlf−/−:Paxx−/−:MGINV abl pre-B cells (Fig. 5A and B). Expression of wild type PAXX, which interacts with Ku70 (Fig. 5C), rescued the repair of RAG DSBs to nearly normal levels in Xlf−/−:Paxx−/−:MGINV abl pre-B cells (Fig. 5D-F). In contrast, PAXXVF, in which the evolutionarily conserved valine and phenylalanine in the C-terminal Ku-binding motif have been mutated to alanine (V200A and F202A), failed to bind Ku70 (Fig. 5A and C) and was unable to rescue V(D)J recombination (Fig. 5D–F). PAXXNmut, in which we mutated the 4 conserved leucine and isoleucine residues (I98D, L100D, L107D and L111D) in the N-terminal head domain previously mutated in human PAXX, was also unable to rescue V(D)J recombination in Xlf−/−:Paxx−/−:MGINV abl pre-B cells (Fig. 5A and D-F).29 However, the PAXXNmut mutations appear to destabilize the murine PAXX protein leading to low levels of PAXXNmut expression (Fig. 5B and C). Thus, it is not possible for us to make any conclusions about requirements for the N-terminus of PAXX in mediating NHEJ in murine lymphocytes. However, these findings clearly implicate a function for the Ku-binding C-terminal domain of PAXX in the NHEJ-mediated repair of RAG DSBs in murine lymphocytes.
Figure 5.

Function of PAXX mutants in NHEJ: (A) The 3 major domains of the PAXX protein: N-terminal head region, coiled coil domain (CC) and C-terminal domain (CTD). The mutations in the are PAXXNmut, and PAXXVF also specified. (B) Western blot analysis of lysates from Xlf−/−:Paxx−/−:MGINV abl pre-B cells reconstituted with Flag-HA-tagged PAXXWT, PAXXNmut, and PAXXVF. Antibodies to HA (Paxx) and GAPDH were used. (C) Western blot analysis of Ku70 association with Paxx and Paxx mutants in WT:MGINV abl pre-B cells. WT:MGINV abl pre-B cells expressing Flag-HA-tagged PAXXWT, PAXXNmut, and PAXXVF were subjected to immunoprecipitation using anti-HA followed by western blotting with anti-HA (Paxx) or anti-Ku70. Input lysates are also shown. (D) Xlf−/−:Paxx−/−:MGINV abl pre-B cells expressing PAXXWT, PAXXNmut, and PAXXVF were treated with imatinib for 0, 48, or 96 hours (h). The percentage of GFP-positive cells is indicated. (E, F) Southern blot analyses of NheI-digested (E) and XbaI-digested (F) genomic DNA isolated from imatinib-treated abl pre-B cells in (D) and hybridized with the Thy1 probe. Hybridizing bands for different pMGINV rearrangements are shown as described in Fig. 1A. Molecular weight markers are indicated (kb).
Discussion
The XLF protein is required for normal NHEJ in non-lymphoid cells but is dispensable for NHEJ in lymphocytes. PAXX also functions in NHEJ in non-lymphoid cells, and here we show that, like XLF, PAXX is also dispensable for NHEJ in lymphoid cells. NHEJ-mediated repair of RAG DSBs and DNA DSBs generated by the radiomimetic agent bleocin is unperturbed in PAXX-deficient abl pre-B cells. However, the combined deficiency of XLF and PAXX causes a dramatic block in the NHEJ-mediated repair of both RAG and genotoxic DSBs in abl pre-B cells. The defect in NHEJ-mediated DSB repair in Xlf−/−:Paxx−/−:MGINV cells is as profound as the defect observed in DNA Ligase IV-deficient abl pre-B cells. During the revision of this manuscript 2 papers were published that also demonstrate that combined deficiency in PAXX and XLF leads to a significant block in the repair of RAG DSBs.40,41
In lymphoid cells, combined deficiency of XLF and several other DNA damage response factors also results in defects in NHEJ that are more significant than their isolated deficiencies. This includes the DNA damage response kinases ATM and DNA-PKcs, the histone variant H2AX, and the DNA repair protein 53BP1.23-26 These proteins could have redundant functions with XLF in NHEJ-mediated DSB repair, or perhaps XLF deficiency compromises NHEJ in lymphoid cells in a manner that is undetectable in the absence of additional, mechanistically distinct, defects. In this regard, both H2AX and 53BP1 function to protect broken DNA ends from being resected and forming single-strand DNA overhangs that would inhibit NHEJ.42-46 Thus, if XLF deficiency slows the kinetics of NHEJ, it is possible that the concomitant deficiency of either H2AX or 53BP1 could block repair by allowing broken DNA ends to be resected before they can be joined.23-25 Notably, signal and coding ends in abl pre-B cells deficient in XLF and PAXX do not appear to be resected, making it less likely that PAXX deficiency leads to a block in NHEJ due to a requirement for PAXX in protecting broken DNA ends.
PAXX may function in NHEJ in a way that is mechanistically distinct from XLF. However, XLF and PAXX have very similar structures, raising the possibility that they may have overlapping functions. PAXX may exist as homodimer, which could potentially form high-order filaments similar to those formed by XLF-XRCC4 heterodimers that have been implicated in tethering and stabilizing DNA ends prior to repair.28 In addition, PAXX and XLF also both interact with the Ku heterodimer through their C-terminal regions, and their localization to laser-induced damage sites depends on such interaction.17,28,29,47 PAXX exhibits more stable binding to Ku in vitro when Ku is bound to DNA.48 While XLF and PAXX show little or no DNA binding affinity themselves, they stimulate in vitro DNA ligation in a Ku-dependent manner, suggesting that they may act as a scaffold through interaction with Ku to stabilize the XRCC4-DNA Ligase IV complex.28,49 By extension, in vivo, they could both promote the stable assembly of similar sets of repair factors. In this regard, a recent study demonstrated that PAXX-deficient human cells exhibit a substantial defect in the recruitment of several repair proteins to DNA DSBs.28 Whether XLF deficiency leads to the same defect in the recruitment of these factors is not known, and it will be interesting to determine if the combined deficiency of XLF and PAXX results in a more severe defect in the recruitment of these factors to DNA DSBs.
Why is it that NHEJ is compromised in non-lymphoid cells with isolated deficiencies of PAXX or XLF, but in lymphoid cells, defects in NHEJ are observed only when there is a combined deficiency of these proteins? It is possible that while PAXX and XLF have generally overlapping activities in NHEJ, they may also have unique functions in non-lymphoid cells that are not required for NHEJ in lymphoid cells. PAXX and XLF could exist in a complex with each other and several other key NHEJ proteins. The components of this complex could vary qualitatively or quantitatively in a way that leads to independent requirements for PAXX and XLF during NHEJ in different tissues.
Materials and methods
Generation of pMGINV
To generate pMGINV (pMSCV-RSS-GFP-INV), the murine Jk5 RS was cloned into the ClaI site of pBluescript, and the Vk8 RS was cloned into the resulting vector between the EcoRV and NotI sites. The ClaI and NotI sites were destroyed. The RSs were oriented as direct repeats, so that rearrangement between them occurs by inversion. The GFP cDNA was then blunt cloned into the EcoRV site between the 2 RSs. The RS-GFP cassette was removed from pBluescript using SalI and SacII, then blunt cloned into a BglII site upstream of IRES-Thy1.2 in pMSCV-IRES-Thy1.2, with the GFP cDNA in the antisense orientation. The BglII site was recreated upstream of the RS-GFP cassette, but was destroyed downstream, leaving a unique BglII site.
Cell line generation and cell culture
Wild type and Xlf−/− abl pre-B cells were created as previously described.8,12 Briefly, bone marrow cells were harvested from 3–5 week-old mice harboring the Eμ-Bcl2 transgene and infected with the pMSCV-v-abl retrovirus to generate stable v-abl-transformed pre-B cell lines. These cells were sequentially transduced with pMGINV and pCW-Cas9 (Plasmid #50661, Addgene) and selected for Thy1.2 expression and puromycin resistance before sub-cloning by limiting dilution. Clones were treated with 2 µg/mL doxycycline for 2 d and assayed for Cas9 induction by western blotting. WT:MGINV and Xlf−/−:MGINV abl pre-B cells with highly inducible Cas9 expression were further transduced with a lentiviral vector pKLV (Plasmid #50946, Addgene) containing a gRNA that targets the mouse Paxx coding sequence (5′- AGATATCCATTCCCGGTTC-3′), sorted for BFP expression, and treated with doxycycline for a week before sub-cloning by limiting dilution. Paxx−/−:MGINV and Xlf−/−:Paxx−/−:MGINV cells were screened by western blotting. All experiments were repeated in 2 knockout cell lines. Abl pre-B cells were cultured in media with 3 µM imatinib (Novartis) at a density of 106 cells/mL to induce G1 arrest.
Mutated Paxx alleles were amplified with primers PAXX 5′ intron 2S (GTGAGTA ACAGTGCTGGGGATA) and PAXX 3′ intron 3 AS (CTAAGGAGGGAGATGTGT GTTA), with the exception of PCR reaction for Xlf−/−:Paxx−/−:MGINV clone 5, where the PAXX 3′ intron AS primer was replaced with PAXX 3′ NotI. PCR products were cloned to pCRII-TOPO vector and sequences determined by M13 (Reverse) primer.
Protein analyses
Western blotting was performed as previously described.42 The primary antibodies used for western blotting and immunoprecipitation were: mouse anti-Flag (F3165, Sigma), rabbit anti-XLF (A300-729A, Bethyl Laboratories), rabbit anti-PAXX (ab126353, Abcam), rabbit anti-HA [Y-11] (sc805, Santa Cruz), rabbit anti-cytoskeletal actin (A300-491A, Bethyl Laboratories), and mouse anti-GAPDH (G8795, Sigma). Immunoprecipitation (IP) was performed as described in Ciccia et al.50 Cell lysate extracted from 30 million G1-arrested wild type abl pre-B cells transduced with pOZ-FH-PaxxWT, PaxxNmut, or PaxxVF were used for each IP with 5 μg of mouse anti-HA IgG.
Southern blot analysis
Southern blotting were performed as previously described.8 The Thy1 probe is a ∼800-bp Thy1.1 cDNA fragment, while the GFP probe is a ∼700-bp GFP cDNA fragment.
Generation of Paxx mutants
The wild type Paxx coding sequence was amplified from cDNA clone BC029124 with primers PAXX 5′ XhoI (5′-GCCCTCG AGATGGCTCCTCCGTTGTTGTC-3′) and PAXX 3′ NotI (5′-GCCGCGGCCGCT CAGGTCTCATCAAAGTCTA-3′) To generate PAXXNmut, primers PAXX 5′ XhoI and PAXX_NmutAS (5′-GGAGTCGTCAAAGGCGTCGGCAGGGGTATCCCCTG AATCGGTATCCAATGC-3′) were used to generate the 5′ Nmut fragment. Primers PAXX 3′ NotI and PAXX_ NumtS (5′-GCATTGGATACCGATTCAGGGG ATACCCCTGCCGACGCCTTTG ACGACTCC-3′) were used to generate the 3′ Nmut fragment. The mutated sequences for PAXXNmut and PAXXVF (below) are highlighted in bold. Overlapping PCR was used to convert the 2 fragments to full-length PAXXNmut. To generate PAXXVF mutant, primers PAXX 5′ XhoI and PAXX_V,F-A_3′ NotI (5′-GCCGCGGCCGCTCAGGTCTCATCAGCGTCTGCAC CAGCAGCTGGT-3′) were used in PCR to incorporate mutated sequences in the Paxx coding sequences (bold fonts in the primer PAXX_V,F-A_3′ NotI). The resulting DNA fragments PaxxWT, PaxxNmut and PaxxVF were cloned into XhoI and NotI sites of the retroviral vector pOZ-Flag-HA-N (pOZ-FH-N).
