

Abstract
Fifty‐five years after the concept of dopamine replacement therapy was introduced, Parkinson disease (PD) remains an incurable neurological disorder. To date, no disease‐modifying therapeutic has been approved. The inability to predict PD incidence risk in healthy adults is seen as a limitation in drug development, because by the time of clinical diagnosis ≥ 60% of dopamine neurons have been lost. We have designed an incidence prediction model founded on the concept that the pathogenesis of PD is similar to that of many disorders observed in ageing humans, i.e. a complex, multifactorial disease. Our model considers five factors to determine cumulative incidence rates for PD in healthy adults: (i) DNA variants that alter susceptibility (D), e.g. carrying a LRRK2 or GBA risk allele; (ii) Exposure history to select environmental factors including xenobiotics (E); (iii) Gene–environment interactions that initiate pathological tissue responses (I), e.g. a rise in ROS levels, misprocessing of amyloidogenic proteins (foremost, α‐synuclein) and dysregulated inflammation; (iv) sex (or gender; G); and importantly, (v) time (T) encompassing ageing‐related changes, latency of illness and propagation of disease. We propose that cumulative incidence rates for PD (P R) can be calculated in healthy adults, using the formula: P R (%) = (E + D + I) × G × T. Here, we demonstrate six case scenarios leading to young‐onset parkinsonism (n = 3) and late‐onset PD (n = 3). Further development and validation of this prediction model and its scoring system promise to improve subject recruitment in future intervention trials. Such efforts will be aimed at disease prevention through targeted selection of healthy individuals with a higher prediction score for developing PD in the future and at disease modification in subjects that already manifest prodromal signs.
Keywords: aetiology, exposome, genetics, neurodegeneration, parkinsonism, probability, toxin
Introduction
Many neurodegenerative disorders that affect ageing adults, such as Parkinson disease (PD) and Alzheimer disease, remain unpredictable, non‐preventable and incurable. The burden of such brain diseases in our ageing societies is steadily increasing. The ability to estimate their future incidence rates in healthy adults, as is commonly done for other conditions, would help to increase the likelihood of therapeutic success for interventions by enabling subject stratification and to focus on individuals at higher risk for disease. One limitation in our ability to quantify individual PD incidence risk lies in its incompletely understood (‘idiopathic’) pathogenesis. By considering known risk categories as essential contributors to the cumulative incidence rate and by assigning relative values to each chosen factor, we have created a novel tool, i.e. the ‘PREDIGT score’.
PD as an environmentally induced disease
For decades, typical, late‐onset PD was considered to be caused by one or more environmental contributor(s), foremost neurotoxic chemicals, such as pesticides, with relative specificity for dopamine cells in the Substantia nigra pars compacta (S. nigra), but with no significant contribution from heritable factors (Tanner et al., 1999). For over 20 years, epidemiologists have identified increasing numbers of potential environmental candidates reported to be in positive or negative association with PD incidence [reviewed in: (Elbaz et al., 2016; Polito et al., 2016)].
Despite the abundance of reported putative environmental modifiers of PD risk, an umbrella review of 75 meta‐analyses, carried out with the goal of determining the actual effect size of candidate risk modulators (Bellou et al., 2016), identified only four risk factors that met class I (convincing)‐type or class II (highly suggestive)‐level evidence in controlled cohort studies. Both chronic constipation and late‐onset anxiety/depression were associated with elevated risk, whereas smoking and physical activity lowered the risk for PD incidence [reviewed by: (Bellou et al., 2016)]. Constipation and depression have frequently been interpreted by clinicians as prodromal signs of idiopathic PD (Abbott et al., 2001, 2003; Berg et al., 2015); however, their potential roles in a latency stage (i.e. in a non‐clinical phase that could evolve into PD) vs. in the clinically detectable, prodromal stage (i.e. prior to the onset of motoric signs; Fig. 1) have yet to be precisely determined.
Figure 1.
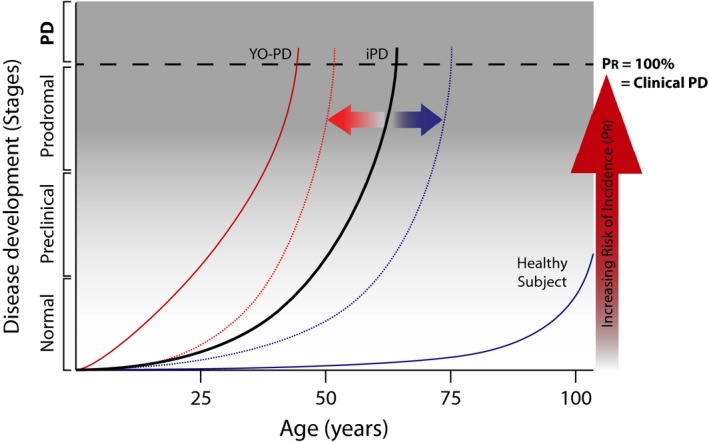
PREDIGT formula calculates incidence rates for Parkinson disease in healthy adults as a function of five variables. The PREDIGT score quantifies Parkinson disease (PD) incidence rates as a function of five variables. Disease projection curves over time are plotted for a representative case of typical, idiopathic PD (iPD; solid black line), young‐onset PD (YO‐PD; solid red line) and for a healthy subject (solid blue line). Two horizontal arrows represent the effects of risk factors that either elevate (orange) or reduce (blue) cumulative PD incidence risk, and therefore, are hypothesized to shift the curve in either direction (dotted lines). Additive risk categories include ‘exposome’ (E), ‘genetics’ (DNA; D), and ‘gene–environment interactions that initiate host responses’ (I), of which the sum is multiplied by risk factors ‘sex/gender’ (G) and ‘time’ (T). Each variable is defined in detail in the text and assigned a value from a score sheet (see Table 1). This is to calculate the total PREDGIT risk score for PD incidence rate at a given time (P R score, %; vertical arrow in red). A PREDIGT score of 100% represents the onset of clinically defined parkinsonism/PD (Postuma et al., 2015) corresponding to a ≥ 60% loss in dopamine (DA) innervation of the striatum; the prodromal stage correlates with ~ 41–59% DA neuron loss; and the preclinical phase corresponds to < 40% of DA neuron loss (Chahine et al., 2016). The curve of a healthy subject is extrapolated from published calculations (Berg et al., 2015) that report the rise in ‘age‐adjusted, prior probability of PD’ to 4% at the age of 80 years. The shape of the curve after age 80 is speculative but informed by the known, age‐dependent loss of DA cells in older individuals (Fearnley & Lees, 1991; Gibb & Lees, 1991; Ma et al., 1999).
Bellou et al. argued that a substantial number of previous studies (and meta‐analyses thereof) contained residual confounders, possibly reflecting reverse causation and/or biases in observed associations (Bellou et al., 2016). Therefore, it seems that in the majority of subjects with typical PD, the ‘missing environmental contributors’ in subjects’ exposure history, collectively referred to as ‘exposome’, have remained unaccounted for (Ritz & Rhodes, 2010; Palacios et al., 2012; Grondin et al., 2016).
There is growing interest in the potential role of the gastrointestinal (GI) tract in disease initiation, in particular in the context of environmental risk factors. This interest largely stems from an autopsy‐based staging system first published by Braak et al. These authors have postulated that the initiation of pathology leading to late‐onset PD starts well over a decade before the occurrence of any motoric deficits, namely in the enteric nervous system and, in parallel, in the olfactory bulb (Braak & Del Tredici, 2008). The missing environmental contributors to PD could thus reside, or have once resided, within the environment‐derived constituents of the GI tract (i.e. from the oropharynx to the rectum) and/or the upper respiratory tract (URT; i.e. beginning in the nasal cavity with its olfactory epithelium) (Duda et al., 1999; Doty, 2008; Del Tredici et al., 2010; Savica et al., 2010; Derkinderen et al., 2014; Gray et al., 2014, 2015; Svensson et al., 2015).
Interestingly, although the independently protective effect of smoking (and less strongly, of coffee consumption) for the incidence risk of PD (Gorell et al., 2004; Driver et al., 2009; Palacios et al., 2012) is thought by many researchers to be mediated pharmacologically in the central nervous system by nicotine (and caffeine), Scheperjans et al., among other investigators, pointed out the multitude of effects by which both compounds can affect GI function including in changing constituents of the gut microbiome (Scheperjans et al., 2015). There is increasing interest in the microbiome itself in PD research with the interrogation of patients’ specimens collected from the GI tract and URT to identify surrogates of an individual's exposure history and candidates for disease‐related markers (Mollenhauer et al., 2013a; Antunes et al., 2016; Felice et al., 2016; Scheperjans, 2016).
In parallel, in exposure science research the standardized collection, cataloguing and integrated analysis of exposome elements (e.g. nutrients, toxicants and xenobiotics) for each individual have become more comprehensive with the aim to better delineate the pathogenesis of late‐onset disorders in humans (Stanberry et al., 2013; Dacks et al., 2014; Patel, 2016; Rappaport, 2016; Siroux et al., 2016). Such investigations encompass large‐scale analyses with exposome measurements in defined populations such as through ‘Environment‐Wide Association Studies’ (Patel & Ioannidis, 2014; Grondin et al., 2016).
PD as a (complex) genetic disease
Since 1997, the pendulum investigating the principal cause of typical PD has swung towards genetic factors owing to the publication by Nussbaum et al. of the first bona fide, heritable form of parkinsonism in the ‘Contursi kindred’ (Polymeropoulos et al., 1997). There, the phenotype of young‐onset PD was found to segregate in affected members with a heterozygous mutation in the α‐synuclein‐encoding gene at the PARK‐SNCA locus (Polymeropoulos et al., 1997) with a penetrance rate of > 80% (Marras et al., 2016). The initial study of this trans‐national and trans‐continental pedigree (Duvoisin & Golbe, 1995) as well as the subsequent identification of a heterozygous p.A53T substitution in α‐synuclein as its cause (Polymeropoulos et al., 1997; Papadimitriou et al., 2016) heralded the beginning of the ‘genetic revolution in PD’ (Klein & Schlossmacher, 2007); it also made possible a more sensitive neuropathological diagnosis of typical, sporadic PD (Spillantini et al., 1997) and the identification of several other, misfolded α‐synuclein‐related disorders (referred to as ‘synucleinopathies’) (Galvin et al., 2001; Irwin et al., 2013) [reviewed in: (Farrer, 2006)].
Together with the advent of high throughput sequencing platforms, the discovery by Nussbaum et al. (Polymeropoulos et al., 1997) also accelerated the pursuit of linkage and genome‐wide association studies to identify additional DNA variants that are associated with an elevated or a reduced incidence rate of PD (e.g. Soto‐Ortolaza et al., 2013; Nalls et al., 2015). Today, genetic markers have been identified at > 26 independent loci in apparent association with altered risk to develop typical PD (Edwards et al., 2010; International Parkinson Disease Genomics Consortium et al., 2011; Nalls et al., 2015). Increasingly large genetic association studies have enabled researchers to estimate that the population attributable fraction in ‘complex genetic disease’ models of PD lies between 11 and 60% (International Parkinson Disease Genomics Consortium et al., 2011; Nalls et al., 2015), which suggests that a sizeable portion of ‘the hidden heritability’ has not yet been accounted for in aetiological models where causality is restricted to genetics (Singleton & Hardy, 2016). Nevertheless, Nalls et al., in attempting to better classify PD patients, developed an 'integrated model of PD’ that combines a cumulative genetic risk score with four variables (i.e. age; sex; sense of smell; and a self‐reported family history of PD). The latter model effectively classified persons as ‘typical PD’ (Nalls et al., 2015); it was validated using five cohorts demonstrating high sensitivity and specificity for the separation of PD (including those with proven loss of dopamine innervation in the striatum) vs. controls, where it achieved a classification accuracy with an area‐under‐the‐curve value of greater than 0.89. Although valuable as a classification tool, their integrative model – by nature of its design – did not yet assess the individual risk of neurologically healthy persons regarding their PD incidence rate later in life. We posit that for this reason, and based on a model that sees ‘idiopathic PD’ as a complex, multifactorial disease rather than a ‘complex genetics‐only’ disease, the integration of environmental exposure – together with genetic risk elements – is essential.
PD as a complex, multifactorial disease: gene–environment interactions
With the rapid progress in genome interrogation platforms and growing list of possible environmental clues for the development of complex, late‐onset disorders (Darabos et al., 2016), several groups have recently begun to conduct putative gene–candidate environment interaction studies. Given the very large number of patients required to carry out non‐candidate‐driven association studies to probe for exposure history in the context of entire genomes to determine true effect size (Grondin et al., 2016; Rappaport, 2016), alternative study designs are being employed, for example through Bayesian classifier models (Li et al., 2012; Elbaz et al., 2016; Patel, 2016).
Our concept of PD as a multifactorial disease that results from interactions between an individual's genetic susceptibility and his/her exposome has been informed by several insights from other late‐onset disorders (e.g. Rappaport, 2016); it has been reinforced by our own studies regarding the in vivo effects of the Leucine‐rich repeat kinase‐2‐encoding gene at the PARK‐LRRK2 locus. LRRK2 variants have been associated with late‐onset, familial PD (Paisan‐Ruiz et al., 2004; Zimprich et al., 2004), late‐onset sporadic PD (Lesage et al., 2006; Ozelius et al., 2006), and two other complex diseases in humans, i.e. Crohn's disease (Barrett et al., 2008) and an inflammation‐rich endophenotype of leprosy (Fava et al., 2016). In our consortium based work, we found that wild‐type LRRK2 plays an important role in the innate immune system co‐regulating the response to virulent, microbial pathogens, and that in this context mutants alter inflammation, thereby affecting neural health following infections in mice and humans (Hakimi et al., 2011; Fava et al., 2016) (B. Shutinoski, J. J. Tomlinson, E. G Brown, M. G. Schlossmacher, unpublished data). From these studies, we concluded that this example of a gene–environment–interaction paradigm (i.e. LRRK2‐xenobiotic‐inflammation) might have direct relevance to idiopathic PD. As a result, we postulated that exposome elements and a subject's tissue responses are essential in contributing to the cumulative incidence rate of PD, and that therefore, both should be included in prediction models.
Classifying stages of PD and predicting prodromal illness
In accordance with the definition of the stages of PD by a task force assembled by the International Parkinson and Movement Disorder Society, we too consider it to evolve in three stages (Fig. 1): during the pre‐clinical stage pathophysiological processes have commenced, but there are no evident symptoms or signs; the prodromal stage reflects a phase when symptoms and signs are present, but are yet insufficient to define parkinsonism; and clinical PD, which equates the diagnosis of parkinsonism, as further supported by bona fide progression, and responsiveness to dopaminergic therapy (Postuma et al., 2015) (Fig.1).
Recently, Berg et al. provided a ‘naïve, Bayesian classifier model‐based prediction formula to identify prodromal PD’ (Berg et al., 2015). Prodromal PD is estimated to span a period of > 10 years in human subjects preceding the expression of the classical parkinsonian phenotype (Braak & Del Tredici, 2008; Hawkes et al., 2010; Berg et al., 2015). In their approach, an individual patient's non‐motoric symptoms and signs were considered to be indicative of neurological dysfunction, and sequentially combined with select items (i.e. epidemiological risk factors; age; sex; and genetic risk markers) to generate probability values (in %) and likelihood ratios (< 1, 1, > 1) for the diagnosis of ‘prodromal PD’ (Berg et al., 2015). This model's strengths include: (i) The description of a first tool attempting to quantify the probability of a prodromal PD diagnosis; (ii) Its multivariate, step‐by‐step assembly of factors previously associated with the incidence of PD; and (iii) Its testability in longitudinal studies. This tool (upon its validation) will likely enable better screening and stratification of those subjects in clinical trials where the disease process has already entered the prodromal stage.
Calculating cumulative incidence risk for PD in healthy adults?
What the field lacks is a tool to quantify the incidence risk for PD in neurologically healthy subjects, and moreover, to predict in each individual the actual time to disease onset. For this, we need to consider the complement of risk factors that functionally interact to initiate a disease process (Rappaport, 2016), i.e. a comprehensive systems‐based focus that Li et al. referred to as ‘Gene‐Environment Wide‐Interaction Studies’ (Li et al., 2012; Go & Jones, 2014; Grondin et al., 2016; Patel, 2016).
In building the PREDIGT score as a platform to calculate future incidence rates of PD, we considered the following properties to be important: (i) To view the development of typical PD and its incidence rate in a hypothesis‐driven manner (rather than a Bayesian classifier approach), i.e. within the framework of a complex disease that has more than one essential contributor; (ii) The ability to prospectively calculate the probability for incident PD in healthy adult subjects; and (iii) To employ an easy‐to‐use formula, as inspired by simple prediction tools developed in other disciplines of medicine, for example: the ‘Wells criteria’ that predict the probability of pulmonary emboli; blood value‐based criteria to help modify the risk of vascular disease and complications; and the ‘CHADS2 score’ to predict the annual likelihood of stroke from atrial fibrillation (Hendriksen et al., 2015; Preiss & Kristensen, 2015; Hsu et al., 2016; Wang et al., 2016).
Our hypothesis that a subject's risk for PD incidence can be calculated rests on three components: (i) Typical PD is not ‘idiopathic’, but should be seen as the outcome of five critical factors; (ii) Genetic susceptibility, exposome, pathological tissue responses, sex, and time interact to promote the progressive degeneration of dopamine neurons in the human S. nigra (Fig. 1); and finally, (iii) An estimate for the cumulative incidence rate of idiopathic PD can be made in healthy subjects by assigning concrete values to each of the five factors, using an easy‐to‐use formula.
Materials and methods
Parkinson disease incidence rate expressed as P R (in %)
In our predictive model, the cumulative incidence rate for PD (P R) is calculated by five variables [(E + D + I) × G × T] and expressed in % values. ‘E’ stands for exposome, ‘D’ for DNA, ‘I’ for the initiation of tissue responses following gene–environment interactions, ‘G’ for sex/gender, and ‘T’ for time. A P R score of 100% (or above) refers to the presence of clinically recognizable parkinsonism, i.e. the diagnosis of the phenotype under consideration (Postuma et al., 2015), which is thought to correlate with the loss of ≥ 60% of striatal innervation (Fig. 1). For model building, we equated a P R score of 0% to the normal complement of dopamine producing cells in the S. nigra, which innervate target cells in the neostriatum, present at birth; of note, we have not yet correlated any P R scores (between 1 and 99%) (Fig. 2) with the actual fraction of dopaminergic cell loss in human autopsy studies.
Figure 2.
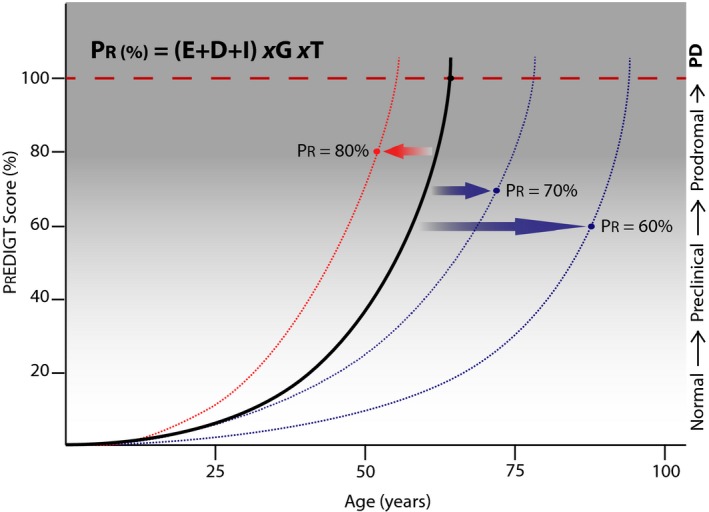
The PREDIGT score quantifies the probability for the risk (PR) of Parkinson disease (PD) as a function of five factors: E (Exposome); D (Genetics); I (Initiation); G (Sex/Gender); and T (Time). The disease projection curve for a case of idiopathic PD is shown (solid line), where a PR of 100% is reached at 66 years of age. The curve shifts based on values for the five factors, thus changing the incidence risk at a given age, for example, to a PR = 80% at 52 years, 70% at 72 years and 60% at 88 years.
Variable ‘E’: exposome
In our model, variable ‘E’ represents the exposure history by an individual to environmental pathogens from the prenatal period onward. These include, but not limited to, nutrients, chemicals, xenobiotics as well as recreational and occupational hazards (Wild, 2009). ‘E’ captures the subject's exposure history to one or more environmental modifier (exposed once, recurrently, or chronically) that has been convincingly associated with elevation (i.e. a risk factor) or reduction (a protective factor) in the incidence rate for typical PD. To date, few factors, apart from constipation, anxiety, depression, smoking and physical activity (Bellou et al., 2016), have been unequivocally linked to the modification of incidence rate for idiopathic PD. Candidate factors that have not met class I or II type evidence in meta‐analyses, but that have been identified in strong association with the development of parkinsonism in smaller cohort studies, include recurrent (sub)concussive head traumas, cumulative exposure to neurotoxic agents (e.g. pesticides, manganese), reduced levels of serum uric acid (related in part to diet), and a history of select microbial illnesses (see below).
For building our model and calculating P R scores, a value for variable ‘E’ is entered. In lieu of the often unavailable details to capture an individual's entire exposure history, we also allowed surrogates to stand in for where an environmental pathogen may play (or could have played) a role, for example: 0.0–0.005, for the paucity of any identifiable, significant environmental exposure history that would modulate PD risk (before age 50 years; Table 1); 0.25–1 reflecting the duration of constipation in the subject's past as an independent risk factor or as surrogate for a previous illness of the GI tract; 0.25–1, one or multiple (sub)concussive head traumas; 0.5–1, a reduction in the sense of smell as an independent risk factor or as surrogate for previous URT infections that led to hyposmia and anosmia, respectively; and on the protective side, for example, values from −0.25 to −1 to account for a subject's smoking history. A list of select examples for environmental risk modifiers and their assigned values under factor ‘E’ (for model building purposes; maximum value, 3) is shown in Table 1.
Table 1.
Select Values for five factors entered into the PREDIGT score formula
| Factor E: Exposome | |||
|---|---|---|---|
| Type of modifier (or surrogate) | Nature of risk modifier (if known) | Assigned value | Select ref(s) used to create value |
| Association with elevated risk | |||
| Neurotoxin | i.v. MPTP exposure (each event) | 1 | Langston et al., Science 1983 |
| i.v. Mn2+ exposure (each event) | 0.5 | Stepens et al., NEJM 2008 | |
| Pesticide exposure (cumulative) | 0.25 | Bellou et al., Parkins Rel Dis 2016 | |
| Farm life before age 20 years | 0.25 | Bellou et al., Parkins Rel Dis 2016 | |
| Head trauma | Concussive events (cumulative) | 1 | Mez et al., Alz Res Therap 2015 |
| (Sub)concussive events (cumulative) | 0.5 | Mez et al., Alz Res Therap 2015 | |
| Xenobiotic exposure | Encephalitis (select pathogens) | 2 | Jang et al., Biochim Biophys Acta 2008 |
| Chronic infection (e.g. H. pylori) | 1 | Bu et al., Park Rel Dis 2015 | |
| Chronic constipation | Lasting for ≥ 20 years | 1 | Ross et al., Park Rel Dis 2012 |
| Lasting for 10–19 years | 0.5 | Ross et al., Park Rel Dis 2012 | |
| Lasting for 5–9 years | 0.25 | Ross et al., Park Rel Dis 2012 | |
| Reduced olfaction | Anosmia (UPSIT score ≤ 28/40) | 1 | Muirhead et al., The Otolaryngol 2013 |
| Hyposmia (UPSIT score 29–33/40) | 0.5 | Muirhead et al., The Otolaryngol 2013 | |
| No known association with risk modulator | Little cumulative pathogen exposure | ||
| Age of proband | |||
| ≤ 50 years | 0 | ||
| 51–59 years | 0.005 | ||
| 60–69 years | 0.0075 | ||
| 70–79 years | 0.02 | ||
| ≥ 80 years | 0.03 | ||
| Association with lower risk | |||
| Smoking history | Current smoker for ≥ 20 years | −0.75 | Ritz et al., Arch Neurol 2007 |
| Current smoker for 11–19 years | −0.5 | Ritz et al., Arch Neurol 2007 | |
| Past smoker for ≥ 20 years | −0.25 | Ritz et al., Arch Neurol 2007 | |
| Past smoker for 11–19 years | −0.125 | Ritz et al., Arch Neurol 2007 | |
| Any smoking history ≤ 10 years | −0.0625 | Ritz et al., Arch Neurol 2007 | |
| Caffeine intake | ≥ 2 cups/day (recent) | −0.25 | Palacios et al., Mov Dis 2012 |
| ≥ 1 cup/day (recent) | −0.125 | Palacios et al., Mov Dis 2012 | |
| Physical exercise | Regular for ≥ 20 years | −0.25 | Bellou et al., Parkins Rel Dis 2016 |
| Irregular for ≥ 20 years | −0.125 | Bellou et al., Parkins Rel Dis 2016 | |
| Regular for ≤ 19 years | −0.125 | Bellou et al., Parkins Rel Dis 2016 | |
| Factor D: DNA (Genetics) | |||
|---|---|---|---|
| Gene (locus)/Family history | Type of genetic variant | Assigned value | Select ref(s) used to create value |
| Association with elevated risk | |||
| SNCA | Gene triplication (n = 4 alleles) | 1 | Trinh et al., JAMA Neurol 2014 |
| Gene duplication (n = 3 alleles) | 0.75 | Trinh et al., JAMA Neurol 2014 | |
| Mutation (e.g. p.A53T; p.A30P) | 0.75 | Trinh et al., JAMA Neurol 2014 | |
| Rep1 repeat expansion (5′) | 0.5 | Markopoulou et al., Parkins Rel Dis 2014 | |
| Other risk variants as per GWAS | 0.25 | Nalls et al., Lancet Neurol 2014 | |
| PARKIN or DJ‐1 or PINK1 | Point mutation (het) | 1 | Kitada et al., Nature 1998 |
| Copy number variant (het) | 1 | Pankratz et al., PLOS One 2011 | |
| Exon deletion (het) | 1 | Kitada et al., Nature 1998 | |
| GBA | Point mutation (het; homo) | 0.5 | Alcalay et al., JAMA Neurol 2014 |
| LRRK2 | Point mutation (het; homo) | 0.5 | Trinh et al., JAMA Neurol 2014 |
| Other risk loci identified by GWAS | Single‐nucleotide polym. (SNPs) | 0.1–0.25 | Nalls et al., Lancet Neurol 2015 |
| Family history of disease | |||
| No known family history | Overall low genetic risk | 0.01 | Elbaz et al., Neurology 2003 |
| Positive family history | 1st degree relative with bona fide PD | 0.5 | Sveinbjoernsdottir et al., NEJM 2000 |
| Positive family history | 2nd degree relative with bona fidePD | 0.25 | Sveinbjoernsdottir et al., NEJM 2000 |
| Positive family history | 3rd degree relative with bona fide PD | 0.125 | Sveinbjoernsdottir et al., NEJM 2000 |
| Association with lower risk | |||
| LRRK2 | Bona fide protective SNPs | −0.5 | Ross et al., Lancet Neurol 2011 |
| Factor I: Initiation of tissue response | |||
|---|---|---|---|
| Type of pathophysiological effect | Outcome(s) of effect in cells/tissue | Assigned value | Select ref(s) used to create value |
| Pathophysiological response | |||
| α‐synuclein dysregulation | Accumulation (n = 4 SNCA alleles) | 1 | Kuo et al., Hum Mol Gen 2010 |
| Accumulation (e.g. p.A53T; p.A30P) | 0.5 | Kuo et al., Hum Mol Gen 2010 | |
| Accumulation (n = 3 SNCA alleles) | 0.5 | Kuo et al., Hum Mol Gen 2010 | |
| Accumulation (Rep1 repeat expansion) | 0.25 | Cronin et al., Hum Mol Gen 2009 | |
| Accumulation (GBA1 mutation) | 0.25 | Cullen et al., Ann Neurol 2011 | |
| Accumulation (select LRRK2 mutant) | 0.25 | Zimprich et al., Neuron 2004 | |
| Tau dysregulation | Accumulation (MAPT mutation) | 1 | Kumar et al., J Biol Chem 2014 |
| Accumulation (encephalitis) | 0.5 | Jang et al., Biochim Biophys 2008 | |
| Accumulation (select LRRK2 mutation) | 0.25 | Zimprich et al., Neuron 2004 | |
| Accumulation (concussive traumas) | 0.5 | Mez et al., Alz Res Therap 2015 | |
| Accumulation (subconcuss. traumas) | 0.25 | Mez et al., Alz Res Therap 2015 | |
| Parkin deficiency | Redox change; mitoch. dysfunction | 1 | Palacino et al., J Biol Chem 2004 |
| DJ‐1 deficiency | Redox change; mitoch. dysfunction | 1 | Rousseaux et al., PNAS 2012 |
| Pink1 deficiency | Redox change; mitoch. dysfunction | 1 | Glasl et al., Exp Neurol 2012 |
| Neurotoxicant (e.g. MPTP) | Mitochondria degeneration; ROS rise | 1 | Fornai et al, PNAS 2005 |
| Chronic inflammation | Cytokine/immune cell dysregulation | 0.25 | Dzamko et al., Mov Dis 2016 |
| Presence of anxiety/depression | Surrogate of disease process in CNS | 0.25 | Bellou et al., Parkins Rel Dis 2016 |
| Presence of REM sleep disorder | Surrogate of disease process in CNS | 0.25 | Postuma et al., Sleep Med 2016 |
| Paucity of pathophysiological response | Adjusting for age: | ||
| ≤ 50 years | 0 | ||
| 51–59 years | 0.001 | ||
| 60–69 years | 0.002 | ||
| 70–79 years | 0.003 | ||
| ≥ 80 years | 0.004 | ||
| Factor G: Sex (Gender) | Sex | Assigned value | Select ref(s) used to create value |
|---|---|---|---|
| General population | |||
| LRRK2 wild‐type | Male | 1.2 | Berg et al., Mov Dis 2015 |
| Female | 0.8 | Berg et al., Mov Dis 2015 | |
| Genotyped subjects | |||
| Bona fide LRRK2 mutation carrier | Male | 0.8 | Marder et al., Neurology 2015 |
| Female | 1.2 | Marder et al., Neurology 2015 | |
| Factor T: Time | |||
|---|---|---|---|
| Measurement of time | Years lived | Assigned value | Select ref(s) used to create value |
| Capturing ageing, latency, progression | Subject's actual age | 1–100 | Driver et al., Neurology 2009 |
Variable ‘D’: an individual's genetic susceptibility (DNA)
Parkinsonism is in part characterized as the shared clinical appearance of genetic heterogeneity (‘phenocopies’). Variants at individual PD‐associated loci can lead to pleiotropic outcomes of parkinsonism itself, as is the case, for example, with mutant LRRK2 alleles (Zimprich et al., 2004; Kalia et al., 2015); perplexingly, genotypic variants at one specific locus can also lead to other disease phenotypes, such as Gaucher disease and dementia with Lewy bodies in the case of GBA mutations (Alcalay et al., 2014). Here, the variable ‘D’ (for DNA) represents the overall contribution of a person's genetic risk. When choosing the related values for model building purposes, we included select genomic variants that confer distinct susceptibility to the incidence rate of PD, for example, based on their known penetrance rate and/or the age‐of‐disease onset. Although from an evolutionary perspective, these genetic variants were likely not selected for, or against, the development of a brain disease that occurs later in adulthood, rare allelic variants at individual loci can confer a large effect size in generating young‐onset parkinsonism. In contrast, commonly found allelic variants at one locus (or multiple loci) may confer relatively small contributions, either individually or collectively, in generating the more common, late‐onset PD phenotype (Kitada et al., 1998, 2012; Nalls et al., 2015; Marras et al., 2016).
Under factor D, the value of 0.01 represents the relative paucity of genetic susceptibility to develop PD; 0.125 and 0.25, for example, the presence of one or more allelic variant, respectively, with a relatively low contribution to the overall development of PD (such as of a non‐coding, small nucleotide polymorphism [SNP] at the SNCA or MAPT locus); or, values from 0.125–0.5 to incorporate a confirmed diagnosis of PD in relatives, as a surrogate for not‐yet‐identified genetic risk (Sveinbjornsdottir et al., 2000; Elbaz et al., 2003). [Of note, we are cognizant of a possible family information bias (i.e. the over‐ or under‐reporting of parkinsonism in relatives of subjects being interviewed) (Elbaz et al., 2003)]; 0.5, an established susceptibility allele of moderate effect size (e.g. a disease‐associated mutation in LRRK2; duplication of wild‐type SNCA); 1, the triplication of wild‐type SNCA, or alternatively, the presence of a disease‐causing mutation in an allele encoding a recessive gene linked to early‐onset parkinsonism (e.g. at PARK‐Parkin; PARK‐DJ‐1; or PARK‐PINK1) (Marras et al., 2016). A list of examples for genetic risk candidates used to build the model and their assigned values under factor ‘D’ (maximum value, 3) are shown in Table 1.
Variable ‘I’: gene–environment interactions initiating a tissue response
In our model of disease development, interactions between genes expressed by the host and elements in his/her exposome are postulated to be transient, recurrent or chronic. There, each interaction between an individual gene and a concrete environmental factor is considered to have the potential for initiating long‐lasting tissue effects. However, few individual interactions will be followed by sustained, pathological tissue responses due to modulation by a network of changes generated by the genome and exposome (Darabos et al., 2016). For example, a dominantly inherited risk allele of high penetrance could be epigenetically silenced in a carrier (or a toxicant not effectively absorbed despite recurrent exposure) owing to a network of interactions; if so, the mutant allele (or the noxious chemical) would fail to promote relevant tissue changes in the host. Based on our hypothesis, factor ‘I’ reflects the initiation of a sustained biological response in vivo stemming from gene–environment interactions that are essential in the development of PD. We postulated that quantifying it will inform incidence risk.
For a disease phenotype to be expressed in vivo, metabolic functions, signalling mechanisms, cellular integrity and/or extracellular matrices have to be compromised. While the reasons for the progressive nature of variants for young‐onset and late‐onset PD in humans are diverse and many remain unknown, several pathways have been delineated in human tissue and animal experimentation that help explain neural injury and cell death during disease initiation and/or its progression. The output of gene–environment interactions can initiate a spectrum of pathological responses, for example, a rise in ROS production, dysregulation of inflammation, and/or the start of amyloidosis (Hirsch et al., 2012; Crunkhorn, 2016). For building our prediction model and to calculate P R scores, factor ‘I’ was included in our formula.
Today, there is limited availability of easily accessible markers to signal in real‐time the ‘state, rate and fate’ of tissue changes that occur in the development of PD (Schlossmacher & Mollenhauer, 2010; Mollenhauer et al., 2013b; Kang et al., 2016); hence, values for ‘I’ were assigned based on published insights regarding the in vivo effects of variables E and D. In addition, we used clinically detectable surrogate markers for inferred tissue responses. For example: 0.001, paucity of evidence for any initiation of a lasting host response; 0.25–1, elevated ROS production resulting from mitochondrial dysfunction (i.e. as informed by results from parkin and pink1 deficiency models); 0.25–1, enhanced pro‐amyloidogenic protein production (or its dysregulation), foremost of α‐synuclein, leading to the onset of neural proteinopathy; 0.25–1, sustained dysregulation of inflammation; and 0.25–1, activation of cell death pathways. A list of examples for tissue responses and their assigned ‘I’ values for model building purposes is shown in Table 1.
Note, the development of new‐onset REM sleep behaviour, anxiety or depression in heretofore healthy probands was interpreted as evidence of a disease process that has involved structures within the central nervous system. For modelling purposes the presence of such prodromal changes were also scored to reflect a more advanced, pathological tissue response; therefore, we added them to the score sheet under factor ‘I’ (maximum value, 3; Table 1).
Variable ‘G’: sex (gender)
Variable G is informed by incidence rates for PD in both sexes. Age‐adjusted prevalence and incidence rates for typical PD were recently reviewed by Berg et al. on behalf of an International Parkinson and Movement Disorder Society‐appointed task force. Table 1 contains modifications of their ‘prior probability of PD estimates’ according to sex, thereby reflecting the overall male preponderance in idiopathic PD cases in cohort studies (e.g. Locascio et al., 2015) and a likely reversed sex bias in LRRK2‐associated illnesses. The latter was recently identified in human PD cohort studies (Cilia et al., 2014; Trinh et al., 2014; Marder et al., 2015). In our formula, the variable ‘sex’ is entered as the independent factor ‘G’; it is expected to reflect the contribution of genetic elements (biological sex) and their interaction with environmental elements that may differ between the sexes (gender). For model building purposes, the value entered under ‘G’ is either 0.8 or 1.2; these numbers reflect prevalence ratios among the sexes in the general population (e.g. Driver et al., 2009; Berg et al., 2015; Locascio et al., 2015), which – based on recent findings in the literature – were corrected for in the context of mutant LRRK2 carrier status (Table 1).
Variable ‘T’: age(ing) of the subject and clinical latency
Ageing is considered to be the most important risk factor for the incidence of PD (e.g. Driver et al., 2009). We view ageing predominantly as the ‘passage of time’ and expressed it as factor ‘T’ (standing for Time) in years. Although in our model, factor ‘T’ is defined by the subject's age (values 1–100), it accommodates three key elements in the development of PD: (i) Physiological, ageing‐associated changes, such as in gene expression rates, immune function, mitochondrial integrity, and metabolic efficiency; (ii) A clinical latency phase, i.e. the period between critical gene–environment interactions that have initiated the disease process and the actual onset of prodromal (pre‐motoric) changes; and (iii) The progression of the disease process in an individual that transitions from the prodromal stage to the clinically diagnosed phase of PD (Fig. 1).
Calculating P R: a six‐step process
The generation of a PREDIGT score to calculate the incidence rate for the development of PD in an individual is determined based on a six‐step process. The first five steps correspond to entering values for each factor from a pre‐populated list into a score sheet and the subject's age (E, D, I, G, T; Table 1); step six corresponds to the calculation of the actual score (in %) following the formula: P R = (E + D + I) × G × T.
For developing and refining the model, we examined cohorts of genetic vs. sporadic cases of parkinsonism published in the literature (between January 1986 and July 2016) and from our own studies. Presented herein are six paradigmatic clinical scenarios for demonstration of the PREDIGT score model. These reflect three rare cases leading to young‐onset parkinsonism and highlight three scenarios with elevated incidence risk for the development of late‐onset, typical PD.
Patient cohorts
Patient characteristics that informed our case scenarios were gleaned from the published literature and available data bases for subjects previously enrolled in case–controlled biomarker studies, e.g. single centre cohorts, such as the Kassel Cohort (Mollenhauer et al., 2011), Ottawa Biomarker Protocol (Bidinosti et al., 2012), DeNoPa Cohort (Mollenhauer et al., 2013a) and Harvard Biomarker Study (Ding et al., 2013; Locascio et al., 2015) as well as the multi‐site PROBE and PPMI cohort (Locascio et al., 2015; Kang et al., 2016). Studies were approved by the respective institutional review boards at all participating hospitals and clinics.
Results
Prototypes of early‐onset parkinsonism
Case 1: SNCA gene mutation‐linked, young‐onset parkinsonism
We first examined the scenario of a 39‐year‐old, male carrier of a wild‐type SNCA triplication from the ‘Iowa kindred’ (Singleton et al., 2003; Farrer et al., 2004; Gwinn et al., 2011; Trinh et al., 2014).
Step 1 (factor E)
In this heritable case of parkinsonism with high penetrance, we assumed that for carriers of such a rare genotype, which leads to dominantly inherited disease beginning in early adulthood [e.g. with age‐of‐disease onset (AOO) frequently before 40 years], the role of exposome may be rather low regarding its overall contribution to incidence risk (Kitada et al., 2012). Under this particular circumstance and the proband's age, factor E was assigned the value 0.00 (Table 1).
Step 2 (factor D)
We assigned the value of 1 for factor D, because the subject carries the rare but significant genetic predisposition to develop dominantly inherited, young‐onset parkinsonism accompanied by dysautonomia and dementia (Singleton et al., 2003; Gwinn et al., 2011).
Step 3 (factor I)
As shown in numerous vertebrate studies of transgenic over‐expression of SNCA cDNA in neurons [reviewed by (Chesselet & Richter, 2011)], we anticipated that this man will develop insoluble α‐synuclein aggregation‐associated disease throughout his nervous system, widely viewed as of 2016 to lead to a progressive proteinopathy [‘synucleinopathy’ (Irwin et al., 2013)]; therefore, the value of 1 was entered for factor I.
Step 4 (factor G)
In the few published kindreds with inheritance of a SNCA triplication (n = 4 SNCA alleles) mutation, no noticeable sex contribution has been reported in affected family members for the incidence of parkinsonism; therefore, we entered the value of 1.2, which was informed by the ‘prior probability of PD in the general population’, under factor G for this male subject (Berg et al., 2015; Locascio et al., 2015) (see Table 1).
Step 5 (factor T)
His current age, 39, was entered as the value for factor T.
Step 6 (score calculation)
The P R score [(0.00(E) + 1(D) + 1(I)) × (1.2)G × 39(T)] was calculated to be 93.6% at his current age. Accounting for changes in age (T), we can thus predict the time of clinically diagnosed parkinsonism in this proband at age 41.7 years (as defined by P R = 100%; Fig. 1), which would be consistent with insights from a recently published meta‐analysis for the AOO in pathogenic SNCA genotypes (Trinh et al., 2014).
Comments
In this man, variant genotypes present at other loci could change the value for factor D in both directions; for example, a cumulative value of 1.25 for D (resulting from carrier status of an additional risk allele of overall smaller effect size) would mean that the P R score of 100% could be reached at an earlier age, whereas a total value of 0.75 for D (i.e. co‐inheritance of a protective allele) would lower the P R score to 81.9% at age 39 years.
Similarly, a 70‐year‐old Japanese man carrying a duplication of the wild‐type SNCA allele on one sister chromatid (n = 3 SNCA alleles; D = 0.75; I = 0.5) (Nishioka et al., 2006) with no known significant exposure to an environmental risk factor associated with PD (E = 0.02 adjusted for age; Table 1) will have a P R score of (0.02(E) + 0.75(D) + 0.5(I)) × 1.2(G) × 70(T) = 106.68%. According to the formula, this man is expected to have expressed his typical PD phenotype by the age of 65.62 years (P R = 100%) (Trinh et al., 2014). Entering a higher value for E (if he had also been exposed to identifiable, PD‐associated risk factors during adulthood) would increase his P R score, and thus, measurably lower this man's AOO, and by inference, the time of diagnosis. In contrast, epigenetic silencing of SNCA alleles in this man (which could be deduced from biomarker studies in vivo) would lower the value entered for ‘I’, reduce the final score, and would thus increase the predicted AOO. Examples for select genotypes that underlie dominantly inherited PD/parkinsonism (Marras et al., 2016) and their assigned values under factor D (and I) can be found in Table 1.
Case 2: bi‐allelic PARKIN mutation‐linked, young‐onset parkinsonism
To further build our model, we next examined the scenario of a 34 year‐old female, compound heterozygote mutation carrier at the PARK‐Parkin locus from the ‘South Tyrolean kindred LA’ in Northern Italy, which is known for the occurrence of young‐onset parkinsonism (Pramstaller et al., 2005; Marras et al., 2016).
Step 1 (factor E)
As with case #1, we first assumed that no major environmental risk factor played a role in the expression of her parkinsonian phenotype at a young age (E = 0.00; Table 1).
Step 2 (factor D)
Given that both of her PARKIN mutations result in the truncation (and thus inactivation) of encoded Parkin proteins, we assigned the total value of 2 (2 × 1) for D (Table 1).
Step 3 (factor I)
As suggested by evidence collected at autopsy from human patients and generated in animal models of parkin deficiency (e.g. Itier et al., 2003; Palacino et al., 2004; Periquet et al., 2005; Kitada et al., 2009), loss of the protein's function promotes the progressive degeneration of dopamine neurons due to rather cell‐selective mitochondrial dysfunction, a steady rise in ROS levels and the reduction in axonal maintenance; hence, we assigned a value of 1 for factor I in this genotypic scenario (Table 1).
Step 4 (factor G)
The value for factor G was entered as 0.8 for this woman (Table 1).
Step 5 (factor T)
Her age, 34, was entered as the value for variable T.
Step 6 (score calculation)
The P R score was calculated as (0.00(E) + 2(D) + 1(I)) × 0.8(G) × 34(T) = 81.6%, which indicates that she is expected to meet criteria for the clinical diagnosis of parkinsonism at the age of 41.7 years (P R score = 100%) (Lucking et al., 2000; Pramstaller et al., 2005; Klein et al., 2007) (Fig. 1).
Comments
As with case #1, carrying additional variant genotypes at other loci would change the cumulative values for factor D and I (up‐ or downward), which is predicted to move the AOO and the subsequent time of diagnosis for her parkinsonism into the 20s, or as high as above 60 years, as suggested by the published literature of PARK‐Parkin mutation carriers (Lucking et al., 2000; Pramstaller et al., 2005; Klein et al., 2007). Similar scenarios could be drawn for carriers of pathological mutations in both alleles of genes encoding for DJ‐1 and PINK1, which are known to lead to an indistinguishable phenotype of recessive, young‐onset parkinsonism (Lucking et al., 2000; Bonifati et al., 2003; Valente et al., 2004).
Over the years, researchers have vigorously debated whether carrier status of only one mutant allele linked to recessive parkinsonism (e.g. PARK‐Parkin; PARK‐PINK1) represents a rare ‘single hit’ leading to PD, such as via a possible dominant‐negative gain‐of‐function effect under exceptional circumstances, or alternatively, whether it could serve as a substantial risk factor for late‐onset PD [reviewed in: (Klein et al., 2007)]. Upon validation, our P R score model could serve as a platform to quantitatively assess the contribution of other genetic factors in the expression of a parkinsonian phenotype in the latter scenario. Examples for select genotypes underlying recessively inherited parkinsonism and their assigned values for factor D (and I) can be found in Table 1.
Case 3: neurotoxin‐associated parkinsonism in a young adult
Strictly for modeling purposes, we next examined bona fide, environmental pathogen‐linked cases of secondary parkinsonism with ‘high penetrance’. There, we assumed, for example, that victims of a neurotoxicant exposure carry a relatively small (but not necessarily, negligible), genetic burden to their overall disease risk (Kitada et al., 2012). Although an exclusively environmental contribution to generate parkinsonism in humans is exceedingly rare (Langston et al., 1983), such variants have been described as the result of accidental intravenous (i.v.) administration of toxicant‐laced, illicit drugs prepared in home laboratories [e.g. meperidine contaminated by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), or methcathinone contaminated by high manganese levels] (Langston et al., 1983; Stepens et al., 2008). Based on their chemical nature, route of exposure and cumulative dose, select compounds may have exceptional tropism to confer toxicity towards neurons of the basal ganglia circuitry in vertebrates, thereby leading to rapid‐onset parkinsonism (Langston et al., 1983; Stepens et al., 2008).
We therefore considered the scenario of a 28‐year‐old man, who self‐administered MPTP‐containing meperidine by i.v. injection twice.
Step 1 (factor E)
We scored his value for factor E as 2 reflecting his total neurotoxin exposure burden (Speciale, 2002; McCormack et al., 2008) (Table 1).
Step 2 (factor D)
Variable D was assigned the value of 0.01 reflecting the smaller (but not insignificant) contribution by genetics to his overall susceptibility to MPTP (Dauer et al., 2002; Fornai et al., 2005; Pattarini et al., 2007) (Table 1).
Step 3 (factor I)
From many toxicological studies of MPTP exposure in rodents and primates (e.g. Dauer et al., 2002; Fornai et al., 2005; McCormack et al., 2008; Fox & Brotchie, 2010), we concluded that this man will have developed mitochondrial impairment with a marked rise in ROS levels, which underlies the degeneration of dopamine cells in the S. nigra of MPTP‐treated mammals. We therefore entered the value of 1.0 under factor I (Table 1).
Step 4 (factor G)
The value for his sex was entered as 1.2 (Table 1).
Step 5 (factor T)
His age, 28, was entered as the value for factor T.
Step 6 (score calculation)
Calculation of his P R score (2(E) + 0.01(D) + 1.0(I)) × 1.2(G) × 28(T) amounted to 101.1%, which reflects the fact that he has met criteria for parkinsonism. In the absence of any neurotoxicant exposure, his P R score would have been 0.3%, which is consistent with the published literature regarding the proband's a priori probability to develop PD at that age (Berg et al., 2015) (Fig. 1). Note, a lower total value for variable E, for example, due to a lesser cumulative dose of neurotoxin exposure, would have reduced his P R score, and thus, markedly delayed the predicted AOO and time of diagnosis.
Comments
Experiments in mice have shown that the targeted deletion of both dj‐1 alleles worsens the rise of ROS in MPTP‐treated animals (and thus, further lowers neuronal survival) (Kim et al., 2005), thereby highlighting the fact that variants in genes encoding modifiers for neurotoxicant uptake, its import into mitochondria, and cellular ROS responses could further alter the cumulative values for D and I, and thus change the outcome of an acute (or chronic) neurotoxin exposure in humans.
In addition to the cases listed above and for building the model, we also examined other environmental factors that have previously been linked to the development of secondary parkinsonism in humans to inform our prediction paradigm; for example, we considered cases of recurrent, (sub)concussive head traumas, such as those sustained by athletes during years of practicing contact sports (Mez et al., 2013), or illnesses following host invasion by select xenobiotics, such as a viral infection leading to an encephalitis (Litvan et al., 1998; Reid et al., 2001; Jang et al., 2009; Tappe & Alquezar‐Planas, 2014) (Table 1). Of note, autopsies of patients with parkinsonism as a result of systemic viral illness or due to recurrent (sub)concussive events have shown, among other neuropathological findings, a dysregulation of MAPT‐encoded tau protein (i.e. hyper‐phosphorylation and insolubility of aggregates) (Jellinger, 2009; Mez et al., 2013). In accordance, and for further consideration, we have entered value estimates for the related factors E and I in Table 1.
Scenarios for typical, late‐onset Parkinson disease
Case 4: GBA mutation‐linked risk to develop late‐onset Parkinson disease
As of 2016, heterozygous variants encoding point mutations at the PARK‐GBA locus (Marras et al., 2016) represent the commonest, known genetic risk factor for typical, late‐onset PD in non‐Ashkenazi subjects (frequency rate, > 8%) and Ashkenazi Jews (frequency of carrier status, > 20%) vs. control groups without PD (~ 1 and > 3%, respectively) (Sidransky et al., 2009; Gan‐Or et al., 2015). According to a report regarding risk‐to‐phenotype conversion rates, the penetrance rate of heterozygous GBA mutations (and even of homozygous mutation carriers that have developed Gaucher disease, type‐I) is < 10% by the age of 80 years (Alcalay et al., 2014). These results strongly suggested to us that additional, ‘hidden factors’ are essential to express the PD phenotype in GBA risk allele carriers.
We present the case of a 40 year‐old, Caucasian, non‐Ashkenazi man without any neurological symptoms (or signs), who carries a heterozygous p.L444P mutation at the PARK‐GBA locus. We entered the following parameters into the PREDIGT formula.
Step 1 (factor E)
During the interview, he informs the examiner that he has no history of constipation and no known, previous exposure to other, PD‐associated environmental risk modifiers. In accordance with his age, the value 0.00 is entered under factor E (Table 1).
Step 2 (factor D)
Because of his sequencing‐confirmed GBA mutation carrier status (and in the absence of any other known, risk modifying allele), the value 0.5 is entered under factor D (Table 1).
Step 3 (factor I)
Multiple laboratories including ours have demonstrated that the expression of mutant GBA1 protein leads to an elevated total concentration for α‐synuclein of up to 25% in select compartments of neural cells in vivo (e.g. Cullen et al., 2011; Sardi et al., 2011). The effect of GBA1 protein mutations in promoting α‐synuclein pathology has also been confirmed in human autopsy studies (e.g. Eblan et al., 2005). For this reason, under factor I, we assigned the value of 0.25 (Table 1).
Step 4 (factor G)
The proband's sex is male; thus, we entered 1.2 for factor G.
Step 5 (factor T)
The value 40 for the age of the subject was entered under variable T.
Step 6 (score calculation)
The P R score was calculated as 36.0%. In other words, at the current age of 40 years, the cumulative PD incidence rate in this individual amounts to 36%; according to the formula, at age 60 years, the rate would be 46.35%.
Comments
If the same man reported a > 20 year history of constipation (as a surrogate marker for past encounters in the GI tract of one or more environmental pathogen with relevance to PD), the value of 1 would be entered for factor E (Table 1), and his P R score would increase substantially. Of note, an interaction between constipation and the dysregulation of human α‐synuclein has been reported in animal models of PD (Kuo et al., 2010) and may occur throughout the human colon including the appendix (Gray et al., 2014). At age 40 years, this man's P R score would then be calculated as 84.0%. For demonstration purposes, at the age of 60 years his cumulative PD incidence rate would amount to a P R of 126%, which – according to our formula – would correspond to the clinical diagnosis of typical PD at the age of 47.61 years. In contrast, in the absence of any GBA mutation (even with constipation as a substantial risk factor present), this man's P R score would have decreased to 81.72% [(1(E) + 0.01(D) + 0.125(I)) × 1.2(G) × 60(T)] at the age of 60 years. Of note, the effects of a mutant GBA allele in significantly lowering the AOO (and thus, the related time of formal diagnosis) for typical PD in affected carriers have now been published in multiple ethnicities (e.g. Alcalay et al., 2014).
In the latter scenario, the bona fide PD risk factor of constipation could have been substituted with, for example, anosmia, as confirmed by a validated smell test (and which would have been entered as value 1 for E), or hyposmia (entered as value 0.5 for E). In our model, the gradual loss of a subject's sense of smell serves as a surrogate for past encounters in the nasal cavity of one or more significant environmental pathogen with relevance to PD (as constipation did for the GI tract), leading to the initiation of a disease process (factor I) beginning in the olfactory epithelium (Duda et al., 1999; Saito et al., 2016) (Table 1). The bi‐directional effects of individual risk modifiers in the exposome (factor E) and genome (factor D) of a person as well as the corresponding tissue effects, [which the network of interactions (captured by factor I) has on the P R score] underlie the predicted time of PD diagnosis (P R score = 100%); the dynamic nature of these changes is graphically displayed in Fig. 1 and Fig. 2.
Case 5: LRRK2 mutation‐linked, late‐onset Parkinson disease with reversed sex bias
Carrier status of a heterozygous mutation at the PARK‐LRRK2 locus at few select codons (Marras et al., 2016) is currently considered the second most frequent, genetic risk factor for late‐onset, sporadic PD (after GBA variants) with an estimated, mean prevalence rate of 1–2% in North America (Lesage et al., 2006; Ozelius et al., 2006; Heckman et al., 2013; Bozi et al., 2014; Trinh et al., 2014; Kang & Marto, 2016). Mutations in the LRRK2 gene had been initially identified as the cause of rare, dominantly inherited, familial PD in multiple pedigrees (Paisan‐Ruiz et al., 2004; Zimprich et al., 2004), but many questions surrounding the causative mechanisms by which bona fide LRRK2 mutants promote brain pathology (either directly, or indirectly) at an overall lower than the expected penetrance rate have remained unanswered.
We therefore examined the scenario of a 51‐year‐old woman with a 10‐year history of reduced sense of smell (i.e. hyposmia), but no other neurological findings. She carries a heterozygous p.G2019S mutation in the LRRK2 gene (Gaig et al., 2014). Therefore, we entered the following parameters into our formula to calculate the cumulative PD incidence rate.
Step 1 (factor E)
We entered the value 0.5 under factor E because of her hyposmia (she had no other known PD‐associated environmental risk modifier in her history). Her reduced olfaction, as confirmed by standardized testing, had been attributed to recurrent congestions and the presence of nasal polyps, and was interpreted by us as surrogate marker for past encounters of one or more environmental pathogen with relevance to PD within her URT (Table 1),
Step 2 (factor D)
For her sequencing‐confirmed LRRK2 mutation carrier status, the value 0.5 was entered under factor D (Table 1).
Step 3 (factor I)
Given the fact that select point mutants of LRRK2 have recently been shown to dysregulate inflammation in humans and mice downstream of infections (e.g. Halliday et al., 2011; Daher et al., 2015; Dzamko & Halliday, 2012; Fava et al., 2016) (B. Shutinoski et al., manuscript in preparation), we entered the value of 0.5 under factor I; it comprises 0.25 for dysregulation of inflammation, and 0.25 for the suspected upregulation of α‐synuclein (Beatman et al., 2016), or tau (Table 1). Note, accumulation of intracellular, insoluble α‐synuclein or tau is seen at autopsy in the majority of patients with mutant LRRK2‐associated parkinsonism (Zimprich et al., 2004; Rajput et al., 2006; Kalia et al., 2015).
Step 4 (factor G)
Intriguingly, in recent studies on the conversion rate of risk‐to‐phenotype in mutant LRRK2 carriers between sexes in select ethnicities (and geographies), researchers have documented a female rather than male sex bias (e.g. Cilia et al., 2014; Marder et al., 2015). Therefore, we entered the sex/gender‐adjusted value of 1.2 under factor G (Table 1).
Step 5 (factor T)
The value 51 was imputed under variable T.
Step 6 (score calculation)
The P R score for this woman was calculated as 91.8%. According to our formula, she is predicted to convert her elevated disease risk into the diagnosis of typical PD by the age of 55.56 years (P R score, 100%). If this carrier were a man, the model predicts that the time of his PD diagnosis would occur at age 76.5 years (P R score, 100%) because his value for G would be imputed as 0.8 instead (based on the observed reversal of sex bias) (Table 1). The effects by a mutant LRRK2 allele on significantly lowering the AOO (and thus, the related time of diagnosis) for typical PD in affected women have been published in at least two cohorts (e.g. Cilia et al., 2014; Marder et al., 2015).
Comments
Intriguingly, in a study of monozygotic twins carrying a heterozygous p.G2019S mutation in LRRK2 with confirmed identity of their genome (Xiromerisiou et al., 2012), the authors described discordance for the expression of the PD phenotype by over 10 years.
Let us therefore examine the theoretical case of our proband's identical twin sister, who has no history of recurrent nasal infections and no hyposmia on formal testing (confirmed by an UPSIT score of > 34/40). The twin's score card for cumulative incidence risk includes the following assigned values: 0.005 for factor E (given her age and without a known, PD‐relevant exposure history to xenobiotics in the URT or GI tract); 0.5 for factor D; 0.001 for factor I given the inferred paucity of tissue response(s) in the absence of a known pathogen at her age (Table 1); 1.2 for G; and 51 for variable T. The twin's P R score would be 30.97% at 51 years and 36.50% at age 60 years. According to our model, the normosmic twin (provided she continues to lack identifiable, PD‐relevant factors in her exposome that would initiate a disease process later; Table 1) would still not have developed an LRRK2‐associated brain disorder at the age of 80 years (P R = 51.26%). Therefore, the network of interactions between the identical genome of the twin sisters (identical value for D) with variable elements in their exposome (variable values for E) likely results in distinct tissue responses. We currently score these with different values for factor I; if true, this concept could explain the marked discordance for PD in the monozygotic twins reported by Xeromerisiou et al. The dynamic, bi‐directional effects of interacting risk modifiers on the P R score, and thus on the predicted AOO and diagnosis of PD, are graphically shown in Fig. 1 and Fig. 2.
The single publication to date of discordant twins with the same p.G2019S point mutant‐carrying allele –when taken together with our recent experimental findings regarding LRRK2's role in complex diseases‐ places the emphasis on necessary environmental triggers and related host response(s) to express a disease phenotype (Hakimi et al., 2011; Hawkes, 2013). Our model therefore may shed light on the three vexing issues in LRRK2 biology mentioned above: First, the interdependence of gene–environment interactions triggering tissue responses also accommodates the fact that brains from patients with mutant LRRK2 genotypes have been associated with pleomorphic neuropathology, such as with evidence of dysregulated tau but no accumulation of misfolded α‐synuclein, or vice versa (Zimprich et al., 2004; Rajput et al., 2006; Kalia et al., 2015). We speculate that distinct environmental triggers, such as recurrent (sub)concussive head traumas or systemic infections (variable factor E) that trigger encephalitis‐type changes, could be responsible for promoting tau dysregulation in some carriers of a LRRK2 mutation, even within the same pedigree (shared risk factor D). There, other affected members might develop α‐synuclein‐positive pathology (variable factor I) (Zimprich et al., 2004; Rajput et al., 2006; Hakimi et al., 2011). We have therefore entered head traumas (Taylor et al., 2016) and infections (Masliah et al., 1994; Bu et al., 2015) as PD risk‐associated environmental triggers into our first draft for a score sheet (Table 1).
As demonstrated, our PREDIGT model also helps explain the intriguingly variable penetrance rates that have been recorded for the same LRRK2 mutation across ethnicities, possibly due to an even higher exposome burden in some regions, or the presence of genetic modifiers of LRRK2 in select geographies (Trinh et al., 2014, 2016). Third, this interdependence between gene–environment interactions and tissue‐specific response(s) (factor I) could possibly explain the association of certain LRRK2 genotypes with the risk of developing Crohn's disease and leprosy. Why? Because without chronic exposure to a dysregulated gut microbiome and without infection by M. leprae, respectively, these two complex diseases would not be expressed (Hakimi et al., 2011; Rocha et al., 2015; Fava et al., 2016).
Furthermore, the interdependence of several essential factors may also explain why mutant Lrrk2‐expressing animals show no discernible PD‐like phenotype in the absence of exposure to a microbial trigger (e.g. Herzig et al., 2011; Moehle et al., 2012; Ness et al., 2013; Daher et al., 2015). Last but not least, our model also serves as a platform to examine conjugal cases of PD, i.e. the concordance of parkinsonism between partners that live together over decades (shared factor E), but are biologically unrelated (variable, but positive, values for factor D) (Willis et al., 2010; Rajput et al., 2016).
Case 6: probability of PD in a 50‐year‐old smoker with a positive family history
We posed the following question relevant to discussions in doctors’ offices and the planning of future clinical trials aimed at preventing PD: In a healthy adult, would the limited availability of concrete information regarding genetic risk and exposure history still permit the calculation of overall incidence rate for developing PD later in life? Let us therefore consider the scenario of a 50‐year‐old smoker, who was medically and neurologically healthy at that age. The proband has a first‐degree relative (i.e. his father) with late‐onset PD (Fig. 3).
Figure 3.
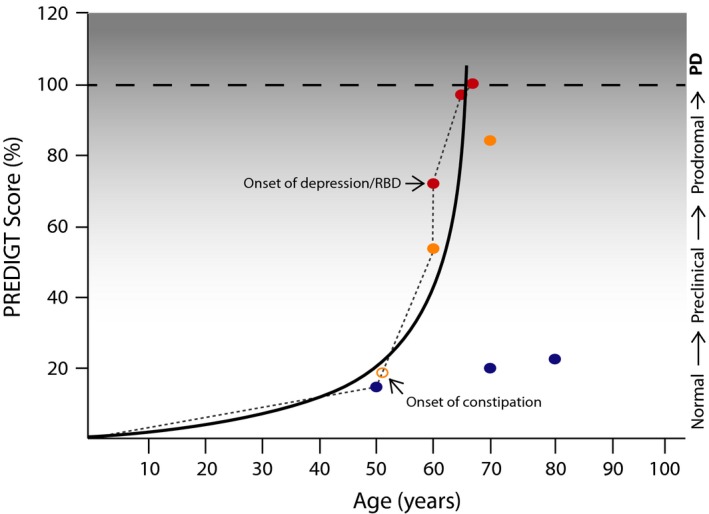
Graphic representation of cumulative incidence rate for PD in a 50‐year‐old man. The prediction of Parkinson disease (PD) in a 50‐year‐old smoker is depicted (see scenario #6 in the text). Blue dots: Incidence rates are calculated based on the individual's current smoking status, a confirmed, positive family history of PD, the inferred tissue response(s), sex/gender, and age: PREDIGT scores of 15% at age 50; 21% at age 70; and 24% at age 80 years are shown. Yellow dots: The calculated cumulative incidence rate for PD has changed after cessation of smoking at age 50 years and the onset of constipation shortly thereafter (open yellow circle): PREDIGT scores, 54% at age 60, and 84% at age 70 years. Red dots: The diagnosis of new‐onset depression (or a REM sleep behaviour disorder; RBD) at age 60 years changes the cumulative incidence rate for PD, as represented by a PREDIGT score of 72% at age 60, of 97.5% at age 65, and the projected diagnosis of typical PD by 66.67 years (score, 100%). The course of disease development (solid black line) was inferred based on PREDIGT scores generated at multiple time points. Clinical stages are as described in the text and in Fig. 1.
Step 1 (factor E)
During the interview, he denied any consumption of caffeinated beverages. A value of −0.5 was imputed under factor E given his less than 20‐year history of cigarette smoking. As an environmental modifier, ‘current smoking’ status has been consistently shown to be associated with reduced PD risk (e.g. Driver et al., 2009) [reviewed in: (Bellou et al., 2016)] (Table 1).
Step 2 (factor D)
For his not yet determined genetic status but elevated theoretical risk given his positive, independently confirmed family history (Sveinbjornsdottir et al., 2000; Elbaz et al., 2003; Nalls et al., 2015), the value of 0.5 was entered under factor D (Table 1).
Step 3 (factor I)
Given the lesser likelihood for the initiation of a tissue response that promotes PD resulting from altered interactions between the subject's genome and exposome (which we postulate may be due to the protective effects of smoking), we entered the total value of 0.25 under factor I (i.e. we equated his possible tissue response(s) to that of a proband carrying a mutant GBA allele or an expanded, Rep1‐positive SNCA allele (Chung et al., 2014); Table 1).
Step 4 (factor G)
We entered the value of 1.2 for his sex as per the a priori probability of PD in the general population (Berg et al., 2015) under factor G (Table 1).
Step 5 (factor T)
The value 50 corresponding to his age was imputed under factor T.
Step 6 (score calculation)
The computed P R score at age 50 years was 15% (Fig. 3). When asked about his probability to develop parkinsonism at age 70 years, which was the age of diagnosis for his father's typical PD, the theoretical incidence rate would be calculated as 21.0%; this, provided the subject indeed carried the same risk allele that played a role in his father's illness and that he continued to smoke (see Fig. 3, where blue dots indicate calculated P R scores).
Comments
When the subject was seen again in the clinic at age 60 years, he reported the cessation of smoking at age 50 years and the presence of chronic constipation for now nearly a decade. A revised incidence risk assessment would incorporate an updated value for E as 0.0 (i.e. −0.25 for the former smoking status; +0.25 for constipation of < 10 years; Table 1), thereby leading to a P R score of 54.0% at the age of 60 years. Based on the formula, we calculated a projected score of P R = 84.0% at age 70 years [−0.25 + 0.5 (the latter value reflecting constipation for > 10 years) (E) + 0.5(D) + 0.25(I)) × 1.2(G) × 70(T) (Fig. 3, where yellow dots indicate revised P R scores).
With inclusion of this proband in sequencing studies of his family to identify the genetic nature (and the presence or absence) of any risk allele, and his participation in future biomarker studies that could accurately assess his degree of an initiated host response (such as through the quantification of analytes in CSF or blood, or based on markers from radiographic/nuclear medicine studies) (Mollenhauer et al., 2011; Berg et al., 2015; Chahine et al., 2016), his P R score could be updated to impute better informed values for factors D and I. By inference, a practitioner's ability to forecast this man's likelihood of PD would be more accurate.
However, let us assume that even without further knowledge of his genetic variation risk and fluid analysis‐based biomarker status, this man was being followed regularly by his doctors. At age 60 years, he now also developed new‐onset anxiety and depression (or, alternatively, he was diagnosed with REM sleep behaviour disorder). We interpreted this change as reflection of a disease process that has reached the central nervous system (i.e. the prodromal stage). Therefore, we updated the score sheet under factor I to the total value of 0.5 (Table 1). This would lead to a revised P R score of 72.0% at age 60 years [−0.25 (remote smoking status) + 0.25 (constipation of < 10 years)(E) + 0.5(D) + 0.5(I) × 1.2(G) × 60(T)] (Fig. 3). The cumulative P R score predicted during a follow up visit at age 65 years would be 97.5%, and would climb to 105% at age 70 years [−0.25 + 0.5 (accounting for constipation for 10+ years)(E) + 0.5(D) + 0.5(I) × 1.2(G) × 70(T)]. Thus, according to our formula, we would predict the diagnosis of PD in this man at the age of 66.67 years (P R score = 100%) (Fig. 3; where red dots indicate scores based on updated information at the time of follow up visits).
Case #6 therefore highlights a scenario by which a multi‐component formula, which is based on clinically available information and pre‐determined surrogate markers, was employed in the context of a hypothesis driven model; it was then applied to the theoretical encounters between a subject without a neurological disorder and his/her health care workers. The PREDIGT score model could thus inform investigators as to the relative incidence rate of PD in a specific individual several years prior to the diagnosis of parkinsonism (Fig. 3). Case #6 also highlights the three crucial opportunities for possible interventions in the future to change the trajectory of such disease development: (i) For neuroprevention at age 50 years with the recognition of a possible genetic risk; as well as (ii) At around age 55 following the onset of chronic constipation; and (iii) For neuroprotection after age 60 years with the development of previously absent anxiety and depression (or a recently diagnosed REM sleep behaviour disorder).
Discussion
Risk stratification for decision‐making has evolved into important tools for medical practitioners in both clinical and research settings, where they are applied to the prevention of illness, avoidance of complications, and slowing of disease progression. This has led to the development of validated and well‐established scoring systems, such as those employed in the risk assessment for annual embolic stroke from atrial fibrillation (Hsu et al., 2016), of coronary events associated with dyslipidaemia (Preiss & Kristensen, 2015), and of deep vein thrombosis and its complications, such as pulmonary emboli (Modi et al., 2016).
Here, we created the PREDIGT score model to calculate the cumulative PD incidence rates in healthy adults where a P R score of 100% (or greater) corresponds to the presence of clinically diagnosed, typical PD (Postuma et al., 2015) (Figs. 1, 3). While the formula‐based scoring system was principally designed to accommodate our view for the pathogenesis of PD/parkinsonism, it could also be employed in future research settings, such as in observational and interventional studies. For this purpose, the PREDIGT score model needs to be transformed into a formula that turns cumulative incidence rates into a probability based risk algorithm. There, final calculations are restricted to approximate predictive values that range between 0 and 1 (with 0 indicating the complete absence of neurological signs, and with 1 equating the clinical diagnosis of PD) and that assign relative measures, such as through proportional hazards, for prediction outcomes. Recently, such transformations were successfully completed in concrete applications, e.g. in the prediction of outcomes related to cognitive function in PD (Locascio et al., 2015) and to mortality in a large North American population (Manuel et al., 2016). Most importantly, the PREDIGT score model has to be validated, and likely be further refined, in real‐life settings, both retrospectively, using cross‐sectional, case–controlled cohorts, as well as prospectively, by testing it in longitudinal population cohorts (Mollenhauer et al., 2011, 2013a; Locascio et al., 2015; Chahine et al., 2016; Kang et al., 2016).
While some disease progression models exist (Locascio et al., 2015; Nalls et al., 2015; Chahine et al., 2016; Venuto et al., 2016) and a risk assessment tool for prodromal PD has recently been published (Berg et al., 2015), the PREDIGT score is unique in that it represents the first platform to quantify cumulative incidence rates for PD in neurologically healthy adults. We consider the strengths of this tool to include the following 6 aspects: (i) It was founded on conservative interpretations of concrete epidemiological evidence, established genetic insights, well‐documented sex differences as well as accepted pathophysiological mechanisms from in vivo studies; (ii) The PREDIGT model, while hypothesis‐driven, functions within the recently revised criteria for the diagnosis of prodromal illness as well as for typical PD (Berg et al., 2015; Postuma et al., 2015); (iii) Identifying relevant risk categories can be done by history‐taking from probands, careful review of their family history (Elbaz et al., 2003) and clinical examination. Once identified, these factors are assigned values in a ready‐to‐use score card system (Table 1). Therefore, the PREDICT formula itself is easy to use; (iv) The model does not favour one variant of parkinsonism (e.g. cases #1–3) vs. another type of PD (e.g. cases of #4–6); rather, because of the integration of multiple risk modalities, it accommodates each one, thereby complementing future efforts to personalize diagnosis and facilitate specific interventions; (v) If validated, it will fill a void, because currently there is no inexpensive, non‐invasive and practical research tool to measure cumulative incidence rates for PD in neurologically healthy individuals; and (vi) Our PREDIGT score model may signal a demystification process for ‘idiopathic PD’ by delineating its pathogenesis in accordance with other late‐onset, complex illnesses. Several, multifactorial ailments in adults evolve from an innocuous start in the periphery earlier in life, only to fully manifest as disorders of the central nervous system many years later (e.g. multiple sclerosis; stroke due to chronic hypertension; viral encephalitis).
Our tool is still hypothetical in nature. The PREDIGT score model is based on our current understanding of risk factors associated with PD incidence, but it could be argued that we presented all six case scenarios in an overly simplified manner because each of the five factors may encompass more nuances. Among them, factor ‘I’ may represent the most controversial risk category to justify and quantify. Until future biomarkers better inform us as to tissue changes in real‐time, the variable is largely inferred from experimental findings in animal studies and autopsy results regarding the effects of both D and E. However, the incomplete penetrance of highly pathogenic mutations at the SNCA locus (e.g. p.A53T substitution) in bona fide carriers [together with the discordance for PD in the (albeit single) case of LRRK2 G2019S‐positive, identical twins] suggested to us that the network of genome–exposome interactions accounts for the initiation of PD in an affected individual vs. an unaffected carrier. Therefore, factor ‘I’ was included in the formula to serve as a surrogate best suited to reflect the outcome of all network interactions that initiate and propagate pathology. Hence, we considered its inclusion as an independent risk category superior to the option of simply increasing the values for E or D.
Our first version of a score card system (Table 1) was designed for easy updates in the future in order to reflect more individualized risk components and to be properly transformed. The PREDIGT score model will be able to incorporate newly discovered elements in accordance with our growing understanding of disease development and the emergence of objective, validated disease markers [see: (Mollenhauer & Trenkwalder, 2009; Schlossmacher & Mollenhauer, 2010; Calabresi et al., 2016) and references therein] by converting solid evidence into revised values.
Another potential criticism of our approach is that it is based on the idea that the course to typical PD is truly interdependent upon 5 factors (E, D, I, G, T). This concept was informed by our understanding of several well‐known examples of complex diseases in humans (e.g. diabetes, multiple sclerosis, lung cancer and stroke) and is strengthened by recent insights into the range of LRRK2‐associated illnesses; however, this rationale has not yet been widely accepted as an explanation for ‘idiopathic PD’. This issue could be addressed in longitudinal cohort studies of healthy adults and at risk persons [such as in the PARS cohort (Chahine et al., 2016)] and in better designed animal experiments of disease development.
As pointed out in the introduction section, there is currently no widespread consensus regarding the relative importance of the exposome in PD pathogenesis. In our model, it is essential and contributes to the values entered under factors E and I. It encompasses –among others – microbiological agents within a person's exposure history, of which interactions with genes expressed by the host have shown to underlie many late‐onset disorders in humans (Pittman et al., 2016). There, we have also been informed by expression patterns and emerging insights into the role of PD‐linked genes outside the nervous system, such as for LRRK2 (Ness et al., 2013; Daher et al., 2015) and SNCA (Beatman et al., 2016) and Alzheimer disease‐linked APP, which play roles in host defences against virulent, microbial pathogens (Ness et al., 2013; Kumar et al., 2016). Moreover, SNPs at several other loci in the human genome (Nalls et al., 2014) have been linked to immunity in genome‐wide association studies of multiple PD cohorts, and dysregulated inflammation has been associated with PD risk including in LRRK2 mutation carriers (e.g. Dzamko et al., 2016). Although a Koch's principles‐based cause and effect relation between xenobotics and PD has not been demonstrated, we and others consider the infectious exposure history of an individual to be a critical factor when assessing a subject's overall risk to develop PD (Bu et al., 2015); we have thus included such events (and surrogates thereof) as a quantifiable risk modulator into our score sheet.
Moreover, given the suggested progression routes of PD from the gut and nasal cavity to the brain (Braak et al., 2004; Braak & Del Tredici, 2008), exposure history by an individual to infectious organisms and other environmental modifiers within mucosal membranes of the URT and GI tract (Doty, 2009; Bu et al., 2015) may be the most important modulator of PD risk; therefore, we grouped chronic constipation and hyposmia/anosmia under the risk category E. Indeed, differences in the microbiome of PD patients relative to normal controls have recently been reported (Scheperjans et al., 2015; Felice et al., 2016; Ghaisas et al., 2016). Although the molecular mechanisms for and consequences of these differences have not yet been delineated, the inclusion of chronic constipation and impaired olfaction into risk models has been pursued by other investigators (Berg et al., 2015; Nalls et al., 2015).
Furthermore, whether xenobiotic‐, nutrient‐, or toxicant‐based in nature, our model has not yet made a more nuanced distinction between monophasic vs. intermittent (recurrent) vs. chronic exposure to environmental pathogens considered under factor E. The more detailed assessment and quantification of each subject's exposome from before birth to infancy and on to young, middle‐aged as well as late adulthood (e.g. Chen et al., 2012) will allow the more accurate differentiation and quantification of this variable. Of note, the same criticism (i.e. of the need for a more nuanced quantification) could be made regarding factor D. Fortuitously, through the employment of standardized procedures clinicians and epidemiologists can now increase our sensitivity and specificity (and their related predictive values) when obtaining family histories of PD; this, to exclude both false positives and false negatives (Elbaz et al., 2003). In parallel, geneticists and bioinformatics experts are actively mining large data sets to delineate individual risk scores into new algorithms to build better DNA‐based prediction models (e.g. Nalls et al., 2015).
In conclusion, the PREDIGT score model evolved from our concept of typical PD as a complex, multifactorial disease. This assessment stems from our interpretation of the published literature regarding insights into other late‐onset disorders, clues for ‘PD as an environmental disease’, clues for ‘PD as a genetic disease’ as well as from our own experimental evidence. By carefully weighing individual risk factors with previously established associations to PD in the context of our disease model, and by fitting them into a simple‐to‐use, mathematical formula, we have created a new prediction tool. The PREDIGT score system represents – to our knowledge – the first, non‐invasive method to possibly predict the cumulative incidence rate for PD in the future. As stated above, the next critical steps are to transform the formula and validate the model in multiple cohorts. Thereafter, our goal would be to test the PREDIGT prediction model in clinical research settings for stratification and population enrichment in intervention trials. In the future, health care practitioners could employ the PREDIGT score system (or an improved derivative thereof) to better counsel and care for healthy adults in order to delay the onset of PD and to prevent its full clinical manifestation during the prodromal stage.
Competing interests
The authors declare no conflict of interest.
Author contributions
M.G.S. conceived and designed the project; all authors contributed to the creation of figures, tables, literature searches and analyses; M.G.S., J.J.T. and T.M. wrote the first draft; all authors reviewed and edited the two versions of the manuscript submitted for peer review.
Abbreviations
- AOO
Age of onset
- D
DNA
- E
exposome
- G
gender
- GI
gastrointestinal
- I
initiation of tissue response
- PD
Parkinson disease
- PREDIGT
Predicting the incidence risk for Parkinson disease
- REM
rapid eye movement
- S. nigra
Substantia nigra pars scompacta
- SNP
single‐nucleotide polymorphism
- T
time
- URT
upper respiratory tract
Acknowledgements
This work has been supported by the: Government of Canada (CIHR; to M.G.S., D.M.), Weston Brain Institute (J.J.T., M.G.S.), Michael J. Fox Foundation (J.J.T., E.G.B., M.G.S.), Department of Medicine at The Ottawa Hospital (T.M., M.G.S.), Uttra and Sam Bhargava Family (M.G.S.) and Parkinson's Research Consortium of Ottawa (M.G.S., T.M.). We are grateful to our patients and their family members for inspiration and support. We thank Drs. Alberto Ascherio, Erwin Schurr and Bill Cameron for discussions regarding complex diseases. We express gratitude to Drs. Lew R. Sudarsky and John Woulfe for critical comments and suggestions on earlier versions of the manuscript. This paper is dedicated to Dr. Oleh Hornykiewcz on the occasion of his 90th birthday celebrated on 5 September at the ‘Dopamine 2016’ conference in Vienna, Austria.
References
- Abbott, R.D. , Petrovitch, H. , White, L.R. , Masaki, K.H. , Tanner, C.M. , Curb, J.D. , Grandinetti, A. , Blanchette, P.L. et al (2001) Frequency of bowel movements and the future risk of Parkinson's disease. Neurology, 57, 456–462. [DOI] [PubMed] [Google Scholar]
- Abbott, R.D. , Ross, G.W. , White, L.R. , Sanderson, W.T. , Burchfiel, C.M. , Kashon, M. , Sharp, D.S. , Masaki, K.H. et al (2003) Environmental, life‐style, and physical precursors of clinical Parkinson's disease: recent findings from the Honolulu‐Asia Aging Study. J. Neurol., 250(Suppl 3), III30–III39. [DOI] [PubMed] [Google Scholar]
- Alcalay, R.N. , Dinur, T. , Quinn, T. , Sakanaka, K. , Levy, O. , Waters, C. , Fahn, S. , Dorovski, T. et al (2014) Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol, 71, 752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes, L. , Frasquilho, S. , Ostaszewski, M. , Weber, J. , Longhino, L. , Antony, P. , Baumuratov, A. , Buttini, M. et al (2016) Similar alpha‐Synuclein staining in the colon mucosa in patients with Parkinson's disease and controls. Movement Disord., 31, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Barrett, J.C. , Hansoul, S. , Nicolae, D.L. , Cho, J.H. , Duerr, R.H. , Rioux, J.D. , Brant, S.R. , Silverberg, M.S. et al (2008) Genome‐wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet., 40, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatman, E.L. , Massey, A. , Shives, K.D. , Burrack, K.S. , Chamanian, M. , Morrison, T.E. & Beckham, J.D. (2016) Alpha‐synuclein expression restricts RNA viral infections in the brain. J. Virol., 90, 2767–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellou, V. , Belbasis, L. , Tzoulaki, I. , Evangelou, E. & Ioannidis, J.P. (2016) Environmental risk factors and Parkinson's disease: an umbrella review of meta‐analyses. Parkinsonism Relat. Disord., 23, 1–9. [DOI] [PubMed] [Google Scholar]
- Berg, D. , Postuma, R.B. , Adler, C.H. , Bloem, B.R. , Chan, P. , Dubois, B. , Gasser, T. , Goetz, C.G. et al (2015) MDS research criteria for prodromal Parkinson's disease. Movement Disord., 30, 1600–1611. [DOI] [PubMed] [Google Scholar]
- Bidinosti, M. , Shimshek, D.R. , Mollenhauer, B. , Marcellin, D. , Schweizer, T. , Lotz, G.P. , Schlossmacher, M.G. & Weiss, A. (2012) Novel one‐step immunoassays to quantify alpha‐synuclein: applications for biomarker development and high‐throughput screening. J. Biol. Chem., 287, 33691–33705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati, V. , Rizzu, P. , van Baren, M.J. , Schaap, O. , Breedveld, G.J. , Krieger, E. , Dekker, M.C. , Squitieri, F. et al (2003) Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset Parkinsonism. Science, 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Bozi, M. , Papadimitriou, D. , Antonellou, R. , Moraitou, M. , Maniati, M. , Vassilatis, D.K. , Papageorgiou, S.G. , Leonardos, A. et al (2014) Genetic assessment of familial and early‐onset Parkinson's disease in a Greek population. Eur. J. Neurol., 21, 963–968. [DOI] [PubMed] [Google Scholar]
- Braak, H. & Del Tredici, K. (2008) Invited Article: nervous system pathology in sporadic Parkinson disease. Neurology, 70, 1916–1925. [DOI] [PubMed] [Google Scholar]
- Braak, H. , Ghebremedhin, E. , Rub, U. , Bratzke, H. & Del Tredici, K. (2004) Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res., 318, 121–134. [DOI] [PubMed] [Google Scholar]
- Bu, X.L. , Wang, X. , Xiang, Y. , Shen, L.L. , Wang, Q.H. , Liu, Y.H. , Jiao, S.S. , Wang, Y.R. et al (2015) The association between infectious burden and Parkinson's disease: a case‐control study. Parkinsonism Relat. Disord., 21, 877–881. [DOI] [PubMed] [Google Scholar]
- Calabresi, P. , Standaert, D.G. , Chiasserini, D. & Parnetti, L. (2016) Biomarkers in Parkinson's disease: from pathophysiology to early diagnosis. Movement Disord., 31, 769–770. [DOI] [PubMed] [Google Scholar]
- Chahine, L.M. , Weintraub, D. , Hawkins, K.A. , Siderowf, A. , Eberly, S. , Oakes, D. , Seibyl, J. , et al (2016) Cognition in individuals at risk for Parkinson's: Parkinson associated risk syndrome (PARS) study findings. Mov. Disord., 31, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R. , Mias, G.I. , Li‐Pook‐Than, J. , Jiang, L. , Lam, H.Y. , Chen, R. , Miriami, E. , Karczewski, K.J. et al (2012) Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell, 148, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet, M.F. & Richter, F. (2011) Modelling of Parkinson's disease in mice. Lancet Neurol., 10, 1108–1118. [DOI] [PubMed] [Google Scholar]
- Chung, S.J. , Biernacka, J.M. , Armasu, S.M. , Anderson, K. , Frigerio, R. , Aasly, J.O. , Annesi, G. , Bentivoglio, A.R. et al (2014) Alpha‐synuclein repete variants and survival in Parkinson's disease. Movement Disord., 29, 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilia, R. , Siri, C. , Rusconi, D. , Allegra, R. , Ghiglietti, A. , Sacilotto, G. , Zini, M. , Zecchinelli, A.L. et al (2014) LRRK2 mutations in Parkinson's disease: confirmation of a gender effect in the Italian population. Parkinsonism Relat. Disord., 20, 911–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunkhorn, S. (2016) Alzheimer disease: antimicrobial role of amyloid‐beta. Nat. Rev. Drug Discov., 15, 456. [DOI] [PubMed] [Google Scholar]
- Cullen, V. , Sardi, S.P. , Ng, J. , Xu, Y.H. , Sun, Y. , Tomlinson, J.J. , Kolodziej, P. , Kahn, I. et al (2011) Acid beta‐glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha‐synuclein processing. Ann. Neurol., 69, 940–953. [DOI] [PubMed] [Google Scholar]
- Dacks, P.A. , Andrieu, S. , Blacker, D. , Carman, A.J. , Green, A.M. , Grodstein, F. , Henderson, V.W. , James, B.D. et al (2014) Dementia Prevention: optimizing the use of observational data for personal, clinical, and public health decision‐making. J. Prev. Alzheimers Dis., 1, 117–123. [PMC free article] [PubMed] [Google Scholar]
- Daher, J.P. , Abdelmotilib, H.A. , Hu, X. , Volpicelli‐Daley, L.A. , Moehle, M.S. , Fraser, K.B. , Needle, E. , Chen, Y. et al (2015) Leucine‐rich repeat kinase 2 (LRRK2) pharmacological inhibition abates alpha‐synuclein gene‐induced neurodegeneration. J. Biol. Chem., 290, 19433–19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabos, C. , Qiu, J. & Moore, J.H. (2016) An integrated network approach to identifying biological pathways and environmental exposure interactions in complex diseases. Pacific Symp. Biocomput., 21, 9–20. [PMC free article] [PubMed] [Google Scholar]
- Dauer, W. , Kholodilov, N. , Vila, M. , Trillat, A.C. , Goodchild, R. , Larsen, K.E. , Staal, R. , Tieu, K. et al (2002) Resistance of alpha ‐synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA, 99, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Tredici, K. , Hawkes, C.H. , Ghebremedhin, E. & Braak, H. (2010) Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson's disease. Acta Neuropathol., 119, 703–713. [DOI] [PubMed] [Google Scholar]
- Derkinderen, P. , Shannon, K.M. & Brundin, P. (2014) Gut feelings about smoking and coffee in Parkinson's disease. Movement Disord., 29, 976–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, H. , Dhima, K. , Lockhart, K.C. , Locascio, J.J. , Hoesing, A.N. , Duong, K. , Trisini‐Lipsanopoulos, A. , Hayes, M.T. et al (2013) Unrecognized vitamin D3 deficiency is common in Parkinson disease: Harvard Biomarker Study. Neurology, 81, 1531–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doty, R.L. (2008) The olfactory vector hypothesis of neurodegenerative disease: is it viable? Ann. Neurol., 63, 7–15. [DOI] [PubMed] [Google Scholar]
- Doty, R.L. (2009) The olfactory system and its disorders. Semin. Neurol., 29, 74–81. [DOI] [PubMed] [Google Scholar]
- Driver, J.A. , Logroscino, G. , Gaziano, J.M. & Kurth, T. (2009) Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology, 72, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda, J.E. , Shah, U. , Arnold, S.E. , Lee, V.M.Y. & Trojanowski, J.Q. (1999) The expression of α‐, β‐, and γ‐synucleins in olfactory mucosa from patients with and without neurodegenerative diseases. Exp. Neurol., 160, 515–522. [DOI] [PubMed] [Google Scholar]
- Duvoisin, R.C. & Golbe, L.I. (1995) Kindreds of dominantly inherited Parkinson's disease: keys to the riddle. Ann. Neurol., 38, 355–356. [DOI] [PubMed] [Google Scholar]
- Dzamko, N. & Halliday, G.M. (2012) An emerging role for LRRK2 in the immune system. Biochem. Soc. T., 40, 1134–1139. [DOI] [PubMed] [Google Scholar]
- Dzamko, N. , Rowe, D.B. & Halliday, G.M. (2016) Increased peripheral inflammation in asymptomatic leucine‐rich repeat kinase 2 mutation carriers. Mov Disord., 31, 889–897. [DOI] [PubMed] [Google Scholar]
- Eblan, M.J. , Walker, J.M. & Sidransky, E. (2005) The glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. New Engl. J. Med., 352, 728–731; author reply 728‐731. [DOI] [PubMed] [Google Scholar]
- Edwards, T.L. , Scott, W.K. , Almonte, C. , Burt, A. , Powell, E.H. , Beecham, G.W. , Wang, L. , Zuchner, S. et al (2010) Genome‐wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet., 74, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz, A. , McDonnell, S.K. , Maraganore, D.M. , Strain, K.J. , Schaid, D.J. , Bower, J.H. , Ahlskog, J.E. & Rocca, W.A. (2003) Validity of family history data on PD: evidence for a family information bias. Neurology, 61, 11–17. [DOI] [PubMed] [Google Scholar]
- Elbaz, A. , Carcaillon, L. , Kab, S. & Moisan, F. (2016) Epidemiology of Parkinson's disease. Rev. Neurol. (Paris), 172, 14–26. [DOI] [PubMed] [Google Scholar]
- Farrer, M.J. (2006) Genetics of Parkinson disease: paradigm shifts and future prospects. Nat. Rev. Genet., 7, 306–318. [DOI] [PubMed] [Google Scholar]
- Farrer, M. , Kachergus, J. , Forno, L. , Lincoln, S. , Wang, D.S. , Hulihan, M. , Maraganore, D. , Gwinn‐Hardy, K. et al (2004) Comparison of kindreds with Parkinsonism and alpha‐synuclein genomic multiplications. Ann. Neurol., 55, 174–179. [DOI] [PubMed] [Google Scholar]
- Fava, V.M. , Manry, J. , Cobat, A. , Orlova, M. , Van Thuc, N. , Ba, N.N. , Thai, V.H. , Abel, L. et al (2016) A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Neglect Trop Dis., 10, e0004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley, J.M. & Lees, A.J. (1991) Ageing and Parkinson's disease: Substantia nigra regional selectivity. Brain, 114, 2283–2301. [DOI] [PubMed] [Google Scholar]
- Felice, V.D. , Quigley, E.M. , Sullivan, A.M. , O'Keeffe, G.W. & O'Mahony, S.M. (2016) Microbiota‐gut‐brain signalling in Parkinson's disease: implications for non‐motor symptoms. Parkinsonism Relat. D., 27, 1–8. [DOI] [PubMed] [Google Scholar]
- Fornai, F. , Schluter, O.M. , Lenzi, P. , Gesi, M. , Ruffoli, R. , Ferrucci, M. , Lazzeri, G. , Busceti, C.L. et al (2005) Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin‐proteasome system and alpha‐synuclein. Proc. Natl. Acad. Sci. USA, 102, 3413–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, S.H. & Brotchie, J.M. (2010) The MPTP‐lesioned non‐human primate models of Parkinson's disease. Past, present, and future. Prog. Brain Res., 184, 133–157. [DOI] [PubMed] [Google Scholar]
- Gaig, C. , Vilas, D. , Infante, J. , Sierra, M. , Garcia‐Gorostiaga, I. , Buongiorno, M. , Ezquerra, M. , Marti, M.J. et al (2014) Nonmotor symptoms in LRRK2 G2019S associated Parkinson's disease. PLoS ONE, 9, e108982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin, J.E. , Lee, V.M. & Trojanowski, J.Q. (2001) Synucleinopathies: clinical and pathological implications. Arch. Neurol., 58, 186–190. [DOI] [PubMed] [Google Scholar]
- Gan‐Or, Z. , Amshalom, I. , Kilarski, L.L. , Bar‐Shira, A. , Gana‐Weisz, M. , Mirelman, A. , Marder, K. , Bressman, S. et al (2015) Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology, 84, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaisas, S. , Maher, J. & Kanthasamy, A. (2016) Gut microbiome in health and disease: linking the microbiome‐gut‐brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Therapeut., 158, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb, W.R. & Lees, A.J. (1991) Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J. Neurol. Neurosurg. Psychiatry, 54, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go, Y.M. & Jones, D.P. (2014) Redox biology: interface of the exposome with the proteome, epigenome and genome. Redox. Biol., 2, 358–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell, J.M. , Peterson, E.L. , Rybicki, B.A. & Johnson, C.C. (2004) Multiple risk factors for Parkinson's disease. J. Neurol. Sci., 217, 169–174. [DOI] [PubMed] [Google Scholar]
- Gray, M.T. , Munoz, D.G. , Gray, D.A. , Schlossmacher, M.G. & Woulfe, J.M. (2014) Alpha‐synuclein in the appendiceal mucosa of neurologically intact subjects. Movement Disord., 29, 991–998. [DOI] [PubMed] [Google Scholar]
- Gray, M.T. , Munoz, D.G. , Schlossmacher, M.G. , Gray, D.A. & Woulfe, J.M. (2015) Protective effect of vagotomy suggests source organ for Parkinson disease. Ann. Neurol., 78, 834–835. [DOI] [PubMed] [Google Scholar]
- Grondin, C.J. , Davis, A.P. , Wiegers, T.C. , King, B.L. , Wiegers, J.A. , Reif, D.M. , Hoppin, J.A. & Mattingly, C.J. (2016) Advancing exposure science through chemical data curation and integration in the comparative toxicogenomics database. Environ. Health Persp., 124, 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn, K. , Devine, M.J. , Jin, L.W. , Johnson, J. , Bird, T. , Muenter, M. , Waters, C. , Adler, C.H. et al (2011) Clinical features, with video documentation, of the original familial lewy body Parkinsonism caused by alpha‐synuclein triplication (Iowa kindred). Movement Disord., 26, 2134–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi, M. , Selvanantham, T. , Swinton, E. , Padmore, R.F. , Tong, Y. , Kabbach, G. , Venderova, K. , Girardin, S.E. et al (2011) Parkinson's disease‐linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural. Transm., 118, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday, G.M. , Holton, J.L. , Revesz, T. & Dickson, D.W. (2011) Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol., 122, 187–204. [DOI] [PubMed] [Google Scholar]
- Hawkes, C.H. (2013) Comment to letter: ‘Identical twins with LRRK2 mutation discordant for Parkinson's disease’. Movement Disord., 28, 561. [DOI] [PubMed] [Google Scholar]
- Hawkes, C.H. , Del Tredici, K. & Braak, H. (2010) A timeline for Parkinson's disease. Parkinsonism Relat. D., 16, 79–84. [DOI] [PubMed] [Google Scholar]
- Heckman, M.G. , Soto‐Ortolaza, A.I. , Aasly, J.O. , Abahuni, N. , Annesi, G. , Bacon, J.A. , Bardien, S. , Bozi, M. et al, Genetic Epidemiology of Parkinson's Disease Consoritum (2013) Population‐specific frequencies for LRRK2 susceptibility variants in the Genetic Epidemiology of Parkinson's disease (GEO‐PD) Consortium. Movement Disord., 28, 1740–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen, J.M. , Geersing, G.J. , Lucassen, W.A. , Erkens, P.M. , Stoffers, H.E. , van Weert, H.C. , Buller, H.R. , Hoes, A.W. et al (2015) Diagnostic prediction models for suspected pulmonary embolism: systematic review and independent external validation in primary care. BMJ, 351, h4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig, M.C. , Kolly, C. , Persohn, E. , Theil, D. , Schweizer, T. , Hafner, T. , Stemmelen, C. , Troxler, T.J. et al (2011) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet., 20, 4209–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, E.C. , Vyas, S. & Hunot, S. (2012) Neuroinflammation in Parkinson's disease. Parkinsonism Relat. D., 18(Suppl 1), S210–S212. [DOI] [PubMed] [Google Scholar]
- Hsu, J.C. , Maddox, T.M. , Kennedy, K.F. , Katz, D.F. , Marzec, L.N. , Lubitz, S.A. , Gehi, A.K. , Turakhia, M.P. et al (2016) Oral anticoagulant therapy prescription in patients with atrial fibrillation across the spectrum of stroke risk: insights from the NCDR PINNACLE registry. JAMA Cardiol, 1, 55–62. [DOI] [PubMed] [Google Scholar]
- International Parkinson Disease Genomics Consortium , Nalls, M.A. , Plagnol, V. , Hernandez, D.G. , Sharma, M. , Sheerin, U.M. , Saad, M. , Simon‐Sanchez, J. , Schulte, C. , Lesage, S. , Sveinbjornsdottir, S. , Stefansson, K. , Martinez, M. , Hardy, J. , Heutink, P. , Brice, A. , Gasser, T. , Singleton, A.B. & Wood, N.W. (2011) Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet, 377, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin, D.J. , Lee, V.M. & Trojanowski, J.Q. (2013) Parkinson's disease dementia: convergence of alpha‐synuclein, tau and amyloid‐beta pathologies. Nat. Rev. Neurosci., 14, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier, J.M. , Ibanez, P. , Mena, M.A. , Abbas, N. , Cohen‐Salmon, C. , Bohme, G.A. , Laville, M. , Pratt, J. et al (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet., 12, 2277–2291. [DOI] [PubMed] [Google Scholar]
- Jang, H. , Boltz, D.A. , Webster, R.G. & Smeyne, R.J. (2009) Viral Parkinsonism. Biochim. Biophys. Acta, 1792, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger, K.A. (2009) Absence of alpha‐synuclein pathology in postencephalitic Parkinsonism. Acta Neuropathol., 118, 371–379. [DOI] [PubMed] [Google Scholar]
- Kalia, L.V. , Lang, A.E. , Hazrati, L.N. , Fujioka, S. , Wszolek, Z.K. , Dickson, D.W. , Ross, O.A. , Van Deerlin, V.M. et al (2015) Clinical correlations with Lewy body pathology in LRRK2‐related Parkinson disease. JAMA Neurol, 72, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, U.B. & Marto, J.A. (2016) Leucine‐rich repeat kinase 2 (LRRK2) and Parkinson's disease. Proteomics, doi: 10.1002/pmic.201600092. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Kang, J.H. , Mollenhauer, B. , Coffey, C.S. , Toledo, J.B. , Weintraub, D. , Galasko, D.R. , Irwin, D.J. , Van Deerlin, V. et al (2016) CSF biomarkers associated with disease heterogeneity in early Parkinson's disease: the Parkinson's Progression Markers Initiative study. Acta Neuropathol., 131, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, R.H. , Smith, P.D. , Aleyasin, H. , Hayley, S. , Mount, M.P. , Pownall, S. , Wakeham, A. , You‐Ten, A.J. et al (2005) Hypersensitivity of DJ‐1‐deficient mice to 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA, 102, 5215–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, T. , Asakawa, S. , Hattori, N. , Matsumine, H. , Yamamura, Y. , Minoshima, S. , Yokochi, M. , Mizuno, Y. et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile Parkinsonism. Nature, 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Kitada, T. , Pisani, A. , Karouani, M. , Haburcak, M. , Martella, G. , Tscherter, A. , Platania, P. , Wu, B. et al (2009) Impaired dopamine release and synaptic plasticity in the striatum of parkin‐/‐ mice. J. Neurochem., 110, 613–621. [DOI] [PubMed] [Google Scholar]
- Kitada, T. , Tomlinson, J.J. , Ao, H.S. , Grimes, D.A. & Schlossmacher, M.G. (2012) Considerations regarding the etiology and future treatment of autosomal recessive versus idiopathic Parkinson disease. Curr. Treat Option N., 14, 230–240. [DOI] [PubMed] [Google Scholar]
- Klein, C. & Schlossmacher, M.G. (2007) Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology, 69, 2093–2104. [DOI] [PubMed] [Google Scholar]
- Klein, C. , Lohmann‐Hedrich, K. , Rogaeva, E. , Schlossmacher, M.G. & Lang, A.E. (2007) Deciphering the role of heterozygous mutations in genes associated with Parkinsonism. Lancet Neurol., 6, 652–662. [DOI] [PubMed] [Google Scholar]
- Kumar, D.K. , Choi, S.H. , Washicosky, K.J. , Eimer, W.A. , Tucker, S. , Ghofrani, J. , Lefkowitz, A. , McColl, G. et al (2016) Amyloid‐beta peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med., 8, 340ra372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, Y.M. , Li, Z. , Jiao, Y. , Gaborit, N. , Pani, A.K. , Orrison, B.M. , Bruneau, B.G. , Giasson, B.I. et al (2010) Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease‐associated alpha‐synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet., 19, 1633–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston, J.W. , Ballard, P. , Tetrud, J.W. & Irwin, I. (1983) Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science, 219, 979–980. [DOI] [PubMed] [Google Scholar]
- Lesage, S. , Durr, A. , Tazir, M. , Lohmann, E. , Leutenegger, A.L. , Janin, S. , Pollak, P. & Brice, A. (2006) LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. New Engl. J. Med., 354, 422–423. [DOI] [PubMed] [Google Scholar]
- Li, R. , Conti, D.V. , Diaz‐Sanchez, D. , Gilliland, F. & Thomas, D.C. (2012) Joint analysis for integrating two related studies of different data types and different study designs using hierarchical modeling approaches. Hum. Hered., 74, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan, I.I. , Jankovic, J. , Goetz, C.G. , Wenning, G.K. , Sastry, N. , Jellinger, K. , McKee, A. , Lai, E.C. et al (1998) Accuracy of the clinical diagnosis of postencephalitic Parkinsonism: a clinicopathologic study. Eur. J. Neurol., 5, 451–457. [DOI] [PubMed] [Google Scholar]
- Locascio, J.J. , Eberly, S. , Liao, Z. , Liu, G. , Hoesing, A.N. , Duong, K. , Trisini‐Lipsanopoulos, A. , Dhima, K. et al (2015) Association between alpha‐synuclein blood transcripts and early, neuroimaging‐supported Parkinson's disease. Brain, 138, 2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking, C.B. , Durr, A. , Bonifati, V. , Vaughan, J. , De Michele, G. , Gasser, T. , Harhangi, B.S. , Meco, G. et al (2000) Association between early‐onset Parkinson's disease and mutations in the parkin gene. New Engl. J. Med., 342, 1560–1567. [DOI] [PubMed] [Google Scholar]
- Ma, S.Y. , Ciliax, B.J. , Stebbins, G. , Jaffar, S. , Joyce, J.N. , Cochran, E.J. , Kordower, J.H. , Mash, D.C. , et al (1999) Dopamine transporter‐immunoreactive neurons decrease with age in the human substantia nigra. J. Comp. Neurol., 409, 25–37. [DOI] [PubMed] [Google Scholar]
- Manuel, D.G. , Perez, R. , Sanmartin, C. , Taljaard, M. , Hennessy, D. , Wilson, K. , Tanuseputro, P. , Manson, H. , et al (2016) Measuring burden of unhealthy behaviours using a multivariable predictive approach: life expectancy lost in Canada attributable to smoking, alcohol, physical inactivity, and diet. PLoS Med., 13, e1002082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder, K. , Wang, Y. , Alcalay, R.N. , Mejia‐Santana, H. , Tang, M.X. , Lee, A. , Raymond, D. , Mirelman, A. et al (2015) Age‐specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology, 85, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras, C. , Lang, A. , van de Warrenburg, B.P. , Sue, C.M. , Tabrizi, S.J. , Bertram, L. , Mercimek‐Mahmutoglu, S. , Ebrahimi‐Fakhari, D. et al (2016) Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Movement Disord., 31, 436–457. [DOI] [PubMed] [Google Scholar]
- Masliah, E. , Achim, C.L. , Ge, N. , De Teresa, R. & Wiley, C.A. (1994) Cellular neuropathology in HIV encephalitis. Res. Publ. Assoc. Res. N., 72, 119–131. [PubMed] [Google Scholar]
- McCormack, A.L. , Mak, S.K. , Shenasa, M. , Langston, W.J. , Forno, L.S. & Di Monte, D.A. (2008) Pathologic modifications of alpha‐synuclein in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐treated squirrel monkeys. J. Neuropath. Exp. Neur., 67, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez, J. , Stern, R.A. & McKee, A.C. (2013) Chronic traumatic encephalopathy: where are we and where are we going? Curr. Neurol. Neurosci., 13, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi, S. , Deisler, R. , Gozel, K. , Reicks, P. , Irwin, E. , Brunsvold, M. , Banton, K. & Beilman, G.J. (2016) Wells criteria for DVT is a reliable clinical tool to assess the risk of deep venous thrombosis in trauma patients. World J. Emergen. Surg., 11, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle, M.S. , Webber, P.J. , Tse, T. , Sukar, N. , Standaert, D.G. , DeSilva, T.M. , Cowell, R.M. & West, A.B. (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci., 32, 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollenhauer, B. & Trenkwalder, C. (2009) Neurochemical biomarkers in the differential diagnosis of movement disorders. Movement Disord., 24, 1411–1426. [DOI] [PubMed] [Google Scholar]
- Mollenhauer, B. , Locascio, J.J. , Schulz‐Schaeffer, W. , Sixel‐Doring, F. , Trenkwalder, C. & Schlossmacher, M.G. (2011) alpha‐Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol., 10, 230–240. [DOI] [PubMed] [Google Scholar]
- Mollenhauer, B. , Trautmann, E. , Sixel‐Doring, F. , Wicke, T. , Ebentheuer, J. , Schaumburg, M. , Lang, E. , Focke, N.K. et al (2013a) Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology, 81, 1226–1234. [DOI] [PubMed] [Google Scholar]
- Mollenhauer, B. , Trautmann, E. , Taylor, P. , Manninger, P. , Sixel‐Doring, F. , Ebentheuer, J. , Trenkwalder, C. & Schlossmacher, M.G. (2013b) Total CSF alpha‐synuclein is lower in de novo Parkinson patients than in healthy subjects. Neurosci. Lett., 532, 44–48. [DOI] [PubMed] [Google Scholar]
- Nalls, M.A. , Pankratz, N. , Lill, C.M. , Do, C.B. , Hernandez, D.G. , Saad, M. , DeStefano, A.L. , Kara, E. et al (2014) Large‐scale meta‐analysis of genome‐wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet., 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls, M.A. , McLean, C.Y. , Rick, J. , Eberly, S. , Hutten, S.J. , Gwinn, K. , Sutherland, M. , Martinez, M. et al (2015) Diagnosis of Parkinson's disease on the basis of clinical and genetic classification: a population‐based modelling study. Lancet Neurol., 14, 1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness, D. , Ren, Z. , Gardai, S. , Sharpnack, D. , Johnson, V.J. , Brennan, R.J. , Brigham, E.F. & Olaharski, A.J. (2013) Leucine‐rich repeat kinase 2 (LRRK2)‐deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS ONE, 8, e66164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka, K. , Hayashi, S. , Farrer, M.J. , Singleton, A.B. , Yoshino, H. , Imai, H. , Kitami, T. , Sato, K. et al (2006) Clinical heterogeneity of alpha‐synuclein gene duplication in Parkinson's disease. Ann. Neurol., 59, 298–309. [DOI] [PubMed] [Google Scholar]
- Ozelius, L.J. , Senthil, G. , Saunders‐Pullman, R. , Ohmann, E. , Deligtisch, A. , Tagliati, M. , Hunt, A.L. , Klein, C. et al (2006) LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. New Engl. J. Med., 354, 424–425. [DOI] [PubMed] [Google Scholar]
- Paisan‐Ruiz, C. , Jain, S. , Evans, E.W. , Gilks, W.P. , Simon, J. , van der Brug, M. , Lopez de Munain, A. , Aparicio, S. et al (2004) Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron, 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Palacino, J.J. , Sagi, D. , Goldberg, M.S. , Krauss, S. , Motz, C. , Wacker, M. , Klose, J. & Shen, J. (2004) Mitochondrial dysfunction and oxidative damage in parkin‐deficient mice. J. Biol. Chem., 279, 18614–18622. [DOI] [PubMed] [Google Scholar]
- Palacios, N. , Gao, X. , McCullough, M.L. , Schwarzschild, M.A. , Shah, R. , Gapstur, S. & Ascherio, A. (2012) Caffeine and risk of Parkinson's disease in a large cohort of men and women. Movement Disord., 27, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadimitriou, D. , Antonelou, R. , Miligkos, M. , Maniati, M. , Papagiannakis, N. , Bostantjopoulou, S. , Leonardos, A. , Koros, C. et al (2016) Motor and nonmotor features of carriers of the p. A53T alpha‐synuclein mutation: a longitudinal study. Movement Disord., 31, 1226–1230. [DOI] [PubMed] [Google Scholar]
- Patel, C.J. (2016) Analytical complexity in detection of gene variant‐by‐environment exposure interactions in high‐throughput genomic and exposomic research. Curr. Environ. Health Rep., 3, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, C.J. & Ioannidis, J.P. (2014) Studying the elusive environment in large scale. JAMA, 311, 2173–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattarini, R. , Smeyne, R.J. & Morgan, J.I. (2007) Temporal mRNA profiles of inflammatory mediators in the murine 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyrimidine model of Parkinson's disease. Neuroscience, 145, 654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periquet, M. , Corti, O. , Jacquier, S. & Brice, A. (2005) Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J. Neurochem., 95, 1259–1276. [DOI] [PubMed] [Google Scholar]
- Pittman, K.J. , Glover, L.C. , Wang, L. & Ko, D.C. (2016) The legacy of past pandemics: common human mutations that protect against infectious disease. PLoS Pathog., 12, e1005680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polito, L. , Greco, A. & Seripa, D. (2016) Genetic profile, environmental exposure, and their interaction in Parkinson's disease. Parkinsons Dis., 2016, 6465793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos, M.H. , Lavedan, C. , Leroy, E. , Ide, S.E. , Dehejia, A. , Dutra, A. , Pike, B. , Root, H. et al (1997) Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Postuma, R.B. , Berg, D. , Stern, M. , Poewe, W. , Olanow, C.W. , Oertel, W. , Obeso, J. , Marek, K. et al (2015) MDS clinical diagnostic criteria for Parkinson's disease. Movement Disord., 30, 1591–1601. [DOI] [PubMed] [Google Scholar]
- Pramstaller, P.P. , Schlossmacher, M.G. , Jacques, T.S. , Scaravilli, F. , Eskelson, C. , Pepivani, I. , Hedrich, K. , Adel, S. et al (2005) Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann. Neurol., 58, 411–422. [DOI] [PubMed] [Google Scholar]
- Preiss, D. & Kristensen, S.L. (2015) The new pooled cohort equations risk calculator. Can. J. Cardiol., 31, 613–619. [DOI] [PubMed] [Google Scholar]
- Rajput, A. , Dickson, D.W. , Robinson, C.A. , Ross, O.A. , Dachsel, J.C. , Lincoln, S.J. , Cobb, S.A. , Rajput, M.L. et al (2006) Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology, 67, 1506–1508. [DOI] [PubMed] [Google Scholar]
- Rajput, A.H. , Ferguson, L.W. , Robinson, C.A. , Guella, I. , Farrer, M.J. & Rajput, A. (2016) Conjugal Parkinsonism – clinical, pathology and genetic study. No evidence of person‐to‐person transmission. Parkinsonism Relat. D., 31, 87–90. [DOI] [PubMed] [Google Scholar]
- Rappaport, S.M. (2016) Genetic factors are not the major causes of chronic diseases. PLoS ONE, 11, e0154387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, A.H. , McCall, S. , Henry, J.M. & Taubenberger, J.K. (2001) Experimenting on the past: the enigma of von Economo's encephalitis lethargica. J. Neuropath. Exp. Neur., 60, 663–670. [DOI] [PubMed] [Google Scholar]
- Ritz, B. & Rhodes, S.L. (2010) After half a century of research on smoking and PD, where do we go now? Neurology, 74, 870–871. [DOI] [PubMed] [Google Scholar]
- Rocha, J.D. , Schlossmacher, M.G. & Philpott, D.J. (2015) LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat. Immunol., 16, 898–900. [DOI] [PubMed] [Google Scholar]
- Ross, O.A. & Rademakers, R. (2016) Modifiers of LRRK2 Parkinsonism: new therapeutic targets. Lancet Neurol., 15, 1200–1201. [DOI] [PubMed] [Google Scholar]
- Saito, Y. , Shioya, A. , Sano, T. , Sumikura, H. , Murata, M. & Murayama, S. (2016) Lewy body pathology involves the olfactory cells in Parkinson's disease and related disorders. Movement Disord., 31, 135–138. [DOI] [PubMed] [Google Scholar]
- Sardi, S.P. , Clarke, J. , Kinnecom, C. , Tamsett, T.J. , Li, L. , Stanek, L.M. , Passini, M.A. , Grabowski, G.A. et al (2011) CNS expression of glucocerebrosidase corrects alpha‐synuclein pathology and memory in a mouse model of Gaucher‐related synucleinopathy. Proc. Natl. Acad. Sci. USA, 108, 12101–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savica, R. , Rocca, W.A. & Ahlskog, J.E. (2010) When does Parkinson disease start? Arch. Neurol., 67, 798–801. [DOI] [PubMed] [Google Scholar]
- Scheperjans, F. (2016) Can microbiota research change our understanding of neurodegenerative diseases? Neurodegener. Dis. Manag., 6, 81–85. [DOI] [PubMed] [Google Scholar]
- Scheperjans, F. , Pekkonen, E. , Kaakkola, S. & Auvinen, P. (2015) Linking smoking, coffee, urate, and Parkinson's disease – a role for gut microbiota? J Parkinsons Dis, 5, 255–262. [DOI] [PubMed] [Google Scholar]
- Schlossmacher, M.G. & Mollenhauer, B. (2010) Biomarker research in Parkinson's disease: objective measures needed for patient stratification in future cause‐directed trials. Biomark Med., 4, 647–650. [DOI] [PubMed] [Google Scholar]
- Sidransky, E. , Nalls, M.A. , Aasly, J.O. , Aharon‐Peretz, J. , Annesi, G. , Barbosa, E.R. , Bar‐Shira, A. , Berg, D. et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. New Engl. J. Med., 361, 1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, A. & Hardy, J. (2016) The evolution of genetics: Alzheimer's and Parkinson's diseases. Neuron, 90, 1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, A.B. , Farrer, M. , Johnson, J. , Singleton, A. , Hague, S. , Kachergus, J. , Hulihan, M. , Peuralinna, T. et al (2003) alpha‐Synuclein locus triplication causes Parkinson's disease. Science, 302, 841. [DOI] [PubMed] [Google Scholar]
- Siroux, V. , Agier, L. & Slama, R. (2016) The exposome concept: a challenge and a potential driver for environmental health research. Eur. Respir. Rev., 25, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto‐Ortolaza, A.I. , Heckman, M.G. , Labbe, C. , Serie, D.J. , Puschmann, A. , Rayaprolu, S. , Strongosky, A. , Boczarska‐Jedynak, M. et al (2013) GWAS risk factors in Parkinson's disease: LRRK2 coding variation and genetic interaction with PARK16. Am. J. Neurodegener. Dis., 2, 287–299. [PMC free article] [PubMed] [Google Scholar]
- Speciale, S.G. (2002) MPTP: insights into parkinsonian neurodegeneration. Neurotoxicol. Teratol., 24, 607–620. [DOI] [PubMed] [Google Scholar]
- Spillantini, M.G. , Schmidt, M.L. , Lee, V.M. , Trojanowski, J.Q. , Jakes, R. & Goedert, M. (1997) Alpha‐synuclein in Lewy bodies. Nature, 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Stanberry, L. , Mias, G.I. , Haynes, W. , Higdon, R. , Snyder, M. & Kolker, E. (2013) Integrative analysis of longitudinal metabolomics data from a personal multi‐omics profile. Metabolites, 3, 741–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepens, A. , Logina, I. , Liguts, V. , Aldins, P. , Eksteina, I. , Platkajis, A. , Martinsone, I. , Terauds, E. et al (2008) A Parkinsonian syndrome in methcathinone users and the role of manganese. New Engl. J. Med., 358, 1009–1017. [DOI] [PubMed] [Google Scholar]
- Sveinbjornsdottir, S. , Hicks, A.A. , Jonsson, T. , Petursson, H. , Gugmundsson, G. , Frigge, M.L. , Kong, A. , Gulcher, J.R. et al (2000) Familial aggregation of Parkinson's disease in Iceland. New Engl. J. Med., 343, 1765–1770. [DOI] [PubMed] [Google Scholar]
- Svensson, E. , Horvath‐Puho, E. , Thomsen, R.W. , Djurhuus, J.C. , Pedersen, L. , Borghammer, P. & Sorensen, H.T. (2015) Vagotomy and subsequent risk of Parkinson's disease. Ann. Neurol., 78, 522–529. [DOI] [PubMed] [Google Scholar]
- Tanner, C.M. , Ottman, R. , Goldman, S.M. , Ellenberg, J. , Chan, P. , Mayeux, R. & Langston, J.W. (1999) Parkinson disease in twins: an etiologic study. JAMA, 281, 341–346. [DOI] [PubMed] [Google Scholar]
- Tappe, D. & Alquezar‐Planas, D.E. (2014) Medical and molecular perspectives into a forgotten epidemic: encephalitis lethargica, viruses, and high‐throughput sequencing. J. Clin. Virol., 61, 189–195. [DOI] [PubMed] [Google Scholar]
- Taylor, K.M. , Saint‐Hilaire, M.H. , Sudarsky, L. , Simon, D.K. , Hersh, B. , Sparrow, D. , Hu, H. & Weisskopf, M.G. (2016) Head injury at early ages is associated with risk of Parkinson's disease. Parkinsonism Relat. D., 23, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh, J. , Gustavsson, E.K. , Guella, I. , Vilarino‐Guell, C. , Evans, D. , Encarnacion, M. , Sherman, H. , Hentati, F. et al (2014) The role of SNCA and MAPT in Parkinson disease and LRRK2 Parkinsonism in the Tunisian Arab‐Berber population. Eur. J. Neurol., 21, e91–e92. [DOI] [PubMed] [Google Scholar]
- Trinh, J. , Gustavsson, E.K. , Vilarino‐Guell, C. , Bortnick, S. , Latourelle, J. , McKenzie, M.B. , Tu, C.S. , Nosova, E. et al (2016) DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser Parkinsonism: a genome‐wide linkage and association study. Lancet Neurol., 15, 1248–1256. [DOI] [PubMed] [Google Scholar]
- Valente, E.M. , Abou‐Sleiman, P.M. , Caputo, V. , Muqit, M.M. , Harvey, K. , Gispert, S. , Ali, Z. , Del Turco, D. et al (2004) Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science, 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Venuto, C.S. , Potter, N.B. , Ray Dorsey, E. & Kieburtz, K. (2016) A review of disease progression models of Parkinson's disease and applications in clinical trials. Movement Disord., 31, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R.C. , Rodriguez, R.M. , Moghadassi, M. , Noble, V. , Bailitz, J. , Mallin, M. , Corbo, J. , Kang, T.L. et al (2016) External validation of the STONE score, a clinical prediction rule for ureteral stone: an observational multi‐institutional study. Ann. Emerg. Med., 67, 423–432. e422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild, C.P. (2009) Environmental exposure measurement in cancer epidemiology. Mutagenesis, 24, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, A.W. , Sterling, C. & Racette, B.A. (2010) Conjugal Parkinsonism and Parkinson disease: a case series with environmental risk factor analysis. Parkinsonism Relat. D., 16, 163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiromerisiou, G. , Houlden, H. , Sailer, A. , Silveira‐Moriyama, L. , Hardy, J. & Lees, A.J. (2012) Identical twins with Leucine rich repeat kinase type 2 mutations discordant for Parkinson's disease. Movement Disord., 27, 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich, A. , Biskup, S. , Leitner, P. , Lichtner, P. , Farrer, M. , Lincoln, S. , Kachergus, J. , Hulihan, M. et al (2004) Mutations in LRRK2 cause autosomal‐dominant Parkinsonism with pleomorphic pathology. Neuron, 44, 601–607. [DOI] [PubMed] [Google Scholar]