

Abstract
Background
Succinate, in addition to its role as an intermediary of the citric acid cycle, acts as an alarmin, initiating and propagating danger signals resulting from tissue injury or inflammatory stimuli. The contribution of this immune sensing pathway to the development of allergic and inflammatory responses is unknown.
Methods
Ear thickness of wild‐type (wt) and Sucnr1‐deficient (Sucnr1 −/−) mice, sensitized and challenged with oxazolone, was used as a criterion to assess the relevance of SUCNR1/GPR91 expression mediating allergic contact dermatitis (ACD). Results obtained in this system were contrasted with data generated using passive cutaneous anaphylaxis, ovalbumin‐induced asthma and arthritis models.
Results
We found augmented ACD reactions in Sucnr1 −/− mice. This observation correlated with increased mast cell activation in vitro and in vivo. However, exacerbated mast cell activation in Sucnr1 −/− mice did not contribute to the enhancement of asthma or arthritis and seemed to be due to alterations during mast cell development as augmented mast cell responses could be recapitulated in wt mast cells differentiated in the absence of succinate.
Conclusions
A deficiency in succinate sensing during mast cell development confers these cells with a hyperactive phenotype. Such a phenomenon does not translate into exacerbation of asthma or mast cell‐dependent arthritis. On the contrary, the fact that Sucnr1 −/− mice developed reduced arthritic disease, using two different in vivo models, indicates that GPR91 antagonists may have therapeutic potential for the treatment of allergic and autoimmune diseases.
Keywords: alarmins, allergy, autoimmunity, immunometabolism, succinate
Succinate is an intermediate of the citric acid cycle, which is generated in mitochondria following hydrolytic release of CoA from succinyl‐CoA. Succinate can accumulate in mitochondria and gets exported to the extracellular milieu when there is an imbalance between nutrients and/or oxygen and energy demands 1. Extracellular succinate binds to its specific receptor, SUCNR1/GPR91 2, on the plasma membrane of hepatic, renal, retinal and immune cells, instructing them to secrete hormones, growth factors or cytokines involved in multiple normal and pathologic processes 3. There is evidence indicating that triggering GPR91 prevents premature subretinal age‐related macular degeneration‐like lesions 4 and facilitates the recovery from cerebral hypoxic–ischemic events 5. On the other hand, the sensory function of GPR91 detecting succinate following ischemic stress has been implicated in the mechanisms governing diabetic retinopathy 6, 7, causing cardiac hypertrophy 8, 9 and eliciting hepatic stellate cell activation 10 and hepatic fibrosis 11. In addition to hypoxia, elevated glucose levels, as observed in diabetes, also result in the export of succinate to the extracellular environment 12, 13. In this case, increased interstitial levels of succinate are also sensed by GPR91 in the kidney juxtaglomerular apparatus 14 and macula densa cells 15, causing the release of renin, which ultimately lead to hypertension and renal tissue injury 13. Furthermore, our laboratory has previously shown that succinate can be released as a consequence of necrosis following tissue damage. We demonstrated that immature dendritic cells (DC) express GPR91 and utilize this receptor to sense immunologic danger and to enhance antigen‐presenting functions required for optimal T‐cell activation 16. We hypothesized that GPR91 plays a broader role in immune regulation, and we therefore aimed to examine its pathophysiologic or protective role in the context of allergic and inflammatory challenges.
Methods
Animals
Sucnr1‐deficient (Sucnr1 −/−) mice were generated as previously described 16. The original model was generated in the C57BL/6N background. These mice featured the single‐nucleotide deletion in the Crb1 gene reported by Mattapallil et al. 17, which is present in all C57BL/6N substrains and can lead to ocular phenotypes. Thus, Sucnr1 −/− mice were backcrossed (>10 generations) onto the C57BL/6J background (Janvier Labs, Le Genest‐Saint‐Isle, France), which does not carry this mutation. The immune phenotype and functions of the Sucnr1 −/− mice on C57BL/6N and C57BL/6J backgrounds were equivalent. Furthermore, the immune phenotype of Sucnr1 −/− mice was normal and comparable to that of wild‐type (wt) mice (Fig. S1). All experiments were performed using 8‐ to 16‐week‐old mice. All animal studies described in this report were performed according to Swiss animal protection laws issued by the Cantonal Veterinary Office Basel‐Stadt, Switzerland, by the Austrian Animal Experimentation Law and by the Novartis Animal Welfare Policy.