Nuclear foci
Abl pre-B cells were cultured in media with 3 µM imatinib for 2 d to induce G1 arrest and then treated with 10 µg/mL bleocin (203408, Millipore) for 2 hours, or 2 ug/mL etoposide (E1383, Sigma) for 1 hour, after which the cells were washed in PBS and rested in fresh imatinib media for 24 hours. For immunofluorescent staining of γH2AX, abl pre-B cells were plated onto glass coverslips coated with Cell-Tak (354240, Corning), fixed in 4% paraformaldehyde for 10 minutes, permeabilized with 0.1% Triton X-100 for 5 minutes, blocked with 3% BSA/PBS (w/v) for 1 hour at room temperature, and incubated with mouse anti-γH2AX (05–636 Millipore) at a 1:1,000 dilution overnight at 4°C. The coverslips were then washed in PBS, incubated with donkey anti-mouse IgG (H + L) Alexa Fluor 594 at 1:1,000 dilution (A-21203, ThermoFisher) for 1 hour at room temperature, washed again in PBS, and mounted onto microscope slides (12-550-343, Fisher Scientific) in ProLong Gold Anti-Fade Mountant with DAPI (P36931, ThermoFisher). Images were acquired at 60X magnification using an Olympus BX53 fluorescence microscope.
Supplementary Material
Abbreviations
- ATM
ataxia–telangiectasia mutated
- CE
coding end
- CJ
coding join
- CRISPR
clustered regularly interspaced short palindromic repeats
- DNAPKcs
DNA-dependent protein kinase catalytic subunit
- DSB
double-strand breaks
- HR
homologous recombination
- NHEJ
non-homologous end joining
- PAXX
paralog of XLF and XRCC4
- pMGINV
pMSCV-RSS-GFP-INV
- RAG
recombination activation gene
- RS
recombination signal sequence
- SE
signal end
- SJ
signal join
- V(D)J
variable diversity and joining
- XLF
XRCC4-like factor
Disclosure of potential conflicts of interest
The authors declare no conflicts of interest..
Acknowledgments
We thank Drs. John Petrini and Wytse Bruinsma for help carrying out foci experiments, and Dr. Frederick Alt for XLF+/− mice and Mr. Ryan Irwin for managing mouse colonies.
Funding
This work was in part supported by NIAID grant 7RO1AI074953 and NCI grant 5R01CA177759 to BPS, and NCI grant 5F32CA183271 to BC.
References
- [1].Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010; 79:181-211; http://dx.doi.org/ 10.1146/annurev.biochem.052308.093131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kumar V, Alt FW, Oksenych V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst) 2014; 16:11-22; PMID:24674624; http://dx.doi.org/ 10.1016/j.dnarep.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet 2013; 47:433-55; PMID:24050180; http://dx.doi.org/ 10.1146/annurev-genet-110711-155540 [DOI] [PubMed] [Google Scholar]
- [5].Tonegawa S. Somatic generation of antibody diversity. Nature 1983; 302:575-81; PMID:25790189; http://dx.doi.org/ 10.1038/302575a0 [DOI] [PubMed] [Google Scholar]
- [6].Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol 2000; 18:495-527; http://dx.doi.org/ 10.1146/annurev.immunol.18.1.495 [DOI] [PubMed] [Google Scholar]
- [7].Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol 2012; 30:175-202; http://dx.doi.org/ 10.1146/annurev-immunol-030409-101320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al.. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006; 442:466-70; http://dx.doi.org/ 10.1038/nature04866 [DOI] [PubMed] [Google Scholar]
- [9].Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, Mahowald GK, Omi KQ, Walker LM, Bednarski JJ, McKinnon PJ, et al.. Ataxia telangiectasia mutated (Atm) and DNA-PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci U S A 2011; 108:2022-7; PMID:21245316; http://dx.doi.org/ 10.1073/pnas.1013295108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zha S, Jiang W, Fujiwara Y, Patel H, Goff PH, Brush JW, Dubois RL, Alt FW. Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proc Natl Acad Sci U S A 2011; 108:2028-33; PMID:21245310; http://dx.doi.org/ 10.1073/pnas.1019293108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zha S, Alt FW, Cheng HL, Brush JW, Li G. Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci U S A 2007; 104:4518-23; http://dx.doi.org/ 10.1073/pnas.0611734104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li G, Alt FW, Cheng HL, Brush JW, Goff PH, Murphy MM, Franco S, Zhang Y, Zha S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol Cell 2008; 31:631-40; http://dx.doi.org/ 10.1016/j.molcel.2008.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fattah FJ, Kweon J, Wang Y, Lee EH, Kan Y, Lichter N, Weisensel N, Hendrickson EA. A role for XLF in DNA repair and recombination in human somatic cells. DNA Repair (Amst) 2014; 15:39-53; http://dx.doi.org/ 10.1016/j.dnarep.2013.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006; 124:301-13; http://dx.doi.org/ 10.1016/j.cell.2005.12.031 [DOI] [PubMed] [Google Scholar]
