

Abstract
Chloromethylgold(I) complexes of phosphine, phosphite, and N‐heterocyclic carbene ligands are easily synthesized by reaction of trimethylsilyldiazomethane with the corresponding gold chloride precursors. Activation of these gold(I) carbenoids with a variety of chloride scavengers promotes reactivity typical of metallocarbenes in solution, namely homocoupling to ethylene, olefin cyclopropanation, and Buchner ring expansion of benzene.
Keywords: Buchner reaction, carbenes, carbenoids, cyclopropanation, gold
Carbene complexes of transition metals are reactive intermediates1 frequently invoked in a wide variety of C−C bond‐forming processes that range from industrial scale olefin metathesis2 to natural product synthesis.3 Despite their central role in gold catalysis,4 few non‐heteroatom‐stabilized gold(I) carbene complexes have been structurally characterized.5 The reduced steric shielding provided by commonly used ligands and the intrinsically high electrophilicity6 exhibited by simple gold(I) carbenes preclude their isolation in condensed phase. Such species are however of high interest as demonstrated by gas‐phase studies by Chen,7 Schwarz,8 and others.9 The isolation of this type of compounds, or their functional equivalents, is therefore of great importance for the fundamental understanding of the reactivity of electrophilic gold carbenes. Transition‐metal complexes of the type [M(CHXR)], formally defined as carbenoids,10 show similar reactivity to their carbene counterparts11 and offer an attractive alternative to otherwise non‐isolable species. To date, however, very little is known about gold(I) carbenoids and their reactivity.12 Herein we report the synthesis of easily accessible gold carbenoids bearing bulky ligands as gold carbene equivalents in solution.
Previous synthesis of chloromethylgold(I) complexes used either toxic and potentially explosive diazomethane12a or low‐temperature in situ formation of Mg(CH2Cl)Cl.12d We have developed a more convenient approach that utilizes the methanol promoted decomposition of trimethylsilyldiazomethane13, 14 at room temperature (Scheme 1). Treatment of phosphine, phosphite, and NHC gold chloride complexes with trimethylsilyldiazomethane in benzene solution in presence of methanol afforded gold carbenoids 1 a–d within minutes. Complexes 1 a–c could be easily purified by column chromatography and have been fully characterized. Remarkably, these gold(I) carbenoids can be stored indefinitely when protected from air, and only decompose slowly when left under ambient conditions.
Scheme 1.
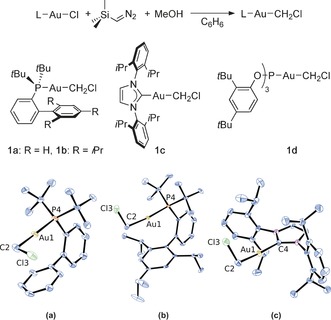
Synthesis of chloromethylgold(I) complexes 1 a–d and structures of complexes a) 1 a, b) 1 b, c) and 1 c.38 ORTEP plots with ellipsoids set at 50 % probability; hydrogen atoms and solvent molecules are omitted for clarity.
Measured Au−C distances (Table 1) are close to 2.088(9) Å observed for the previously reported [(PPh3)AuCH2Cl] complex (2).12d On the other hand, C−Cl distances (1.828–1.830 Å) are markedly longer than found in 2 (1.68(1) Å), and in fact are the longest among all 79 published crystal structures containing a [MCH2Cl] motif.15 The longest previously reported C−Cl distance in a (chloromethyl)metal fragment was 1.826(2) Å in a RhIII complex.16 On the other hand, the Au1‐C2‐Cl3 angles are closer to the ideal tetrahedron as opposed to the majority of other structures in which the M‐C‐Cl angle is about 116°.17 Chemical shifts for the chloromethyl moiety are found between 2.94 and 3.63 ppm in 1H NMR and 47.2 and 53.9 ppm in 13C NMR spectra.
Table 1.
Selected bond distances [Å] and angles [°] from solid‐state molecular structures and chemical shifts[a] [δ, ppm] for the new carbenoid complexes.
| Complex | Au1−C2 | C2−Cl3 | Au1‐C2‐Cl3 | δ 1H[a] | δ 13C[a] |
|---|---|---|---|---|---|
| 1 a | 2.058(9) | 1.828(1) | 110.3(5) | 2.96 | 53.6 |
| 1 b | 2.088(5) | 1.829(5) | 111.1(3) | 2.94 | 53.9 |
| 1 c | 2.060(2) | 1.830(2) | 110.4(1) | 3.34 | 47.2 |
| 1 d | – | – | – | 3.63 | 49.1 |
[a] Chemical shifts corresponding to the chloromethyl moiety measured in CD2Cl2 solution at 23 °C.
When solutions of complexes 1 a–d were treated with chloride scavengers possessing weakly coordinating counterions, for example, TMSX and AgX (X=Tf2N−, TfO−, MeSO3 −, CF3CO2 −) as well as AgSbF6, formation of ethylene (3) and simple gold salts 6 was observed (Scheme 2). Analysis of the volatile products by GC‐MS confirmed the identity of ethylene and excluded the formation of any additional organic compounds (Supporting Information, Figure S5). The 1H NMR spectrum obtained upon chloride abstraction on the partially monodeuterated carbenoid 1 a‐d 1 (Supporting Information, Figure S1) exhibits signals with characteristic deuterium couplings arising from the formation of the mono‐ and bi‐deuterated ethylenes 3‐d 1 18 and 3‐d 2 19 alongside 3, as expected from the bimolecular ethylene formation. Furthermore, intermediates of the type 7, in which the chloride has been replaced by the counterion of the scavenger, could be detected in all cases except when using AgSbF6 (Figure 1). The chemical shift for the methylidene resonance of 7 a–d strongly depends on the counterion used and ranged from 3.45 to 5.01 ppm. Despite the apparently counterintuitive trend (chemical shift does not follow the acidity of the counterion), a similar behavior has been observed on the methyl compounds MeX.20
Scheme 2.
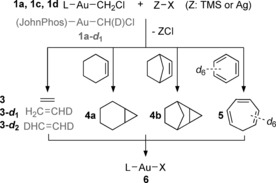
General reactivity of gold carbenoids after activation by chloride scavengers.
Figure 1.
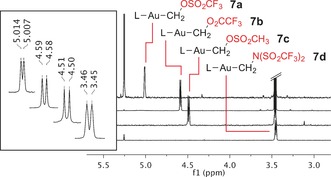
1H NMR spectra of intermediates 7 a–d obtained by reaction of 1 a with different TMSX reagents in [D8]toluene solution.
The reaction of 1 a with an excess of AgOTf (Scheme 3) could be conveniently monitored by 1H NMR spectroscopy to obtain accurate kinetic concentration profiles (Supporting Information, Figure S15) of carbenoid 1 a, triflate substituted species 7 a, and the final gold salt 6 a. Even though ethylene was detected by 1H NMR, its signal cannot be accurately integrated owing to its rapid equilibration with the gas phase. For this reason, ethylene was quantified by means of pressure measurements in a closed system (Supporting Information, Figures S3 and S4).
Scheme 3.
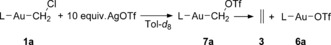
Reaction of 1 a with excess AgOTf used for the determination of the corresponding activation parameters by 1H NMR spectroscopy.
The consumption of carbenoid 1 a follows a first‐order decay as expected from the pseudo first‐order conditions employed. Surprisingly, the subsequent decay of intermediate 7 a into complex 6 a and ethylene also follows a first‐order kinetic regime contrary to what would be expected for a dimerization process and the reported second order homocoupling of well‐characterized methylidene complexes of Ta21 and Re.22 A final yield of 60 % ethylene could be quantified independently in the closed system. More importantly, at room temperature the pressure increase, that is, the formation of ethylene, also follows a first‐order regime in good agreement with the decay of 7 a observed by NMR. Variable‐temperature measurements allowed the determination of the activation parameters for both processes. The initial substitution of chloride for triflate (1 a to 7 a) exhibits values of ΔH ≠=12.9±0.5 kcal mol−1 and ΔS ≠=−19.2±2 cal K−1 mol−1, whereas the conversion of 7 a to 3 and ethylene shows values of ΔH ≠=15.0±0.7 kcal mol−1 and ΔS ≠=−21.5±2 cal K−1 mol−1.
Activation of 1 a in THF suppressed the formation of ethylene and yielded instead a gold‐stabilized oxonium ylide 8 (Figure 2). Trapping is irreversible and no ethylene is formed even upon heating of the sample. Despite the lack of reactivity of 8, its formation hinted at the possibility of an intermolecular trapping of the methylene moiety with a suitable substrate. When the activation of the new carbenoid complexes was carried out in the presence of olefins, methylene transfer to form cyclopropanes 4 alongside ethylene was observed (Scheme 2). This process closely resembles the gold‐catalyzed cyclopropanation of olefins with diazo compounds.23 Thus, complexes 1 a and 1 c could methylenate cyclohexene and norbornene, yielding norcarene 4 a and exo‐tricyclo[3.2.1.02,4]octane 4 b in moderate to good yields when activated with either a silver salt or a TMS reagent (Table 2, entries 1a,b–4a,b). Phosphite complex 1 d could only generate traces of cyclopropanes with cyclohexene (Table 2, entries 5a,b and 6a,b). These results highlight the aptitude of good σ‐donor and poor π‐acceptor ligands (phosphines and N‐heterocyclic carbenes) to enhance the stabilization of carbene fragments by gold.4 Cycloheptatriene 5 was formed when the activation of carbenoids 1 a and 1 c was performed in benzene23a (Table 2, entries 1c–4c). In this case, initial methylenation to form norcaradiene is followed by electrocyclic opening to yield 5.24 Carbenoid 1 d yielded only traces of 5 in parallel with its low reactivity towards cyclopropanation (Table 2, entries 5c, 6c).
Figure 2.
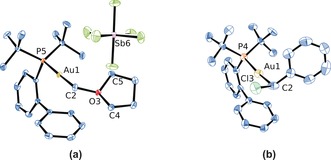
Molecular structures of complexes a) 8 and b) 9.38 ORTEP plots with ellipsoids set at 50 % probability; solvent molecules and hydrogen atoms are omitted for clarity. Selected bond distances [Å] and angles [°] for 8: Au1–C2 2.065(2), C2–O3 1.517(2), C4–O3 1.479(2), C5–O3 1.486(2), Au1–P5 2.3069(5); C2‐O3‐C4 117.8(1), C2‐O3‐C5 115.4 (1), C4‐O3‐C5 108.8(1); and 9: Au1–C2 2.056(1), C2–Cl3 1.838(1), Au1–P4 2.287(2); Au1‐C2‐Cl3 106.7(5).
Table 2.
Yields[a] for methylene transfer from complexes 1 a,c,d to olefins[b] to form cyclopropanes 4 a,b and to benzene[c] to form cycloheptatriene 5 (see Scheme 2).
| Entry | Complex | Chloride scavenger | a) | b) | c) |
|---|---|---|---|---|---|
| 4 a | 4 b | 5 | |||
| 1 | 1 a | TMSNTf2 | 51 | 97 | 12 |
| 2 | 1 a | AgNTf2 | 60 | 74 | 7 |
| 3 | 1 c | TMSNTf2 | 67 | 75 | 28 |
| 4 | 1 c | AgNTf2 | 45 | 51 | 12 |
| 5 | 1 d | TMSNTf2 | 0 | 0 | 2 |
| 6 | 1 d | AgNTf2 | 4 | 0 | 1 |
[a] Yield determined from 1H NMR spectroscopy (average of two runs). [b] Reactions performed with 0.017 mmol of carbenoid complexes and 20 equiv of olefin in CD2Cl2 solution. [c] Same scale as cyclopropanation reactions using [D6]benzene as solvent and substrate.
The formation of gold carbenoids is not limited to the insertion of diazomethane. Reaction of phenyldiazomethane with [(JohnPhos)AuCl] provided chloro(phenyl)methylgold carbenoid 9 (Figure 3). In comparison with its chloromethyl analogues, complex 9 exhibits a longer C−Cl bond at 1.838(1) Å and a more acute Au‐C‐Cl angle of 106.7(5) degrees.25 Activation of carbenoid complex 9 with TMSOTf in CD2Cl2 solution showed complete consumption of 9 and formation of homocoupling products cis‐ and trans‐stilbene 10 together with their cyclopropanation product 1,2,3‐triphenylcyclopropane 11 among other not identified products (Scheme 4). The observed reactivity closely resembles that previously reported for the transition‐metal‐catalyzed decomposition of phenyldiazomethane26 and the cyclopropanation of stilbenes by gold carbenes generated by retro‐Buchner reaction (decarbenation).27
Figure 3.
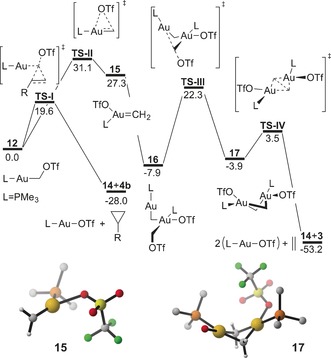
Calculated reaction profiles for the cyclopropanation of norbornene and formation of ethylene on the model system. DFT calculations were performed at B3LYP‐D3/6‐31G(d,p)+SDD(f,g) on Au. Toluene was represented with the PCM. Free energies are in kcal mol−1. Optimized structure of the proposed intermediates 15 and 17 are shown. Non‐CH2 H atoms as well as front facing TfO− in 17 are omitted for clarity. Selected bond distances [Å] and angles [°] for 15: Au–CH2 1.906, Au–O 2.281,30 Au–P 2.403; H2C‐Au‐O 144.2, H2C‐Au‐P 139.7, O‐Au‐P 76.1; and 17: Au–CH2 2.079, Au–Au 2.983, CH2–CH2 2.461, Au–O 2.222, Au–P 2.406; H2C‐Au‐O 100.5, H2C‐Au‐P 98.4, O‐Au‐P 87.9.
Scheme 4.
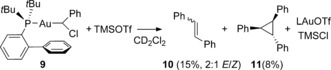
Activation of complex 9 with TMSOTf. Yields determined by GC‐FID.
The possible reaction mechanisms leading to cyclopropanes and the kinetically unusual formation of ethylene were examined on a model system by means of DFT calculations (Figure 3). Initially, a purely dissociative mechanism analogous to the proposed formation of carbenes from carbenoids of Pd28 and Ni29 was considered. Complete dissociation of the triflate anion from the activated carbenoid 12 leads to the formation of a gold methylidene 13 from which cyclopropanation of norbornene can occur without an apparent barrier (Supporting Information, Figure S31). This scenario is however unlikely as the energy required to split a neutral molecule into two charged species is prohibitively high (70.9 kcal mol−1). Nevertheless, a species corresponding to cationic [(JohnPhos)AuCH2]+ could be experimentally detected upon ESI‐MS of 1 a, suggesting that in sufficiently energetic conditions gold(I) carbenes can be accessed from stable carbenoids.
Cyclopropanation can alternatively occur via a three‐centered transition state TS‐I, in analogy to the Simmons–Smith reaction,31 leading to [LAuOTf] (14) and cyclopropane 4 b (Figure 3). This pathway is energetically accessible and can be considered competent for the formation of cyclopropanes. Alternative mechanisms involving reductive elimination from metallacyclobutane32 structures were also considered but found to be unlikely (Supporting Information, Figure S33). No path for the formation of ethylene from two carbenoids 12 could be located. It should be noted that, to the best of our knowledge, no homocoupling of a Simmons–Smith carbenoid has been reported to date. Ethylene most probably arises from the coupling of two bridging methylene units in a dimeric structure as originally proposed for Re22 and Sc33 methylidenes and found experimentally in well‐characterized Co34 and Rh35 complexes (Scheme 5).
Scheme 5.
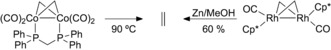
Reported formation of ethylene from well‐characterized dimeric complexes of Co34 and Rh35 containing two bridging methylene units.
To reach the analogous dimeric structure 17 (Figure 3), from which ethylene can be generated with a low energy barrier over transition state TS‐IV, we propose the involvement of the neutral gold carbene 15. This species can be formed by migration of the triflate anion from the carbon atom to the gold center via TS‐II. Intermediate 15 exhibits a distorted trigonal planar geometry around gold center and a short Au−CH2 bond length (1.906 vs. 2.087 Å in 12). Triflate remains coordinated to gold as evidenced by the Au−O distance (2.281 vs. 2.101 Å in 14). Three‐gold(I) complexes bearing two neutral ligands and one halogen are known and have been structurally characterized.36 The analogous species 15 b could be located using the complete JohnPhos ligand and despite the increased steric demand imposed by the full ligand 15 b lies only 28.8 kcal mol−1 above the parent carbenoid 7 a (Supporting Information, Figure S35). Carbene 15 can react with one molecule of carbenoid 12 to yield the AuI−AuIII dimer 16 without an apparent barrier by a formal oxidative addition.37 The AuI center in this intermediate can perform an intramolecular SN2 type attack on the CH2OTf moiety bound to the AuIII center over TS‐III, yielding 17 in a second oxidative addition step. Since the formation of 15 (RDS according to our calculations) and the ethylene extrusion are both unimolecular reactions, the experimentally observed first order decay of 7 a can be rationalized in terms of our proposed mechanism. The difference in activation energies for cyclopropanation and ethylene formation processes accounts for the observed reactivity pattern, while cyclopropanations can be performed at very low temperatures, ethylene generation only reaches completion after more than one hour at room temperature.
In summary, we have developed a simple method for the preparation of well‐defined gold(I) carbenoids [LAuCH2Cl] that, upon activation with a chloride scavenger, exhibit the reactivity expected from gold carbenes in solution, that is, homocoupling, olefin cyclopropanation, and Buchner reaction. We expect these complexes to become a useful tool for the examination and mechanistic understanding of processes involving gold carbenes in solution, particularly elusive methylidenes, which have been so far primarily studied in the gas phase.
In memory of José Barluenga
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge funding from MINECO (CTQ2013‐42106‐P, Severo Ochoa Excellence Accreditation 2014–2018, SEV‐2013‐0319, and FPI fellowship to C.G.M.), the European Research Council (Advanced Grant No. 321066), the AGAUR (2014 SGR 818), Swiss National Science Foundation (Early Postdoc. Mobility fellowship to J.M.S.T.), and CERCA Programme/ Generalitat de Catalunya. We thank Prof. Pedro J. Pérez and Dr. Manuel R. Fructos at CIQSO (University of Huelva) for their help in the quantification of gaseous ethylene. We also thank the reviewers of this manuscript for their valuable suggestions, Dr. Dirk Spiegel for additional work, and the help of the ICIQ X‐ray and NMR research support units.
J. M. Sarria Toro, C. García-Morales, M. Raducan, E. S. Smirnova, A. M. Echavarren, Angew. Chem. Int. Ed. 2017, 56, 1859.