Animal models
Allergic and inflammatory responses were conducted in parallel on wt and Sucnr1 −/− mice. Elicitation and sensitization for allergic contact dermatitis (ACD) was performed with 1% oxazolone (Sigma‐Aldrich, Buchs, Switzerland) in acetone as previously described 18. RNA was extracted from ear tissue and subjected to real‐time RT‐PCR analysis for mouse EF‐1α, IL‐1β, IL‐4, IL‐5, IL‐13, IL‐17, IFN‐γ and TNF‐α. Irritant contact dermatitis (ICD) was induced by a single topical application of 10 μl of 0.005% 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) (wt/vol in acetone; Sigma‐Aldrich) on the inner and outer surfaces of the left ears of wt and Sucnr1 −/− mice. The right ears were treated with vehicle alone (acetone). After 6 h, animals were euthanized and right and left ears were removed. Treatment‐induced swelling was calculated as percentage increase in ear weight as described by Sheu et al. 19. Passive cutaneous anaphylaxis (PCA) tests, using PBS or 50 ng IgE (anti‐DNP antibody; BD Biosciences, Allschwil, Switzerland) and subsequent (48 h) i.v. injection with 100 μg DNP‐BSA (Sigma‐Aldrich, Buchs, Switzerland) diluted in 0.5% Evans blue (in 0.9% NaCl; AppliChem, Darmstadt, Germany), were conducted as described elsewhere 20. The ovalbumin (OVA)‐induced allergic asthma model was performed as described by Mojtabavi et al. 21. Briefly, wt and Sucnr1 −/− mice were sensitized i.p. on day 0 with PBS or 10 μg OVA (A550; Sigma) and were boosted i.p. after 21 days. On day 28 and 29, mice were challenged twice a day with either 1% PBS aerosol or 1% OVA aerosol for 1 h. On day 31, mice were killed and bronchoalveolar lavage (BAL) was performed using 2 ml of PBS. Cytospins were made, and slides were fixed with methanol for 5 min. After staining with May–Grünwald–Giemsa solution (Merck, Zug, Switzerland), cellularity of neutrophils, eosinophils, lymphocytes and macrophages in the BAL was analyzed using a hemacytometer. IgE and IgG concentrations were analyzed from BAL and serum by standard sandwich ELISA using specific antibodies (BD Biosciences). Antigen‐induced arthritis (AIA) using methylated bovine serum albumin (mBSA) and K/BxN serum transfer‐induced arthritis were conducted as described previously 22.
Real‐time RT‐PCR
Total cellular RNA was extracted from cells using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. Real‐time RT‐PCR was performed on an ABI7500 Thermocycler using Platinum ThermoScript One‐Step system (Invitrogen, Zug, Switzerland). All mouse gene‐specific mRNA levels were determined using commercial TaqMan GeneAssays (Applied Biosystems, Zug, Switzerland): Eif1a (Mm00456651_m1), Sucnr1 (Mm00519024_m1), Tnf (Mm00443258_m1), Il1b (Mm00434228_m1), Il4 (Mm00445259_m1), Il5 (Mm01290072_g1), Il13 (Mm00434206_g1), Il17 (Mm00521423_m1) and Ifng (Mm99999071_m1). For measurement of human EF1A mRNA, primers and probes (Eurogentec, Liege, Belgium) were designed with primerexpress software (Applied Biosystems, Zug, Switzerland) (forward, 5′‐TTTGAGACCAGCAAGTACTATGTGACT‐3′; reverse 5′‐TCAGCCTGAGATGTCCCTGTAA‐3′; probe 5′‐TCATTGATGCCCCAGGACACAGAGAC‐3′). The expression of gene‐specific human SUCNR1 mRNA was measured with commercial TaqMan GeneAssays kits (Applied Biosystems, Hs00263701_m1).
Mass cytometry by time of flight
Mass cytometry by time of flight (CyTOF) analysis on spleens, inguinal lymph nodes, blood and bone marrow of wt and Sucnr1 −/− mice was performed as previously described 23 using a cocktail of monoclonal antibodies, conjugated to monoisotopic lanthanides (Table S1). All samples (three wt and three Sucnr1 −/−) isolated from a given tissue were barcoded using the Cell‐ID 20‐Plex Pd Barcoding Kit (Fluidigm, SanFrancisco, CA, USA), and acquired simultaneously as a mixture to correct for fluctuations in signal strength during acquisition time. Data analysis was performed using cytobank (Mountain View, CA, USA), spotfire (Palo Alto, CA, USA) software and R‐based algorithms.
Human cell isolation and differentiation
Peripheral blood mononuclear cells were isolated using standard Ficoll density gradients from peripheral blood of healthy volunteers obtained from the Swiss Red Cross after signing informed consent. Human mast cells were differentiated from hematopoietic precursors (Lonza, Switzerland) as described by Rådinger et al. 24.
Generation and activation of mouse bone marrow‐derived mast cells
Bone marrow (BM) cells were obtained by flushing the humeri, femurs and tibias of wt and Sucnr1 −/− mice with HBSS (Gibco, Zug, Switzerland) and subsequently cultured for 4–8 weeks using RPMI‐1640/glutaMAX medium supplemented with 10% FCS, 1% penicillin/streptomycin, 1% MEM essential amino acids, 50 μM β‐ME, 1 mM sodium pyruvate (all from Gibco) and 20 ng/ml IL‐3 (Novartis, Basel, Switzerland). To investigate the role of GPR91 on mast cell development, BM cells were cultured in X‐Vivo 15 medium (Gibco) supplemented with 1% MEM essential amino acids, 50 μM β‐ME, 1 mM sodium pyruvate and 20 ng/ml IL‐3 in the presence or absence of 50 μM succinate. After 4 weeks of cultivation, BM‐derived mast cells (BMMC) were analyzed by flow cytometry using CD11b and CD117 antibodies (BD Biosciences). The purity of the BMMC was always >98%. BMMC were generally stimulated for 1–4 h in 96‐well flat‐bottom plates at a density of 2 × 105 cells/well using supplemented RPMI‐1640/glutaMAX medium and 2 μg/ml IgE (BD Biosciences) and 0.1 μg/ml DNP‐BSA as antigen (Sigma) in the absence or presence of succinate. At indicated time points, cells were harvested and processed for mRNA expression using cytokine‐specific primers and probes (as described above). To monitor mast cell degranulation by the measurement of the release of the granule component, β‐hexosaminidase, BMMC were washed with RPMI‐1640/glutaMAX (without phenol red) supplemented with 25 mM HEPES, 0.1% BSA, 1% penicillin/streptomycin, 1× sodium pyruvate, 1× MEM essential amino acids and 50 μM β‐mercaptoethanol and cross‐linked for 1 h with 2 μg/ml IgE and 0.1 μg/ml DNP‐BSA as the antigen. Cells were centrifuged, and supernatants were incubated for 30 min with a substrate 4‐methylumbelliferyl‐N‐acetyl‐β‐d‐glucosaminide (Sigma‐Aldrich) diluted in citrate buffer and measured using white OptiPlate (PerkinElmer, Schwarzenbach, Switzerland). In some cases, BMMC were transfected using specific siRNA against random or mouse Sucnr1 (Ambion, Zug, Switzerland, s96542, s96541, s96543) gene for 24 h. Cells were then stimulated with either vehicle or 2 μg/ml IgE and 0.1 μg/ml DNP‐BSA as antigen for 1 h before RNA was isolated and Sucnr1 and Tnf mRNA expression were amplified by RT‐PCR.
Statistics
One‐tailed Mann–Whitney U‐test or two‐tailed Student's t‐test was used to analyze results. Significant differences with P < 0.05, P < 0.01 and P < 0.001 are illustrated as *, ** and ***, respectively.
Results and discussion
The skin is an effective barrier used by all vertebrates as an interface with the environment to provide the first line of defense from external factors. Infectious agents, toxins or trauma can harm the skin, causing the necrotic secretion of internal cell metabolites, which contribute to the activation of DC and the initiation of immune responses 25. In this context, we aimed to test the hypothesis that GPR91 expressed on epidermal Langerhans and dermal DC potentiates the development of ACD by sensing succinate release following tissue damage in the skin. We compared the reactions of wt and Sucnr1 −/− mice to a classical ACD challenge (i.e. using oxazolone as the inducing hapten and measuring ear thickness as the primary readout). We hypothesized that hypersensitive responses to oxazolone would be lowest in Sucnr1 −/− mice. Surprisingly, the extent of the responses observed in Sucnr1 −/− mice following oxazolone challenge was consistently higher than that displayed by the wt counterparts (Fig. 1A,B). This result was confounding, as we had postulated earlier that antagonizing GPR91 would be beneficial for the treatment of several immune disorders 16 and such a reaction could compromise the safety of putative GPR91‐based therapeutics. On the other hand, skin hyperreactivity was not augmented during the sensitization procedure with oxazolone and was reduced, rather than enhanced, following an ICD challenge (Fig. S2) of naïve Sucnr1 −/− mice with TPA. Furthermore, the strong hypersensitivity of Sucnr1 −/− mice to oxazolone did not correlate with an underlying increased T‐cell response, either measured as T‐cell infiltration (not shown) or measured as cutaneous transcript expression for classical Th2 cytokines like IL‐4 and IL‐13, which were rather reduced in the challenged gene‐deficient mice (Fig. 1C). Of note, mRNA expression for the innate inflammatory cytokines IL‐1β and TNF‐α was not elevated in the ears of challenged Sucnr1 −/− mouse group (Fig. S3A), which also did not display signs for exacerbated Th1, Th2 or Th17 cell differentiation, as assessed by the transcript expression of signature cytokines in the ear draining lymph nodes (Fig. S3B).
Figure 1.
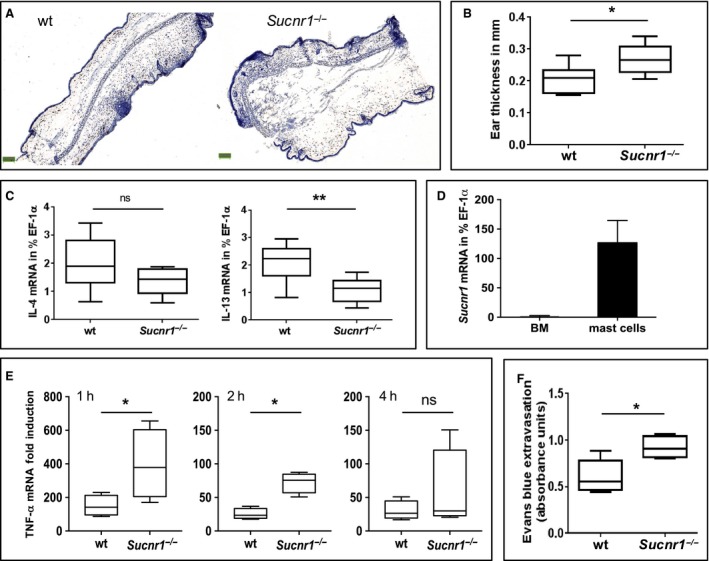
Sucnr1 −/− mice display exacerbated mast cell responses in vitro and in vivo. (A) Representative hematoxylin staining of ear sections from wt and Sucnr1 −/− mice after oxazolone challenge. (B) Comparative ear thickness from eight wt and eight Sucnr1 −/− oxazolone‐challenged mice. (C) Cytokine mRNA expression in oxazolone‐challenged ears from eight wt and eight Sucnr1 −/− mice. (D) Sucnr1 mRNA expression from bone marrow (BM) cells and BM‐derived mast cells isolated from two and eight, respectively, different mice. (E) TNF‐α mRNA expression by BM‐derived mast cells from wt and Sucnr1 −/− mice stimulated for indicated period of time with 2 μg/ml IgE plus 0.1 μg/ml DNP‐BSA. Boxplots represent fold increases in TNF‐α mRNA by stimulated mast cells related to corresponding unstimulated controls (pooled data from three independent experiments). (F) Evans Blue extravasation after passive cutaneous anaphylaxis (PCA) of five wt and five Sucnr1 −/− mice. Analysis using one‐tailed Mann–Whitney U‐test.
A closer look at the hematoxylin staining presented in Fig. 1A revealed a severe intradermal and subcutaneous edema in the ears of the hapten‐challenged Sucnr1 −/− mice, which is indicative for mast cell degranulation. RT‐PCR analysis of both mouse and human mast cells differentiated in vitro from corresponding bone marrow precursors revealed copious transcripts encoding GPR91 (Figs 1D and S4). These observations prompted us to compare the in vitro activation of mast cells differentiated from the bone marrow of wt and Sucnr1 −/− mice. As shown in Fig. 1E, mast cells from both, wt and Sucnr1 −/− mice, express TNF‐α mRNA, following in vitro activation with IgE cross‐linked by a specific antigen. TNF‐α expression was maximal at 1 h after activation and decreased progressively in the subsequent 4 h. However, at all time points examined, the magnitude of the response displayed by the mast cells derived from the Sucnr1 −/− mice was higher than that observed from wt counterparts. To further assess the in vivo biological relevance of the enhanced activation displayed by Sucnr1 −/− mast cells, we monitored blood vessel permeability following a PCA test. As demonstrated in Figs 1F and S5, Sucnr1 −/− mice displayed exacerbated blood vessel leakage, as shown by the release of the Evans blue following cross‐link of i.d. injected DNP‐specific IgE by the specific hapten. The responses were specific, as they were not elicited at the dermal sites injected with PBS (Fig. S5). On the other hand, the PCA test is an artificial setting that simply recapitulates the mechanistic mast cell activation induced in vitro, and thus, the potential risk for allergic hypersensitization associated with Sucnr1 deficiency was further assessed using an OVA‐specific sensitization and challenge procedure, which mimics the allergic response associated with asthma. The macrophages present in the BAL of OVA‐aerosol‐challenged mice were less abundant in the Sucnr1 −/− mouse group (Fig. 2A). Lymphocytes, neutrophils and eosinophils, as well as overall cell counts, in the BAL of OVA‐aerosol‐challenged Sucnr1 −/− mice were always reduced with respect to BAL from wt mice (Fig. 2A,B), even though the results did not reach statistical significance. By contrast, the BAL levels of IgE (Fig. 2C), but not IgG (not shown), were significantly reduced in the Sucnr1 −/− group. Concordantly, serum IgE but not IgG was also reduced in Sucnr1 −/− mice (not shown). These results are in agreement with our previous report 16 implicating succinate as an endogenous adjuvant for T‐cell‐mediated responses.
Figure 2.
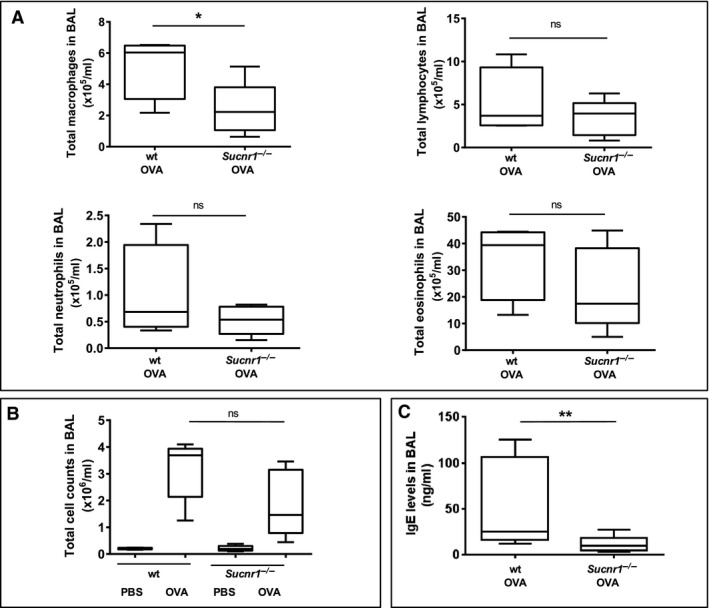
Sucnr1 −/− mice do not have increase cellularity in bronchoalveolar lavage (BAL) and mount limited IgE responses after challenge with aerosolized ovalbumin (OVA). (A) Macrophage, lymphocyte, neutrophil and eosinophil infiltration in BAL of four wt and five Sucnr1 −/− mice upon sensitization and challenge with OVA. (B) Total cell counts of BAL described in A. (C) IgE in BAL of mice challenged with OVA as described in A. Data were analyzed using one‐tailed Mann–Whitney U‐test.
The above data indicate that the absence of GPR91 signaling during the sensitization phase of immediate type I hypersensitivity responses reduces the development of T‐cell‐dependent IgE production and thus minimizes the risk of anaphylaxis upon subsequent encounter with the allergen. However, these experiments will not predict the risk of exacerbated mast cell activation mediated by GPR91 antagonists in an environment where preexisting immunocomplexes can drive pathology. Furthermore, bronchial hyperreactivity is not necessarily driven by mast cells in the C57BL/6 background, particularly if the challenge procedure is not repeated over a lengthy period of time 26. Thus, elucidating the threat of anaphylaxis would require specific and bioavailable low molecular weight antagonists of GPR91, which, to the best of our knowledge, are not yet available. We envisioned an alternative experimental condition in the mouse arthritis model induced by the transfer of the autoantibodies, directed to glucose‐6‐phosphatase isomerase, that develop in K/BxN mice 27. Passive transfer of serum from K/BxN mice to healthy wt recipient induces arthritis without the need to involve the generation of an adaptive response 27. The mechanism of this pathogenic antibody‐induced disease has been investigated in detail, revealing major roles for IL‐1β, TNF‐α 28, FcγRIII/CD16 29, 30 and C5aR/CD88 29. In addition, the observation that mice with functional mast cell deficiency are protected from K/BxN serum induced arthritis and that disease susceptibility is restored in these mice following reconstitution with normal mast cells has been used as a strong argument implicating mast cells in this pathologic process 30, 31. Thus, we anticipated that if the lack of Sucnr1 predisposes for exacerbated mast cell function, the arthritic disease induced by pathogenic autoreactive antibodies derived from the serum of K/BxN mice should manifest more intensely in Sucnr1 −/− mice than in wt counterparts. Concomitantly, arthritis induced by immunization with methylated bovine serum albumin (mBSA), which is mechanistically an antigen‐specific and T‐cell‐dependent 32 pathology, should be reduced in Sucnr1 −/− mice, in accordance with our previous observations 16. Unexpectedly, the swelling of the paws from Sucnr1 −/− mice transferred with K/BxN serum was significantly reduced in comparison with the paw swelling in wt mice (Fig. 3A). Disease amelioration in Sucnr1 −/− mice correlated with prevention of joint inflammation and with reduction in IL‐1β mRNA expression in popliteal lymph nodes draining the inflamed knees (not shown). Conversely, the results obtained in the AIA model were as predicted. Sucnr1 −/− mice mounted diminished adaptive responses, and hence, they were less susceptible than wt animals to develop arthritis following challenge with mBSA (Fig. 3B). Together, these results indicate that the potential exacerbation of mast cell activation, which results from deletion of Sucnr1 gene, does not worsen mast cell‐dependent arthritis. This apparent paradox could be explained by the observation that mast cells are crucial for the initiation of arthritis in the K/BxN serum transfer model but they are dispensable for disease progression, which requires a subsequent recruitment and effector function of macrophages 33.
Figure 3.
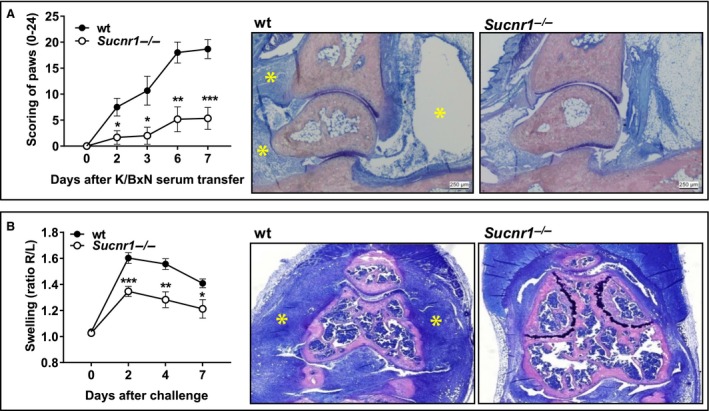
Sucnr1 −/− mice develop milder K/BxN serum and mBSA‐induced arthritis. (A) Inflammatory arthritic response in paws from wt (black circles, n = 6) and Sucnr1 −/− (white circles, n = 6) mice after injection of K/BxN serum. Analysis using Welch‐corrected, unpaired Student's t‐test. Representative Giemsa staining of hind paws from wt (severe edema and inflammation; see asterisks) and Sucnr1 −/− mice (no edema and reduced inflammation). (B) Inflammatory arthritic response in knees (ratios of antigen challenged vs control) from wt (black circles, n = 5) and Sucnr1 −/− (white circles, n = 5) mice after mBSA challenge. Analysis using unpaired Student's t‐test. Representative Giemsa staining of knees from wt (severe inflammation; see asterisks) and Sucnr1 −/− (reduced inflammation) mice.
Classic pathway activation of macrophages is associated with a metabolic switch from oxidative phosphorylation to glycolysis 34, similar to the changes occurring in tumors 35. Succinate dehydrogenase (SDH) catalyzes the conversion of succinate and flavin adenine dinucleotide quinone (FAD) into fumarate plus FAD hydroquinone (FADH2), which is coupled to the reduction of ubiquinone in the mitochondrial electron transport chain. Reduced oxygen or increase nutrients, as observed during hypoxia or in obesity, limits the activity of SDH resulting in the accumulation of succinate 36, which is ultimately exported to the cytosol and subsequently to the extracellular environment. Succinate inhibits hypoxia‐inducible factor‐1α (HIF‐1α) prolyl hydroxylase and therefore stabilizes HIF‐1α 37, which in turns enhances IL‐1β production. Recently, Tannahill et al. 38 have shown that IL‐1β production by activated macrophages is linked to the accumulation of succinate in the cytoplasm of these cells and we have observed that this mechanism is reinforced by the autocrine and paracrine re‐uptake of secreted succinate by GPR91 expressed on macrophages 39. Thus, although Sucnr1 −/− mast cells could potentially augment initiation of arthritis, these effects are counteracted by the reduced IL‐1β production by Sucnr1 −/− macrophages.
The above findings are in line with our previous report, describing succinate as an alarmin 16. The only apparent contradiction was the observed hyperreactivity of Sucnr1 −/− mast cells, which, although it did not translate into augmented disease, must have a physiological significance. Currently, we can only speculate that mast cells use GPR91 as a homeostatic receptor during their development. Indeed, mast cells from Sucnr1 −/− mice displayed hypomorphic aspects when compared to equivalent cells from wt mice (Fig. 4A). To search for evidence of the presumed developmental defect, we differentiated mast cells from bone marrow precursors of wt mice in the absence of succinate using serum‐free medium. Such differentiation process was very challenging in comparison with standard protocols performed in FCS‐containing medium. Nevertheless, mast cells differentiated under these conditions and became stimulated in the presence of allergen and specific IgE. We found that the amount of TNF‐α or β‐hexosaminidase secreted by activated mast cells differentiated in the absence of succinate was higher than that produced by mast cells derived from the same precursors differentiated in the presence of 50 μM succinate (Fig. 4B). Thus, the absence of succinate during in vitro mast cell development recapitulated Sucnr1 deficiency. On the other hand, we could not exacerbate cytokine release by wt mast cells differentiated in normal culture medium, which were subsequently devoid of GPR91 signaling by exposure to Sucnr1‐specific siRNA (Fig. 4C), further suggesting that the abnormal mast cell response was linked to the development of these cells.
Figure 4.
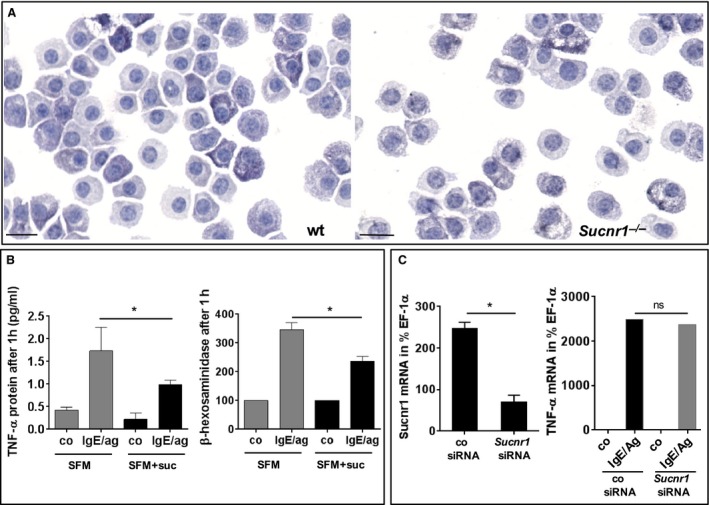
Mast cells differentiate abnormally in the absence of succinate signaling. (A) Mouse bone marrow‐(BM)‐derived mast cells from Sucnr1 −/− mice (right) are hypomorphic compared to wt counterparts (left). Toluidine blue staining (40× magnification). (B) TNF‐α and β‐hexosaminidase secretion by wt mast cells differentiated from BM precursors in serum‐free medium (SFM) alone or supplemented with 50 μM succinate (SFM+suc). Cells were stimulated with vehicle (co) or 2 μg/ml IgE plus 0.1 μg/ml DNP‐BSA (IgE/ag). Average results from four (TNF‐α) or seven (β‐hexosaminidase) independent experiments. (C) Expression of Sucnr1 mRNA by mast cells derived from wt BM after knockdown using specific Sucnr1 siRNA or scrambled control (left). TNF‐α mRNA expression by control or Sucnr1 knockeddown mast cells stimulated as indicated in B (right). Average results from four experiments. Analysis using one‐tailed Mann–Whitney U‐test or unpaired Student's t‐test.
Here, we have confirmed the role of GPR91 augmenting immune responses, and simultaneously, we have discovered that mast cell development in the absence of GPR91 signaling could lead to an hyperreactive reaction. This effect was apparent in the ACD model but not appreciated in the OVA‐aerosol challenge or in the arthritis models, particularly in the K/BxN model, which is driven by pathogenic autoantibodies. While these results do not eliminate completely the risk of adverse effects mediated by mast cells developing with little GPR91 agonism, they suggest that the maintenance and progression of antibody mediated diseases are not crucially dependent on mast cell function. Of note, the phenotype imposed by genetic deletion of Sucnr1 is expected to be more penetrant than that enforced by pharmacologic antagonism of GPR91, which could be transient and affecting predominantly DC‐ and macrophage‐mediated enhancement of adaptive effector responses.
Conclusion
This study reports the expression of GPR91 on mouse and human mast cells and reveals a hyperactive behavior of mouse Sucnr1 −/− mast cells in a mechanistic in vivo model of skin inflammation. While such an effect potentially poses a threat for the development of therapeutic antagonists aiming to interfere with adaptive immune responses, subsequent investigations using animal models of asthma and arthritis suggest that the abnormal reactions of Sucnr1 −/− mast cells are manifestations linked to their development in the absence of succinate signaling and do not exacerbate autoimmune pathology. Overall, our data support the use of GPR91 antagonists for the treatment of autoimmune diseases and particularly for rheumatoid arthritis.
Author contributions
All authors contributed to the execution and analysis of the experiments as well as to the writing of the manuscript and approved its final version. TR‐S and JMC designed, analyzed and interpreted all experiments. NC‐P, GL and SJ performed ACD, asthma and PCA in vivo experiments. CRe, LR, BC and RK performed in vitro mast cell differentiation and activation experiments. CRa and LR performed the CyTOF experiments. JD, GW and AL‐E performed arthritis in vivo experiments.
Conflicts of interest
All authors listed in this manuscript are (or were) employees of NIBR and are engaged in drug development activities.
Supporting information
Figure S1. CyTOF analysis of wt and Sucnr1 −/− mice.
Figure S2. ICD challenge of wt and Sucnr1 −/− mice.
Figure S3. mRNA expression in oxazolone‐challenged ears and draining lymph nodes from wt and Sucnr1 −/− mice.
Figure S4. SUCNR1 mRNA expression in human hematopoietic stem cell‐derived mast cells.
Figure S5. Macroscopic PCA response in wt and Sucnr1 −/− mice.
Table S1. List of metal‐tagged mAbs used in CyTOF experiments.
Acknowledgments
We thank Michaela Hahn, Mario Hernusz and Werner Höllriegl for animal husbandry, Susanne Huber for mouse genotyping, Geraldine Greiner‐Wacker for microscopy, Joseph Meingassner for ACD, Julia Kund, Sonja Hinteregger, Sophie Sarret, Deborah Garcia, Rachel Cuttat, Annick Waldt and Bernhard Jost for technical assistance and Christoph Schwärzler for helpful discussions.
Rubić‐Schneider T, Carballido‐Perrig N, Regairaz C, Raad L, Jost S, Rauld C, Christen B, Wieczorek G, Kreutzer R, Dawson J, Lametschwandner G, Littlewood‐Evans A, Carballido JM. GPR91 deficiency exacerbates allergic contact dermatitis while reducing arthritic disease in mice. Allergy 2017; 72:444–452.
Edited by: Hans‐Uwe Simon
References
- 1. Krebs HA. Rate control of the tricarboxylic acid cycle. Adv Enzyme Regul 1970;8:335–353. [DOI] [PubMed] [Google Scholar]
- 2. He W, Miao FJ‐P, Lin DC‐H, Schwandner RT, Wang Z, Gao J et al. Citric acid cycle intermediates as ligands for orphan G‐protein‐coupled receptors. Nature 2004;429:188–193. [DOI] [PubMed] [Google Scholar]
- 3. Ariza AC, Deen PMT, Robben JH. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress‐related conditions. Front Endocrinol 2012;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Favret S, Binet F, Lapalme E, Leboeuf D, Carballido JM, Rubic T et al. Deficiency in the metabolite receptor SUCNR1 (GPR91) leads to outer retinal lesions. Aging 2013;5:427–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hamel D, Sanchez M, Duhamel F, Roy O, Honoré J‐C, Noueihed B et al. G‐protein‐coupled receptor 91 and succinate are key contributors in neonatal postcerebral hypoxia‐ischemia recovery. Arterioscler Thromb Vasc Biol 2014;34:285–293. [DOI] [PubMed] [Google Scholar]
- 6. Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal J‐S, Cho J‐H et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med 2008;14:1067–1076. [DOI] [PubMed] [Google Scholar]
- 7. Gnana‐Prakasam JP, Ananth S, Prasad PD, Zhang M, Atherton SS, Martin PM et al. Expression and iron‐dependent regulation of succinate receptor GPR91 in retinal pigment epithelium. Invest Ophthalmol Vis Sci 2011;52:3751–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang L, Yu D, Fan H‐H, Feng Y, Hu L, Zhang W‐Y et al. Triggering the succinate receptor GPR91 enhances pressure overload‐induced right ventricular hypertrophy. Int J Clin Exp Pathol 2014;7:5415–5428. [PMC free article] [PubMed] [Google Scholar]
- 9. Aguiar CJ, Rocha‐Franco JA, Sousa PA, Santos AK, Ladeira M, Rocha‐Resende C et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun Signal 2014;12:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Correa PRAV, Kruglov EA, Thompson M, Leite MF, Dranoff JA, Nathanson MH. Succinate is a paracrine signal for liver damage. J Hepatol 2007;47:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li YH, Woo SH, Choi DH, Cho E‐H. Succinate causes α‐SMA production through GPR91 activation in hepatic stellate cells. Biochem Biophys Res Commun 2015;463:853–858. [DOI] [PubMed] [Google Scholar]
- 12. Sadagopan N, Li W, Roberds SL, Major T, Preston GM, Yu Y et al. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens 2007;20:1209–1215. [DOI] [PubMed] [Google Scholar]
- 13. Peti‐Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int 2010;78:1214–1217. [DOI] [PubMed] [Google Scholar]
- 14. Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest 2008;118:2526–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vargas SL, Toma I, Kang JJ, Meer EJ, Peti‐Peterdi J. Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J Am Soc Nephrol 2009;20:1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido‐Perrig N et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 2008;9:1261–1269. [DOI] [PubMed] [Google Scholar]
- 17. Mattapallil MJ, Wawrousek EF, Chan C‐C, Zhao H, Roychoudhury J, Ferguson TA et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 2012;53:2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meingassner JG, Fahrngruber H, Bavandi A. Pimecrolimus inhibits the elicitation phase but does not suppress the sensitization phase in murine contact hypersensitivity, in contrast to tacrolimus and cyclosporine A1. J Invest Dermatol 2003;121:77–80. [DOI] [PubMed] [Google Scholar]
- 19. Sheu MY, Fowler AJ, Kao J, Schmuth M, Schoonjans K, Auwerx J et al. Topical peroxisome proliferator activated receptor‐α activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol 2002;118:94–101. [DOI] [PubMed] [Google Scholar]
- 20. Graf C, Zemann B, Rovina P, Urtz N, Schanzer A, Reuschel R et al. Neutropenia with impaired immune response to Streptococcus pneumoniae in ceramide kinase‐deficient mice. J Immunol 2008;180:3457–3466. [DOI] [PubMed] [Google Scholar]
- 21. Mojtabavi N, Dekan G, Stingl G, Epstein MM. Long‐lived Th2 memory in experimental allergic asthma. J Immunol 2002;169:4788–4796. [DOI] [PubMed] [Google Scholar]
- 22. Koziczak‐Holbro M, Littlewood‐Evans A, Pöllinger B, Kovarik J, Dawson J, Zenke G et al. The critical role of kinase activity of interleukin‐1 receptor‐associated kinase 4 in animal models of joint inflammation. Arthritis Rheum 2009;60:1661–1671. [DOI] [PubMed] [Google Scholar]
- 23. Högenauer K, Arista L, Schmiedeberg N, Werner G, Jaksche H, Bouhelal R et al. G‐protein‐coupled bile acid receptor 1 (GPBAR1, TGR5) agonists reduce the production of proinflammatory cytokines and stabilize the alternative macrophage phenotype. J Med Chem 2014;57:10343–10354. [DOI] [PubMed] [Google Scholar]
- 24. Rådinger M, Jensen BM, Kuehn HS, Kirshenbaum A, Gilfillan AM. Generation, isolation, and maintenance of human mast cells and mast cell lines derived from peripheral blood or cord blood. Curr Protoc Immunol 2010;Chapter 7:Unit 7.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- 26. Pae S, Cho JY, Dayan S, Miller M, Pemberton AD, Broide DH. Chronic allergen challenge induces bronchial mast cell accumulation in BALB/c but not C57BL/6 mice and is independent of IL‐9. Immunogenetics 2010;62:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T et al. From systemic T cell self‐reactivity to organ‐specific autoimmune disease via immunoglobulins. Immunity 1999;10:451–461. [DOI] [PubMed] [Google Scholar]
- 28. Ji H, Pettit A, Ohmura K, Ortiz‐Lopez A, Duchatelle V, Degott C et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody‐induced arthritis. J Exp Med 2002;196:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FMA, Boackle SA et al. Arthritis critically dependent on innate immune system players. Immunity 2002;16:157–168. [DOI] [PubMed] [Google Scholar]
- 30. Corr M, Crain B. The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J Immunol 2002;169:6604–6609. [DOI] [PubMed] [Google Scholar]
- 31. Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 2002;297:1689–1692. [DOI] [PubMed] [Google Scholar]
- 32. Brackertz D, Mitchell GF, Mackay IR. Antigen‐induced arthritis in mice. I. Induction of arthritis in various strains of mice. Arthritis Rheum 1977;20:841–850. [DOI] [PubMed] [Google Scholar]
- 33. Solomon S, Rajasekaran N, Jeisy‐Walder E, Snapper SB, Illges H. A crucial role for macrophages in the pathology of K/B x N serum‐induced arthritis. Eur J Immunol 2005;35:3064–3073. [DOI] [PubMed] [Google Scholar]
- 34. Rodríguez‐Prados J‐C, Través PG, Cuenca J, Rico D, Aragonés J, Martín‐Sanz P et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010;185:605–614. [DOI] [PubMed] [Google Scholar]
- 35. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 2006;25:4675–4682. [DOI] [PubMed] [Google Scholar]
- 36. Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci USA 2000;97:2826–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF‐alpha prolyl hydroxylase. Cancer Cell 2005;7:77–85. [DOI] [PubMed] [Google Scholar]
- 38. Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G et al. Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature 2013;496:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Littlewood‐Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 2016;213:1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CyTOF analysis of wt and Sucnr1 −/− mice.
Figure S2. ICD challenge of wt and Sucnr1 −/− mice.
Figure S3. mRNA expression in oxazolone‐challenged ears and draining lymph nodes from wt and Sucnr1 −/− mice.
Figure S4. SUCNR1 mRNA expression in human hematopoietic stem cell‐derived mast cells.
Figure S5. Macroscopic PCA response in wt and Sucnr1 −/− mice.
Table S1. List of metal‐tagged mAbs used in CyTOF experiments.