- [15].Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, et al.. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 2006; 124:287-99; http://dx.doi.org/ 10.1016/j.cell.2005.12.030 [DOI] [PubMed] [Google Scholar]
- [16].Vera G, Rivera-Munoz P, Abramowski V, Malivert L, Lim A, Bole-Feysot C, Martin C, Florkin B, Latour S, Revy P, et al.. Cernunnos deficiency reduces thymocyte life span and alters the T cell repertoire in mice and humans. Mol Cell Biol 2013; 33:701-11; http://dx.doi.org/ 10.1128/MCB.01057-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yano K, Morotomi-Yano K, Wang SY, Uematsu N, Lee KJ, Asaithamby A, Weterings E, Chen DJ. Ku recruits XLF to DNA double-strand breaks. EMBO Rep 2008; 9:91-6; http://dx.doi.org/ 10.1038/sj.embor.7401137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Mol Cell 2007; 28:1093-101; http://dx.doi.org/ 10.1016/j.molcel.2007.10.024 [DOI] [PubMed] [Google Scholar]
- [19].Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, et al.. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem 2011; 286:32638-50; http://dx.doi.org/ 10.1074/jbc.M111.272641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, Faure G, Marquez JA, Pietrement O, Guerois R, Callebaut I, et al.. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc Natl Acad Sci U S A 2011; 108:12663-8; PMID:21768349; http://dx.doi.org/ 10.1073/pnas.1100758108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet 2011; 45:247-71; PMID:21910633; http://dx.doi.org/ 10.1146/annurev-genet-110410-132435 [DOI] [PubMed] [Google Scholar]
- [22].Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, Melo AJ, Zhang H, Normanno D, Modesti M, Peterman EJ, et al.. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature 2016; 535(7613):566-9; PMID:27437582. [DOI] [PubMed] [Google Scholar]
- [23].Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, Patel H, Su A, Guo C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A 2012; 109:2455-60; http://dx.doi.org/ 10.1073/pnas.1121458109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, et al.. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011; 469:250-4; http://dx.doi.org/ 10.1038/nature09604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z, Zha S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci U S A 2012; 109:3903-8; http://dx.doi.org/ 10.1073/pnas.1120160109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Oksenych V, Kumar V, Liu X, Guo C, Schwer B, Zha S, Alt FW. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc Natl Acad Sci U S A 2013; 110:2234-9; PMID:23345432; http://dx.doi.org/ 10.1073/pnas.1222573110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lescale C, Abramowski V, Bedora-Faure M, Murigneux V, Vera G, Roth DB, Revy P, de Villartay JP, Deriano L. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat Commun 2016; 7:10529; http://dx.doi.org/ 10.1038/ncomms10529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, et al.. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015; 347:185-8; http://dx.doi.org/ 10.1126/science.1261971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Ning S, Yan Z, Li W, Wang Q, et al.. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun 2015; 6:6233; http://dx.doi.org/ 10.1038/ncomms7233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Craxton A, Somers J, Munnur D, Jukes-Jones R, Cain K, Malewicz M. XLS (c9orf142) is a new component of mammalian DNA double-stranded break repair. Cell Death Differ 2015; 22:890-7; http://dx.doi.org/ 10.1038/cdd.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science 2013; 339:823-6; http://dx.doi.org/ 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014; 343:80-4; http://dx.doi.org/ 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 2014; 32:267-73; http://dx.doi.org/ 10.1038/nbt.2800 [DOI] [PubMed] [Google Scholar]
- [34].Li Y, Chirgadze DY, Bolanos-Garcia VM, Sibanda BL, Davies OR, Ahnesorg P, Jackson SP, Blundell TL. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. EMBO J 2008; 27:290-300; PMID:18046455; http://dx.doi.org/ 10.1038/sj.emboj.7601942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Macrae CJ, McCulloch RD, Ylanko J, Durocher D, Koch CA. APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst) 2008; 7:292-302; PMID:18077224; http://dx.doi.org/ 10.1016/j.dnarep.2007.10.008 [DOI] [PubMed] [Google Scholar]
- [36].Grundy GJ, Rulten SL, Zeng Z, Arribas-Bosacoma R, Iles N, Manley K, Oliver A, Caldecott KW. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J 2013; 32:112-25; http://dx.doi.org/ 10.1038/emboj.2012.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hsu HL, Yannone SM, Chen DJ. Defining interactions between DNA-PK and ligase IV/XRCC4. DNA Repair (Amst) 2002; 1:225-35; http://dx.doi.org/ 10.1016/S1568-7864(01)00018-0 [DOI] [PubMed] [Google Scholar]
- [38].Mani RS, Yu Y, Fang S, Lu M, Fanta M, Zolner AE, Tahbaz N, Ramsden DA, Litchfield DW, Lees-Miller SP, et al.. Dual modes of interaction between XRCC4 and polynucleotide kinase/phosphatase: implications for nonhomologous end joining. J Biol Chem 2010; 285:37619-29; PMID:20852255; http://dx.doi.org/ 10.1074/jbc.M109.058719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P, Brenner C, Moreira MC, Taylor AM, Caldecott KW. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst) 2004; 3:1493-502; PMID:15380105; http://dx.doi.org/ 10.1016/j.dnarep.2004.06.017 [DOI] [PubMed] [Google Scholar]
- [40].Kumar V, Alt FW, Frock RL. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc Natl Acad Sci U S A 2016; 113:10619-24; PMID: 27601633; http://dx.doi.org/ 10.1073/pnas.1611882113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lescale C, Lenden Hasse H, Blackford AN, Balmus G, Bianchi JJ, Yu W, Bacoccina L, Jarade A, Clouin C, Sivapalan R, et al.. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep 2016; 16:2967-79; http://dx.doi.org/ 10.1016/j.celrep.2016.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tubbs AT, Dorsett Y, Chan E, Helmink B, Lee BS, Hung P, George R, Bredemeyer AL, Mittal A, Pappu RV, et al.. KAP-1 promotes resection of broken DNA ends not protected by gamma-H2AX and 53BP1 in G(1)-phase lymphocytes. Mol Cell Biol 2014; 34:2811-21; http://dx.doi.org/ 10.1128/MCB.00441-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, et al.. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature 2011; 469:245-9; PMID:21160476; http://dx.doi.org/ 10.1038/nature09585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al.. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 2011; 42:319-29; http://dx.doi.org/ 10.1016/j.molcel.2011.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al.. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; http://dx.doi.org/ 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 2010; 207:855-65; http://dx.doi.org/ 10.1084/jem.20100244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yano K, Morotomi-Yano K, Lee KJ, Chen DJ. Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS Lett 2011; 585:841-6; http://dx.doi.org/ 10.1016/j.febslet.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tadi SK, Tellier-Lebegue C, Nemoz C, Drevet P, Audebert S, Roy S, Meek K, Charbonnier JB, Modesti M. PAXX Is an Accessory c-NHEJ Factor that Associates with Ku70 and Has Overlapping Functions with XLF. Cell Rep 2016; 17:541-55; PMID: 27705800; http://dx.doi.org/ 10.1016/j.celrep.2016.09.026 [DOI] [PubMed] [Google Scholar]
- [49].Tsai CJ, Chu G. Cooperative assembly of a protein-DNA filament for nonhomologous end joining. J Biol Chem 2013; 288:18110-20; PMID: 23620595; http://dx.doi.org/ 10.1074/jbc.M113.464115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev 2009; 23:2415-25; PMID: 19793862; http://dx.doi.org/ 10.1101/gad.1832309 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.