

Abstract
Formylglycine‐generating enzyme (FGE) is an O2‐utilizing oxidase that converts specific cysteine residues of client proteins to formylglycine. We show that CuI is an integral cofactor of this enzyme and binds with high affinity (K D=of 10−17 m) to a pair of active‐site cysteines. These findings establish FGE as a novel type of copper enzyme.
Keywords: copper, EPR, formylglycine, oxidases, oxygen activation, protein engineering
Formylglycine‐generating enzymes (FGEs) catalyze the O2‐dependent conversion of specific cysteine residues on client proteins to formylglycine (fGly; Scheme 1). This post‐translational modification is essential for the catalytic activity of phosphatases and sulfatases.1 Reduced FGE activity in human cells leads to sulfatase deficiency.2 In addition, FGE has emerged as a versatile tool for protein engineering, because it can introduce unique aldehyde functions into recombinant proteins.1a, 3 Initial biochemical and structural characterization of this enzyme raised an interesting mechanistic question: how does this enzyme activate O2? None of the published crystal structures of this catalyst revealed any known redox cofactor.4 The only redox‐active features in the active site are two conserved cysteine residues, which by themselves can hardly activate O2.5, 6 One‐electron transfers between thiols and O2 are prohibited by mismatched redox potentials, and ionic mechanisms are spin forbidden.
Scheme 1.
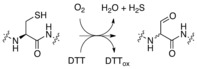
FGE catalyzes O2‐dependent conversion of cysteine residues to formylglycine (fGly), H2S, and water. The enzyme requires an auxiliary reducing agent such as DTT to complete the four‐electron reduction of O2
Recently we and others found that copper salts increase the in vitro activity of FGE up to 20‐fold.7 Although copper is a plausible agent in O2 activation,8 the interaction between FGE and transition metals remained mysterious. The general absence of copper in published crystal structures suggests that a potential FGE:copper complex would not be very stable.4 On the other hand, CuI‐dependent FGE activity is not affected by millimolar concentrations of EDTA,7b which is a strong ligand for CuII (pK D=18.8) or DTT (a strong ligand for CuI, pK D=15.3).9 Hence, either FGE is an even stronger copper ligand, or it does not require direct metallation. In the later scenario copper might serve as an artificial source of electrons or activated oxygen species in the in vitro assay.10
Here we address this puzzle. We show that FGE from Thermomonospora curvata (FGEcurvata) binds CuI with an affinity similar to that of known high‐affinity copper proteins.9 We found that CuI binds to both active‐site cysteines and remains bound throughout multiple catalytic cycles. We have previously shown that the active‐site cysteines of FGEcurvata readily form a disulfide bond under aerobic conditions.7b For unambiguous determination of the redox state of these two residues, we constructed an FGE variant in which all cysteines outside the active site were mutated to either serine or alanine (FGE4C).7b This variant proved sevenfold more active than wild‐type,7b and therefore was used in most of our subsequent experiments. FGE4C also contained a disulfide bond after purification in the absence of reducing agents.7b
We used a published titration assay to estimate the CuI affinities of FGEcurvata and FGE4C.9 A solution containing the 1:2 complex between CuI and bathocuproine disulfonate (CuI:(BCS)2) was titrated with FGE4C (Figure S1 in the Supporting Information). The transfer of CuI from the complex to FGE was monitored by measuring the decreasing absorption of CuI:(BCS)2 at 483 nm. This qualitative experiment revealed that oxidized FGE4C has no greater affinity for CuI than the control protein (BSA). In contrast, FGE4C purified in reduced form showed significant CuI affinity (Figure S1). Because the oxidized and reduced forms of FGE4C differ only by the redox state of the two active‐site cysteines, we concluded that these thiols must be essential for CuI binding.
For a more quantitative estimation of the copper affinity of FGE (K D) we recorded the absorption at 483 nm as a function of FGE4C or FGEcurvata concentration and fitted the resulting curves to an equation describing the equilibrium between the CuI:(BCS)2 and FGE:CuI complexes (Supporting Information).9 All titration buffers contained 2 mm cysteamine to keep the enzymes in reduced form. Because cysteamine is a comparably weak CuI binder (K D=10−14.1 m; Table 1), its presence should not affect the apparent CuI affinity of FGE dramatically. As a test of this assumption we determined the apparent CuI affinity of DTT in the absence (K D=10−15.1 m) and in the presence (K D=10−15.6 m) of cysteamine. Both values are in fair agreement with a published value (K D=10−15.3 m).9 With this assay we determined an apparent dissociation constant (K D, Table 1) of the CuI complexes with FGE4C or FGEcurvata. As both proteins bind CuI with similar strengths we concluded that none of the Cys residues outside the active site contributes to copper binding. Similar complex stabilities have been reported for copper chaperones from human (Atox1, K D=10−17 m),9 Saccharomyces cerevisiae (Atx1, K D=10−17 m)9 and Bacillus subtilis (CopZ, K D=10−17 m),11 thus suggesting that FGE should be well equipped to procure copper in a cellular context.12
Table 1.
Kinetic parameters and CuI affinities of FGE variants and auxiliary thiols.[a]
| k cat [min−1] | K M [μm] | k cat/K M [min−1 m −1] | pK | |
|---|---|---|---|---|
| FGEcurvata | 1.6±0.1 | 580±40 | 2 900±50 | 17.1[b] |
| FGE4C | 4.2±0.5 | 230±40 | 20 000±4 000 | 17.1[b] |
| FGES266A | 0.006 | 520±240 | 49±8 | 17.7[b] |
| FGES290K | n.d. | n.d. | 6.6±0.2 | 16.7[b] |
| FGEC269S | n.d. | n.d. | ≤1 | n.d. |
| FGEC274S | n.d. | n.d. | ≤1 | n.d. |
| cysteamine | 14.1[c] | |||
| DTT | 15.1,[c] 15.6[b] | |||
| DTBA | 15.8[c] |
[a] Michaelis–Menten parameters for FGE‐catalyzed oxidation of a Cys‐containing peptide to the fGly‐containing product (Supporting Information). Apparent dissociation constants (K D) of CuI complexes with FGE variants or low‐molecular‐weight thiols were determined by using a published titration assay.9 [b] Values determined in the presence of 2 mm cysteamine. [c] Values determined in the absence of additional thiols. n.d.: low specific activities prevented accurate determination of these parameters.
Despite the remarkable copper affinity of FGE, it remains puzzling that this enzyme is fully active in a millimolar DTT solution. The apparent CuI affinity of DTT is 100 times lower than that of FGE. A 1000‐fold excess of DTT should therefore destabilize the FGE:CuI complex. This is not what we observed. Reactions containing 2 μm FGE, 2 μm CuI, and 2 mm of either cysteamine (K D=10−14.0 m), DTT (K D=10−15.1 m), or dithiobutylamine (DTBA, K D=10−15.8 m)13 displayed approximately the same rate of product formation (Figure S2), thus showing that the CuI affinity of the redox buffer does not influence catalytic activity.
One possible explanation for this behavior could be that the CuI affinity of FGE in the presence of substrate is at least two orders of magnitude higher than for the resting enzyme. We could not directly measure the CuI affinity of the enzyme:substrate complex because the substrate peptide (Abz‐SALCSPTRA‐NH2) is a proficient CuI binder in its own right, thus saturating concentrations of this peptide are incompatible with the titration assay. A substrate analogue containing Ser in place of Cys (Abz‐SALSSPTRA‐NH2) did not interfere with this assay, but also proved a poor FGE ligand (Figure S3). Consequently, the presence of this peptide did not change the CuI affinity of either FGE4C or FGES266A.
As an alternative strategy to gauge the influence of the substrate on CuI binding by FGE, we analyzed the ability of FGE4C and inactive FGE variants to exchange CuI during catalysis. For this, we designed four variants of FGE4C by using a structural model based on the crystal structure of human FGE (Figure 1). We produced two variants, each of which had one of the active site cysteines mutated to serine (FGEC269S and FGEC274S). Both were inactive and unable to bind CuI (Table 1); this suggested that both thiols are important for CuI binding and for catalysis. The third variant had a conserved active‐site serine at position 266 substituted to alanine (FGES266A). This reduced k cat 270‐fold, but did not affect K M, and did not interfere with copper binding (K D=10−17.6 m). The mutation in the fourth variant (FGES290K) was designed to impair substrate binding. Ser290 is at the bottom of the substrate binding groove, more than 15 Å from the catalytic site (Figure 1).4c We mutated this residue to lysine in order to block substrate binding through steric and coulombic repulsion. As expected, the corresponding protein could not be saturated with substrate, and the catalytic efficiency (k cat/K M) was reduced 440‐fold. CuI affinity reduced only 2.5‐fold (K D=10−16.7 m; Table 1).
Figure 1.
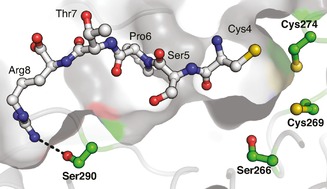
Structural model of FGEcurvata based on the structure of human FGE (PDB code: 2AIJ).4c, 14 Residues 4–8 of the substrate were modeled according to a similar substrate bound to the human enzyme. This model suggests that Ser290 makes a 3.0 Å hydrogen bond to Arg8 on the substrate (dashed line).
We tested the ability of these variants to compete with FGE4C for CuI during catalysis in reactions containing 0.5 μm FGE4C, 0.5 μm CuSO4, 2 mm DTT, 50 mm EDTA, and 200 μm substrate (Figure 2). The reactions were started by addition of FGE4C. After one minute the reaction mixtures were supplemented with a ninefold excess of FGEC269S, FGEC274S, FGES266A, FGES290K, or BSA (Figure 2). FGES266A reduced FGE4C activity approximately 15‐fold (m 2/m 1, Figure 2 A), consistent with redistribution of limiting CuI among 0.5 μm FGE4C and 4.5 μm FGES266A. Addition of more CuI immediately restored full FGE4C activity (Figure S4), thus confirming CuI as the limiting factor.
Figure 2.
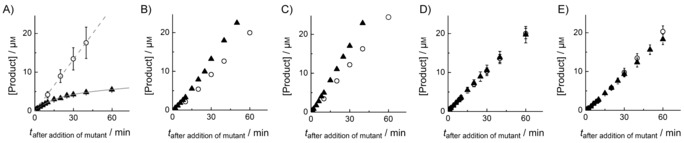
FGE4C‐catalyzed product formation as a function of time (○) in reactions containing 0.5 μm FGE4C, 200 μm substrate, 0.5 μm CuSO4, 2 mm DTT, 50 mm EDTA, 50 mm NaCl and 50 mm Tris (pH 8). The reaction mixtures were supplemented with 5 μm A) FGES266A, B) FGEC274S, C) FGEC269S, D) BSA, or E) FGES290K, one minute after initiation with FGE4C (▴). In A), ▴ data were fitted to the function [P]=A(1−exp(−k t))+m 1 t (—), and the ○ data were fitted to [P]=m 2 t (‐ ‐ ‐ ‐), where A is the concentration of product formed until CuI redistribution between FGE4C and FGES266A is complete (4.0 μm), k is the rate at which the FGE4C:CuI complex decays (0.07 min−1), m 1 is the residual activity after CuI redistribution (m 1/[FGE4C]=0.05 min−1), and m 2 is the activity of CuI‐complemented FGE4C (m 2/[FGE4C]=0.8 min−1). The data are averaged values from two or more independent experiments.
The rate at which CuI redistributed between FGE4C and FGES266A (0.07±0.02 min−1, Figure 2) provides an estimation of how fast the FGE4C:CuI complex decays (k ; Figure 2). This rate is eleven times slower than the catalytic turnover (m 2/[FGE4C]=0.8 min−1), thus suggesting that metal binding and unbinding cannot be part of the catalytic cycle. The same competition experiment showed that neither BSA, FGEC269S, FGEC274S, nor FGES290K can extract CuI from FGE4C. For BSA and the two cysteine variants this result is consistent with their complete lack of CuI affinity (Table 1). FGES290K, however, is a strong CuI binder (Table 1), but its ternary complex with copper and substrate is weak. The observation that FGES290K cannot sequester CuI in the FGE4C‐catalyzed reaction is consistent with the idea that substrate‐binding increases the apparent CuI affinity of FGE. This finding, in combination with the observation that the Ser‐containing substrate analogue is a poor FGE ligand, indicates that the thiol function of the substrate might be the third copper ligand in the active site.
In a final experiment we used EPR spectroscopy to probe the redox state of copper bound to FGE4C. Freeze‐quenched reactions containing FGE4C, CuI, EDTA, DTT, and substrate yielded a featureless EPR spectrum, not significantly different from that measured with a control sample (without added copper; Figure 3). Apparently, the accumulating copper species during catalysis is EPR silent. In contrast, a sample without DTT showed the clear EPR signal of CuII, whereas a control reaction containing no DTT and no added copper was again EPR silent (Figure 3). Accumulation of a CuII species is consistent with the previous observation that FGE4C in absence of DTT oxidizes to the disulfide form, which does not bind copper.
Figure 3.
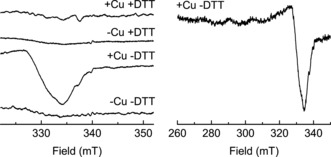
X‐band (∼9.52 GHz) continuous‐wave EPR spectra of 50 μm FGE4C and 0.5 mm substrate in the absence or presence of 50 μm CuSO4 and/or 5 mm DTT. Temperature: 150 K; modulation amplitude: 1 mT; microwave attenuation: 15 dB (6.346 mW); 160 scans each. Spectra were acquired on a Bruker Elexsys 500 spectrometer equipped with a super‐high Q resonator. Left: detail of the region of the strongest CuII signal component. Right: full spectrum of the CuII species in the absence of DTT.
The combination of catalytic and structural analyses of FGE described here and elsewhere4a–4c, 7, 15 strongly implicates FGE as a copper‐metalloenzyme: the active form strongly binds one equivalent of CuI in the active site;7b the CuI:protein complex remains intact throughout the entire catalytic cycle (Figure 2); other transition metals cannot complement FGE,7 thus suggesting that the cofactor engages in redox chemistry;8 FGE reduces O2 by using two electrons from the substrate and two electrons from an auxiliary reducing agent such as DTT;4a, 7b in the presence of DTT the rate‐limiting step is hydrogen‐atom abstraction from the substrate;7b the accumulating species during catalysis is an EPR‐silent species (Figure 3); in the absence of an appropriate reducing agent turnover is much slower,4a, 4b, 7 and a CuII‐containing species accumulates (Figure 3); this oxidized species can slowly turn over by using the substrate thiol as an electron source;7b and finally, addition of a proper reducing agent to this slow reaction immediately reactivates the enzyme.7b, 15
In our view these observations are best explained the following mechanistic proposal (Scheme 2). FGE in the cuprous state (A) binds substrate (B) and O2 to form a cupric superoxo intermediate (C); hydrogen atom transfer (HAT) and electron transfer (ET) from the substrate reduce this intermediate to a CuI hydroperoxo complex (D); the resulting thioaldehyde hydrolyzes to form the fGly‐containing product and hydrogen sulfide, and the hydroperoxo complex collapses into a stable but oxidized form of FGE (E). In the presence of DTT this species is quickly reduced to the active resting state (A). In absence of a proper reducing agent, species e decays to the disulfide form of FGE (F), which does not bind copper. Therefore CuI leaves the active site and oxidizes to CuII. A much slower three‐electron process restores the reduced FGE:CuI complex (A).
Scheme 2.
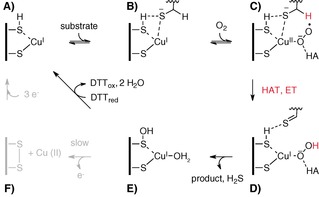
Plausible catalytic mechanism of FGE‐catalyzed formylglycine formation.
The absence of any EPR signature during turnover may be explained by the following scenarios: 1) the cupric superoxo species (C) is not formed or does not accumulate to a significant extent, even though the following HAT is rate‐limiting; 2) species c does accumulate but has a diamagnetic singlet ground‐state because of a highly covalent interaction between CuII and superoxide;16 or 3) the accumulating CuII species has a triplet ground‐state that does not produce an EPR signature in the X‐band spectrum.17 The reactivity of previously characterized CuII superoxo complexes shows that diamagnetic species generally do not cleave C−H bonds,16, 18 whereas paramagnetic species do.19 Based on this, we predict that FGE forms an EPR‐silent paramagnetic CuII superoxo species that mediates homolytic C−H bond cleavage.
A similar sequence of events has been implicated in the catalytic mechanisms of the copper enzymes polysaccharide monooxygenase (PMO)20 and peptidylglycine α‐hydroxylating monooxygenase (PHM).8a, 21 In PHM a CuII superoxo species has been shown to cleave the Cα−H bond of a C‐terminal glycine residue. Electron transfer from a neighboring CuI center forms the CuI hydroperoxo species, which immediately eliminates water to form CuII‐oxyl, which in turn hydroxylates the substrate radical. In PMOs a CuII superoxo species has been proposed to extract a hydrogen atom from the anomeric carbon (C1) in polysaccharides. Electron transfer from an auxiliary reducing agent forms a CuI hydroperoxo complex, followed by CuII‐oxyl formation, and by hydroxylation of the substrate radical. FGE, PMO, and PHM oxidize their substrates by two electrons, and therefore depend on a reducing agent to fully reduce oxygen. PHM activity depends on ascorbate,8a in vitro FGE activity depends on thiols, and PMOs seem to accept electron donors such as gallic acid or the reduced form of cellobiose dehydrogenase.20a Future investigations will investigate to what extent these three reactions follow analogous catalytic mechanisms.
In conclusion, the data show that FGE is a copper‐dependent oxidase. Although the reduced enzyme:CuI complex is very stable, it is highly sensitive to oxidation. The apparent instability under aerobic conditions might explain the previous difficulties in observing CuI‐containing FGE by crystallography. Our discovery raises novel questions about the in vivo copper delivery to FGE, and highlights a potential connection between oxidative stress, copper homeostasis, and sulfatase deficiency in humans.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Laetitia Misson for ESI measurements, and Ken Woycechowsky for valuable comments. This project was supported by the Swiss National Foundation (NRP66) and an ERC starting grant. F.P.S. is supported by the “Professur für Molekulare Bionik”.
M. Knop, T. Q. Dang, G. Jeschke, F. P. Seebeck, ChemBioChem 2017, 18, 161.
References
- 1.
- 1a. Appel M. J., Bertozzi C. R., ACS Chem. Biol. 2015, 10, 72–84; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Schmidt B., Selmer T., Ingendoh A., von Figura K., Cell 1995, 82, 271–278; [DOI] [PubMed] [Google Scholar]
- 1c. Jonas S., van Loo B., Hyvönen M., Hollfelder F., J. Mol. Biol. 2008, 384, 120–136. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Cosma M. P., Pepe S., Annunziata I., Newbold R. F., Grompe M., Parenti G., Ballabio A., Cell 2003, 113, 445–456; [DOI] [PubMed] [Google Scholar]
- 2b. Dierks T., Schmidt B., Borissenko L. V., Peng J., Preusser A., Mariappan M., von Figura K., Cell 2003, 113, 435–444. [DOI] [PubMed] [Google Scholar]
- 3. Carrico I. S., Carlson B. L., Bertozzi C. R., Nat. Chem. Biol. 2007, 3, 321–322. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Dierks T., Dickmanns A., Preusser-Kunze A., Schmidt B., Mariappan M., von Figura K., Ficner R., Rudolph M. G., Cell 2005, 121, 541–552; [DOI] [PubMed] [Google Scholar]
- 4b. Carlson B. L., Ballister E. R., Skordalakes E., King D. S., Breidenbach M. A., Gilmore S. A., Berger J. M., Bertozzi C. R., J. Biol. Chem. 2008, 283, 20117–20125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Roeser D., Preusser-Kunze A., Schmidt B., Gasow K., Wittmann J. G., Dierks T., von Figura K., Rudolph M. G., Proc. Natl. Acad. Sci. USA 2006, 103, 81–86; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Roeser D., Schmidt B., Preusser-Kunze A., Rudolph M. G., Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 621–627. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. G. W. Luther III , Findlay A. J., MacDonald D. J., Owings S. M., Hanson T. E., Beinart R. A., Girguis P. R., Front. Microbiol. 2011, 2, 1–9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Huxtable R. J., Biochemistry of the Elements, Vol. 6: Biochemistry of Sulfur, Plenum, New York, 1986, pp. 199–268. [Google Scholar]
- 6. Toscano M. D., Woycechowsky K. J., Hilvert D., Angew. Chem. Int. Ed. 2007, 46, 3212–3236; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3274–3300. [Google Scholar]
- 7.