References
- 1. Carpenter B. K., Harvey J. N., Orr-Ewing A. J., J. Am. Chem. Soc. 2016, 138, 4695–4705. [DOI] [PubMed] [Google Scholar]
- 2. Rouhi A. M., Chem. Eng. News 2002, 80, 29–38. [Google Scholar]
- 3.
- 3a. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Egger J., Carreira E. M., Nat. Prod. Rep. 2014, 31, 449–455; [DOI] [PubMed] [Google Scholar]
- 3c. Andrew G. H. W., Curr. Org. Synth. 2006, 3, 499–555. [Google Scholar]
- 4.
- 4a. Harris R. J., Widenhoefer R. A., Chem. Soc. Rev. 2016, 45, 4533–4551; [DOI] [PubMed] [Google Scholar]
- 4b. Wang Y., Muratore M. E., Echavarren A. M., Chem. Eur. J. 2015, 21, 7332–7339; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Fructos M. R., Díaz-Requejo M. M., Pérez P. J., Chem. Commun. 2016, 52, 7326–7335. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Harris R. J., Widenhoefer R. A., Angew. Chem. Int. Ed. 2014, 53, 9369–9371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9523–9525; [Google Scholar]
- 5b. Hussong M. W., Rominger F., Krämer P., Straub B. F., Angew. Chem. Int. Ed. 2014, 53, 9372–9375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9526–9529; [Google Scholar]
- 5c. Joost M., Estévez L., Mallet-Ladeira S., Miqueu K., Amgoune A., Bourissou D., Angew. Chem. Int. Ed. 2014, 53, 14512–14516; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14740–14744. [Google Scholar]
- 6. Benitez D., Shapiro N. D., Tkatchouk E., Wang Y., Goddard W. A., Toste F. D., Nat. Chem. 2009, 1, 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Ringger D. H., Kobylianskii I. J., Serra D., Chen P., Chem. Eur. J. 2014, 20, 14270–14281; [DOI] [PubMed] [Google Scholar]
- 7b. Fedorov A., Batiste L., Bach A., Birney D. M., Chen P., J. Am. Chem. Soc. 2011, 133, 12162–12171; [DOI] [PubMed] [Google Scholar]
- 7c. Fedorov A., Chen P., Organometallics 2010, 29, 2994–3000; [Google Scholar]
- 7d. Fedorov A., Chen P., Organometallics 2009, 28, 1278–1281; [Google Scholar]
- 7e. Fedorov A., Moret M.-E., Chen P., J. Am. Chem. Soc. 2008, 130, 8880–8881. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Zhou S., Li J., Schlangen M., Schwarz H., Chem. Eur. J. 2016, 22, 3073–3076; [DOI] [PubMed] [Google Scholar]
- 8b. Zhou S., Li J., Wu X.-N., Schlangen M., Schwarz H., Angew. Chem. Int. Ed. 2016, 55, 441–444; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 452–455; [Google Scholar]
- 8c. Brown J. R., Schwerdtfeger P., Schröder D., Schwarz H., J. Am. Soc. Mass Spectrom. 2002, 13, 485–492. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Swift C. A., Gronert S., Organometallics 2014, 33, 7135–7140; [Google Scholar]
- 9b. Bertani R., Michelin R. A., Mozzon M., Traldi P., Seraglia R., Busetto L., Cassani M. C., Tagliatesta P., D'Arcangelo G., Organometallics 1997, 16, 3229–3233; [Google Scholar]
- 9c. Chowdhury A. K., Wilkins C. L., J. Am. Chem. Soc. 1987, 109, 5336–5343. [Google Scholar]
- 10.IUPAC, Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”), Blackwell Scientific Publications, Oxford, 1997. [Google Scholar]
- 11.
- 11a. Lebel H., Marcoux J.-F., Molinaro C., Charette A. B., Chem. Rev. 2003, 103, 977–1050; [DOI] [PubMed] [Google Scholar]
- 11b.V. H. Gessner, Chem. Commun 2016, DOI: 10.1039/C1036CC05524A.
- 12.
- 12a. Nesmeyanov A. N., Perevalova É. G., Smyslova E. I., Dyadchenko V. P., Grandberg K. I., Russ. Chem. Bull. 1977, 26, 2417–2419; [Google Scholar]
- 12b. Perevalova E. G., Smyslova E. I., Grandberg K. I., Russ. Chem. Bull. 1982, 31, 2506–2506; [Google Scholar]
- 12c. Perevalova É. G., Struchkov Y. T., Dyadchenko V. P., Smyslova E. I., Slovokhotov Y. L., Grandberg K. I., Russ. Chem. Bull. 1983, 32, 2529–2536; [Google Scholar]
- 12d. Steinborn D., Becke S., Herzog R., Günther M., Kircheisen R., Stoeckli-Evans H., Bruhn C., Z. Anorg. Allg. Chem. 1998, 624, 1303–1307. [Google Scholar]
- 13.
- 13a. Kühnel E., Laffan D. D. P., Lloyd-Jones G. C., Martínez del Campo T., Shepperson I. R., Slaughter J. L., Angew. Chem. Int. Ed. 2007, 46, 7075–7078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7205–7208; [Google Scholar]
- 13b. Aoyama T., Shioiri T., Tetrahedron Lett. 1990, 31, 5507–5508. [Google Scholar]
- 14.A similar strategy had been employed for the synthesis of analogous platinum(II) complexes: Bergamini P., Bortolini O., Costa E., Pringle P. G., Inorg. Chim. Acta 1996, 252, 33–37. [Google Scholar]
- 15. Groom C. R., Bruno I. J., Lightfoot M. P., Ward S. C., Acta Crystallogr. Sect. B 2016, 72, 171–179. For a detailed analysis of bond lengths and angles in these structures see supporting information. [Google Scholar]
- 16. Vetter A. J., Rieth R. D., Brennessel W. W., Jones W. D., J. Am. Chem. Soc. 2009, 131, 10742–10752. [DOI] [PubMed] [Google Scholar]
- 17.The statistical mode of the M-C-Cl angle in all reported structures is 116.2 degrees.
- 18. Reddy G. S., Goldstein J. H., J. Mol. Spectrosc. 1962, 8, 475–484. [Google Scholar]
- 19. Chetcuti M. J., Chisholm M. H., Folting K., Haitko D. A., Huffman J. C., J. Am. Chem. Soc. 1982, 104, 2138–2146. [Google Scholar]
- 20.
- 20a. Ravenscroft M., Roberts R. M. G., Tillett J. G., J. Chem. Soc. Perkin Trans. 2 1982, 1569–1572; [Google Scholar]
- 20b. Zefirov N. S., Zhdankin V. V., Makhon'kova G. V., Dan'kov Y. V., Koz'min A. S., J. Org. Chem. 1985, 50, 1872–1876; [Google Scholar]
- 20c. Abraham R. J., Tormena C. F., Rittner R., J. Chem. Soc. Perkin Trans. 2 2001, 815–820; [Google Scholar]
- 20d. Kübler P., Sundermeyer J., Dalton Trans. 2014, 43, 3750–3766. [DOI] [PubMed] [Google Scholar]
- 21. Schrock R. R., Sharp P. R., J. Am. Chem. Soc. 1978, 100, 2389–2399. [Google Scholar]
- 22. Merrifield J. H., Lin G. Y., Kiel W. A., Gladysz J. A., J. Am. Chem. Soc. 1983, 105, 5811–5819. [Google Scholar]
- 23.
- 23a. Fructos M. R., Belderrain T. R., de Frémont P., Scott N. M., Nolan S. P., Díaz-Requejo M. M., Pérez P. J., Angew. Chem. Int. Ed. 2005, 44, 5284–5288; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5418–5422; [Google Scholar]
- 23b. Prieto A., Fructos M. R., Díaz-Requejo M. M., Pérez P. J., Pérez-Galán P., Delpont N., Echavarren A. M., Tetrahedron 2009, 65, 1790–1793. [Google Scholar]
- 24. Dzhemilev U. M., Dokichev V. A., Sultanov S. Z., Sadykov R. A., Tolstikov G. A., Nefedov O. M., Russ. Chem. Bull. 1991, 40, 945–950. [Google Scholar]
- 25.Only one crystallographically characterized Os compound contains a similar chloro(methyl)phenyl moiety as a part of a metallacycle resulting from the activation of PPh3 ligand: Hoskins S. V., Rickard C. E. F., Roper W. R., J. Chem. Soc. Chem. Commun. 1984, 1000–1002. [Google Scholar]
- 26. Shankar B. K. R., Shechter H., Tetrahedron Lett. 1982, 23, 2277–2280. [Google Scholar]
- 27. Solorio-Alvarado C. R., Wang Y., Echavarren A. M., J. Am. Chem. Soc. 2011, 133, 11952–11955. [DOI] [PubMed] [Google Scholar]
- 28. Fillion E., Taylor N. J., J. Am. Chem. Soc. 2003, 125, 12700–12701. [DOI] [PubMed] [Google Scholar]
- 29. Künzi S. A., Sarria Toro J. M., den Hartog T., Chen P., Angew. Chem. Int. Ed. 2015, 54, 10670–10674; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10817–10821. [Google Scholar]
- 30.For comparison, the Au−O distance in [(JhonPhos)AuOTf] is 2.113(2) Å. Homs A., Escofet I., Echavarren A. M., Org. Lett. 2013, 15, 5782–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.
- 31a. Nakamura M., Hirai A., Nakamura E., J. Am. Chem. Soc. 2003, 125, 2341–2350; [DOI] [PubMed] [Google Scholar]
- 31b. Bernardi F., Bottoni A., Miscione G. P., J. Am. Chem. Soc. 1997, 119, 12300–12305; [Google Scholar]
- 31c. Fang W.-H., Phillips D. L., Wang D.-q., Li Y.-L., J. Org. Chem. 2002, 67, 154–160. [DOI] [PubMed] [Google Scholar]
- 32.A. A. S. W. Tchawou, M. Raducan, P. Chen, Organometallics 2016, DOI: 10.1021/acs.organomet.6b00531.
- 33.Homocoupling of scandium methylidenes: D. S. Levine, T. D. Tilley, R. A. Andersen, Organometallics 2016, DOI: 10.1021/acs.organomet.6b00394.
- 34. Laws W. J., Puddephatt R. J., J. Chem. Soc. Chem. Commun. 1984, 116–117. [Google Scholar]
- 35. Saez I. M., Andrews D. G., Maitlis P. M., J. Organomet. Chem. 1987, 334, C17–C19. [Google Scholar]
- 36.
- 36a. Gimeno M. C., Laguna A., Chem. Rev. 1997, 97, 511–522; [DOI] [PubMed] [Google Scholar]
- 36b.For the structure of [(PPh3)2AuI] see: Fränkel R., Kniczek J., Ponikwar W., Nöth H., Polborn K., Fehlhammer W. P., Inorg. Chim. Acta 2001, 312, 23–39; [Google Scholar]
- 36c.for the structure of [(Xantphos)AuCl], see: Saito H., Miyahara T., Zhong C., Sawamura M., Organometallics 2009, 28, 4829–4840. [Google Scholar]
- 37.
- 37a. Tamaki A., Kochi J. K., J. Organomet. Chem. 1972, 40, C81–C84; [Google Scholar]
- 37b. Johnson A., Puddephatt R. J., Inorg. Nucl. Chem. Lett. 1973, 9, 1175–1177. [Google Scholar]
- 38. CCDC 1504818 (1 a), 1504819 (1 b), 1504820 (1 c), 1504821 (8), and 1504822 (9) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary