

ABSTRACT
To further enhance the intensity of deep tumor drug delivery and integrate a combined therapy, we herein report on a core-shell nanocarrier that could simultaneously overcome the double barriers of the extracellular matrix (ECM) and multiple layers of tumor cells (MLTC). A pH-triggered reversible swelling-shrinking core and an MMP2 (matrix metallopeptidase 2) degradable shell were developed to encapsulate chemotherapeutics and macroautophagy/autophagy inhibitors, respectively. MMP2 degraded the shell, which was followed by the autophagy inhibitors' release. The exposed core could diffuse along the pore within the ECM to deliver chemotherapeutics into deep tumors, and it was able to swell in lysosomes and shrink back in the cytoplasm or ECM. The swelling of the core resulted in the rapid release of chemotherapeutics to kill autophagy-inhibited cells. After leaving the dead cells, the shrinking core could act on neighboring cells that were closer to the center of the tumor. The core thus could also cross MLTC layer by layer to deliver chemotherapeutics into the deep tumor.
KEYWORDS: autophagy inhibition, deep tumor penetration, multidrug delivery, multilayers tumor cells crossing, multistage size changeable nanocarrier
Introduction
Currently, 2 major challenges blunt the nonsurgical treatment of malignant solid tumors. First, the physiological barriers of the tumor restrict the effective distribution of the therapeutics to all tumor cells.1,2 Second, the acquired drug resistance resulting from monotherapy cripples the antitumor efficiency of available drugs.3,4 Codelivery nanocarriers have shown the potential to overcome these 2 challenges or reduce their effects.5-7
Many new types of nanocarriers have been developed to optimize solid tumor therapy.8-12 However, traditional nanocarriers generally cannot effectively overcome the physiological barriers of the solid tumor itself; thus, the major nanocarriers often simply distribute around the tumor vessels.13-15 The tumor extravascular barrier, which is composed of tumor extracellular matrix (ECM) and multiple layers of tumor cells (MLTC), represents the most formidable barrier.16-19 Drug resistance and recurrence of the tumor develop sequentially because the nanocarriers could not effectively traverse this barrier and deliver therapeutics to deep tumor tissues where cancer stem cells may hide.20-24 Generally, controlling the size of nanocarriers can solve this problem, at least to a certain extent. Nanocarriers are required to have sufficiently large sizes (∼100-200 nm) to reduce the clearance and improve the retention within the tumor by an enhanced permeability and retention (EPR) effect. However, smaller particle sizes (< 100 nm) are required for better tumor penetration.25-28 To balance this contradiction, large-to-small size variable nanocarriers have been designed.29 The small-sized nanocarrier can penetrate into deep tumors based partly on passive physical diffusion along the pore within the ECM, but the MLTC still limits its penetration capability. Thus, a nanocarrier that can simultaneously overcome the double barriers of the ECM and MLTC via size controlling is urgently required.
Multidrug codelivery can act on multiple essential pathways of tumor growth and is usually considered as a promising strategy for reversing drug resistance.30,31 Among these pathways, autophagy plays a significant role for cancer growth. Autophagy not only provides an energy source, but can also provide the ability to address various stresses for cancer cells.32-34 As a cellular defense mechanism, autophagy lowers the antitumor efficiency of docetaxel-loaded poly(lactic-co-glycolic acid)/PLGA nanoparticles.35 Thus, the combination of autophagy inhibitors and chemotherapeutics is suggested to obtain desirable antitumor efficacy.36-37 3-Methyladenine (3-MA), which inhibits autophagy by blocking autophagosome formation via the inhibition of the class III phosphatidylinositol 3-kinases,38 was found to be effective in cancer treatments in combination with anticancer drugs.39,40 Thus, we adopted 3-MA to codeliver with doxorubicin (DOX) to explore their combined antitumor effect.
We herein report on a core-shell nanocarrier that could achieve “double deep” tumor penetration by ECM and MLTC crossing (Fig. 1A). The nanocarrier was designed as follows: A reversible swelling-shrinking core (named “PSP”) for loading DOX (D/PSP), which had an acid-triggered charge reversal capability in lysosomes (pH 4.0–5.0); an MMP2 (matrix metallopeptidase 2) degradable gelatin shell was developed to encapsulate 3-MA (M/GNP). During blood circulation (Fig. 1B), this nanocarrier (named “D/PSP@M/GNP”) could passively accumulate and remain around abnormal tumor vasculatures because of the EPR effect and its larger size. MMP2 then degraded the gelatin and was followed by the 3-MA release and the exposure of D/PSP. The released 3-MA preferentially entered cancer cells to inhibit their autophagy level, thereby potentially weakening the defense of cancer cells against D/PSP ahead of time. The exposed smaller D/PSP could diffuse along the interstitial space within the ECM to deliver DOX into the deep tumor (Fig. 1C). After being endocytosed by tumor cells, D/PSP was able to swell in lysosomes and shrink when back in the cytoplasm or ECM. The swelling of D/PSP resulted in the rapid release of DOX to kill autophagy-inhibited tumor cells. After leaving the dead cells, D/PSP could act on neighboring tumor cells that were closer to the center of the tumor. D/PSP thus could also cross the MLTC layer by layer to deliver DOX into the deep tumor (Fig. 1D).
Figure 1.
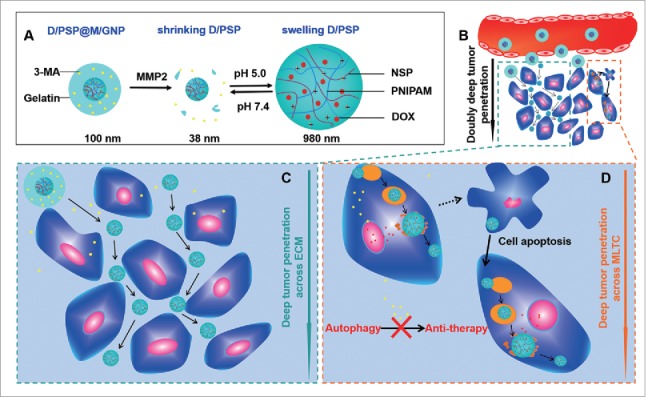
Schematic design of the nanocarrier. (A) The main components of D/PSP@M/GNP. (B) The effective retention of D/PSP@M/GNP at the tumor site and the double deep-tumor penetration effect. (C) The ECM crossing process depends on the smaller size of D/PSP. (D) The MLTC crossing process is based on the reversible swelling-shrinking size of D/PSP triggered by lysosomes.
Results
Fabrication and characterization of D/PSP@M/GNP
The branched polyethylenimine (bPEI) was chemically altered via covalently bonding with succinic anhydride to develop N-succinyl branched polyethylenimine (NSP) with different isoelectric points (pIs) (Fig. S1). Compared with the bPEI, the number average molecular weight (Mn) of NSP with a pI of 6.1, whose structure and pI was respectively identified by NMR spectra and nephelometry (Fig. S2 and S3), increased by 4589 (Fig. S4). PSP was then fabricated by polymerizing N-isopropylacrylamide (NIPAM) forming poly(N-isopropylacrylamide [PNIPAM]) in the presence of NSP. DOX was physically encapsulated in PSP (form “D/PSP”) and its payload was 18.6%. To demonstrate the pH-triggered swelling of D/PSP, we measured its particle size in various pHs. When the pH cycled between 7.4 and 5.0 (close to the pH value of endosomes or lysosomes),41-43 D/PSP showed a reversible swelling (to ∼1000 nm) and shrinking (to ∼40 nm) (Fig. 2A, 2B and 2C). The zeta potential of D/PSP also displayed a reversible change from ∼-10 mV at pH 7.4 to ∼+25 mV at pH 5.0 (Fig. 2D). This high electropositivity of D/PSP at pH 5.0 could contribute to its endo-lysosomal escape.44-46 As a control nanoparticle, PP was fabricated by polymerizing N-isopropylacrylamide in the presence of bPEI. In the same manner, DOX was physically encapsulated in PP (form “D/PP”) and its payload was 15.1%. As a result, D/PP exhibited irrevocable changes both in its size and zeta potential under the corresponding conditions for D/PSP.