- 7a. Holder P. G., Jones L. C., Drake P. M., Barfield R. M., Bañas S., de Hart G. W., Baker J., Rabuka D., J. Biol. Chem. 2015, 290, 15730–15745; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Knop M., Engi P., Lemnaru R., Seebeck F. P., ChemBioChem 2015, 16, 2147–2150. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Klinman J. P., J. Biol. Chem. 2006, 281, 3013–3016; [DOI] [PubMed] [Google Scholar]
- 8b. Prigge S. T., Eipper B. A., Mains R. E., Amzel L. M., Science 2004, 304, 864–867. [DOI] [PubMed] [Google Scholar]
- 9. Xiao Z., Brose J., Schimo S., Ackland S. M., La Fontaine S., Wedd A. G., J. Biol. Chem. 2011, 286, 11047–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Speisky H., Gómez M., Carrasco-Pozo C., Pastene E., Lopez-Alarcon C., Olea-Azar C., Bioorg. Med. Chem. 2008, 16, 6568–6574. [DOI] [PubMed] [Google Scholar]
- 11. Badarau A., Dennison C., J. Am. Chem. Soc. 2011, 133, 2983–2988. [DOI] [PubMed] [Google Scholar]
- 12. Rae T. D., Schmidt P. J., Pufahl R. A., Culotta V. C., O'Halloran T. V., Science 1999, 284, 805–808. [DOI] [PubMed] [Google Scholar]
- 13. Lukesh J. C., Palte M. J., Raines R. T., J. Am. Chem. Soc. 2012, 134, 4057–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnold K., Bordoli L., Kopp J., Schwede T., Bioinformatics 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- 15. Peng J. H., Alam S., Radhakrishnan K., Mariappan M., Rudolph M. G., May C., Dierks T., von Figura K., Schmidt B., FEBS J. 2015, 282, 3262–3274. [DOI] [PubMed] [Google Scholar]
- 16. Chen P., Root D. E., Campochiaro C., Fujisawa K., Solomon E. I., J. Am. Chem. Soc. 2003, 125, 466–474. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lanci M. P., Smirnov V. V., Cramer C. J., Gauchenova E. V., Sundermeyer J., Roth J. P., J. Am. Chem. Soc. 2007, 129, 14697–14709; [DOI] [PubMed] [Google Scholar]
- 17b. Donoghue P. J., Gupta A. K., Boyce D. W., Cramer C. J., Tolman W. B., J. Am. Chem. Soc. 2010, 132, 15869–15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Spencer D. J. E., Aboelella N. W., Reynolds A. M., Holland P. L., Tolman W. B., J. Am. Chem. Soc. 2002, 124, 2108–2109; [DOI] [PubMed] [Google Scholar]
- 18b. Aboelella N. W., Lewis E. A., Reynolds A. M., Brennessel W. W., Cramer C. J., Tolman W. B., J. Am. Chem. Soc. 2002, 124, 10660–10661; [DOI] [PubMed] [Google Scholar]
- 18c. Aboelella N. W., Kryatov S. V., Gherman B. F., Brennessel W. W., V. G. Young, Jr. , Sarangi R., Rybak-Akimova E. V., Hodgson K. O., Hedman B., Solomon E. I., Cramer C. J., Tolman W. B., J. Am. Chem. Soc. 2004, 126, 16896–16911; [DOI] [PubMed] [Google Scholar]
- 18d. Aboelella N. W., Gherman B. F., Hill L. M. R., York J. T., Holm N., V. G. Young, Jr. , Cramer C. J., Tolman W. B., J. Am. Chem. Soc. 2006, 128, 3445–3458; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18e. Fujisawa K., Tanaka M., Moro-oka Y., Kitajima N., J. Am. Chem. Soc. 1994, 116, 12079–12080. [Google Scholar]
- 19.
- 19a. Peterson R. L., Himes R. A., Kotani H., Suenobu T., Tian L., Siegler M. A., Solomon E. I., Fukuzumi S., Karlin K. D., J. Am. Chem. Soc. 2011, 133, 1702–1705; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Kunishita A., Kubo M., Sugimoto H., Ogura T., Sato K., Takui T., Itoh S., J. Am. Chem. Soc. 2009, 131, 2788–2789; [DOI] [PubMed] [Google Scholar]
- 19c. Kim S., lee J. Y., Cowley R. E., Ginsbach J. W., Siegler M. A., Solomon E. I., Karlin K. D., J. Am. Chem. Soc. 2015, 137, 2796–2799; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Sánchez-Eguía B. N., Flores-Alamo M., Orio O., Castillo I., Chem.Commun. 2015, 51, 11134–11137. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Beeson W. T., Vu V. V., Span E. A., Phillips C. M., Marletta M. A., Annu. Rev. Biochem. 2015, 84, 923–946; [DOI] [PubMed] [Google Scholar]
- 20b. Phillips C. M., Beeson W. T., Cate J. H., Marletta M. A., ACS Chem. Biol. 2011, 6, 1399–1406. [DOI] [PubMed] [Google Scholar]
- 21. Evans J. P., Blackburn N. J., Klinman J. P., Biochemistry 2006, 45, 15419–15429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary