

Abstract
Regulators of G protein signalling (RGS) proteins are celebrating the 20th anniversary of their discovery. The unveiling of this new family of negative regulators of G protein signalling in the mid‐1990s solved a persistent conundrum in the G protein signalling field, in which the rate of deactivation of signalling cascades in vivo could not be replicated in exogenous systems. Since then, there has been tremendous advancement in the knowledge of RGS protein structure, function, regulation and their role as novel drug targets. RGS proteins play an important modulatory role through their GTPase‐activating protein (GAP) activity at active, GTP‐bound Gα subunits of heterotrimeric G proteins. They also possess many non‐canonical functions not related to G protein signalling. Here, an update on the status of RGS proteins as drug targets is provided, highlighting advances that have led to the inclusion of RGS proteins in the IUPHAR/BPS Guide to PHARMACOLOGY database of drug targets.
Abbreviations
- DEP
Disheveled Egl‐10 Pleckstrin
- GAP
GTPase‐activating protein
- GGL
G protein γ‐like
- PPI
protein–protein interaction
- RGS
regulator of G protein signalling
- R7BP
R7 binding protein
- R9AP
RGS9 associated protein
Tables of Links
| TARGETS | |
|---|---|
| R4 family | R7 family |
| RGS1 | RGS6 |
| RGS2 | RGS7 |
| RGS3 | RGS9 |
| RGS4 | RGS11 |
| RGS5 | R12 family |
| RGS8 | RGS10 |
| RGS13 | RGS12 |
| RGS16 | RGS14 |
| RGS18 | RZ family |
| RGS21 | RGS17 |
| RGS19 | |
| RGS20 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
G protein‐mediated signalling pathways have played a pivotal role in drug discovery and development for many decades. The large family of GPCRs or their downstream effectors are the target of 40% of clinically used drugs and thus represent a multi‐billion‐dollar industry (Wise et al., 2002). Interestingly, of the more than 300 non‐olfactory GPCRs known, only a fraction of them are targeted by drugs. Thus, there is a large untapped area of drug development still available. Moreover, many GPCR drugs are associated with low efficacy and/or side effects. More targeted therapies are therefore required, and as we learn more about the structure and function of GPCRs and their regulators, these goals will be achievable.
All biological signals are tightly regulated and for every on‐switch there is usually an off‐switch. GPCRs are activated by ligands, transmitting signalling information to Gα subunits of heterotrimeric G proteins by enhancing the exchange of GDP for GTP in the Gα nucleotide binding site, which results in the dissociation of Gα from Gβγ dimers and activation of both G protein components. Deactivation of G proteins does not occur by simple reversal of nucleotide exchange, but rather by an independently regulated GTPase activity, hydrolyzing GTP to GDP. Although Gα proteins possess an intrinsic ability to hydrolyze GTP, this process is very slow and cannot account for the transient nature of intracellular signalling cascades in vivo. Hence, additional kinetic mechanisms are required for the physiological timing of signals. One of the most critical of these kinetic mechanisms is mediated through regulator of G protein signalling (RGS) proteins, which have received increasing interest as novel drug targets in the past two decades. As a result, RGS proteins have now been added as the most recent addition to the International Union of Basic and Clinical Pharmacology/British Pharmacology Society (IUPHAR/BPS) Guide to PHARMACOLOGY database of drug targets (www.guidetopharmacology.org) (Alexander et al., 2015; Sjögren et al., 2016a).
RGS proteins all share a common RGS domain that directly interacts with active, GTP‐bound Gα subunits of heterotrimeric G proteins. RGS proteins stabilize the transition state for GTP hydrolysis on Gα (Berman et al., 1996a; Tesmer et al., 1997) and thus induce a conformational change in the Gα subunit that accelerates GTP hydrolysis, thereby effectively turning off signalling cascades mediated by GPCRs (Figure 1). To date, there have been many excellent reviews published on the structure and function of RGS proteins as well as on their role in drug discovery. The purpose of this review is not to give a comprehensive summary of the RGS literature, but rather to serve as a guide to current advances and ways of thinking in the field of RGS protein drug discovery. For more extensive reviews on RGS proteins and their potential as therapeutic targets, see for example, Ross and Wilkie, 2000; Zhong and Neubig, 2001; Hollinger and Hepler, 2002; Druey, 2003; Cho et al., 2004; Siderovski and Willard, 2005; Blazer and Neubig, 2009; Gu et al., 2009; Sjögren et al., 2010; Sjögren, 2011; Zhang and Mende, 2011.
Figure 1.
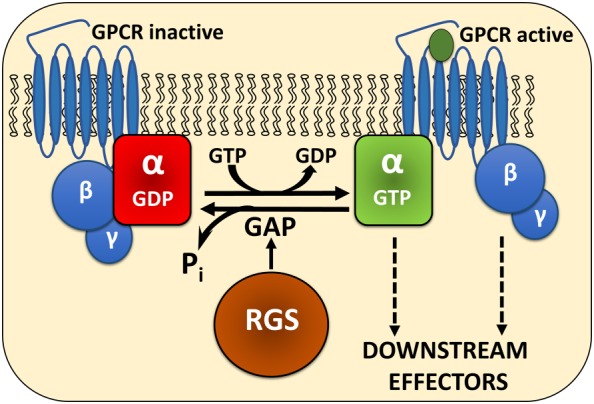
The canonical action of RGS proteins. In its inactive state, Gα is bound to GDP. Upon receptor activation, GDP is exchanged for GTP, Gα dissociates from Gβγ and both can mediate signalling cascades. RGS proteins bind to the transition state of GTP‐bound Gα, accelerate GTP hydrolysis and effectively reduce the amplitude and duration of GPCR signalling.
RGS proteins – a brief history
The existence and characterization of negative regulators of G protein activity was almost simultaneously demonstrated in Saccharomyces cerevisiae (S. cerevisiae), Caenorhabditis elegans (C. elegans) and mammalian cells in key publications in the mid‐1990s, and the specific identification of the RGS proteins in each of these systems followed soon after (Siderovski et al., 1994; Dohlman et al., 1995; Wu et al., 1995; De Vries et al., 1996; Druey et al., 1996; Koelle and Horvitz, 1996; Siderovski et al., 1996; Watson et al., 1996; Koelle, 1997). Within the span of a few short years, a new family of G protein regulators was established as a critical piece of the G protein regulation cycle.
As early as 1982, a novel factor regulating pheromone sensitivity and G1 cell cycle arrest was identified in yeast (Chan and Otte, 1982a,b). This factor, Sst2, was subsequently identified as a negative regulator of the G protein Gpa1 in S. cerevisiae (Dohlman et al., 1995; 1996) and later demonstrated to be a GTPase‐activating protein (GAP) for the yeast G protein Gpa1 (Apanovitch et al., 1998). This feature is the hallmark of all RGS proteins, and this work established Sst2 and Gpa1 as the cognate G protein‐RGS pair in yeast.
Around the same time, Koelle and Horvitz (1996) demonstrated that loss‐of‐function mutations in the egl‐10 gene led to reduced egg‐laying behaviour and locomotion behaviour in C. elegans (Koelle and Horvitz, 1996). This effect was the opposite of loss‐of‐function mutations in the C. elegans G protein GOA‐1, and the authors postulated that the two proteins might function in a common signalling pathway, one with positive and one with negative regulation. They subsequently demonstrated that EGL‐10 shows high sequence similarity to the yeast protein Sst2 as well as several mammalian proteins that we now know as RGS proteins, including RGS1 (formally known as BL34 and 1R20), RGS2 (formally known as G0S8) and, most closely related, RGS7 (Koelle and Horvitz, 1996).
Finally, the Gilman lab described the first biochemical function of mammalian RGS proteins, demonstrating that the proteins RGS4 and GAIP (now known as RGS19) could serve as GAPs at certain Gα subtypes in vitro, including all members of the Gαi subfamily (Berman et al., 1996b). The following year, in 1997, Doupnik et al. demonstrated that heterologous expression of RGS4 in Xenopus oocytes could replicate the temporal characteristics of G protein‐coupled inward rectifying potassium channel deactivation following GPCR activation observed in endogenous systems, such as atrial myocytes (Doupnik et al., 1997). This demonstrated functionality of mammalian RGS proteins in a biologically relevant setting and established that RGS proteins account for physiological GTPase kinetics.
It is now recognized that RGS proteins make up a large family of proteins containing a common ~120 residue RGS domain, responsible for their GAP activity towards Gα subunits of heterotrimeric G proteins. The 20 classical RGS proteins are divided into four subfamilies (R4, R7, R12 and RZ) based on sequence and domain homology (Figure 2). In addition, several other families of proteins have been identified containing an RGS homology domain. These include GPCR kinases (GRK1–7), ankyrin, AKAPs, Rho‐GEFs, and sorting nexin proteins (SNX13, 14 and 25) (Siderovski and Willard, 2005). For the purpose of this review, the focus will be limited to the 20 classical RGS proteins.
Figure 2.
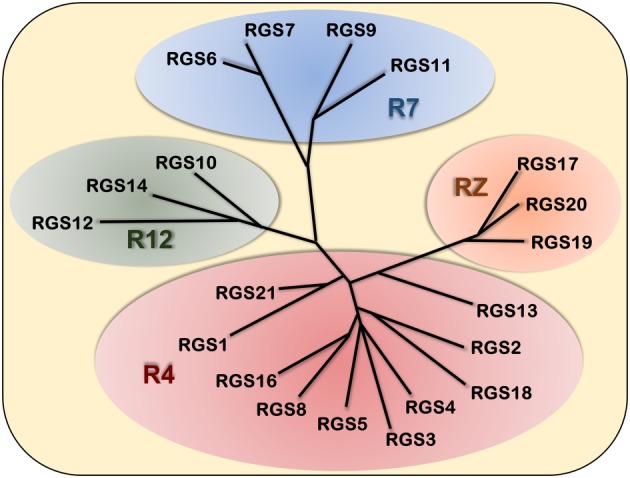
Classification of the 20 canonical human RGS proteins. The classical RGS proteins are divided into four families based on sequence and domain homology. The largest, the R4 family, contains RGS1, 2, 3, 4, 5, 8, 13, 16, 18 and 21. The R7 family consists of RGS6, 7, 9 and 11. The R12 family members are RGS10, 12 and 14. Finally the RZ family consists of RGS17, 19 and 20. This unrooted dendrogram was created using ClustalW alignment of the full‐length RGS protein sequences and Dendroscope (Huson and Scornavacca, 2012) was used for visualization.
To understand the importance of RGS proteins in vivo, numerous genetic models have been produced, such as global knockouts, as well as conditional and/or tissue specific knockout or transgenic models. While some of these display a clear phenotype, as will be exemplified in later sections, some RGS protein knockout models have produced little to no effect, most likely due to redundancy where one RGS protein can substitute for another. Early work from the Dohlman lab had identified a point mutation in the yeast Gα protein Gpa1 that made it insensitive to RGS protein action (DiBello et al., 1998). Subsequently, the corresponding mutations were identified in mammalian Gαo (G184S) and Gαi1 (G183S) (Lan et al., 1998). This glycine to serine mutation prevents binding of all RGS proteins to Gα, thus enabling studies of global disabling of RGS protein GAP activity. Transgenic animal models of these mutated Gα subunits have since been created, and have proven valuable tools to study the effects of global RGS protein action towards specific Gα subunits. In addition, they have provided key insights into the role of individual Gα subtypes. The phenotypes of these mice are extensively discussed in a recent review (Neubig, 2015) and reveal major roles for RGS proteins in regulating key physiological functions.
Why target RGS proteins in drug discovery?
In the past two decades, RGS proteins have received increasing interest as potential drug targets in numerous therapeutic areas, including cardiovascular disease, multiple CNS disorders and several types of cancer (Mittmann et al., 2002; Riddle et al., 2005; Hurst and Hooks, 2009; Sjögren et al., 2010). While GPCR signalling has been a major focus in drug development, traditional GPCR agonists and antagonists are often associated with side effects due to widespread expression of many receptors and lack of receptor selectivity of drugs. Furthermore, many receptors can couple to more than one Gα subunit as well as initiate β‐arrestin‐mediated signalling pathways, resulting in several different signalling pathways being activated by the same receptor. This raises the possibility that while activation of one pathway may lead to a desired therapeutic effect, another might result in an unwanted side effect. It is therefore clear that drugs or drug combinations that are able to fine‐tune cellular responses by selectively modulating a subset of downstream pathways are desirable over a simple receptor on/off switch. At the level of the receptor, great progress is being made in the field of biased signalling, as well as the development of positive and negative modulators of receptor activity (see e.g. Kenakin, 2012; Khoury et al., 2014; Shukla et al., 2014; Bertekap et al., 2015; Bisignano et al., 2015; Burford et al., 2015). There is also significant potential for RGS proteins to serve a similar signalling pathway‐specific role at the level of the G protein in order to improve the selectivity and efficacy of GPCR‐targeted approaches.
Many RGS proteins have selectivity towards different Gα subtypes and thus can affect one pathway over another. In the early days of RGS proteins, the Gilman lab demonstrated that RGS4 has high affinity for all members of the Gαi/o subtypes, while showing lower affinity for Gαq and no activity towards Gαs and Gα12 (Berman et al., 1996a). RGS2, on the other hand, was demonstrated to be selective for Gαq over all other Gα subtypes tested (Heximer et al., 1997), although later studies demonstrated that RGS2 can also inhibit Gαi‐mediated signalling in vivo (Chakir et al., 2011). Furthermore, all members of the R7 family of RGS proteins (RGS6, 7, 9 and 11) are selective for Gαi/o proteins (Anderson et al., 2009) while other RGS proteins are more promiscuous in their selectivity, for example, RGS1, RGS8, RGS13, RGS16 (Johnson and Druey, 2002; Soundararajan et al., 2008). To date, no RGS proteins have been shown to have GAP activity towards Gαs, although RGS2 has been demonstrated to associate with Gαs in cells (Roy et al., 2006). These differences in G protein selectivity among RGS proteins could enable signalling pathway‐specific regulation of GPCRs in drug development.
A second opportunity for enhanced GPCR selectivity via RGS regulation is based on expression patterns. The tissue distribution of RGS proteins is often more discrete than the G proteins they regulate, and thus, an RGS protein modulator would enable tissue‐specific regulation of GPCR signalling. One example is RGS9‐2, which is specifically enriched in striatum (Mancuso et al., 2009) [an alternative isoform, RGS9‐1, is exclusively expressed in photoreceptor cells in the retina (He et al., 1998)]. This selective distribution matches the critical site of L‐DOPA‐induced dyskinesia, and the Gαi/o‐protein selectivity of RGS9 GAP activity corresponds to the Gαi/o coupling of the dopamine D2 receptor, which is critical in mediating L‐DOPA effects in the striatum (Gold et al., 2007; Blundell et al., 2008). The broad distribution of D2 receptor expression limits the use of D2 receptor targeted approaches to regulate striatal signalling pathways. However, the overlapping G protein selectivity and expression of RGS9‐2 and D2 receptors in striatum suggests that RGS9‐2 may be a useful complementary target for the treatment of involuntary movements following L‐DOPA treatment in Parkinson's disease (PD). In this approach, an RGS modulator would result in selective regulation of GPCR signalling only in locations where the RGS protein is expressed. The effect of a receptor‐targeted drug would then be selectively altered in these tissues, enabling the use of lower doses for an effective therapeutic effect (see Blazer and Neubig, 2009 for an expanded discussion on this topic). Thus, dual targeting of RGS proteins and GPCRs can add selectivity to GPCR targeting strategies by virtue of their ability to modulate a subset of downstream pathways and their unique distribution in tissues.
Additional domains and non‐canonical functions of RGS proteins
Apart from the canonical GAP activity encoded by the common RGS domain, many RGS proteins also possess additional domains and functions that provide additional potential targets for drug discovery. In many cases, GAP‐independent domains and regulatory elements serve to target or regulate the RGS domain GAP functionality, while in some cases these additional domains possess signalling functionality of their own. Numerous non‐canonical functions have been demonstrated for RGS proteins; due to space limitations, only a few will be discussed here. For a more comprehensive review see Sethakorn et al., 2010.
Non‐canonical functions of RGS proteins are typically mediated through protein–protein interactions (PPI), many of which result from additional domains present in many RGS proteins. A classic example of domain‐mediated targeting is found in the R7 family of RGS proteins. The interaction between the R7 family members and binding partners Gβ5 and R7BP/R9AP are mediated by the G protein γ‐like (GGL), Disheveled, Egl‐10, pleckstrin (DEP) and DEP helical extension domains present in these proteins (Anderson et al., 2009). R7BP (for R7 binding protein) and R9AP (RGS9 associated protein, specifically in photoreceptor cells) are membrane tethered proteins that mediate targeting of R7 family RGS proteins to the plasma membrane, thereby enhancing proximity to the G protein target (Drenan et al., 2005; Grabowska et al., 2008). The functionality of all R7 family members is enhanced in the presence of R7BP (Drenan et al., 2006; Jayaraman et al., 2009). Gβ5 interaction with the GGL domain of R7 protein regulates protein stability and is described below. Additional interactions between the DEP domain and intracellular regions of GPCRs have also been demonstrated (Sandiford and Slepak, 2009) providing additional mechanisms whereby this family of RGS proteins can regulate GPCR signalling in a non‐canonical manner.
Another example of a GAP‐independent domain that mediates independent functionality is the Gαi/o‐Loco (GoLoco) motif present in the R12 family members RGS12 and 14 (Kimple et al., 2001; Siderovski and Willard, 2005). Like the RGS domain, the GoLoco motif binds Gα, but this interaction inhibits GTP exchange, thereby preventing G protein activation. It also blocks the association of Gα with Gβγ, potentially leading to prolonged Gβγ signalling. This enables dual regulation of Gα by RGS12 and RGS14 (Traver et al., 2004). RGS12 and RGS14 are two of the largest classical RGS proteins with additional domains apart from the RGS and GoLoco domains. Notably, the Ras binding domain(s) present in these proteins has been demonstrated to integrate GPCR and Ras/MAPK signalling pathways (Shu et al., 2010; Zhao et al., 2013; Brown et al., 2015). Furthermore, through an additional domain, the phosphotyrosine binding (PTB) domain, RGS12 can interact with, and modulate the activity of, N‐type calcium channels in a phosphorylation‐dependent manner (Schiff et al., 2000). Altogether, these examples shed light on the important role additional protein domains can play in mediating non‐canonical functions of multidomain RGS proteins.
The presence of additional domains is not always necessary for an RGS protein to exert GAP‐independent PPIs and non‐canonical functions. The R4 family member RGS2 is one of the smallest RGS proteins with only a small N‐ and C‐terminus flanking the RGS domain. Despite this, several functions have been attributed to RGS2 that are not related to its GAP activity. Firstly, RGS2 can suppress Gαs signalling through direct interactions with adenylate cyclase I, II and VI (Salim et al., 2003; Roy et al., 2006). Secondly, RGS2 suppresses general protein translation through interaction with eukaryotic initiation factor 2Bε (eIF2Bε) (Nguyen et al., 2009; Chidiac et al., 2014). Thirdly, RGS2 has been shown to interact directly with several GPCRs, including the α1A adrenoceptor (Hague et al., 2005) and the M1 muscarinic receptor (Bernstein et al., 2004b). Another member of the R4 family, RGS13, can bind directly to the transcription factor CREB and act as a transcriptional repressor (Xie et al., 2008). Like RGS2, RGS13 does not contain any additional protein domains.
Together, these examples of mechanisms of regulation and non‐canonical functions described above and elsewhere (Sethakorn et al., 2010) reveal the complexity of RGS protein biology and contribute to their diverse potential as drug targets.
Regulation of function, localization and expression
Mechanisms regulating RGS protein levels and function range from posttranslational modifications, such as phosphorylation and palmitoylation, to control of degradation, transcription and subcellular localization. Correct function of RGS proteins requires rapid spatial and temporal regulation. Post‐translational modifications, such as phosphorylation, can either enhance or inhibit RGS protein function in a rapid, cell state‐specific manner. Phosphorylation of RGS14 by PKA at Thr494, adjacent to the GoLoco motif, enhances its guanine nucleotide dissociation inhibitory activity towards Gαi, while having no effect on GAP activity (Hollinger et al., 2003).In contrast, GPCR ligand‐dependent phosphorylation of RGS16 at Ser53 has been shown to inhibit GAP activity (Chen et al., 2001), while Src‐mediated phosphorylation at Tyr168 protects RGS16 from degradation leading to enhanced GAP activity in cells (Derrien and Druey, 2001; Derrien et al., 2003). Another member of the R4 family, RGS2, was demonstrated to be phosphorylated by PKC, which inhibited GAP activity of RGS2 in vitro (Cunningham et al., 2001). In contrast, we recently demonstrated that activation of PKC enhances RGS2 protein levels, leading to increased RGS2‐mediated suppression of GPCR signalling in HEK‐293 cells (Raveh et al., 2014). Although it is not clear whether this effect is due to direct phosphorylation of RGS2 by PKC, it provides a clear example of the importance of the experimental context in which RGS protein function is studied and the complexity of RGS protein biology.
Canonical RGS domain GAP functionality requires localization to the plasma membrane, the site of action of G proteins. For several RGS proteins, palmitoylation of the N‐terminus provides this targeting mechanism. Both RGS4 and RGS16 are palmitoylated at their amino‐terminal, anchoring them to the plasma membrane and the GPCR‐G protein complex (Chen et al., 1999; Druey et al., 1999; Hiol et al., 2003; Bastin et al., 2012). Palmitoylation within the RGS domain of these and other RGS proteins can also modulate GAP activity (Tu et al., 1999; Castro‐Fernandez et al., 2002; Hiol et al., 2003; Osterhout et al., 2003; Jones, 2004; Bernstein et al., 2004a; Ni et al., 2006). For the members of the R7 family, as mentioned above, this function is accomplished through PPI‐mediated interaction with R7BP. The protein stability of these RGS proteins is also regulated through the formation of obligatory dimers with Gβ5 (Anderson et al., 2009). In the absence of this Gβ subunit, as in Gβ5 −/− mice, all members of the R7 family (RGS6, 7, 9 and 11) are also absent due to robust protein degradation (Chen et al., 2003). While R7BP binding is not necessary for protein stability of all members of the R7 family, the exception is RGS9, which in the absence of R7BP has been shown to be degraded by cysteine proteases (Anderson et al., 2007a,b).
The expression of RGS proteins is not only spatially regulated per cell type and subcellular localization but also temporally regulated by mechanisms that induce or suppress RGS expression in response to specific cues or during pathological conditions. Examples include RGS2 down‐regulation in androgen‐independent prostate cancer (Cao et al., 2006) and hypertension (Semplicini et al., 2006), the down‐regulation of RGS10 and RGS17 in models of chemoresistance in ovarian cancer (Hooks et al., 2010), as well as the up‐regulation of RGS17 in lung and prostate cancer (James et al., 2009; Bodle et al., 2013). Importantly, RGS transcript and protein levels may be independently regulated. Xie et al. (2009) demonstrated that RGS4 mRNA levels were greatly enhanced in human breast cancer tumours. In contrast, protein levels of RGS4 from the same tissues were virtually absent, due to enhanced proteasomal degradation of RGS4 protein (Xie et al., 2009). Furthermore, re‐expression of RGS4 in invasive cancer cell lines in which RGS4 protein is down‐regulated suppresses cancer cell invasion and migration (Xie et al., 2009). Apart from RGS4, several other members of the R4 family, including RGS2 and RGS5 are substrates for the ubiquitin‐proteasomal pathway and are rapidly and constitutively degraded (Davydov and Varshavsky, 2000; Lee et al., 2005; Bodenstein et al., 2007; Lee et al., 2011; Sjögren et al., 2015). This mechanism may be a way for physiological systems to very rapidly adapt to new environments. In the study by Xie et al. (2009), mentioned above, inhibition of proteasome activity could restore RGS4 protein levels in invasive breast cancer cells and thereby suppress invasion and migration. Altogether, this suggests that stabilizing RGS4 protein could be a promising strategy in the treatment of invasive breast cancer. In contrast, inhibiting RGS4 could also have therapeutic merit. In animal models of PD, several groups found that RGS4 mRNA is increased and contributes to the development of involuntary movement disorders following L‐DOPA treatment, an effect that could be blocked by silencing RGS4 by RNAi (Lerner and Kreitzer, 2012; Ko et al., 2014).
The notion that one might seek to inhibit or enhance RGS protein function depending on the therapeutic indication is further highlighted by the R7 family member RGS6 (reviewed in Ahlers et al., 2016). Prolonged alcohol exposure in mice leads to increased levels of both RGS6 mRNA and protein in the ventral tegmental area (VTA), a brain region strongly associated with drug addiction. Furthermore, RGS6−/− mice display a reduction in alcohol seeking behaviour compared to wild‐type mice, as well as diminished symptoms of conditioned reward and withdrawal (Stewart et al., 2015). Inhibition of RGS6 has also been implicated as a promising therapeutic strategy in depression and anxiety (Stewart et al., 2014). In contrast, RGS6 protects against dopaminergic neuron loss in the VTA, indicating that enhancing RGS6 could be beneficial in the treatment of PD (Bifsha et al., 2014). This would suggest that an RGS6 modulator would have broad implications in CNS diseases. An RGS6 enhancer could also be beneficial as a novel cancer therapeutic. RGS6 has been proposed as a tumour suppressor in several types of cancer, including bladder, lung and breast cancer (Berman et al., 2004; Gu et al., 2006; Maity et al., 2011; Maity et al., 2013). While the effects of RGS6 in the CNS seem to mainly be mediated through is canonical GAP activity, its action as a tumour suppressor is mediated through non‐G protein mechanisms (Maity et al., 2011).
These and other studies not only further demonstrate the potential for RGS proteins as potential targets in drug development for a wide range of therapeutic indications but also highlights the complexity and challenges facing investigators that wish to pursue this avenue. While enhancement of an RGS protein may be beneficial in one disease model, other indications might benefit from an RGS protein inhibitor. Furthermore, the specific function to be targeted – GAP versus non‐canonical function – may also differ between therapeutic areas.
Advances in RGS protein drug discovery – from biochemical activity to in vivo efficacy
Based on the non‐canonical activities described above, successful RGS targeted drug discovery efforts will ultimately have to take into account that RGS proteins are not only GAPs for active, GTP‐bound Gα subunits. Nevertheless, the early efforts to target RGS proteins have focused on this feature, which is the common structural element for all RGS protein family members. More recent efforts are starting to elucidate other strategies for targeting non‐canonical functions and mechanisms that control expression and localization.
RGS proteins are challenging targets for small molecules. Firstly, because they are intracellular proteins, a potential RGS‐modulating drug needs to be both cell permeable as well as stable in the intracellular environment. However, this is not a particularly high obstacle to overcome, and advances have been made in the drug discovery of many other intracellular protein families, such as kinases, phosphatases and nuclear receptors (Rask‐Andersen et al., 2011; He et al., 2014; Barnes, 2016; Shang et al., 2016). Indeed, small molecules have recently emerged that are active as RGS inhibitors both in cells and in vivo (see below).
The second, and more daunting, challenge for the development of small molecule RGS inhibitors is the task of inhibiting a PPI. The canonical mode of action of RGS proteins is through a transient PPI with active, GTP‐bound Gα, a flat surface with an area of more than 2000 Å2. PPIs are receiving increasing interest in drug discovery and this mechanism, that historically has been considered ‘un‐druggable’, is now one of the fastest expanding areas in drug development (Arkin et al., 2014). Thus, while these obstacles are significant, they have not prevented several efforts to identify inhibitors of RGS proteins, with growing success. Early work on identifying RGS protein inhibitors used yeast two‐hybrid and biochemical methods to detect peptides that could serve as inhibitors of the RGS‐Gα interaction. These studies led to several peptides that effectively blocked RGS protein activity in vitro (e.g. YJ34 and 5nd; Jin et al., 2004; Young et al., 2004; Roof et al., 2006; 2008; 2009; Wang et al., 2008).
The first published small molecule RGS inhibitor, CCG‐4986, was identified by the group of Richard Neubig, using a novel flow cytometry‐based PPI assay (Roman et al., 2007). Subsequent work from the same group used biochemical time‐resolved FRET (TR‐FRET) (Leifert et al., 2006) to identify CCG‐63802, and analogues thereof, as the first reversible RGS protein inhibitor (Blazer et al., 2010). Like CCG‐4986, CCG‐63802 showed selectivity for RGS4 over other RGS proteins studied. A third series of small molecule RGS4 inhibitors is represented by CCG‐50014, which is the first RGS inhibitor that has shown activity in cells (Blazer et al., 2011). A derivative of CCG‐50014, CCG‐203769, was also demonstrated to have effects in vivo. In a mouse model of Parkinson's disease (PD), CCG‐203769 was able to reverse raclopride‐induced akinesia and bradykinesia. It also potentiated Gαi‐dependent muscarinic bradycardia (Blazer et al., 2015), thereby replicating a phenotype previously demonstrated in RGS4−/− mice to be dependent on RGS4 (Cifelli et al., 2008). This shows that RGS protein inhibitors may be used in a clinical setting alone or in conjunction with other therapies.
Although CCG‐50014 and CCG‐203769 have been shown to be active in biological systems, many early inhibitors identified in biochemical screens failed to move forward due to lack of cellular activity. We attempted to overcome this problem by developing a cell‐based high‐throughput assay for RGS4 inhibitors. We used the FlpIn‐TREx system (Invitrogen™) to develop a cell line with stable expression of the Gαq‐coupled M3 muscarinic receptor and doxycycline‐inducible RGS4 expression and screened for compounds that could reverse RGS4‐mediated suppression of Ca2+ signalling (Storaska et al., 2013). This screen identified several novel inhibitors and studies are ongoing to characterize them further.
Apart from RGS4, RGS17 has been a focus for high‐throughput screening for small molecule inhibitors. As discussed above, RGS17 is one of several RGS proteins that are up‐regulated in different cancers. In both lung and prostate cancer, RGS17 mRNA is significantly increased and this contributes to tumour progression (Bodle et al., 2013). This led the lab of David Roman to develop an Alpha Screen assay to screen for small molecule inhibitors of the RGS17‐Gαo interaction (Mackie and Roman, 2011). The hits identified from this screening campaign inhibited this interaction with micro molar potency. Future development of these or other RGS17 inhibitors could serve as novel cancer therapeutics.
Although much of RGS protein drug discovery efforts have been focused on identifying inhibitors of GAP activity, identifying enhancers of RGS protein function may be equally important from a clinical perspective. However, this is a more daunting task, since enhancing a PPI is far more difficult than blocking it. Several RGS proteins are down‐regulated during pathological insults so finding ways to increase their expression, and thereby function, is an attractive alternative strategy to achieve this goal. This encouraged us to develop a cell based enzyme complementation assay to screen for small molecule stabilizers of RGS2 (Sjögren et al., 2012; Raveh et al., 2014). As discussed above, RGS2 is one of several RGS proteins that is rapidly degraded through the ubiquitin‐proteasomal pathway. Low RGS2 protein levels are associated with hypertension and other cardiovascular pathologies (Heximer et al., 2003; Takimoto et al., 2009; Tsang et al., 2010) and could also be involved in the progression of prostate and breast cancer (Cao et al., 2006; Lyu et al., 2015). In our initial screen, we identified digoxin and other cardiotonic steroids as selective stabilizers of RGS2 protein levels (Sjögren et al., 2012). In subsequent work, we demonstrated that digoxin is protective in a murine model of cardiac injury, an effect that was lost in RGS2−/− mice (Sjögren et al., 2016b). This is the first study demonstrating that pharmacological enhancement of an RGS protein has effects in vivo and opens up new avenues for RGS protein drug discovery.
RGS protein drug discovery – what does the future hold?
Although great progress has been made in the field of RGS protein biology, many mechanisms still need to be elucidated. What has become clear is that members of this family are more than just GAPs for G proteins, and the emerging plethora of non‐canonical functions may become a more prominent focus in the future. Given the important role of GPCRs in physiology and drug discovery, however, the canonical G protein regulatory role of RGS proteins is likely to remain a focus in future drug development efforts. Early drug discovery efforts focused solely on the inhibition of RGS proteins interacting with Gα subunits, but other functions, as well as dynamic regulation of expression, were ignored. Future efforts may investigate these regulatory mechanisms further, especially for the development of RGS protein enhancers.
The RGS proteins that have been targeted in drug discovery thus far (RGS2, 4 and 17 as described above) all have in common that they are small, containing no additional domains apart from the RGS domain. This makes them ‘easier’ to work with from an experimental stand point. Although other RGS proteins might also have great therapeutic potential, such as RGS9 in PD drug development, the additional non‐canonical functions of additional protein domains present in these RGS proteins make drug discovery efforts less straight‐forward. In some cases, targeting GAP activity may not be the primary goal when developing small molecules to target these larger, multi‐domain, RGS proteins. Thus, although many RGS proteins could have great therapeutic potential, more studies are required to determine their physiological function and how best to target them. These may depend on a detailed knowledge of the mechanisms of RGS protein regulation that control their expression, posttranslational modifications and other mechanisms that have yet to be elucidated. After all, the existence of RGS proteins has only been acknowledged in the last 20 years. What will the next 20 years bring?
Conflict of interest
The author declares no conflicts of interest.
Acknowledgements
The author is funded by The American Heart Association [15SDG21630002]. The author would like to thank Drs. Shelley B. Hooks and Richard R. Neubig for providing constructive feedback on the manuscript.
Sjögren, B. (2017) The evolution of regulators of G protein signalling proteins as drug targets – 20 years in the making: IUPHAR Review 21. British Journal of Pharmacology, 174: 427–437. doi: 10.1111/bph.13716.
This article is an IUPHAR review contributed by members of the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC‐IUPHAR) subcommittee for the RGS proteins.
References
- Ahlers KE, Chakravarti B, Fisher RA (2016). RGS6 as a novel therapeutic target in CNS diseases and cancer. AAPS J 18: 560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GR, Lujan R, Semenov A, Pravetoni M, Posokhova EN, Song JH et al. (2007a). Expression and localization of RGS9‐2/G 5/R7BP complex in vivo is set by dynamic control of its constitutive degradation by cellular cysteine proteases. J Neurosci 27: 14117–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GR, Posokhova E, Martemyanov KA (2009). The R7 RGS protein family: multi‐subunit regulators of neuronal G protein signaling. Cell Biochem Biophys 54: 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GR, Semenov A, Song JH, Martemyanov KA (2007b). The membrane anchor R7BP controls the proteolytic stability of the striatal specific RGS protein, RGS9‐2. J Biol Chem 282: 4772–4781. [DOI] [PubMed] [Google Scholar]
- Apanovitch DM, Slep KC, Sigler PB, Dohlman HG (1998). Sst2 is a GTPase‐activating protein for Gpa1: purification and characterization of a cognate RGS‐Galpha protein pair in yeast. Biochemistry 37: 4815–4822. [DOI] [PubMed] [Google Scholar]
- Arkin MR, Tang Y, Wells JA (2014). Small‐molecule inhibitors of protein–protein interactions: progressing toward the reality. Chem Biol 21: 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2016). Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol Rev 68: 788–815. [DOI] [PubMed] [Google Scholar]
- Bastin G, Singh K, Dissanayake K, Mighiu AS, Nurmohamed A, Heximer SP (2012). Amino‐terminal cysteine residues differentially influence RGS4 protein plasma membrane targeting, intracellular trafficking, and function. J Biol Chem 287: 28966–28974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DM, Kozasa T, Gilman AG (1996a). The GTPase‐activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J Biol Chem 271: 27209–27212. [DOI] [PubMed] [Google Scholar]
- Berman DM, Wang Y, Liu Z, Dong Q, Burke LA, Liotta LA et al. (2004). A functional polymorphism in RGS6 modulates the risk of bladder cancer. Cancer Res 64: 6820–6826. [DOI] [PubMed] [Google Scholar]
- Berman DM, Wilkie TM, Gilman AG (1996b). GAIP and RGS4 are GTPase‐activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86: 445–452. [DOI] [PubMed] [Google Scholar]
- Bernstein LS, Linder ME, Hepler JR (2004a). Analysis of RGS protein palmitoylation. Methods Mol Biol 237: 195–204. [DOI] [PubMed] [Google Scholar]
- Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI et al. (2004b). RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem 279: 21248–21256. [DOI] [PubMed] [Google Scholar]
- Bertekap RL Jr, Burford NT, Li Z, Alt A (2015). High‐throughput screening for allosteric modulators of GPCRs. Methods Mol Biol 1335: 223–240. [DOI] [PubMed] [Google Scholar]
- Bifsha P, Yang J, Fisher RA, Drouin J (2014). Rgs6 is required for adult maintenance of dopaminergic neurons in the ventral substantia nigra. PLoS Genet 10: e1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisignano P, Burford NT, Shang Y, Marlow B, Livingston KE, Fenton AM et al. (2015). Ligand‐based discovery of a new scaffold for allosteric modulation of the mu‐opioid receptor. J Chem Inf Model 55: 1836–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer LL, Neubig RR (2009). Small molecule protein–protein interaction inhibitors as CNS therapeutic agents: current progress and future hurdles. Neuropsychopharmacology 34: 126–141. [DOI] [PubMed] [Google Scholar]
- Blazer LL, Roman DL, Chung A, Larsen MJ, Greedy BM, Husbands SM et al. (2010). Reversible, allosteric small‐molecule inhibitors of regulator of G protein signaling proteins. Mol Pharmacol 78: 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM et al. (2015). Selectivity and anti‐Parkinson's potential of thiadiazolidinone RGS4 inhibitors. ACS Chem Nerosci 6: 911–919. [DOI] [PubMed] [Google Scholar]
- Blazer LL, Zhang H, Casey EM, Husbands SM, Neubig RR (2011). A nanomolar‐potency small molecule inhibitor of regulator of G‐protein signaling proteins. Biochemistry 50: 3181–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Hoang CV, Potts B, Gold SJ, Powell CM (2008). Motor coordination deficits in mice lacking RGS9. Brain Res 1190: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenstein J, Sunahara RK, Neubig RR (2007). N‐terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol Pharmacol 71: 1040–1050. [DOI] [PubMed] [Google Scholar]
- Bodle CR, Mackie DI, Roman DL (2013). RGS17: an emerging therapeutic target for lung and prostate cancers. Future Med Chem 5: 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NE, Goswami D, Branch MR, Ramineni S, Ortlund EA, Griffin PR et al. (2015). Integration of G protein alpha (Galpha) signaling by the regulator of G protein signaling 14 (RGS14). J Biol Chem 290: 9037–9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Livingston KE, Canals M, Ryan MR, Budenholzer LM, Han Y et al. (2015). Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the delta‐opioid receptor. J Med Chem 58: 4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Qin J, Xie Y, Khan O, Dowd F, Scofield M et al. (2006). Regulator of G‐protein signaling 2 (RGS2) inhibits androgen‐independent activation of androgen receptor in prostate cancer cells. Oncogene 25: 3719–3734. [DOI] [PubMed] [Google Scholar]
- Castro‐Fernandez C, Janovick JA, Brothers SP, Fisher RA, Ji TH, Conn PM (2002). Regulation of RGS3 and RGS10 palmitoylation by GnRH. Endocrinology 143: 1310–1317. [DOI] [PubMed] [Google Scholar]
- Chakir K, Zhu W, Tsang S, Woo AY, Yang D, Wang X et al. (2011). RGS2 is a primary terminator of beta(2)‐adrenergic receptor‐mediated G(i) signaling. J Mol Cell Cardiol 50: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RK, Otte CA (1982a). Isolation and genetic analysis of Saccharomyces cerevisiae mutants supersensitive to G1 arrest by a factor and alpha factor pheromones. Mol Cell Biol 2: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RK, Otte CA (1982b). Physiological characterization of Saccharomyces cerevisiae mutants supersensitive to G1 arrest by a factor and alpha factor pheromones. Mol Cell Biol 2: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Seow KT, Guo K, Yaw LP, Lin SC (1999). The membrane association domain of RGS16 contains unique amphipathic features that are conserved in RGS4 and RGS5. J Biol Chem 274: 19799–19806. [DOI] [PubMed] [Google Scholar]
- Chen C, Wang H, Fong CW, Lin SC (2001). Multiple phosphorylation sites in RGS16 differentially modulate its GAP activity. FEBS Lett 504: 16–22. [DOI] [PubMed] [Google Scholar]
- Chen CK, Eversole‐Cire P, Zhang H, Mancino V, Chen YJ, He W et al. (2003). Instability of GGL domain‐containing RGS proteins in mice lacking the G protein beta‐subunit Gbeta5. Proc Natl Acad Sci U S A 100: 6604–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidiac P, Sobiesiak AJ, Lee KN, Gros R, Nguyen CH (2014). The eIF2B‐interacting domain of RGS2 protects against GPCR agonist‐induced hypertrophy in neonatal rat cardiomyocytes. Cell Signal 26: 1226–1234. [DOI] [PubMed] [Google Scholar]
- Cho H, Harrison K, Kehrl JH (2004). Regulators of G protein signaling: potential drug targets for controlling cardiovascular and immune function. Curr Drug Targets Immune Endocr Metabol Disord 4: 107–118. [DOI] [PubMed] [Google Scholar]
- Cifelli C, Rose RA, Zhang H, Voigtlaender‐Bolz J, Bolz SS, Backx PH et al. (2008). RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ Res 103: 527–535. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Waldo GL, Hollinger S, Hepler JR, Harden TK (2001). Protein kinase C phosphorylates RGS2 and modulates its capacity for negative regulation of Galpha 11 signaling. J Biol Chem 276: 5438–5444. [DOI] [PubMed] [Google Scholar]
- Davydov IV, Varshavsky A (2000). RGS4 is arginylated and degraded by the N‐end rule pathway in vitro. J Biol Chem 275: 22931–22941. [DOI] [PubMed] [Google Scholar]
- De Vries L, Elenko E, Hubler L, Jones TL, Farquhar MG (1996). GAIP is membrane‐anchored by palmitoylation and interacts with the activated (GTP‐bound) form of G alpha i subunits. Proc Natl Acad Sci U S A 93: 15203–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien A, Druey KM (2001). RGS16 function is regulated by epidermal growth factor receptor‐mediated tyrosine phosphorylation. J Biol Chem 276: 48532–48538. [DOI] [PubMed] [Google Scholar]
- Derrien A, Zheng B, Osterhout JL, Ma YC, Milligan G, Farquhar MG et al. (2003). Src‐mediated RGS16 tyrosine phosphorylation promotes RGS16 stability. J Biol Chem 278: 16107–16116. [DOI] [PubMed] [Google Scholar]
- DiBello PR, Garrison TR, Apanovitch DM, Hoffman G, Shuey DJ, Mason K et al. (1998). Selective uncoupling of RGS action by a single point mutation in the G protein alpha‐subunit. J Biol Chem 273: 5780–5784. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Apaniesk D, Chen Y, Song J, Nusskern D (1995). Inhibition of G‐protein signaling by dominant gain‐of‐function mutations in Sst2p, a pheromone desensitization factor in Saccharomyces cerevisiae . Mol Cell Biol 15: 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Song J, Ma D, Courchesne WE, Thorner J (1996). Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G‐protein alpha subunit). Mol Cell Biol 16: 5194–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA, Kofuji P (1997). RGS proteins reconstitute the rapid gating kinetics of gbetagamma‐activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A 94: 10461–10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Doupnik CA, Boyle MP, Muglia LJ, Huettner JE, Linder ME et al. (2005). Palmitoylation regulates plasma membrane‐nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J Cell Biol 169: 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Doupnik CA, Jayaraman M, Buchwalter AL, Kaltenbronn KM, Huettner JE et al. (2006). R7BP augments the function of RGS7*Gbeta5 complexes by a plasma membrane‐targeting mechanism. J Biol Chem 281: 28222–28231. [DOI] [PubMed] [Google Scholar]
- Druey KM (2003). Regulators of G protein signalling: potential targets for treatment of allergic inflammatory diseases such as asthma. Expert Opin Ther Targets 7: 475–484. [DOI] [PubMed] [Google Scholar]
- Druey KM, Blumer KJ, Kang VH, Kehrl JH (1996). Inhibition of G‐protein‐mediated MAP kinase activation by a new mammalian gene family. Nature 379: 742–746. [DOI] [PubMed] [Google Scholar]
- Druey KM, Ugur O, Caron JM, Chen CK, Backlund PS, Jones TL (1999). Amino‐terminal cysteine residues of RGS16 are required for palmitoylation and modulation of Gi‐ and Gq‐mediated signaling. J Biol Chem 274: 18836–18842. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Hoang CV, Potts BW, Porras G, Pioli E, Kim KW et al. (2007). RGS9‐2 negatively modulates L‐3,4‐dihydroxyphenylalanine‐induced dyskinesia in experimental Parkinson's disease. J Neurosci 27: 14338–14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowska D, Jayaraman M, Kaltenbronn KM, Sandiford SL, Wang Q, Jenkins S et al. (2008). Postnatal induction and localization of R7BP, a membrane‐anchoring protein for regulator of G protein signaling 7 family‐G[beta]5 complexes in brain. Neuroscience 151: 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wu X, Dong Q, Romeo MJ, Lin X, Gutkind JS et al. (2006). A nonsynonymous single‐nucleotide polymorphism in the PDZ‐Rho guanine nucleotide exchange factor (Ser1416Gly) modulates the risk of lung cancer in Mexican Americans. Cancer 106: 2716–2724. [DOI] [PubMed] [Google Scholar]
- Gu S, Cifelli C, Wang S, Heximer SP (2009). RGS proteins: identifying new GAPs in the understanding of blood pressure regulation and cardiovascular function. Clin Sci (Lond) 116: 391–399. [DOI] [PubMed] [Google Scholar]
- Hague C, Bernstein LS, Ramineni S, Chen Z, Minneman KP, Hepler JR (2005). Selective inhibition of alpha1A‐adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem 280: 27289–27295. [DOI] [PubMed] [Google Scholar]
- He RJ, Yu ZH, Zhang RY, Zhang ZY (2014). Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin 35: 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Cowan CW, Wensel TG (1998). RGS9, a GTPase accelerator for phototransduction. Neuron 20: 95–102. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N et al. (2003). Hypertension and prolonged vasoconstrictor signaling in RGS2‐deficient mice. J Clin Invest 111: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR (1997). RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci U S A 94: 14389–14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiol A, Davey PC, Osterhout JL, Waheed AA, Fischer ER, Chen CK et al. (2003). Palmitoylation regulates regulators of G‐protein signaling (RGS) 16 function. I. Mutation of amino‐terminal cysteine residues on RGS16 prevents its targeting to lipid rafts and palmitoylation of an internal cysteine residue. J Biol Chem 278: 19301–19308. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR (2002). Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 54: 527–559. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Ramineni S, Hepler JR (2003). Phosphorylation of RGS14 by protein kinase A potentiates its activity toward G alpha i. Biochemistry 42: 811–819. [DOI] [PubMed] [Google Scholar]
- Hooks SB, Callihan P, Altman MK, Hurst JH, Ali MW, Murph MM (2010). Regulators of G‐protein signaling RGS10 and RGS17 regulate chemoresistance in ovarian cancer cells. Mol Cancer 9: 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst JH, Hooks SB (2009). Regulator of G‐protein signaling (RGS) proteins in cancer biology. Biochem Pharmacol 78: 1289–1297. [DOI] [PubMed] [Google Scholar]
- Huson DH, Scornavacca C (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61: 1061–1067. [DOI] [PubMed] [Google Scholar]
- James MA, Lu Y, Liu Y, Vikis HG, You M (2009). RGS17, an overexpressed gene in human lung and prostate cancer, induces tumor cell proliferation through the cyclic AMP‐PKA‐CREB pathway. Cancer Res 69: 2108–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman M, Zhou H, Jia L, Cain MD, Blumer KJ (2009). R9AP and R7BP: traffic cops for the RGS7 family in phototransduction and neuronal GPCR signaling. Trends Pharmacol Sci 30: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zhong H, Omnaas JR, Neubig RR, Mosberg HI (2004). Structure‐based design, synthesis, and pharmacologic evaluation of peptide RGS4 inhibitors. J Pept Res 63: 141–146. [DOI] [PubMed] [Google Scholar]
- Johnson EN, Druey KM (2002). Functional characterization of the G protein regulator RGS13. J Biol Chem 277: 16768–16774. [DOI] [PubMed] [Google Scholar]
- Jones TL (2004). Role of palmitoylation in RGS protein function. Methods Enzymol 389: 33–55. [DOI] [PubMed] [Google Scholar]
- Kenakin T (2012). The potential for selective pharmacological therapies through biased receptor signaling. BMC Pharmacol Toxicol 13: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury E, Clement S, Laporte SA (2014). Allosteric and biased g protein‐coupled receptor signaling regulation: potentials for new therapeutics. Front Endocrinol (Lausanne) 5: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple RJ, De Vries L, Tronchere H, Behe CI, Morris RA, Gist Farquhar M et al. (2001). RGS12 and RGS14 GoLoco motifs are G alpha(i) interaction sites with guanine nucleotide dissociation inhibitor Activity. J Biol Chem 276: 29275–29281. [DOI] [PubMed] [Google Scholar]
- Ko WK, Martin‐Negrier ML, Bezard E, Crossman AR, Ravenscroft P (2014). RGS4 is involved in the generation of abnormal involuntary movements in the unilateral 6‐OHDA‐lesioned rat model of Parkinson's disease. Neurobiol Dis 70: 138–148. [DOI] [PubMed] [Google Scholar]
- Koelle MR (1997). A new family of G‐protein regulators – the RGS proteins. Curr Opin Cell Biol 9: 143–147. [DOI] [PubMed] [Google Scholar]
- Koelle MR, Horvitz HR (1996). EGL‐10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84: 115–125. [DOI] [PubMed] [Google Scholar]
- Lan KL, Sarvazyan NA, Taussig R, Mackenzie RG, DiBello PR, Dohlman HG et al. (1998). A point mutation in Galphao and Galphai1 blocks interaction with regulator of G protein signaling proteins. J Biol Chem 273: 12794–12797. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV et al. (2005). RGS4 and RGS5 are in vivo substrates of the N‐end rule pathway. Proc Natl Acad Sci U S A 102: 15030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PC, Sowa ME, Gygi SP, Harper JW (2011). Alternative ubiquitin activation/conjugation cascades interact with N‐end rule ubiquitin ligases to control degradation of RGS proteins. Mol Cell 43: 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifert WR, Bailey K, Cooper TH, Aloia AL, Glatz RV, McMurchie EJ (2006). Measurement of heterotrimeric G‐protein and regulators of G‐protein signaling interactions by time‐resolved fluorescence resonance energy transfer. Anal Biochem 355: 201–212. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC (2012). RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron 73: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu JH, Park DW, Huang B, Kang SH, Lee SJ, Lee C et al. (2015). RGS2 suppresses breast cancer cell growth via a MCPIP1‐dependent pathway. J Cell Biochem 116: 260–267. [DOI] [PubMed] [Google Scholar]
- Mackie DI, Roman DL (2011). Development of a novel high‐throughput screen and identification of small‐molecule inhibitors of the Galpha‐RGS17 protein–protein interaction using AlphaScreen. J Biomol Screen 16: 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity B, Stewart A, O'Malley Y, Askeland RW, Sugg SL, Fisher RA (2013). Regulator of G protein signaling 6 is a novel suppressor of breast tumor initiation and progression. Carcinogenesis 34: 1747–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity B, Yang J, Huang J, Askeland RW, Bera S, Fisher RA (2011). Regulator of G protein signaling 6 (RGS6) induces apoptosis via a mitochondrial‐dependent pathway not involving its GTPase‐activating protein activity. J Biol Chem 286: 1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso JJ, Qian Y, Long C, Wu GY, Wensel TG (2009). Distribution of RGS9‐2 in neurons of the mouse striatum. J Neurochem 112: 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann C, Chung CH, Hoppner G, Michalek C, Nose M, Schuler C et al. (2002). Expression of ten RGS proteins in human myocardium: functional characterization of an upregulation of RGS4 in heart failure. Cardiovasc Res 55: 778–786. [DOI] [PubMed] [Google Scholar]
- Neubig RR (2015). RGS‐insensitive G proteins as in vivo probes of RGS function. Prog Mol Biol Transl Sci 133: 13–30. [DOI] [PubMed] [Google Scholar]
- Nguyen CH, Ming H, Zhao P, Hugendubler L, Gros R, Kimball SR et al. (2009). Translational control by RGS2. J Cell Biol 186: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Qu L, Yang H, Wang M, Huang Y (2006). Palmitoylation and its effect on the GTPase‐activating activity and conformation of RGS2. Int J Biochem Cell Biol 38: 2209–2218. [DOI] [PubMed] [Google Scholar]
- Osterhout JL, Waheed AA, Hiol A, Ward RJ, Davey PC, Nini L et al. (2003). Palmitoylation regulates regulator of G‐protein signaling (RGS) 16 function. II. Palmitoylation of a cysteine residue in the RGS box is critical for RGS16 GTPase accelerating activity and regulation of Gi‐coupled signalling. J Biol Chem 278: 19309–19316. [DOI] [PubMed] [Google Scholar]
- Rask‐Andersen M, Almen MS, Schioth HB (2011). Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10: 579–590. [DOI] [PubMed] [Google Scholar]
- Raveh A, Schultz PJ, Aschermann L, Carpenter C, Tamayo‐Castillo G, Cao S et al. (2014). Identification of protein kinase C activation as a novel mechanism for RGS2 protein upregulation through phenotypic screening of natural product extracts. Mol Pharmacol 86: 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle EL, Schwartzman RA, Bond M, Insel PA (2005). Multi‐tasking RGS proteins in the heart: the next therapeutic target? Circ Res 96: 401–411. [DOI] [PubMed] [Google Scholar]
- Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR (2007). Identification of small‐molecule inhibitors of RGS4 using a high‐throughput flow cytometry protein interaction assay. Mol Pharmacol 71: 169–175. [DOI] [PubMed] [Google Scholar]
- Roof RA, Jin Y, Roman DL, Sunahara RK, Ishii M, Mosberg HI et al. (2006). Mechanism of action and structural requirements of constrained peptide inhibitors of RGS proteins. Chem Biol Drug Des 67: 266–274. [DOI] [PubMed] [Google Scholar]
- Roof RA, Roman DL, Clements ST, Sobczyk‐Kojiro K, Blazer LL, Ota S et al. (2009). A covalent peptide inhibitor of RGS4 identified in a focused one‐bead, one compound library screen. BMC Pharmacol 9: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof RA, Sobczyk‐Kojiro K, Turbiak AJ, Roman DL, Pogozheva ID, Blazer LL et al. (2008). Novel peptide ligands of RGS4 from a focused one‐bead, one‐compound library. Chem Biol Drug Des 72: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM, Wilkie TM (2000). GTPase‐activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS‐like proteins. Annu Rev Biochem 69: 795–827. [DOI] [PubMed] [Google Scholar]
- Roy AA, Baragli A, Bernstein LS, Hepler JR, Hebert TE, Chidiac P (2006). RGS2 interacts with Gs and adenylyl cyclase in living cells. Cell Signal 18: 336–348. [DOI] [PubMed] [Google Scholar]
- Salim S, Sinnarajah S, Kehrl JH, Dessauer CW (2003). Identification of RGS2 and type V adenylyl cyclase interaction sites. J Biol Chem 278: 15842–15849. [DOI] [PubMed] [Google Scholar]
- Sandiford SL, Slepak VZ (2009). The Gbeta5‐RGS7 complex selectively inhibits muscarinic M3 receptor signaling via the interaction between the third intracellular loop of the receptor and the DEP domain of RGS7. Biochemistry 48: 2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff ML, Siderovski DP, Jordan JD, Brothers G, Snow B, De Vries L et al. (2000). Tyrosine‐kinase‐dependent recruitment of RGS12 to the N‐type calcium channel. Nature 408: 723–727. [DOI] [PubMed] [Google Scholar]
- Semplicini A, Lenzini L, Sartori M, Papparella I, Calo LA, Pagnin E et al. (2006). Reduced expression of regulator of G‐protein signaling 2 (RGS2) in hypertensive patients increases calcium mobilization and ERK1/2 phosphorylation induced by angiotensin II. J Hypertens 24: 1115–1124. [DOI] [PubMed] [Google Scholar]
- Sethakorn N, Yau DM, Dulin NO (2010). Non‐canonical functions of RGS proteins. Cell Signal 22: 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Lu S, Jiang Y, Zhang J (2016). Allosteric modulators of MEK1: drug design and discovery. Chem Biol Drug Des 88: 485–497. [DOI] [PubMed] [Google Scholar]
- Shu FJ, Ramineni S, Hepler JR (2010). RGS14 is a multifunctional scaffold that integrates G protein and Ras/Raf MAPkinase signalling pathways. Cell Signal 22: 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Singh G, Ghosh E (2014). Emerging structural insights into biased GPCR signaling. Trends Biochem Sci 39: 594–602. [DOI] [PubMed] [Google Scholar]
- Siderovski DP, Hessel A, Chung S, Mak TW, Tyers M (1996). A new family of regulators of G‐protein‐coupled receptors? Curr Biol 6: 211–212. [DOI] [PubMed] [Google Scholar]
- Siderovski DP, Heximer SP, Forsdyke DR (1994). A human gene encoding a putative basic helix–loop‐helix phosphoprotein whose mRNA increases rapidly in cycloheximide‐treated blood mononuclear cells. DNA Cell Biol 13: 125–147. [DOI] [PubMed] [Google Scholar]
- Siderovski DP, Willard FS (2005). The GAPs, GEFs, and GDIs of heterotrimeric G‐protein alpha subunits. Int J Biol Sci 1: 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren B (2011). Regulator of G protein signaling proteins as drug targets: current state and future possibilities. Adv Pharmacol 62: 315–347. [DOI] [PubMed] [Google Scholar]
- Sjögren B, Blazer LL, Neubig RR (2010). Regulators of G protein signaling proteins as targets for drug discovery. Prog Mol Biol Transl Sci 91: 81–119. [DOI] [PubMed] [Google Scholar]
- Sjögren B, Druey KM, Fisher RM, Hepler JR, Hooks SB, Martemyanov K , et al. (2016a). Regulators of G protein signaling (RGS) proteins. IUPHAR/BPS Guide to PHARMACOLOGY. http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId = 891.
- Sjögren B, Parra S, Atkins KB, Karaj B, Neubig RR (2016b). Digoxin‐mediated upregulation of RGS2 protein protects against cardiac injury. J Pharmacol Exp Ther 357: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren B, Parra S, Heath LJ, Atkins KB, Xie ZJ, Neubig RR (2012). Cardiotonic steroids stabilize regulator of G protein signaling 2 protein levels. Mol Pharmacol 82: 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren B, Swaney S, Neubig RR (2015). FBXO44‐mediated degradation of RGS2 protein uniquely depends on a cullin 4B/DDB1 complex. PLoS One 10: e0123581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA et al. (2008). Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha‐subunits. Proc Natl Acad Sci U S A 105: 6457–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A, Maity B, Anderegg SP, Allamargot C, Yang J, Fisher RA (2015). Regulator of G protein signaling 6 is a critical mediator of both reward‐related behavioral and pathological responses to alcohol. Proc Natl Acad Sci U S A 112: E786–E795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A, Maity B, Wunsch AM, Meng F, Wu Q, Wemmie JA et al. (2014). Regulator of G‐protein signaling 6 (RGS6) promotes anxiety and depression by attenuating serotonin‐mediated activation of the 5‐HT(1A) receptor‐adenylyl cyclase axis. FASEB J 28: 1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storaska AJ, Mei JP, Wu M, Li M, Wade SM, Blazer LL et al. (2013). Reversible inhibitors of regulators of G‐protein signaling identified in a high‐throughput cell‐based calcium signaling assay. Cell Signal 25: 2848–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Koitabashi N, Hsu S, Ketner EA, Zhang M, Nagayama T et al. (2009). Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J Clin Invest 119: 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR (1997). Structure of RGS4 bound to AlF4‐‐activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell 89: 251–261. [DOI] [PubMed] [Google Scholar]
- Traver S, Splingard A, Gaudriault G, De Gunzburg J (2004). The RGS (regulator of G‐protein signalling) and GoLoco domains of RGS14 co‐operate to regulate Gi‐mediated signalling. Biochem J 379: 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang S, Woo AY, Zhu W, Xiao RP (2010). Deregulation of RGS2 in cardiovascular diseases. Front Biosci (Schol Ed) 2: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Popov S, Slaughter C, Ross EM (1999). Palmitoylation of a conserved cysteine in the regulator of G protein signaling (RGS) domain modulates the GTPase‐activating activity of RGS4 and RGS10. J Biol Chem 274: 38260–38267. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lee Y, Zhang J, Young KH (2008). Identification of peptides that inhibit regulator of G protein signaling 4 function. Pharmacology 82: 97–104. [DOI] [PubMed] [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ (1996). RGS family members: GTPase‐activating proteins for heterotrimeric G‐protein alpha‐subunits. Nature 383: 172–175. [DOI] [PubMed] [Google Scholar]
- Wise A, Gearing K, Rees S (2002). Target validation of G‐protein coupled receptors. Drug Discov Today 7: 235–246. [DOI] [PubMed] [Google Scholar]
- Wu HK, Heng HH, Shi XM, Forsdyke DR, Tsui LC, Mak TW et al. (1995). Differential expression of a basic helix–loop‐helix phosphoprotein gene, G0S8, in acute leukemia and localization to human chromosome 1q31. Leukemia 9: 1291–1298. [PubMed] [Google Scholar]
- Xie Y, Wolff DW, Wei T, Wang B, Deng C, Kirui JK et al. (2009). Breast cancer migration and invasion depend on proteasome degradation of regulator of G‐protein signaling 4. Cancer Res 69: 5743–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Geiger TR, Johnson EN, Nyborg JK, Druey KM (2008). RGS13 acts as a nuclear repressor of CREB. Mol Cell 31: 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KH, Wang Y, Bender C, Ajit S, Ramirez F, Gilbert A et al. (2004). Yeast‐based screening for inhibitors of RGS proteins. Methods Enzymol 389: 277–301. [DOI] [PubMed] [Google Scholar]
- Zhang P, Mende U (2011). Regulators of G‐protein signaling in the heart and their potential as therapeutic targets. Circ Res 109: 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Nunn C, Ramineni S, Hepler JR, Chidiac P (2013). The Ras‐binding domain region of RGS14 regulates its functional interactions with heterotrimeric G proteins. J Cell Biochem 114: 1414–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Neubig RR (2001). Regulator of G protein signaling proteins: novel multifunctional drug targets. J Pharmacol Exp Ther 297: 837–845. [PubMed] [Google Scholar]