Figure 2.
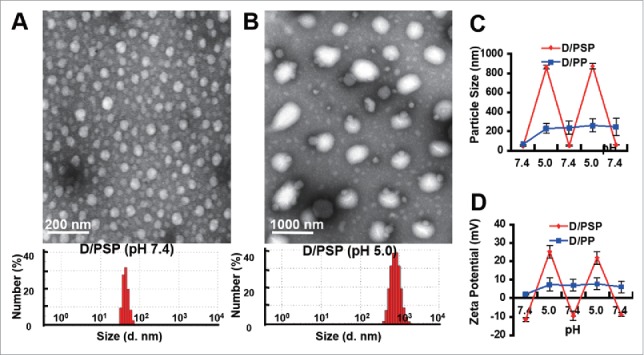
Characterization of D/PSP. The morphology and size distribution (A) at pH 7.4 and (B) at pH 5.0. The change in (C) size and (D) zeta potential of D/PSP and D/PP with different pHs. d. nm, diameter nanometer.
Next, 3-MA was encapsulated in the outer gelatin shell (form “D/PSP@M/GNP”) and its payload in the gelatin was 26.4%. D/PSP@M/GNP also displayed pH-dependent size reversibility, which was ∼100 nm at pH 7.4 and ∼1000 nm at pH 5.0 (Fig. 3A and 3B). Additionally, the gelatin shell could be effectively degraded by MMP2 to expose the smaller D/PSP (Fig. 3C) and facilitate the complete release of 3-MA (Fig. 3D). We constantly alternated the fresh medium of D/PSP between pH 7.4 and pH 5.0 to analyze the residual DOX within D/PSP after several cycles of swelling-shrinking. The DOX released from D/PSP was measured by the HPLC method after a 4-h incubation. The residual DOX was calculated as the formula: (Residual DOX)% = 100% – (Released DOX)%. The results indicated that DOX within D/PSP could not be totally released during the 1st swelling-shrinking cycle, but could last for 5 cycles under our evaluation condition (Fig. S5).
Figure 3.
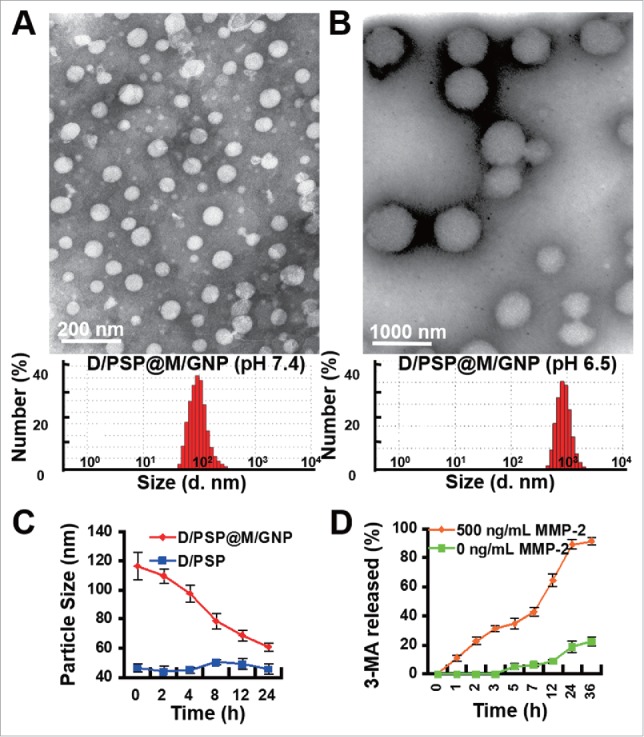
Characterization of D/PSP@M/GNP. The morphology and size distribution (A) at pH 7.4 and (B) at pH 5.0. (C) The change in size and (D) 3-MA release of D/PSP@M/GNP in response to MMP2. d. nm, diameter nanometer.
In vitro intercellular drug delivery ability of D/PSP@M/GNP
In contrast to D/PP, the intracellular yellow large-area fluorescence indicated the successful expansion of D/PSP within endo-lysosomes (Fig. 4A). Importantly, the D/PSP that escaped from the endo-lysosomes could still shrink back to a small size (Fig. 4A, arrows). The DOX amount within cells after several rounds of intercellular delivery was analyzed using the HPLC-MS/MS method. As shown in Fig. 4B, free DOX could not be detected in the third round of treated cells (III), and D/PP hardly delivered DOX to even the second set of treated cells (II), indicating that free DOX and D/PP had no intercellular drug delivery ability. In contrast, D/PSP could deliver DOX to the fifth set of treated cells (V), meaning that D/PSP could sequentially deliver DOX into at least the fifth cell layers. These results indicated that D/PSP displayed the capacity for intercellular delivery.
Figure 4.
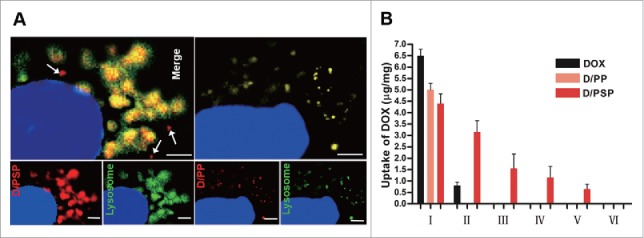
Endo-lysosomal swelling effect and cellular uptake of PSP. (A) The appearance of D/PSP (left) and D/PP (right) in endo-lysosomes observed by CLSM. Endo-lysosomes were stained with LysoTracker Green, while the D/PSP was traced by red DOX. The white arrows indicate the PSP that escaped from endo-lysosomes. The nuclei were stained with Hoechst 33342. Scale bar: 2 μm. (B) Cellular uptake of DOX within cells during sequential intercellular delivery.
We further evaluated the pH sensitivity of nanocarriers that escaped from cells. The morphology of nanocarriers that had passed through the cell monolayer was captured by transmission electron microscopy (TEM). The size of these nanocarriers could still shrink back to ∼40 nm at pH 7.4 and swelled to ∼1000 nm at pH 5.0 (Fig. 5). This result indicated that, after going through a layer of tumor cells, D/PSP still kept its reversible swelling-shrinking ability.
Figure 5.
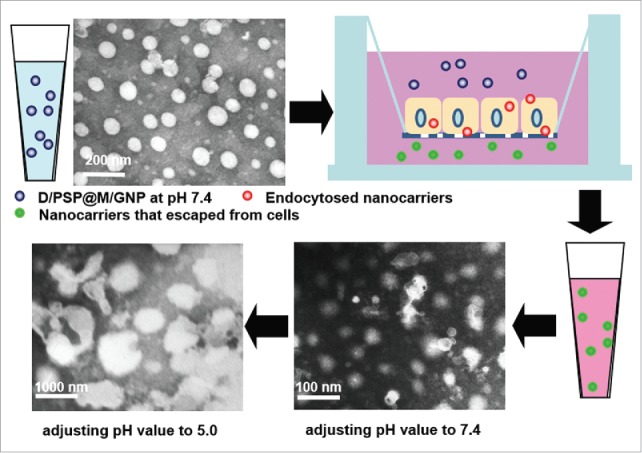
TEM images of nanocarriers that had passed through the cell monolayer at pH 7.4 and pH 6.5.
The enhanced deep tumor penetration of D/PSP@M/GNP was verified on 3-dimensional tumor spheroids. After a 12-h incubation, D/PSP@M/GNP (+MMP2)-treated tumor spheroids showed more remarkable DOX fluorescence than the D/PP@M/GNP (+MMP2) group at different depths (Fig. 6A). At the same time, the total DOX within tumor spheroids treated with D/PSP@M/GNP (+MMP2) was 2.82-fold higher that those treated with D/PP@M/GNP (+MMP2) (Fig. 6B). These data meant that D/PSP@M/GNP exhibited a higher penetration efficiency into the core of tumor spheroids, compare with the D/PP@M/GNP which could not cross MLTC.
Figure 6.
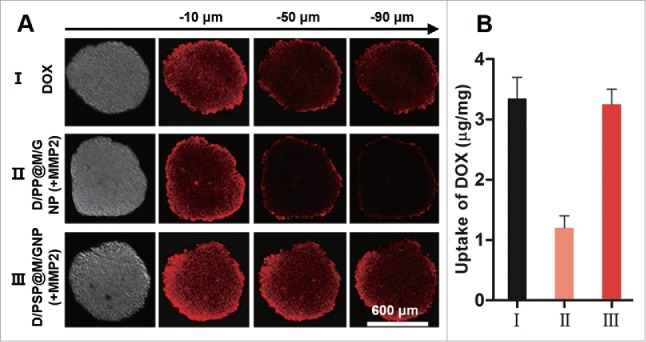
In vitro penetration assay using tumor spheroids. (A) Qualitative and (B) quantitative evaluation of nanocarriers on tumor spheroids.
In vivo tumor penetration
After being injected intravenously into the B16F10 tumor-xenografted mouse model, the in vivo fate of Cy5.5-labeled nanocarriers was monitored using near-infrared fluorescent imaging. As shown in Fig. 7A, the average fluorescence of tumors treated with D/PSP@M/GNP was significantly higher than that of tumors treated with D/PP@M/GNP except for that at 1 h. Notably, the tumor's fluorescence intensity at 36 h in mice treated with D/PSP@M/GNP was even higher than that at 12 h in mice treated with D/PP@M/GNP. This result indicated that D/PSP@M/GNP possessed the higher ability of accumulation and retention within tumors than D/PP@M/GNP, which was caused by the “double deep” tumor penetration effect of D/PSP@M/GNP. Additionally, the different depths of tumors treated with D/PSP@M/GNP were filled with more DOX (Fig. 7B). These data verified that the tumor distribution of the DOX delivered by D/PSP@M/GNP was more even and deeper.
Figure 7.
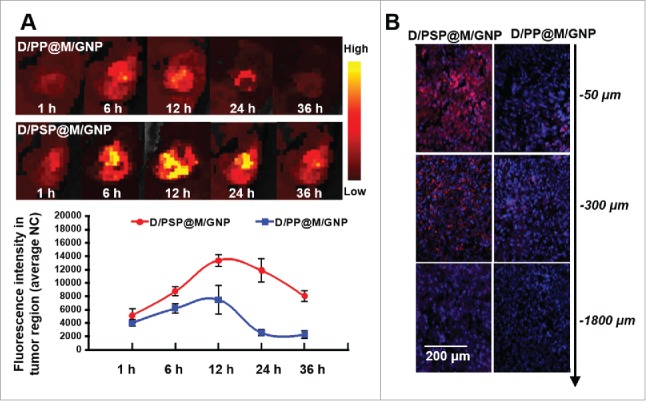
In vivo distribution and tumor penetration assay. (A) In vivo near-infrared fluorescent imaging of whole body and changes of fluorescence intensity in tumor regions (photo counts per mm2). The images were taken at 1, 6, 12, 24 and 36 h after nanocarrier injection via tail vein. (B) Tumor penetration based on different depths of tumor frozen sections. NC, normalized counts.
In vivo distribution of DOX delivered by D/PSP@M/GNP
At 24 h after tail vein injection of nanocarriers into the B16F10 tumor-xenografted mouse model, ex vivo imaging and HPLC-MS/MS analysis were utilized to evaluate the bio-distribution of DOX delivered by D/PSP@M/GNP and D/PP@M/GNP. As shown in Fig. S6A and S6B, nanocarriers could deliver more DOX to tumors than was achieved with free DOX, and significantly decrease heart distribution of DOX. Compared to D/PP@M/GNP, D/PSP@M/GNP could deliver 1.82-fold higher DOX to tumors. Although the difference of DOX distribution in the peripheral tumor was not significant between D/PSP@M/GNP- and D/PP@M/GNP-treated mice, the DOX in the central tumor treated with D/PSP@M/GNP was 4.2-fold higher than that treated with D/PP@M/GNP. These results further proved that D/PSP@M/GNP could effectively penetrate and deliver drug into the deep tumor.
Autophagy inhibition evaluation
The autophagy inhibition activity of D/PSP@M/GNP was then explored. B16F10 eGFP-LC3 cells were cultured as in our previous work.26 LC3 and eGFP-LC3 localized only to the autophagic membrane structures, which indicated autophagic vesicles.47 After 4 h of incubation, PP- or PSP-treated cells exhibited more punctate fluorescence than did PBS-treated cells (Fig. 8A and B). Simultaneously, the amount of punctate fluorescence was significantly reduced with the 3-MA addition, whereas the hydroxychloroquine (HCQ) addition increased the punctate fluorescence. Moreover, LC3 is an autophagy-related protein.48,49 The higher LC3-II:LC3-I ratios caused by the PP or PSP treatment were alleviated by PP@M/GNP or PSP@M/GNP with the help of MMP2 (Fig. 8C). These results indicated that PP and PSP could induce autophagy, which was beneficial for tumor cells. This autophagy-inducing ability of PP and PSP could be explained by the cellular self-protective mechanism against extraneous nanoparticles.36,37 That is, phagophores (autophagosome precursors) could sequester the nanoparticles that escaped from endo-lysosomes into double-membrane vesicles and transport them to lysosomes for degradation, which hinders the advantages of nanoparticles used for intracellular drug delivery.50,51 Fortunately, the 3-MA in the gelatin shell of PP@M/GNP or PSP@M/GNP could reverse this effect.
Figure 8.
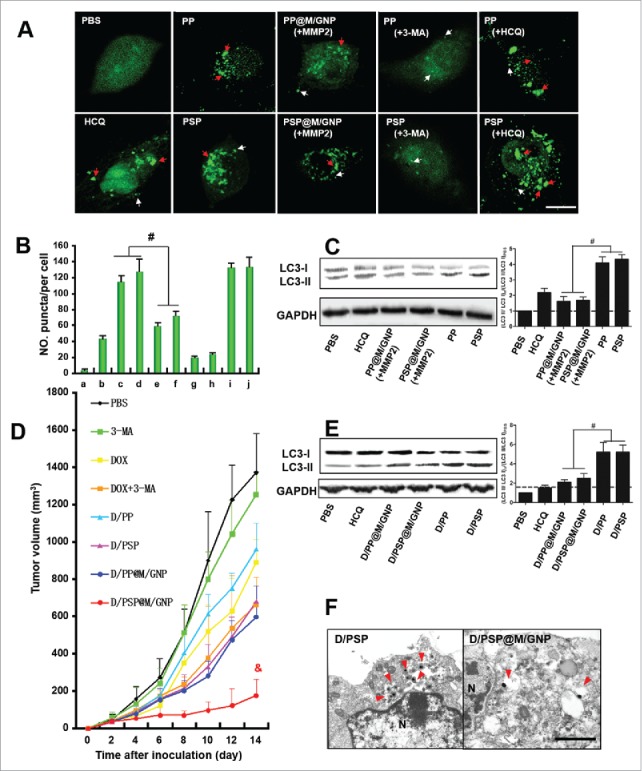
The autophagic flux assessment and antitumor activity. (A) Observation and (B) Quantitative analysis of eGFP-labeled autophagosomes. White arrows indicate the small puncta; red arrows indicate the dense puncta. Scale bar: 5 μm. (C) LC3 expression of cells. (D) Tumor growth curves. (E) LC3 expression of tumors. (F) Electron micrographs of tumors. Red arrows indicate autophagic vesicles. Scale bar: 500 nm. # and and indicates p < 0.005 and p < 0.001 versus the other groups, respectively. HCQ, hydroxychloroquine. [In panel A, change “a” to “i,” “b“ to “ii,” etc. or simply delete these letters. In B, insert a space after “No.” In C and E change to LC3-I“ and “LC3-II.”
In vitro and in vivo antitumor efficiency
We further evaluated the apoptosis-inducing effect of D/PSP@M/GNP on B16F10 cells. With the help of MMP2, the viability of B16F10 cells treated with D/PSP@M/GNP (3.04%) was significantly lower than the cells treated with D/PP@M/GNP (15.13%) (Fig. S7). Additionally, the MTT assay showed that 3-MA + D/PSP display the greatest cytotoxicity (Fig. S8). The IC50 of DOX (D/PSP + 3-MA) decreased by 11.0 fold for B16F10 cells, 13.7-fold for 4T1 cells, and 9.92 fold for MCF-7 cells compared with that of D/PSP only (Table S1). In vivo, D/PSP@M/GNP more effectively inhibited the tumor growth than D/PSP (Fig. 8D), which indicated that a synergistic antitumor activity occurred by combining DOX with 3-MA. Notably, D/PSP@M/GNP showed a 2.84-fold reduction in tumor growth rate and a 9.83-fold reduction of excised tumor weight vs. D/PP@M/GNP (Fig. S9A, B and C). These results substantiated the hypothesis that the sequential delivery of 3-MA and DOX by D/PSP@M/GNP and the deep tumor drug delivery, which was based on the multistage change in the nanocarrier size, enabled a reinforcement of the synergistic antitumor efficiency. The body weight of mice in the DOX group and DOX-3-MA group began to reduce on the 8th d (Fig. S9d), and the mice in these 2 groups began to show signs of lethargy starting from the 5th d, but the mortality of mice was 0% during our short 14-d evaluation. Furthermore, the result of heart tissue histological observation showed that the level of fragmentation of the myocardium caused by free DOX could significantly be alleviated via encapsulating DOX into nanocarricers (Fig. S10). Tumors showed the highest level of cell necrosis and apoptosis after applying D/PSP@M/GNP (Fig. S11 and Fig. S12). Compared with the D/PP or D/PSP treatment group, D/PP@M/GNP or D/PSP@M/GNP could lower the LC3-II:LC3-I ratio(Fig. 8E). Morphologically, the amount of autophagic vesicles in tumors treated with D/PSP@M/GNP were significantly less than that of the D/PSP group (Fig. 8F). These results offered considerable evidence of the in vivo antitumor and autophagy-inhibiting efficiency of D/PSP@M/GNP.
Because components of nanocarriers used in these experiments included bPEI, the related toxicity is an issue. bPEI is known to be toxic when applied at higher doses, which is mainly associated with a strong positive charge of this polycation.52 In vitro, blank PSP/GNP displays a lower toxicity compare with PP/GNP, which could be due to the succinylation of bPEI that may significantly reduce the cytotoxicty of bPEI.53 Even under a high concentration of 2000 μg/ml, cells treated with PSP@GNP could still survive above 80% after 48-h incubation (Fig. S13), which meant having little impact on our short-term in vitro and in vivo experiments. However, the long-term toxicity still needs to be investigated in future work.
Discussion
Nanoparticles manufactured by biodegradable polymers have been considered one of the strategies with the greatest potential as tumor-targeting drug delivery vehicles.54 After accumulating in tumor sites via EPR effect, however, traditional nanoparticles generally cannot effectively overcome the ECM and MLTC.16-19 In this study, to overcome both the ECM and MLTC, we fabricated a MMP2 and low pH sensitive, core-shell, codelivery nanocarrier—D/PSP@M/GNP—for sequential and deep tumor drug delivery. Our experimental results proved that D/PSP@M/GNP was able to respond to MMP2 and then shrink to a smaller D/PSP core. Then the reversibly swelling-shrinking D/PSP could respond to the endo-lysosomal pH value, overcoming the double barriers from ECM and MLTC of solid tumors to help loaded drugs penetrate into the deep tumor area.
Autophagy plays a significant role in cytoplasmic renewal, degradation of intracellular dysfunctional proteins, and clearance of toxic substances.55 An increasing number of nanoparticles have been reported as having the ability to modulate the cellular process of autophagy,37 and it has been considered that autophagy induction may be a potential common cellular reaction toward nanoparticles as an attempt to eliminate the foreign substances.50 Several polymeric nanoparticles such as chitosan-based nanoparticles and poly(lactic-co-glycolic acid)/PLGA-based nanoparticles are internalized into cells through endocytosis, and then transported into the lysosome for degradation and clearance.56 Thus, autophagy of nanoparticles plays an important role in deciding their fate after endocytosis and influencing their therapeutic effects as a drug delivery system. In consideration of these studies, the combination of autophagy inhibitors such as 3-MA and chemotherapeutics is suggested to obtain desirable anti-tumor efficacy.
Thus, we think that the design philosophy via integrating the advantages of enhanced deep tumor drug delivery and combination therapy could effectively lower drug resistance and recurrence of tumors, which are currently intractable in clinical cancer therapy, thus further improving therapeutic efficacy for various kinds of solid tumors.
Materials and methods
Materials
The following reagents were obtained as indicated: Branched polyethylenimine (Sigma-Aldrich, 408727); doxorubicin hydrochloride (Sigma-Aldrich, D1515); 3-methyladenine (Sigma-Aldrich, M9281); succinic anhydride (TCI Development, S0107); N,N’-methylenebisacrylamide (TCI Development, M0506); N-isopropylacrylamide (TCI Development, I0401); ammonium peroxodisulfate (TCI Development, A2098); gelatin type A (MP Biomedicals, 02960102); anti-LC3 (Medical & Biological Laboratories, PM036); LysoTracker Green (Invitrogen, L7526); DAPI (Invitrogen, D1306); Hoechst 33342 (Invitrogen, 62249); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (Invitrogen, M6494); ANXA5/Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (KeyGEN Biotech, KGA512); Dulbecco's modified Eagle's medium (DMEM; Gibco, 88420); Roswell Park Memorial Institute/RPMI 1640 medium (Gibco, 61870036); trypsin (Gibco, 27250018); plastic cell culture dishes (NEST Biotechnology, 704004); plastic cell culture plates (NEST Biotechnology, 703001); HCQ (TCI Development, H1306).
Synthesis of N-succinyl-bPEI (NSP)
The synthetic process is shown in Figure S1. bPEI (1000 mg; Sigma-Aldrich, 408727) was dissolved in 50 mL of deionized water. SA (20 mmol; TCI Development, S0100) was dissolved in DMSO (25 mL). Different volumes of SA solution (8, 5, 4, 3, 2 and 1 mL) were added into 10 mL of the bPEI solution. The reaction solution was then adjusted to pH 8.0 by NaOH (1 M) with subsequent stirring for 36 h at 40°C. After dialysis through a membrane (MWCO 10,000; Spectrum, 131267) to remove unreacted SA and freeze drying, NSP with different degrees of modification were obtained. The structures were confirmed by 1H NMR (D2O, 600 MHz) and 13C NMR (D2O, 600 MHz) spectra. The molecular weight distribution of NSP was measured by gel permeation chromatography (GPC) in dimethyl formamide containing lithium bromide 1 g/L at a flow rate of 1.2 mL/min. The GPC system consisted of a Waters 1515 pump (Waters, WAT1515, Japan), an Ultrahydrogel TM 500 column (Waters, WAT011520, Japan), and a Waters 2417 differential refractive index detector (Waters, WAT2417, Japan) with PEG (Sigma, P3515) as a standard for calibration.
Isoelectric point (pI) measurement of NSP
The turbidity method was used to determine the pI of NSP. The obtained NSP (10 mg) was dissolved in 5 mL of different pH citric acid-sodium citrate buffer solutions at 37°C (pH 3.0, 3.5, 4.0, 4.5, 5.0, 5.2, 5.5, 5.8, 6.0, 6.1, 6.2, 6.5, 7.0, 7.4, 8.2, and 11.0). After stirring for 10 min, the transmittance was detected at 580 nm using an ultraviolet and visible spectrophotometer (UVD-680–1, Shanghai Jinda Biochemical Instrument Co., China). The relative transmittance is the ratio between the transmittance in a specific pH and the transmittance in deionized water.
Preparation of the PSP core
To prepare the reversible swelling-shrinking PSP core, a radical polymerization of NIPAM was introduced. The process was as follows: NSP (with a pI of 6.1) and NIPAM were dissolved in deionized water at the mass ratio of NSP:NIPAM = 3:2 (the w/v of the system was 5%), then ammonium persulfate (TCI Development, A2098) as an initiator and N,N’-methylenebisacrylamide (TCI Development, M0506) as a crosslinker were added to trigger the reaction at 40°C for 12 h. The primary PSP was obtained by dialysis in deionized water to remove unreacted NIPAM, ammonium persulfate and N,N’-methylenebisacrylamide. Uniform-sized PSP was collected by eluting the primary PSP over a Sephadex G-50 column (1 cm × 25 cm; Pharmacia, S8150, USA) with deionized water. The solution was ultracentrifuged at 89,000 g for 20 min to concentrate the PSP core. PP (the control) was prepared identically to PSP, except NSP replaced bPEI. For the DOX-loaded PSP (D/PSP), the purified PSP core was added to 0.2 mL of triethylamine-treated DOX-HCl (10 mg) in DMF drop by drop with constant stirring for 24 h. The mixture solution was then dialyzed for 24 h to remove the residual DMF, triethylamine and nonencapsulated DOX. Finally, the DOX-loaded PSP solution was lyophilized to obtain the dried powders. The preparation method of DOX-loaded PP (D/PP) was identical to that of D/PSP except that PSP was replaced with PP. These procedures were performed in the absence of light.
Preparation of PSP@GNP
A gelatin shell was packed around the core by a 2-step desolvation method as previously described.29 Briefly, PSP (w/v = 5%, 10 mL) was added to the gelatin type A solution (w/v = 5%, 10 mL) and then acetone (10 mL) was added to the system at 6.0 mL/min. After finishing the addition for exactly 1 min, the gel-like precipitate was collected and redissolved in deionized water (10 mL) at 40°C. Acetone was then added drop by drop (1 mL/min) until the solution appeared white milk-like and stirred at 1000 rpm at 40°C for 4 h. Next, a glutaraldehyde solution (25%, 50 μL) in acetone (1 mL) was added at 0.05 mL/min. After stirring at 1000 rpm at 40°C for another 4 h, the acetone was removed by a rotary evaporator and the remaining solution was filtered through a 0.22-μm syringe filter (MiliPore, SLGV033RB) to obtain the primary PSP@GNP. By eluting the primary PSP@GNP over a Sephadex G-50 column (1 cm × 25 cm; Pharmacia, S8150, USA) with deionized water, uniform-sized PSP@GNP was collected. Ultracentrifugation (89,000 g for 20 min) was used to concentrate the PSP@GNP. To encapsulate 3-MA into the gelatin to obtain PSP@M/GNP, an aqueous solution of 3-MA (w/v = 10%, 2 mL) with pH 4.0 adjusted by HCl (1 M) was added to the gelatin type A solution, and the other processes were identical to the PSP@GNP process. The preparation of the codelivery D/PSP@M/GNP and D/PP@M/GNP was identical to that of PSP@M/GNP, except PSP was replaced with D/PSP and D/PP.
Characterization of nanocarriers
The particle size and zeta potential of nanocarriers (PSP, PP, D/PSP, D/PP, PSP@GNP, PP@GNP, D/PSP@M/GNP and D/PP@M/GNP) were measured using a laser particle size analyzer (Malvern Nano ZS, Malvern, UK). The morphology features were measured by a transmission electron microscope (JEOL JEM-1200EX, Japan). Briefly, nanocarriers suspended in pH 5.0 or pH 7.4 buffer solutions were dropped onto a copper grid and then stained with 1% phosphotungstic acid. The samples were observed after being air-dried.
MMP2 and pH sensitivity of PSP@GNP
For MMP2 sensitivity, the particle size of PSP@GNP, PP@GNP, D/PSP@M/GNP and D/PP@M/GNP were recorded (at 0, 2, 4, 8, 12, and 24 h) during the incubation with MMP2 (500 ng/mL) at 37°C. The released amount of 3-MA from the gelatin shell was measured. Briefly, 1 mL of nanocarriers with MMP2 (500 ng/mL) was added into a dialysis bag (MWCO 10,000; Spectrum, 131267). The bag was then immersed in 20 mL of MMP2 containing deionized water at 37°C under stirring at 100 rpm. At planned time points, the total released medium was replaced with 20 mL of fresh medium. The amount of 3-MA was measured by HPLC (Agilent 1200, USA). For the pH sensitivity of the nanocarriers of PSP, D/PSP and D/PSP@M/GNP, the pH of the nanocarrier solutions were reversibly changed between pH 7.4 and pH 5.0. The particle size and zeta potential were then measured. Moreover, the released DOX from D/PSP was measured by HPLC (UVIS-201, Alltech, USA) when the medium was reversibly changed between fresh pH 7.4 and pH 5.0 buffer solution after 1 h of incubation.
Cell culture
B16F10 cells and MCF-7 cells were cultured in DMEM, and 4T1 cells were cultured in RPMI-1640 medium. All cultures were supplemented with 10% fetal bovine serum, 100 U/mL streptomycin, and 100 U/mL penicillin in an atmosphere of 5% CO2 at 37°C. The eGFP-LC3 plasmid was obtained from Addgene (38195; deposited by Noboru Mizushima). The eGFP-LC3-expressing B16F10 cells (B16F10 eGFP-LC3 cells) were constructed by eGFP-LC3 plasmid transfection.57 Briefly, cells were allowed to grow to 0.25–1 × 106 per well (6-well) in 2 mL DMEM without antibiotics and were then transfected using Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer's instructions. For each well, 14 μg pooled eGFP-LC3 plasmid was diluted in 700 μL OptiMEM (Gibco, 31985070). After a 5-min incubation at room temperature, the diluted RNA and Lipofectamine 2000 were combined and incubated for 10 min at room temperature. The 250-μL plasmid-reagent complex was then added to the well. The eGFP-LC3-expressing cells were obtained after 24 h of incubation.
Endo-lysosomal swelling effect of PSP
B16F10 cells were seeded in a well (6-well) at a density of 1 × 104 cells per well and incubated overnight in DMEM containing 10% fetal bovine serum. For dynamically monitoring the endo-lysosomal swelling effect of PSP, the cells were washed 3 times with PBS, and their nuclei were stained with Hoechst 33342 (Beyotime Biotech, C1022) for 20 min at 37°C. After washing the cells 3 times, PP or PSP (3 mg/mL) was added into the wells to be observed by the Live Cell Imaging System (Leica DMI6000B, Germany). For the colocalization of PSP and endo-lysosomes, after the incubation with D/PP or D/PSP (3 mg/mL) for 50 min, LysoTracker Green (50 nM) was added for endo-lysosome staining for 30 min. Cells were washed 3 times with PBS and fixed with 4% (vol/vol) paraformaldehyde. The nuclei were then stained with DAPI for 15 min and washed another 3 times for observation by confocal laser scanning microscopy (CLSM; TCS SP5 AOBS confocal microscopy system, Leica, Germany).
In vitro cellular uptake
B16F10 cells were seeded on 6-well plates at 2 × 105 cells per well and cultured for 24 h. The cells were then treated with various samples (at 8 μg/mL DOX) for 4 h. The cells were then washed 3 times with PBS, trypsinized, and resuspended in PBS for the flow cytometry analysis (CytomicsTM FC 500, Beckman Coulter, Miami, FL, USA).
Intercellular delivery of D/PSP
Intercellular delivery of D/PSP, D/PP and the DOX solution among B16F10 cells was analyzed using an HPLC-MS/MS method. B16F10 cells seeded on coverslips were pretreated with the DOX solution, D/PP or D/PSP at DOX concentration of 8 μg/mL for 8 h. The pretreated cells (I) were washed with PBS and then co-incubated with fresh cells on a coverslip for 20 h in fresh culture medium. The fresh cells on a coverslip after co-incubation with (I) are referred to as (II). Then, (II) was withdrawn and co-incubated with the fresh cells on another new coverslip (III) for 20 h in fresh culture medium. Then, (III) was withdrawn and co-incubated with the fresh cells on another new coverslip (IV) for 20 h in fresh culture medium. In the same way, (IV) was withdrawn and co-incubated with the fresh cells on another new coverslip (V) for 20 h in fresh culture medium, etc. Afterwards, the cells (I,-VI) were washed with ice-cold PBS 3 times and lysed using cell lysis buffer (Beyotime Biotech, C3702) to release DOX in the cells. The amounts of DOX and cell proteins were measured by HPLC-MS/MS and the BCA protein assay kit (ThermoFisher Scientific, 23225), respectively.
In vitro penetration assay using tumor spheroids
To prepare the 3-dimensional tumor spheroids, B16F10T1 cells were seeded in 96-well plates coated with 100 μL 2% low melting point agarose (Invitrogen, 15517014; 1 × 105 cells per well). After 2 d, the tumor spheroids were treated with DOX, D/PP@M/GNP (+MMP2) or D/PSP@M/GNP at a DOX concentration of 12.5 μg/mL for 24 h. The tumor spheroids were then washed 3 times with PBS and fixed with 4% paraformaldehyde for 30 min. The images of the tumor spheroids were captured by tomoscan using Z-axis scanning with 20-μm intervals from the top of the spheroid to the middle by CLSM. The semiquantitative analysis of the mean fluorescence intensity of DOX was obtained by the software used in the CLSM.
In vivo distribution and tumor penetration assay
C57BL/6 mice weighing approximately 20 g were purchased from the experimental animal center of Sichuan University for this study. All animal experiments were performed according to the experimental guidelines of the Animal Experimentation Ethics Committee of Sichuan University. The B16F10 cells were harvested, resuspended in ice-cold PBS and subcutaneously injected into the left flank of mice (1 × 106 cells/flank). DOX, D/PP@M/GNP or D/PSP@M@GNP were intravenously injected into the mice at a dose of 10 mg/kg DOX per mouse after the tumor volume reached 500 mm3. The tumor size and animal body weight were measured every 2 d during the study, and the volume of tumors was calculated by the following formula: volume (mm3) = 1/2 A (length) × B (width)2 After a 24-h administration, the mice were anesthetized with 4% chloral hydrate and sacrificed; the main organs were removed to be observed with an in vivo fluorescence imaging system (CRi Maestro TX, USA). For HPLC-MS/MS analysis (Agilent 1260, USA), tumors or organs were homogenized with 1 mL water, to which 500 μL acetonitrile containing 100 ng/mL daunorubicin (Sigama-Aldrich, D8809) as internal standard was added. The mixture was vortexed and subsequently centrifuged at 13,400 x g for 20 min, then the supernatant fractions were diluted with the corresponding buffer to a protein concentration of 5 μg/μL and prepared in duplicate by protein precipitation before being analyzed by HPLC-MS/MS for analysis according to our previous method.58 DOX distributed in tumors and organs could be completely separated and detected under the selected analytical method, which could be validated by the standard curves (r of all tissues > 0.995), and the recoveries were between 85% and 115% in all tested tissues. For PECAM1/CD31 staining, the collected tumors were washed with PBS and were then subjected to ultracryotomy. FITC-PECAM1/CD31 antibody (Abcam, ab33858) was used to stain the frozen tumor section, and DAPI was used to stain the nuclei. The tumor sections were observed by CLSM. To obtain different deep tumor sections, the frozen tumors were sliced at different layers from the top of the tumors. After staining the cell nuclei with DAPI, the sections were observed by CLSM.
The autophagic flux assessment in vitro and ex vivo
In vitro, B16F10 eGFP-LC3 cells were seeded on 6-well plates at 2 × 105 cells per well and cultured for 24 h. The cells were then treated with various samples (50 μM HCQ, 3 mg/mL blank PP or PSP, 50 μM 3-MA, 8 mg/mL PP@M/GNP or PSP@M/GNP) for 4 h and fixed with 4% (vol/vol) paraformaldehyde. Fluorescence imaging was performed using CLMS. To quantify autophagosomes in the cells, the number of bright green puncta (autophagosomes) was counted in at least 25 cells. The eGFP-LC3 dot quantification was analyzed using the OriginPro 8.0 Software (Origin Lab Corporation, USA). Three independent experiments were performed. Additionally, a western blot was performed to detect the LC3 protein. Briefly, the cells were harvested and lysed in cell lysis buffer (Beyotime Biotech, C3702) after treatment. In total, 20 μg of proteins was loaded onto 10% SDS-PAGE gels for separation and then transferred onto a polyvinylidene difluoride (ThermoFisher Scientific, LC2002) membrane. After incubation with the specific primary antibodies at 4°C overnight, the membranes were washed with TBST (ThermoFisher Scientific, 37571) solution. Subsequently, the membranes were incubated with HRP-labeled secondary antibodies (Detroit R&D, EB 1) and detected with the Immobilon Western HRP Substrate (Millipore, WBKLS0500) on a Bio-Rad ChemiDoc MP system (Bio-Rad Laboratories, USA). ImageJ was used to quantify the band intensity. Ex vivo, a western blot was conducted to detect the LC3 in the total proteins of tumors after extraction. Additionally, to observe the autophagosomes on a transmission electron microscope, freshly excised tumors were fixed in 2.5% glutaraldehyde, pH 7.4 for 2 h at room temperature. The samples were then rinsed and post-fixed. Subsequently, the tissue was dehydrated and embedded in Agar 100 resin (Beyotime Biotech, P2210). Nanometer sections were cut and stained for observation.
In vitro and in vivo antitumor activity
The MTT assay was performed to evaluate the cytotoxicity of bare nanocarriers and the antitumor activity of drug loaded nanocarriers. Briefly, cells (2–5 × 103 per well) were seeded onto a 96-well plate, and 5 wells were included in each group. After the cells were cultured for 24 h, various nanocarriers were added (as indicated in Figure S7). The incubation continued for the planned times. Next, 20 μL of the MTT solution (5 mg/mL in PBS; Cellchipbj, 130005-M) was added to each well and incubated with the cells for 4 h. The medium was then replaced with 150 μL DMSO. The absorbance at 570 nm was measured with a microplate reader (Thermo Scientific Varioskan Flash) using wells without cells as blanks. An ANXA5/annexin V-FITC/PtdIns double-staining method was used to assess the capacity of apoptosis induced by nanocarriers. Cells were treated for 36 h. At the end of the treatment, the cells were trypsinized, washed with PBS and centrifuged at 10,000 g for 5 min. The cells were then resuspended and stained with ANXA5-FITC and PtdIns. After incubation for 15 min, the cells were collected for cytometric analyses.
C57BL/6 mice bearing B16F10 tumors were randomly divided into 8 groups (n = 6). After the size of tumors reached 50 mm3, the mice were randomly assigned into the following 8 groups: PBS group, DOX group, 3-MA group, DOX + 3-MA group, D/PP group, D/PSP group, D/PP@M/GNP group and D/PSP@M/GNP group. The drugs were injected through the tail vein one time every 3 d for a total of 4 times (DOX at 10 mg/kg and 3-MA at 18 mg/kg). By the end of the study, the mice were sacrificed, and the tumors were weighed and collected for further evaluation.
Histological analysis of the tumor tissue and other organs was performed by haematoxylin and eosin (H&E) staining. Briefly, 2-μm sections from at least 3 different planes of the tumors and other organs were cut and used for staining. Sections were evaluated using an optical microscope at various magnifications within different fields. An in situ terminal deoxynucleotidyltransferase- mediated UTP end-labeling (TUNEL) assay was manually processed. Briefly, tumor sections were transferred onto a glass slide and then deparaffinized and washed in TBS (Beyotime Biotech, P0228). The sections were incubated with proteinase for 20 min. After inactivating endogenous peroxidases, the sections were treated with a terminal deoxynucleotidyl transferase. Exposed 3’-OH ends of the breakage DNA fragment in the apoptotic cells were labeled with biotin-labeled deoxynucleotides. Finally, the sections were incubated with streptavidin-horseradish peroxidase conjugate (Prozyme, CJ30H) for 30 min at room temperature for apoptosis and necrosis area observation.
Statistical analysis
When not otherwise stated, all values are presented as the means ± SD. Statistical analysis was performed with 2-tailed Student t tests for 2 groups and one-way ANOVA for multiple groups. Probabilities less than 0.05 were considered significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- bPEI
branched polyethylenimine
- D/PP
DOX encapsulated in PP
- D/PSP
DOX encapsulated in PSP
- DMF
dimethyl formamide
- DOX
doxorubicin
- ECM
extracellular matrix
- EGFP
enhanced green fluorescent protein
- EPR
enhanced permeability and retention
- H&E
hematoxylin and eosin
- LC3
microtubule-associated protein 1 light chain 3
- MLTC
multiple layers of tumor cells
- MMP2
matrix metallopeptidase 2
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NIPAM
N-isopropylacrylamide
- PNIPAM
poly(N-isopropylacrylamide)
- NSP
N-succinyl branched polyethylenimine
- PBS
phosphate-buffered saline
- pI
isoelectric point
- PP
polymerization product of NIPAM in the presence of bPEI
- PSP
polymerization product of NIPAM in the presence of NSP
- TEM
transmission electron microscopy
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by the National Basic Research Program of China (973 Program, 2013CB932504) and the National Natural Foundation of China (81573367, 81373337).
References
- [1].Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol 2010; 7:653-64; PMID:20838415; http://dx.doi.org/ 10.1038/nrclinonc.2010.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li Y, Wang J, Wientjes MG, Au JL. Delivery of nanomedicines to extracellular and intracellular compartments of a solid tumor. Adv Drug Delivery Rev 2012; 64:29-39; http://dx.doi.org/ 10.1016/j.addr.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kerbel RS, Yu J, Tran J, Man S, Viloria-Petit A, Klement G, Coomber BL, Rak J. Possible mechanisms of acquired resistance to anti-angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metastasis Rev 2001; 20:79-86; PMID:11831651; http://dx.doi.org/ 10.1023/A:1013172910858 [DOI] [PubMed] [Google Scholar]
- [4].Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001; 7:987-9; PMID:11533692; http://dx.doi.org/ 10.1038/nm0901-987 [DOI] [PubMed] [Google Scholar]
- [5].Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MM, Decuzzi P, Tour JM, Robertson F. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat Nanotechnol 2008; 3:151-7; PMID:18654487; http://dx.doi.org/ 10.1038/nnano.2008.34 [DOI] [PubMed] [Google Scholar]
- [6].Xiao H, Li W, Qi R, Yan L, Wang R, Liu S, Zheng Y, Xie Z, Huang Y, Jing X. Co-delivery of daunomycin and oxaliplatin by biodegradable polymers for safer and more efficacious combination therapy. J Controlled Release 2012; 163:304-14; http://dx.doi.org/ 10.1016/j.jconrel.2012.06.004 [DOI] [PubMed] [Google Scholar]
- [7].He Q, Shi J. MSN Anti Cancer Nanomedicines: Chemotherapy Enhancement, Overcoming of Drug Resistance, and Metastasis Inhibition. Adv Mater 2014; 26:391-411; PMID:24142549; http://dx.doi.org/ 10.1002/adma.201303123 [DOI] [PubMed] [Google Scholar]
- [8].Park K, Lee S, Kang E, Kim K, Choi K, Kwon IC. New generation of multifunctional nanoparticles for cancer imaging and therapy. Adv Funct Mater 2009; 19:1553-66; http://dx.doi.org/ 10.1002/adfm.200801655 [DOI] [Google Scholar]
- [9].Corti A, Pastorino F, Curnis F, Arap W, Ponzoni M, Pasqualini R. Targeted drug delivery and penetration into solid tumors. Med Res Rev 2012; 32:1078-91. [DOI] [PubMed] [Google Scholar]
- [10].Nakanishi W, Minami K, Shrestha LK, Jia Q, Hill JP, Ariga K. Bioactive nanocarbon assemblies: Nanoarchitectonics and applications. Nano Today 2014; 9:378-94; http://dx.doi.org/ 10.1016/j.nantod.2014.05.002 [DOI] [Google Scholar]
- [11].Biju V. Chemical modifications and bioconjugate reactions of nanomaterials for sensing, imaging, drug delivery and therapy. Chem Soc Rev 2014; 43:744-64; PMID:24220322; http://dx.doi.org/ 10.1039/C3CS60273G [DOI] [PubMed] [Google Scholar]
- [12].Baeza A, Colilla M, Vallet-Regí M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Exp Opin Drug Deliv 2015; 12:319-37; http://dx.doi.org/ 10.1517/17425247.2014.953051 [DOI] [PubMed] [Google Scholar]
- [13].Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci 2011; 108:2909-14; http://dx.doi.org/ 10.1073/pnas.1018892108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, Torchilin VP, Munn LL. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res 2002; 62:6831-6; PMID:12460895. [PubMed] [Google Scholar]
- [15].Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009; 16:510-20; PMID:19962669; http://dx.doi.org/ 10.1016/j.ccr.2009.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev 1990; 9:253-66; PMID:2292138; http://dx.doi.org/ 10.1007/BF00046364 [DOI] [PubMed] [Google Scholar]
- [17].Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007; 99:1441-54; http://dx.doi.org/ 10.1093/jnci/djm135 [DOI] [PubMed] [Google Scholar]
- [18].Aznavoorian S, Stracke ML, Krutzsch H, Schiffmann E, Liotta LA. Signal transduction for chemotaxis and haptotaxis by matrix molecules in tumor cells. J Cell Biol 1990; 110:1427-38; PMID:2324200; http://dx.doi.org/ 10.1083/jcb.110.4.1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tannock IF, Lee CM, Tunggal JK, Cowan DSM, Egorin MJ. Limited Penetration of Anticancer Drugs through Tumor Tissue: A Potential Cause of Resistance of Solid Tumors to Chemotherapy. Clin Cancer Res 2002; 8:878-84; PMID:11895922. [PubMed] [Google Scholar]
- [20].Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8:755-68; PMID:18784658; http://dx.doi.org/ 10.1038/nrc2499 [DOI] [PubMed] [Google Scholar]
- [21].Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007; 1:313-23; PMID:18371365; http://dx.doi.org/ 10.1016/j.stem.2007.06.002 [DOI] [PubMed] [Google Scholar]
- [22].Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5:275-84; PMID:15803154; http://dx.doi.org/ 10.1038/nrc1590 [DOI] [PubMed] [Google Scholar]
- [23].Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29:4741-51; PMID:20531305; http://dx.doi.org/ 10.1038/onc.2010.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol 2009; 14:3-9; http://dx.doi.org/ 10.1007/s10911-009-9109-9 [DOI] [PubMed] [Google Scholar]
- [25].Huang K, Ma H, Liu J, Huo S, Kumar A, Wei T, Zhang X, Jin S, Gan Y, Wang PC. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. Nano ACS 2012; 6:4483-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huo S, Ma H, Huang K, Liu J, Wei T, Jin S, Zhang J, He S, Liang X-J. Superior penetration and retention behavior of 50 nm gold nanoparticles in tumors. Cancer Res 2013; 73:319-30; PMID:23074284; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2071 [DOI] [PubMed] [Google Scholar]
- [27].Ju C, Mo R, Xue J, Zhang L, Zhao Z, Xue L, Ping Q, Zhang C. Sequential Intra Intercellular Nanoparticle Delivery System for Deep Tumor Penetration. Angew Chem Int Ed 2014; 53:6253-8; http://dx.doi.org/ 10.1002/anie.201311227 [DOI] [PubMed] [Google Scholar]
- [28].Serda RE, Godin B, Blanco E, Chiappini C, Ferrari M. Multi-stage delivery nano-particle systems for therapeutic applications. Biochim Biophys Acta, Gen Subj 2011; 1810:317-29; http://dx.doi.org/ 10.1016/j.bbagen.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popović Z, Jain RK, Bawendi MG, Fukumura D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci 2011; 108:2426-31; http://dx.doi.org/ 10.1073/pnas.1018382108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother 2001; 45:105-16; PMID:11120952; http://dx.doi.org/ 10.1128/AAC.45.1.105-116.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel–Liposome Mediated Co Delivery of Anticancer Membrane Associated Proteins and Small Molecule Drugs for Enhanced Therapeutic Efficacy. Adv Funct Mater 2014; 24:2295-304; http://dx.doi.org/ 10.1002/adfm.201303222 [DOI] [Google Scholar]
- [32].Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/ 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zabirnyk O, Yezhelyev M, Seleverstov O. Nanoparticles as a novel class of autophagy activators. Autophagy 2007; 3:278-81; PMID:17351332; http://dx.doi.org/ 10.4161/auto.3916 [DOI] [PubMed] [Google Scholar]
- [34].Lee CM, Huang ST, Huang SH, Lin HW, Tsai HP, Wu JY, Lin CM, Chen CT. C 60 fullerene-pentoxifylline dyad nanoparticles enhance autophagy to avoid cytotoxic effects caused by the β-amyloid peptide. Nanomedicine 2011; 7:107-14. [DOI] [PubMed] [Google Scholar]
- [35].Zhang X, Dong Y, Zeng X, Liang X, Li X, Tao W, Chen H, Jiang Y, Mei L, Feng S-S. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials 2014; 35:1932-43; PMID:24315578; http://dx.doi.org/ 10.1016/j.biomaterials.2013.10.034 [DOI] [PubMed] [Google Scholar]
- [36].Zhang X, Yang Y, Liang X, Tai X, Liu Z, Tao W, Xiao X, Chen H, Huang L, Mei L. Enhancing therapeutic effects of docetaxel-loaded dendritic copolymer nanoparticles by Co-treatment with autophagy inhibitor on breast cancer. Theranostics 2014; 4:1085-95; PMID:25285162; http://dx.doi.org/ 10.7150/thno.9933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mei L, Zhang X, Feng S. Autophagy inhibition strategy for advanced nanomedicine. Nanomedicine 2014; 9:377-80; PMID:24746189; http://dx.doi.org/ 10.2217/nnm.13.218 [DOI] [PubMed] [Google Scholar]
- [38].Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850-61; PMID:20123989; http://dx.doi.org/ 10.1074/jbc.M109.080796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16:761-71; PMID:19116755; http://dx.doi.org/ 10.1245/s10434-008-0260-0 [DOI] [PubMed] [Google Scholar]
- [40].Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol 2011; 28:105-11; PMID:20041317; http://dx.doi.org/ 10.1007/s12032-009-9397-3 [DOI] [PubMed] [Google Scholar]
- [41].Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 2002; 115:599-607; PMID:11861766. [DOI] [PubMed] [Google Scholar]
- [42].Schmaljohann D. Thermo-and pH-responsive polymers in drug delivery. Adv Drug Delivery Rev 2006; 58:1655-70; http://dx.doi.org/ 10.1016/j.addr.2006.09.020 [DOI] [PubMed] [Google Scholar]
- [43].Chen W, Achazi K, Schade B, Haag R. Charge Charge-conversional and reduction-sensitive poly(vinyl alcohol) nanogels for enhanced cell uptake and efficient intracellular doxorubicin release. J Controlled Release 2015; 205:15-24; http://dx.doi.org/ 10.1016/j.jconrel.2014.11.012 [DOI] [PubMed] [Google Scholar]
- [44].Lee Y, Fukushima S, Bae Y, Hiki S, Ishii T, Kataoka K. A protein nanocarrier from charge-conversion polymer in response to endosomal pH. J Am Chem Soc 2007; 129:5362-3; PMID:17408272; http://dx.doi.org/ 10.1021/ja071090b [DOI] [PubMed] [Google Scholar]
- [45].Xu P, Van Kirk EA, Zhan Y, Murdoch WJ, Radosz M, Shen Y. Targeted charge reversal nanoparticles for nuclear drug delivery. Angew Chem Int Ed 2007; 46:4999-5002; http://dx.doi.org/ 10.1002/anie.200605254 [DOI] [PubMed] [Google Scholar]
- [46].Wang Y, Tai X, Zhang L, Liu Y, Gao H, Chen J, Shi K, Zhang Q, Zhang Z, He Q. A novel antitumour strategy using bidirectional autophagic vesicles accumulation via initiative induction and the terminal restraint of autophagic flux. J Controlled Release 2015; 199:17-28; http://dx.doi.org/ 10.1016/j.jconrel.2014.12.005 [DOI] [PubMed] [Google Scholar]
- [47].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- [49].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [50].Peynshaert K, Manshian B, Joris F, Braeckmans K, De Smedt SC, Demeester J, Soenen S. Exploiting Intrinsic Nanoparticle Toxicity: The Pros and Cons of Nanoparticle-Induced Autophagy in Biomedical Research. Chem Rev 2014; 114:7581-609. [DOI] [PubMed] [Google Scholar]
- [51].Liang X, Yang Y, Wang L, Zhu X, Zeng X, Wu X, Chen H, Zhang X, Mei L. pH-Triggered burst intracellular release from hollow microspheres to induce autophagic cancer cell death. J Mater Chem B. 2015; 3:9383-96; http://dx.doi.org/ 10.1039/C5TB00328H [DOI] [PubMed] [Google Scholar]
- [52].Beyerle A, Irmler M, Beckers J, Kissel T, Stoeger T. Toxicity Pathway Focused Gene Expression Profiling of PEI-Based Polymers for Pulmonary Applications. Mol Pharmaceutics 2010; 7:727-37; http://dx.doi.org/ 10.1021/mp900278x [DOI] [PubMed] [Google Scholar]
- [53].Zintchenko A, Philipp A, Dehshahri A, Wagner E. Simple Modifications of Branched PEI Lead to Highly Efficient siRNA Carriers with Low Toxicity. Bioconjugate Chem 2008; 19:1448-55; http://dx.doi.org/ 10.1021/bc800065f [DOI] [PubMed] [Google Scholar]
- [54].Wang Y, Tai X, Zhang L, Liu Y, Gao H, Chen J, Shi K, Zhang Q, Zhang Z, He Q. A novel antitumour strategy using bidirectional autophagic vesicles accumulation via initiative induction and the terminal restraint of autophagic flux. J Control Release 2015; 199:17-28; PMID:25499918. [DOI] [PubMed] [Google Scholar]
- [55].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875. [DOI] [PubMed] [Google Scholar]
- [56].Bae YM, Park YI, Nam SH, Kim JH, Lee K, Kim HM, Yoo B, Choi JS, Lee KT, Hyeon T. Endocytosis, intracellular transport, and exocytosis of lanthanide-doped upconverting nanoparticles in single living cells. Biomaterials 2012; 33:9080-6; PMID:22981077; http://dx.doi.org/ 10.1016/j.biomaterials.2012.08.039 [DOI] [PubMed] [Google Scholar]
- [57].Bae Y, Jang WD, Nishiyama N, Fukushima S, Kataoka K. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Mol BioSyst 2005; 1:242-50; PMID:16880988; http://dx.doi.org/ 10.1039/b500266d [DOI] [PubMed] [Google Scholar]
- [58].Zong T, Mei L, Gao H, Shi K, Chen J, Wang Y, Zhang Q, Yang Y, He Q. Enhanced Glioma Targeting and Penetration by Dual-Targeting Liposome Co-modified with T7 and TAT. J Pharm Sci 2014; 102:3891-901; http://dx.doi.org/ 10.1002/jps.24186 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.