

Abstract
A nanoparticle (NP) was developed to target choroidal neovascularization (CNV) via topical ocular administration. The NPs were prepared through conjugation of internalizing arginine-glycine-aspartic acid RGD (iRGD; Ac-CCRGDKGPDC) and transactivated transcription (TAT) (RKKRRQRRRC) peptide to polymerized ethylene glycol and lactic-co-glycolic acid. The iRGD sequence can specifically bind with integrin αvβ3, while TAT facilitates penetration through the ocular barrier. 1H nuclear magnetic resonance and high-performance liquid chromatography demonstrated that up to 80% of iRGD and TAT were conjugated to poly(ethylene glycol)– poly(lactic-co-glycolic acid). The resulting particle size was 67.0±1.7 nm, and the zeta potential of the particles was −6.63±0.43 mV. The corneal permeation of iRGD and TAT NPs increased by 5.50- and 4.56-fold compared to that of bare and iRGD-modified NPs, respectively. Cellular uptake showed that the red fluorescence intensity of iRGD and TAT NPs was highest among primary NPs and iRGD- or TAT-modified NPs. CNV was fully formed 14 days after photocoagulation in Brown Norway (BN) rats as shown by optical coherence tomography and fundus fluorescein angiography analyses. Choroidal flat mounts in BN rats showed that the red fluorescence intensity of NPs followed the order of iRGD and TAT NPs > TAT-modified NPs > iRGD-modified NPs > primary NPs. iRGD and TAT dual-modified NPs thus displayed significant targeting and penetration ability both in vitro and in vivo, indicating that it is a promising drug delivery system for managing CNV via topical ocular administration.
Keywords: nanoparticles, ocular drug delivery, choroidal neovascularization, RGD, cell-penetrating peptides
Introduction
Choroidal neovascularization (CNV) refers to new blood vessel growth from the choroid that extends into the subretinal pigment epithelium, or the subretinal space, or a combination of both.1 It is frequently accompanied by various ophthalmic diseases in the posterior segment and is a key pathologic change, causing severe vision loss as part of the pathogenesis of several diseases, especially in patients with age-related macular degeneration (AMD).2 As the aging population grows, the number of individuals suffering from AMD is expected to increase significantly over the next few decades.3 The treatment of diseases associated with choroid angiogenesis is a hotspot in ophthalmological research.
The most common drug administration method for treating posterior segment diseases, such as CNV, is intravitreal injection. Frequent administration is required because the medicine has a short half-life, which poses or increases the risks of vitreous hemorrhage, retinal detachment, and endophthalmitis.4–6 Moreover, patients may not comply with such regimens. Systemic administration requires a large quantity of drug to maintain sufficient therapeutic concentrations that will overcome the blood–retinal barrier and enter the retina,7 but this leads to undesirable side effects.8 Topical administration with eyedrops is convenient and noninvasive and has less side effects, but it is less effective because the corneal and conjunctival epithelia, along with the tear film, are biological barriers that block the penetration of drugs. Consequentially, traditional eyedrop formulations enable only low intraocular bioavailability and a low concentration in the posterior segment.9 Therefore, it is worth the effort to develop a new drug system that will overcome ocular barriers and deliver the drug to the posterior segment to target CNV.
There are two significant issues that must be overcome to develop a topical ocular drug delivery. On the one hand, improvement of ocular barrier permeability, such as through the cornea or conjunctiva, is required to enable adequate transport of the drug to the posterior segment by the drug delivery system. On the other hand, improved targeting of the drug delivery system to the posterior segment of the CNV area is required to further improve drug concentrations within the diseased tissue.
Cell-penetrating peptides (CPPs), eg, RKKRRQRRRC (transactivated transcription [TAT]), are short peptides that facilitate cellular uptake of various molecular cargos (from small chemical molecules to nanosized particles).10 Studies have shown that CPPs play an important role in carrying molecules across body barriers, such as the blood–brain barrier, the blood–testis barrier, and the placental barrier.11 More recently, it has been reported12–14 that nucleic acids, proteins, or fluorescent markers mediated by different CPPs can be delivered to the posterior segments of the eyes, such as the retina and choroid, by topical ocular drug delivery. This suggests that CPPs are an effective promoter of ocular barrier penetration, but CPP-mediated drug carrier systems to promote ocular permeability have not been reported to date.
Integrin αvβ3 was found to be selectively expressed on blood vessels in human wound granulation tissue but not in normal skin.15 Some researchers reported that integrin αvβ3 overexpression was observed on blood vessels in ocular tissues with active neovascularization, such as the retina or choroid in AMD patients, diabetic retinopathy, and presumed ocular angiogenesis.16,17 These findings suggest that integrin αvβ3 may be used as a receptor for ocular drug delivery targeting the CNV. The RGD peptide, a type of small peptide containing arginine–glycine–aspartate (Arg–Gly–Asp), is regarded as the recognition site for the interaction between integrin αvβ3 and a ligand. Peptides containing the RGD sequence can specifically bind with integrin αvβ3.18 It was reported that RGD-modified poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) applied for gene targeting in CNV therapy showed satisfactory results through intravenous administration.19 However, topical ocular drug delivery has not yet been reported.
Using the specific binding activity of an internalizing RGD (iRGD, Ac-CCRGDKGPDC) toward integrin αvβ3 and a common type of CPP, TAT, we established a topical ocular NP drug delivery system. This system has high corneal permeability and targets CNV to realize a noninvasive CNV treatment.
Materials and animals
Maleimide–poly(ethylene glycol)-poly(lactic-co-glycolic acid) (Mal–PEG–PLGA (molecular weight [MW] 5,000– 20,000 Da, 50:50 lactic acid [LA]: glycolic acid [GA], w/w) was purchased from PolySciTech (West Lafayette, IN, USA). Methoxy-poly(ethylene glycol)-poly(lactic-co-glycolic acid) (mPEG-PLGA, MW 5,000–20,000 Da, 50:50 LA:GA, w/w) was purchased from Jinan Daigang Biomaterial Co., Ltd (Jinan, People’s Republic of China). TAT peptide (MW =1,442 Da) was purchased from Bank Peptide Ltd (Hefei, People’s Republic of China), and iRGD peptide (Ac-CCRGDKGPDC, MW =1,093 Da) was purchased from ChinaPeptides (Shanghai, People’s Republic of China). Nile red and Hoechst 33342 dyes were purchased from Beijing Fanbo Biochemical Co., Ltd (Beijing, People’s Republic of China). Coumarin-6 was purchased from Aladdin Industrial Corp. (Shanghai, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium (DMEM) and DMEM/Ham’s F12 were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Human umbilical vein endothelial cells (HUVECs) were kindly provided by the Luye Pharma Group (Shandong, People’s Republic of China). The human corneal epithelial (HCE) cell line was purchased from Celltech (Shanghai, People’s Republic of China).
Brown Norway (BN) male rats were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, People’s Republic of China). All animal studies were performed according to the Guide for the Care and Use of Laboratory Animals and the animal study protocols of the China Animal Care and Use Committee, who also gave ethical approval for this study.
Methods
Synthesis and characterization of the conjugated polymers
iRGD-modified PEG–PLGA (iRGD–PEG–PLGA) was synthesized from iRGD and Mal–PEG–PLGA in a single-step reaction that coupled iRGD to PEG–PLGA using a previously reported method.20 Briefly, Mal–PEG–PLGA was dissolved in acetone and the organic solvent then evaporated, dispersing the solute evenly on the flask wall. The flask was replenished with 0.01 M phosphate-buffered saline (PBS, pH 7.4) and left overnight to react with iRGD. The iRGD was conjugated to Mal–PEG–PLGA (at 4°C, at a 1:1 molar ratio of peptide to Mal–PEG–PLGA). A high-performance liquid chromatography (HPLC, LC-20A VP system; Shimadzu, Kyoto, Japan) method was used to quantify the free (unconjugated) iRGD peptide until the concentration of iRGD became almost constant. HPLC was performed on an Inertsil ODS-2 C18 column 4.6×250 mm, 5.0 μm. The mobile phase consisted of a mixture of acetonitrile and water (linear gradient from 5:95 to 40:60, v/v, over 14 min). The flow rate was 1.0 mL/min. The UV detection was 220 nm. The reaction mixture was centrifuged to remove unreacted iRGD. The final solution was lyophilized and stored at −20°C until use. Synthesis of TAT-modified PEG–PLGA (TAT– PEG–PLGA) followed the same method. Free TAT peptide was determined using the same HPLC method as described earlier, except that mobile phase was 23%–48% (v/v) linear gradient acetonitrile in water in 23 min. The UV detection was 214 nm. The 1H nuclear magnetic resonance (NMR) spectrum was used to identify the final product. TAT and iRGD were dissolved in 0.5 mL of D2O, respectively. The Mal–PEG–PLGA, iRGD–PEG–PLGA, and TAT–PEG–PLGA were dissolved in 0.5 mL of CCl3D, respectively. They were subsequently detected via nuclear resonance spectrometry (Advance Bruker 400M; Switzerland Bruker Company, Madison, WI, USA).
Preparation and characterization of NPs
Different NPs, including iRGD and TAT dual-modified NPs labeled with Nile red (iRGD–TAT-Nile red-NP), iRGD-modified NPs labeled with Nile red (iRGD-Nile red-NP), TAT-modified NPs labeled with Nile red (TAT-Nile red-NP), and nonmodified NPs (mPEG–PLGA-Nile red-NP), were each prepared by nanoprecipitation21 as described elsewhere (Table 1). Briefly, mPEG–PLGA, iRGD–PEG–PLGA, TAT–PEG–PLGA, and Nile red were dissolved in acetone and added dropwise into water under stirring at room temperature for 4–5 h to allow complete evaporation of the acetone. The NP suspension was filtered through 0.45 μm filters to remove aggregates and insoluble coumarin-6. The flow through of NPs was concentrated through ultrafiltration and washed with distilled water three times to remove the residual coumarin-6 in the aqueous solution. Fluorescently labeled NPs containing coumarin-6 (1% loading, w/w) were prepared using the same procedure described earlier. The NP size, size distribution, and zeta potential were determined using a dynamic light scattering (DLS) particle size analyzer (Deisa™ Nano C; Beckman Coulter, Brea, CA, USA) equipped with a 50 mV laser at a scattering angle of 90°. The morphology of the NPs was examined using a transmission electron microscope (TEM, JEM-1400; Hitachi, Tokyo, Japan) after staining with a 2% sodium phosphotungstate solution. NPs labeled with Nile red were used for cellular uptake and choroid flat mounts, and NPs labeled with coumarin-6 were used to evaluate corneal permeation.
Table 1.
Formulation of various nanoparticles
| Groups | mPEG–PLGA (mg) | iRGD–PEG–PLGA (mg) | TAT–PEG–PLGA (mg) | Nile red (mg) | Acetone (mL) | Deionized water (mL) |
|---|---|---|---|---|---|---|
| mPEG–PLGA-Nile red-NP | 40 | – | – | 0.4 | 4 | 40 |
| iRGD-Nile red-NP | 30 | 10 | – | 0.4 | 4 | 40 |
| TAT-Nile red-NP | 30 | – | 10 | 0.4 | 4 | 40 |
| iRGD–TAT-Nile red-NP | 20 | 10 | 10 | 0.4 | 4 | 40 |
Notes: iRGD–PEG–PLGA, iRGD-modified PEG–PLGA; TAT–PEG–PLGA, TAT-modified PEG–PLGA; mPEG–PLGA-Nile red-NP, mPEG–PLGA NPs labeled with Nile red; iRGD-Nile red-NP, iRGD-modified NPs labeled with Nile red; TAT-Nile red-NP, TAT-modified NPs labeled with Nile red; iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red.
Abbreviations: iRGD, internalizing RGD; mPEG, methoxy-poly(ethylene glycol); NP, nanoparticle; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
Cellular uptake
Cellular uptake of NPs with different densities of iRGD–PEG–PLGA
NPs labeled with Nile red (Table 2) were prepared with 5, 20, and 40% iRGD–PEG–PLGA, as described in the “Preparation and characterization of NPs” section. Cellular uptake of NPs with variable proportions of iRGD–PEG–PLGA was determined by HUVECs with overexpression of integrin αvβ3.
Table 2.
Composition of NPs used to optimize the densities of iRGD–PEG–PLGA
| Groups | mPEG–PLGA (mg) | iRGD–PEG–PLGA (mg) | TAT–PEG–PLGA (mg) | Nile red (mg) | Acetone (mL) | Deionized water (mL) |
|---|---|---|---|---|---|---|
| 5% iRGD–TAT-Nile red-NP | 27.5 | 2.5 | 10 | 0.4 | 4 | 40 |
| 20% iRGD–TAT-Nile red-NP | 20 | 10 | 10 | 0.4 | 4 | 40 |
| 40% iRGD–TAT-Nile red-NP | 10 | 20 | 10 | 0.4 | 4 | 40 |
Notes: iRGD–PEG–PLGA, iRGD-modified PEG–PLGA; TAT–PEG–PLGA, TAT-modified PEG–PLGA; 5% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 5% iRGD–PEG–PLGA; 20% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 20% iRGD–PEG–PLGA; 40% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 40% iRGD–PEG–PLGA.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; mPEG, methoxy-poly(ethylene glycol); NP, nanoparticle; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
HUVECs were seeded in 24-well plates at a density of 30,000 cells/well in DMEM with 10% heat-inactivated fetal bovine serum (FBS). The cells were cultured in an incubator at 37°C and 5% CO2, and the DMEM was replaced every day. The cells were examined for growth daily using an inverted microscope. In the exponential growth phase, the cell medium was replaced with 500 μL of medium containing three groups (n=3 per group) of the following types of NPs: iRGD–TAT-Nile red-NP with 5% iRGD–PEG–PLGA, iRGD–TAT-Nile red-NP with 20% iRGD–PEG–PLGA, and iRGD–TAT-Nile red-NP with 40% iRGD–PEG–PLGA. The NPs were added to the HUVECs. Nile red was at a concentration of 200 ng/mL. After 1, 3, and 6 h of incubation at 37°C, the cells were washed three times with cold PBS and fixed with 4% paraformaldehyde in PBS for 15 min. The cells were examined under a fluorescence microscope (Vert.A1; Carl Zeiss AG, Oberkochen, Germany) for cellular uptake of NPs. The excitation wavelength for Nile red was 559 nm.
Cellular uptake of NPs at different densities of TAT–PEG–PLGA
NPs labeled with Nile red were prepared with 5, 20, and 40% TAT–PEG–PLGA according to the “Preparation and characterization of NPs” section (Table 3). The size, size distribution, and zeta potential of NPs were determined using a DLS particle size analyzer. Cellular uptake was detected using the following types of NPs: iRGD–TAT-Nile red-NP with 5% TAT–PEG–PLGA, iRGD–TAT-Nile red-NP with 20% TAT–PEG–PLGA, and iRGD–TAT-Nile red-NP with 40% TAT–PEG–PLGA as stated in the “Cellular uptake of NPs with different densities of iRGD–PEG–PLGA” section via fluorescence microscopy.
Table 3.
Composition of NPs used to optimize the densities of TAT–PEG–PLGA
| Groups | mPEG–PLGA (mg) | iRGD–PEG–PLGA (mg) | TAT–PEG–PLGA (mg) | Nile red (mg) | Acetone (mL) | Deionized water (mL) |
|---|---|---|---|---|---|---|
| iRGD–5% TAT-Nile red-NP | 27.5 | 10 | 2.5 | 0.4 | 4 | 40 |
| iRGD–20% TAT-Nile red-NP | 20 | 10 | 10 | 0.4 | 4 | 40 |
| iRGD–40% TAT-Nile red-NP | 10 | 10 | 20 | 0.4 | 4 | 40 |
Notes: iRGD–PEG–PLGA, iRGD-modified PEG–PLGA; TAT–PEG–PLGA, TAT-modified PEG–PLGA; iRGD–5% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 5% TAT–PEG–PLGA; iRGD–20% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 20% TAT–PEG–PLGA; iRGD– 40% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 40% TAT–PEG–PLGA.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; mPEG, methoxy-poly(ethylene glycol); NP, nanoparticle; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
Cellular interactions of NPs
The binding affinity of NPs to integrin αvβ3 was determined by the cellular uptake in HUVECs. HUVECs were seeded as described in the “Cellular uptake of NPs with different densities of iRGD–PEG–PLGA” section. Only cells in the exponential phase were eligible for the subsequent experiments. NPs labeled with Nile red (Table 1) were prepared for this study. The original medium was replaced with 500 μL of medium that contained four groups (n=3 per group) of the following types of NPs (“Preparation and characterization of NPs” section), which were added to the HUVECs: 1) mPEG–PLGA-Nile red-NP, 2) TAT-Nile red-NP, 3) iRGD-Nile red-NP, and 4) iRGD–TAT-Nile red-NP. Nile red was at 200 ng/mL. Following 1, 3, and 6 h of incubation at 37°C, the cells were washed three times with cold PBS and fixed with 4% paraformaldehyde in PBS for 15 min. The cells were then processed in a Hoechst 33342 stain for 15 min. The cells were examined under the fluorescence microscope for cellular uptake of NPs. The excitation wavelengths for Hoechst 33342 and Nile red were 405 and 559 nm, respectively.
In vitro corneal permeation
In vitro corneal permeability studies were performed using the immortalized HCE cell culture model.22 HCE cells were cultured in DMEM/Ham’s F12 containing 10% (v/v) FBS. Then 0.5 mL suspensions of HCE cells were seeded in polyester membrane Transwell®-Clear inserts (1.12 cm2 surface area, with a pore size of 0.4 μm) coated with rat tail collagen, type I, at a concentration of 42,000 cells/well. Culture medium on the basolateral side was replaced with 1.5 mL. The HCE cells were maintained at 37°C under 5% CO2, and the culture medium was changed every other day. After 2–3 weeks, trans-epithelial electrical resistance (TEER) was measured using a Millicell ERS meter (Millipore Corporation, Bedford, MA, USA). HCE cell layers with TEER values of ≥200 Ω cm2 were used for transport studies. Cell layers were washed with balanced salt solution (BSS). Then, four groups (n=3 per group) of the NP types, iRGD and TAT dual-modified NPs labeled with coumarin-6 (iRGD–TAT-coumarin-6-NP), iRGD-modified NPs labeled with coumarin-6 (iRGD-coumarin-6-NP), TAT-modified NPs labeled with coumarin-6 (TAT-coumarin-6-NP), and primary NPs (mPEG–PLGA-Nile red-NP) were diluted with BSS, and 0.5 mL of each NP solution was added to the apical compartments at a coumarin-6 concentration of 2 μg/mL. A total of 1.5 mL of BSS was then added to the basolateral compartment, and 200 μL of samples was collected at 30, 60, and 90 min from the basolateral compartment and immediately replaced with an equal volume of BSS solution to maintain a constant volume. The contents of the collected samples were analyzed by fluorescence detection in HPLC under constant chromatographic conditions. HPLC analysis was equipped with a fluorescence detector and performed on an Inertsil ODS-2 C18 column 4.6×250 mm, 5.0 μm. The mobile phase consisted of 90% methanol and 10% water, pumped at a flow rate of 1.0 mL/min. The excitation wavelength was 466 nm, and the emission wavelength was 504 nm. The apparent permeability of the NPs was calculated using the equation Papp = (ΔQ/Δt)/ACd; where Papp is the apparent permeability (cm/s), ΔQ/Δt is the appearance rate in the basolateral compartment (μg/s), A is the surface area (1.12 cm2) of HCE layers, and Cd expresses the donor substance concentration (μg/mL).
Induction and evaluation of CNV in rats
Laser-induced animal models of CNV were applied to investigate the various treatments. Male BN rats, weighing 180–200 g, were used. All rats were examined by eye before experiments, and no abnormalities were detected in the anterior segment or eyebase. Pupils were dilated using tropicamide eyedrops 15 min pre-experiment. Animals were anesthetized with an intraperitoneal injection of 10% chloral hydrate solution (3.0 mL/kg). CNV was induced in the rats’ eyes with a 532 nm laser (Vision One, Lumenis, CA, USA) with a slit lamp delivery system (180 mW, 100 mm, 100 ms). Six to eight laser spots were created with ~2–3 disk diameters from the optic nerve head. The sign of Bruch’s membrane rupture by the laser was the formation of bubbling at the site of the laser application, with or without hemorrhage. CNV was induced only in the right eye of the rats, whereas the left eye served as a control for each animal.
We used fundus fluorescein angiography (FFA, TRC-50IX; Topcon, Tokyo, Japan) and optical coherence tomography (OCT; Spectralis OCT; Heidelberg Engineering, Heidelberg, Germany) to monitor CNV development and changes in vivo before laser photocoagulation and 7, 14, and 21 days after laser photocoagulation.
In vivo targeting and penetration evaluation
The BN rats were divided into five groups (n=3 per group) to receive one of the following types of Nile red solution and different NPs: 1) Nile red solution, 2) mPEG–PLGA-Nile red-NP, 3) iRGD-Nile red-NP, 4) TAT-Nile red-NP, and 5) iRGD–TAT-Nile red-NP. On day 21 after laser induction, a single dose of Nile red solution and Nile red-labeled NPs were dropped onto the surface of the right eye at the same concentration. The BN rats were then anesthetized after 30 min and perfused with 50 mL of PBS and 20 mL (5 mg/mL) of fluorescein isothiocyanate (FITC)–dextran (MW =2×106 Da) to prepare choroidal flat mounts, as described previously.23 Briefly, the eyes were sectioned at the equator and the anterior segment of the eye and the vitreous layers were removed. The retinas were isolated, and the posterior segment of the eye, including the sclera and the choroid, was dissected into quarters by four radial cuts and mounted on a slide. The fluorescent pictures of the flat mounts were captured with a confocal laser scanning microscope (CLSM; Olympus Corporation, Tokyo, Japan). All sections were scanned in multitrack mode to avoid overlap of the red (excitation 559 nm) and green (excitation 488 nm) channels.
Data analysis
The data are shown as the mean ± standard deviation (SD). Statistical comparisons were evaluated using student’s t-tests and one-way analysis of variance. A P-value of ≤0.05 was considered statistically significant.
Results and discussion
Characterization of the conjugated polymers
iRGD or TAT peptides were conjugated to PEG through reaction with the maleimide on PEG and the sulfhydryl group on the iRGD or TAT peptides in phosphate buffer. An HPLC method was used to monitor the reaction efficiency of iRGD or TAT to MAL–PEG–PLGA. Figure 1 shows that ~80% of iRGD (Figure 1A and B) and TAT (Figure 1C and D) were conjugated to PEG–PLGA. Figure 2A–E shows the 1H NMR spectra of TAT, iRGD, Mal–PEG–PLGA, iRGD–PEG–PLGA, and TAT–PEG–PLGA, respectively. The single peak at δ (ppm) ≈6.7 indicates the double bond of maleimide in Figure 2C, but almost no peak at δ (ppm) ≈6.7 indicates iRGD–PEG–PLGA (Figure 2D) and TAT–PEG–PLGA (Figure 2E). This indicates virtually complete reaction of PEG–PLGA with iRGD and TAT. The characteristic peaks of maleimide disappeared. These results demonstrate the successful synthesis of iRGD–PEG–PLGA and TAT–PEG–PLGA.
Figure 1.

HPLC results for iRGD or TAT coupling to Mal–PEG–PLGA.
Notes: (A) Before the coupling step, free iRGD in the mixture of Mal–PEG–PLGA and iRGD showed a peak with a retention time at ~10.8 min. (B) After the coupling step, free iRGD in the mixture of Mal–PEG–PLGA and iRGD was incorporated onto the Mal–PEG–PLGA and the peak for iRGD was not obvious. (C) Before the coupling step, free TAT in the mixture of Mal–PEG–PLGA and TAT showed a peak with a retention time at ~20.08 min. (D) After the coupling step, free TAT in the mixture of Mal–PEG–PLGA and TAT was incorporated onto the Mal–PEG–PLGA and the peak for TAT was not obvious.
Abbreviations: HPLC, high-performance liquid chromatography; iRGD, internalizing arginine-glycine-aspartic acid; Mal–PEG–PLGA, maleimide-poly(ethylene glycol)-poly(lactic-co-glycolic acid); TAT, transactivated transcription.
Figure 2.

1H NMR spectra of iRGD (A), TAT (B), Mal–PEG–PLGA (C), iRGD–PEG–PLGA (D), and TAT–PEG–PLGA (E). iRGD–PEG–PLGA, iRGD-modified PEG–PLGA; TAT–PEG–PLGA, TAT-modified PEG–PLGA.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; Mal–PEG–PLGA, maleimide-poly(ethylene glycol)-poly(lactic-co-glycolic acid); NMR, nuclear magnetic resonance; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
Characterization of the NPs
The morphology and size distribution of iRGD and TAT dual-modified NPs were determined using TEM and DLS, respectively. The results are presented in Figure 3. iRGD and TAT dual-modified NPs appeared to be spherical (Figure 3B) and were homogeneously distributed (Figure 3A), with an average diameter of ~67 nm. The mean diameter, polydispersity index, and zeta potential of the various NPs are given in Table 4. Four NPs (mPEG–PLGA-Nile red-NP, iRGD-Nile red-NP, TAT-Nile red-NP, and iRGD–TAT-Nile red-NP) had an average particle size of 53.4±2.4, 60.1±2.8, 62.7±1.5, and 67.0±1.7 nm, respectively. A uniform size distribution with a polydispersity index of ≤0.2 suggests that the size distribution of the NPs was narrow. mPEG–PLGA NPs had a strongly negative zeta potential (−18.84±0.57 mV), but dual-modified NPs had a less negative zeta potential (−6.63±0.43 mV). This indicates that continuous modification with positively charged iRGD and TAT resulted in a slight increase in zeta potential.
Figure 3.

DLS image (A) and TEM (B) characterization of iRGD and TAT dual-modified nanoparticles.
Abbreviations: DLS, dynamic light scattering; iRGD, internalizing arginine-glycine-aspartic acid; TAT, transactivated transcription; TEM, transmission electron microscope.
Table 4.
Characterization of NPs
| Groups | Particle size (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| mPEG–PLGA-Nile red-NP | 53.4±2.4 | 0.128±0.011 | −18.84±0.57 |
| iRGD-Nile red-NP | 60.1±2.8 | 0.106±0.052 | −12.63±0.75 |
| TAT-Nile red-NP | 62.7±1.5 | 0.157±0.039 | −10.86±0.39 |
| iRGD–TAT-Nile red-NP | 67.0±1.7 | 0.163±0.017 | −6.63±0.43 |
Notes: Values represent the mean ± SD (n=3). mPEG–PLGA-Nile red-NP, mPEG–PLGA NPs labeled with Nile red; iRGD-Nile red-NP, iRGD-modified NPs labeled with Nile red; TAT-Nile red-NP, TAT-modified NPs labeled with Nile red; iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; mPEG, methoxy-poly(ethylene glycol); NP, nanoparticle; PDI, polydispersity index; PLGA, poly(lactic-co-glycolic acid); SD, standard deviation; TAT, transactivated transcription.
In ophthalmic delivery systems, nanosized particles represent an important surface area available for association between the cornea and the conjunctiva. The above results indicate that iRGD and TAT dual-modified NPs had a size that was suitable for cellular uptake and administration.
Cellular uptake of NPs in HUVECs
Cellular uptake of NPs with different densities of iRGD–PEG–PLGA
HUVECs are a common source of endothelial cell cultures. Because they overexpress integrin αvβ3, HUVECs are commonly used in simulations of pathological neovascularization, such as tumor angiogenesis.24
The amount of iRGD–PEG–PLGA was screened to identify the highest effect on the cellular uptake in HUVECs. The size, polydispersity index, and zeta potential of NPs show an increasing trend with increasing amounts of iRGD–PEG–PLGA, in Table 5. When the NPs contained 5% iRGD–PEG–PLGA, the particle size was 62.4±1.6 nm and the zeta potential was −13.57±0.46 mV. When the NPs were prepared with 40% iRGD–PEG–PLGA, the particle size increased significantly to 192.9±5.7 nm and the zeta potential increased to −3.63±0.43 mV. The increase in particle size might be due to the charge that the peptide carried. The zeta potential approaches neutrality. The change in zeta potential results in the particles’ aggregation because of the smaller repulsion of the near-neutral zeta potential. Fluorescence microscopy images (Figure 4) showed a gradual increase in red fluorescence with time. When the iRGD–PEG–PLGA concentration was increased from 5% to 20%, the cell fluorescence intensity slightly increased, suggesting that increasing the iRGD dosage can promote cellular uptake of NPs, depending on the binding affinity of iRGD to integrin αvβ3. In addition, a low red fluorescence intensity was detected in 40% iRGD–TAT-Nile red-NP. This was probably caused by a larger particle size limiting cellular uptake, whereas competition restrictions occur via saturation when iRGD specifically binds with integrin αvβ3.25 Therefore, the optimal concentration of iRGD–PEG–PLGA was determined to be 20%.
Table 5.
Characterization of NPs with different densities of iRGD–PEG–PLGA
| Groups | Particle size (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| 5% iRGD–TAT-Nile red-NP | 62.4±1.6 | 0.136±0.032 | −13.57±0.46 |
| 20% iRGD–TAT-Nile red-NP | 67.0±1.7 | 0.163±0.017 | −6.63±0.43 |
| 40% iRGD–TAT-Nile red-NP | 192.9±5.7 | 0.389±0.073 | −3.63±0.43 |
Notes: Values represent the mean ± SD (n=3). 5% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 5% iRGD–PEG–PLGA; 20% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 20% iRGD–PEG–PLGA; 40% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 40% iRGD–PEG–PLGA.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; NP, nanoparticle; PEG, poly(ethylene glycol); PDI, polydispersity index; PLGA, poly(lactic-co-glycolic acid); SD, standard deviation; TAT, transactivated transcription.
Figure 4.

Fluorescence microscopy images of the cellular uptake of NPs with 5, 20, and 40% iRGD–PEG–PLGA in HUVECs.
Notes: 5% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 5% iRGD–PEG–PLGA; 20% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 20% iRGD–PEG–PLGA; 40% iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 40% iRGD–PEG–PLGA.
Abbreviations: HUVECs, human umbilical vein endothelial cells; iRGD, internalizing arginine-glycine-aspartic acid; NP, nanoparticle; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
Cellular uptake of NPs with different densities of TAT–PEG–PLGA
It has been reported that TAT peptide may enhance cellular uptake because of its strong adherence to cells, with no dependence on receptors or temperature, or an energy-dependent pathway.26 To achieve the strongest effect on cellular uptake, the density of TAT–PEG–PLGA was screened. When TAT–PEG–PLGA increased from 5% to 40%, the particle size increased from 64.7±1.2 to 232.0±4.1 nm and the zeta potential ranged from −14.28±0.61 to −3.19±0.36 mV because of the positive charge of TAT (Table 6). Fluorescence microscopy images of the cellular uptake in HUVECs (Figure 5) show the highest red fluorescence intensity with 20% TAT–PEG–PLGA, while a lower red fluorescence intensity is evident at 40% TAT–PEG–PLGA because of the larger particle size. Therefore, the optimal density of TAT–PEG–PLGA was determined to be 20%.
Table 6.
Characterization of NPs with different densities of TAT–PEG–PLGA
| Groups | Particle size (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| iRGD–5% TAT-Nile red-NP | 64.7±1.2 | 0.103±0.025 | −14.28±0.61 |
| iRGD–20% TAT-Nile red-NP | 67.0±1.7 | 0.163±0.017 | −6.63±0.43 |
| iRGD–40% TAT-Nile red-NP | 232.0±4.1 | 0.453±0.087 | −3.19±0.36 |
Notes: Values represent the mean ± SD (n=3). iRGD–5% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 5% TAT–PEG–PLGA; iRGD–20% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 20% TAT–PEG–PLGA; iRGD–40% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 40% TAT–PEG–PLGA.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; NP, nanoparticle; PEG, poly(ethylene glycol); PDI, polydispersity index; PLGA, poly(lactic-co-glycolic acid); SD, standard deviation; TAT, transactivated transcription.
Figure 5.

Fluorescence microscopy images of the cellular uptake of NPs with 5, 20, and 40% TAT–PEG–PLGA in HUVECs.
Notes: iRGD–5% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 5% TAT–PEG–PLGA; iRGD–20% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 20% TAT–PEG–PLGA; iRGD–40% TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red with 40% TAT–PEG–PLGA.
Abbreviations: HUVECs, human umbilical vein endothelial cells; iRGD, internalizing arginine-glycine-aspartic acid; NP, nanoparticle; PEG, poly(ethylene glycol); PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
Cellular interactions of NPs
To evaluate the targeting effect of the iRGD and TAT dual-modified NPs, we used HUVEC uptake and the intracellular distribution of NPs to study the interactions between integrin αvβ3 and iRGD.
As shown in Figure 6, the red fluorescence intensity increased with time, showing that the NPs exhibited a clear time-dependent relationship for cellular uptake. NPs loaded with Nile red were distributed around the cell nucleus and gradually moved into the cell nucleus with incubation time. NPs that were not modified by either iRGD or TAT could not easily enter the cells, and the fluorescence intensity was weak at 1 and 3 h. When incubation time was prolonged, the red fluorescence intensity of NPs surface-functionalized with TAT, iRGD peptide, or both, was obviously stronger than that of the nonfunctionalized NP group (mPEG–PLGA-Nile red-NP). The red fluorescence intensity of the NPs at 6 h followed the order of iRGD–TAT-Nile red NP > TAT-Nile red-NP and iRGD-Nile red-NP > mPEG–PLGA-Nile red-NP. The red fluorescence intensity of iRGD-Nile red-NP was stronger than that of the nonfunctionalized NP group, showing that NPs modified with iRGD had increased performance in target and cell penetration. NPs modified by TAT also showed stronger fluorescence intensity than the nonfunctionalized NP group and had increased cell penetration performance. The dual-modified NP group had the strongest red fluorescence intensity of all three groups, reaching the strongest level at 6 h, suggesting that TAT and iRGD may offer synergistic effects on cellular uptake of NPs. The results primarily showed that NPs modified with iRGD and TAT peptides can improve penetration and targeting functions for HUVECs overexpressing integrin αvβ3.
Figure 6.

Cellular uptake images for different NPs after treating HUVECs at 1, 3, and 6 h.
Notes: The blue fluorescence and the red fluorescence belong to Hoechst 33342 and NPs labeled with Nile red, respectively. mPEG–PLGA-Nile red-NP, mPEG–PLGA NPs labeled with Nile red; iRGD-Nile red-NP, iRGD-modified NPs labeled with Nile red; TAT-Nile red-NP, TAT-modified NPs labeled with Nile red; iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red.
Abbreviations: HUVECs, human umbilical vein endothelial cells; iRGD, internalizing arginine-glycine-aspartic acid; mPEG, methoxy-poly(ethylene glycol); NP, nanoparticle; PLGA, poly(lactic-co-glycolic acid); TAT, transactivated transcription.
In vitro corneal permeation
The HCE cell line was established by Araki-Sasaki et al27 by infecting primary HCE cells with a recombinant SV40-adenovirus vector and cloning three times to obtain a continuously growing cell line. The immortalized HCE cell culture model of ocular barriers can provide a powerful system to investigate drug behavior, bioadhesion characteristics, and pharmacokinetic properties of new formulations.28 Accordingly, data obtained using HCE cell lines could be a valid model for studies predicting drug behavior in the human eye.29
The permeabilities of NPs through HCE cells were investigated (Figure 7). The permeability coefficients of TAT-modified NPs labeled with coumarin-6 (TAT-coumarin-6-NP) were 4.83-fold greater than those of primary NPs labeled with coumarin-6 (mPEG–PLGA-coumarin-6-NP). The permeability coefficients of iRGD and TAT dual-modified NPs labeled with coumarin-6 (iRGD–TAT-coumarin-6-NP) were 5.50- and 4.56-fold greater than those of primary NPs and iRGD-modified NPs labeled with coumarin-6 (iRGD-coumarin-6-NP) and exhibited a markedly stronger penetration ability. The permeability of NPs was significantly increased in the presence of TAT peptides compared to that of the control NPs. However, there was no obvious difference between the permeability coefficients of iRGD-coumarin-6-NP and mPEG–PLGA-coumarin-6-NP. Although iRGD peptides were reported to enhance vascularity and tissue permeability in a tumor-specific and neuropilin-1 (NRP-1)-dependent manner,30 permeability of iRGD cannot play a role in the ocular anterior segment, because of the low level of integrin αvβ3 expression in normal tissue. Possible reasons for this are that the permeability of iRGD peptides needs to be triggered by first binding to integrin αvβ3 and then being proteolytically cleaved to produce CRGDK/R, which has an affinity for NRP-1 through the C terminal end R motif.20 Activation of the C-end Rule (Cend R) requires prior binding of the peptide to integrin αvβ3.
Figure 7.
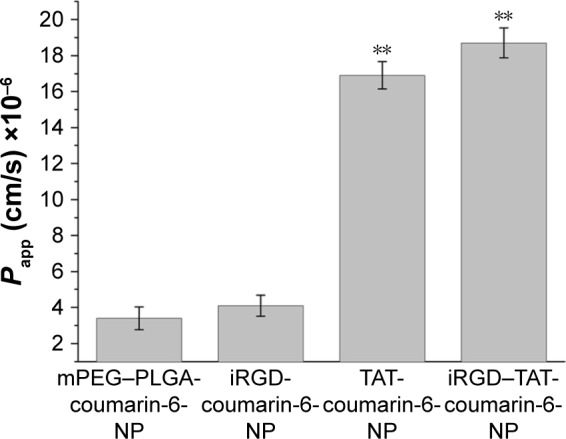
Permeability coefficients of mPEG–PLGA-coumarin-6-NP, iRGD-coumarin-6-NP, TAT-coumarin-6-NP, and iRGD–TAT-coumarin-6-NP.
Notes: Each data point represents the mean ± SD (n=3) of four determinations. **Significant differences from the control with P<0.01. mPEG–PLGA-coumarin-6-NP, mPEG–PLGA NPs labeled with coumarin-6; iRGD-coumarin-6-NP, iRGD-modified NPs labeled with coumarin-6; TAT-coumarin-6-NP, TAT-modified NPs labeled with coumarin-6; iRGD–TAT-coumarin-6-NP, iRGD and TAT dual-modified NPs labeled with coumarin-6.
Abbreviations: iRGD, internalizing arginine-glycine-aspartic acid; mPEG, methoxy-poly(ethylene glycol); NPs, nanoparticles; Papp, apparent permeability; PLGA, poly(lactic- co-glycolic acid); SD, standard deviation; TAT, transactivated transcription.
This result suggests that iRGD and TAT dual-modified NPs can penetrate the corneal barrier and significantly improve permeability. Moreover, the penetration function of iRGD and TAT dual-modified NPs relies mostly on the CPP TAT.
The cell culture model provides a custom culture system in which parameters and conditions can be changed. Compared with isolated animal tissues, the experimental results obtained from the cell model are reproducible, while the adoption of human cell lines avoids species-dependent differences caused by using animal tissues, making it more reliable.
Evaluation of CNV in rats
The laser-induced CNV model is widely applied in basic experimental and treatment research, and it offers the advantage that it is similar to the natural process of CNV disease in human beings. Currently, the use of laser-induced BN rats is a common method for producing a choroid neovascularization model.31,32
FFA is a common method for monitoring CNV formation. The CNV area possesses a high vascular permeability because of its immature neovascularization. The degree of sodium fluorescein leakage can be roughly assessed by the size of the CNV (Figure 8) after intraperitoneal injection of sodium fluorescein. There was no sodium fluorescein leakage (Figure 8A) before laser photocoagulation, but there was irregular fluorescein leakage in the photocoagulation area 7 days (Figure 8B) after laser photocoagulation. Sodium fluorescein leakage was thus seen to increase with time. Compared with day 7, fluorescein leakage was higher and it occurred over a larger area at day 14 (Figure 8C), and the CNV areas showed moderate-to-severe fluorescein leakage. The fluorescein leakage area reached a peak 21 days (Figure 8D) after laser photocoagulation.
Figure 8.

Fundus fluorescein angiography images of the CNV.
Notes: (A) Normal structure of rat eye by intraperitoneal injection of sodium fluorescein before photocoagulation; (B) 7 days after photocoagulation; (C) 14 days after photocoagulation; and (D) 21 days after photocoagulation.
Abbreviation: CNV, choroidal neovascularization.
OCT can display lesions in a cross-sectional location. Intraretinal layers were recognizable in high-resolution OCT images, as shown in Figure 9. At 7 days after photocoagulation, OCT images showed disruption of a highly reflective layer corresponding to the retinal pigment epithelium and choriocapillaries in the lesion. OCT images obtained between days 14 and 21 showed that the highly reflective layer extended to the subretinal space, and its thickness had increased.
Figure 9.

Optical coherence tomography images of the CNV.
Notes: (A) Normal structure of the retina and the choriocapillaries of rats before photocoagulation; (B) 7 days after photocoagulation; (C) 14 days after photocoagulation; and (D) 21 days after photocoagulation.
Abbreviation: CNV, choroidal neovascularization.
OCT and FFA can allow effective, dynamic observations of the formation and changes of CNV and have good synchronicity and relevance: the greater the fluorescein leakage area, the thicker the retina, as shown by OCT. Simultaneous observations by OCT and FFA confirmed that CNV was fully formed 14 days after photocoagulation and reached a peak 21 days after photocoagulation.
In vivo targeting and penetration evaluation
Blood vessels were labeled by vascular perfusion with high molecular weight FITC–dextran using a method similar to that described previously.23 High molecular weight FITC (2×106 Da) can be retained within blood vessels and does not undergo capillary wall leakage. Choroid-sclera flat mounts were examined by confocal microscopy. FITC showed green fluorescence and marked the CNV area. Nile red labeling of the NPs was used to localize the particles in choroidal flat mounts (Figure 10).
Figure 10.


Choroid-sclera flat mounts of laser-treated eyes after eyedrop NP administration.
Notes: (A) Confocal microscope images of posterior segment flat mounts of laser-treated eyes after eyedrop NP administration. The flat mounts were subsequently scanned using a ×10 objective and two channels in multitrack mode: red (NPs containing Nile red as a tracking dye) and green (FITC–dextran accumulation in the CNV). The white arrows indicate the NPs labeled with Nile red. (B) Red fluorescence intensity was measured by FV10-ASW 2.1 Viewer software. *Statistical significance (P<0.05). **Statistical significance (P<0.01). Values represent the mean ± SD (n=3). mPEG–PLGA-Nile red-NP, mPEG–PLGA NPs labeled with Nile red; iRGD-Nile red-NP, iRGD-modified NPs labeled with Nile red; TAT-Nile red-NP, TAT-modified NPs labeled with Nile red; iRGD–TAT-Nile red-NP, iRGD and TAT dual-modified NPs labeled with Nile red.
Abbreviations: CNV, choroidal neovascularization; FITC, fluorescein isothiocyanate; iRGD, internalizing arginine-glycine-aspartic acid; mPEG, methoxy-poly(ethylene glycol); NP, nanoparticle; PLGA, poly(lactic-co-glycolic acid); SD, standard deviation; TAT, transactivated transcription.
As shown in Figure 10A, there was an overlap of FITC-labeled CNV and the Nile red-labeled NPs and solution. Choroidal flat mounts performed after in vivo imaging revealed accumulation of Nile red-labeled NPs in the CNV lesion. Nile red fluorescence intensity occurred in the order of iRGD–TAT-Nile red-NP > TAT-Nile red-NP > iRGD-Nile red-NP > mPEG–PLGA-Nile red-NP > Nile red solution. Nile red was dissolved in dimethyl sulfoxide (DMSO) solution at the same concentration as the NPs because of the extremely low solubility of Nile red in distilled water. No fluorescence was observed in DMSO solutions containing Nile red, in contrast to the NP formulations (Figure 10A). Therefore, Nile red molecules dissolved in DMSO solution cannot deliver to the ocular posterior segment in a molecular state.
The nonmodified NPs group (mPEG–PLGA-Nile red-NP) showed negligible emission of Nile red after eyedrop administration in the CNV area. This suggests that it is difficult to deliver primary mPEG–PLGA NPs to the retina. The blood–retinal barrier, a part of the blood–ocular barrier, protects the eyes from interfering foreign bodies in the blood, and it also prevents drug penetration. However, some findings suggest that the blood–retinal barrier is disrupted in many retinal diseases. The enhanced permeability and retention (EPR) effect occurs in CNV tissues with high selective permeability and retention of macromolecular substances and liposomes, and the CNV can be a target of passive drug targeting.33,34
Obvious fluorescence was exhibited in the iRGD-Nile red-NP and TAT-Nile red-NP groups. There was red fluorescence within the CNV area with the iRGD-Nile red-NP group, suggesting that NPs modified with iRGD could improve the active targeting effect toward CNV. Although it was difficult for iRGD-Nile red-NP to penetrate the corneal barrier, we could not exclude other pathways to the ocular posterior segment. However, stronger red fluorescence was exhibited around the CNV area in the TAT-Nile red-NP group. NPs modified with the TAT peptide could penetrate the ocular barrier and deliver to the retina and choroid, but they were not focused in the CNV regions because of the absence of selectivity by TAT. Note that the strongest Nile red accumulation was observed in the pattern of the iRGD and TAT dual-modified NPs at the periphery of and particularly within the CNV. This might suggest that first, iRGD–TAT-Nile red-NP can be delivered to the posterior segment, relying on the penetration of TAT peptides and iRGD-specific binding with integrin and penetration to target the NPs to the CNV. iRGD-modified nanocarriers can improve the targeting effect, but this is constrained by receptor saturation. TAT-modified nanocarriers can increase the ability to penetrate the cell but lack selectivity. In summary, the experimental results show that iRGD and TAT dual-modified NPs retain the respective functions of iRGD and TAT and, thereby, gain the synergetic functions of both, resulting in improved permeability and targeting efficiency.
Topical instillation of ophthalmic drops is the preferred route of drug administration, primarily because of better patient compliance and cost effectiveness. Productive absorption from topical delivery is assumed to occur primarily through two routes, the corneal and noncorneal (conjunctival/scleral) pathways.35 Topically dosed drugs can transport to the posterior segment and then diffuse through the cornea, entering the vitreous layer or penetrating the aqueous humor, continuing through the uvea–sclera.36 Noncorneal pathways mean that drug delivery mainly infiltrates into the eye tissue through the conjunctiva and sclera. This mechanism of absorption was once thought to be nonproductive, although a few studies claim significant noncorneal absorption for compounds with poor corneal permeability, such as timolol maleate and gentamicin.37,38 However, Amrite and Kompella39 concluded that particulate systems at ≥200 nm are likely to be retained at the periocular site of administration for prolonged periods and can be used as sustained retinal drug delivery devices, using the transscleral mode of delivery. Conjunctival epithelial cells are more prone to leakage than the cornea, and the surface area of the conjunctiva is much larger than that of the cornea. Therefore, we cannot rule out the possibility that the noncorneal pathway for the absorbance of iRGD and TAT dual-modified NPs provided the stronger permeability and targeting effect. Further investigations are planned for future study.
Flat mount images further confirmed that topical ocular drug delivery of iRGD and TAT dual-modified NPs offered high corneal permeability, targeted to the CNV, showing the potential for broad applications in noninvasive CNV treatment. These absorption mechanisms will be investigated further in the future.
The hydrophobic fluorescent dyes, such as Nile red and coumarin-6, have been investigated for cellular uptake and elimination process of different nanocarriers and cellular drug delivery mechanism of PLGA NPs.40,41 We used Nile red and coumarin-6 to label NPs for better visualization in CNV targeting evaluation in vitro and in vivo. PEG–PLGA NPs are being extensively studied to load hydrophobic drugs, such as sorafenib and curcumin.42,43 The hydrophobic fluorescent dyes we selected as model drug were representative for topical ocular drug delivery in safe and effective antineovas-culatural therapy. We will refer to hydrophobic drug loading and drug release in the ocular delivery system, which can inhibit laser-induced CNV, in the future experiments.
Conclusion
In this study, we synthesized nanocarriers modified with iRGD and TAT and developed iRGD and TAT dual-modified NPs for topical ocular drug delivery. Cellular interactions of dual-modified NPs showed significant affinity toward integrin αvβ3, and the dual-modified NPs showed an apparent ability to penetrate the corneal barrier. In the laser-induced CNV model, choroid flat mounts further illustrated that iRGD and TAT confer complementary advantages and that the dual-modified ocular drug delivery method offers high corneal permeability and targeting toward CNV. In conclusion, the topical ocular drug delivery system modified with iRGD and TAT is a potentially efficient targeted drug delivery system that offers a new, safe, and effective treatment for CNV.
Acknowledgments
This study was supported by the Natural Science Foundation of Shandong (number ZR2014HM062) and the 12th Five Years Key Programs for Science and Technology Development of China (2013ZX09402201).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Jian L, Panpan Y, Wen X. Current choroidal neovascularization treatment. Ophthalmologica. 2013;230(2):55–61. doi: 10.1159/000351660. [DOI] [PubMed] [Google Scholar]
- 2.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration – emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38(7):450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kompella UB, Amrite AC, Pacha Ravi R, Durazo SA. Nanomedicines for back of the eye drug delivery, gene delivery, and imaging. Prog Retin Eye Res. 2013;36:172–198. doi: 10.1016/j.preteyeres.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maurice D. Review: practical issues in intravitreal drug delivery. J Ocul Pharmacol Ther. 2001;17(4):393–401. doi: 10.1089/108076801753162807. [DOI] [PubMed] [Google Scholar]
- 5.Lim JI, Wolitz RA, Dowling AH, Bloom HR, Irvine AR, Schwartz DM. Visual and anatomic outcomes associated with posterior segment complications after ganciclovir implant procedures in patients with AIDS and cytomegalovirus retinitis. Am J Ophthalmol. 1999;127(3):288–293. doi: 10.1016/s0002-9394(98)00443-7. [DOI] [PubMed] [Google Scholar]
- 6.Shane TS, Martin DF, Endopthalmitis-Gancioclovir Implant Study Group Endophthalmitis after ganciclovir implant in patients with AIDS and cytomegalovirus retinitis. Am J Ophthalmol. 2003;136(4):649–654. doi: 10.1016/s0002-9394(03)00333-7. [DOI] [PubMed] [Google Scholar]
- 7.Janoria KG, Gunda S, Boddu SH, Mitra AK. Novel approaches to retinal drug delivery. Expert Opin Drug Deliv. 2007;4(4):371–388. doi: 10.1517/17425247.4.4.371. [DOI] [PubMed] [Google Scholar]
- 8.Sahoo SK, Dilnawaz F, Krishnakumar S. Nanotechnology in ocular drug delivery. Drug Discov Today. 2008;13(3–4):144–151. doi: 10.1016/j.drudis.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 9.Inokuchi Y, Hironaka K, Fujisawa T, et al. Physicochemical properties affecting retinal drug/coumarin-6 delivery from nanocarrier systems via eyedrop administration. Invest Ophthalmol Vis Sci. 2010;51(6):3162–3170. doi: 10.1167/iovs.09-4697. [DOI] [PubMed] [Google Scholar]
- 10.Trabulo S, Cardoso AL, Mano M, De Lima MC. Cell-penetrating peptides-mechanisms of cellular uptake and generation of delivery systems. Pharmaceuticals (Basel) 2010;3(4):961–993. doi: 10.3390/ph3040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farkhani SM, Valizadeh A, Karami H, Mohammadi S, Sohrabi N, Badrzadeh F. Cell penetrating peptides: efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides. 2014;57:78–94. doi: 10.1016/j.peptides.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Johnson LN, Cashman SM, Kumar-Singh R. Cell-penetrating peptide for enhanced delivery of nucleic acids and drugs to ocular tissues including retina and cornea. Mol Ther. 2008;16(1):107–114. doi: 10.1038/sj.mt.6300324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Tai L, Zhang W, Wei G, Pan W, Lu W. Penetratin, a potentially powerful absorption enhancer for noninvasive intraocular drug delivery. Mol Pharm. 2014;11(4):1218–1227. doi: 10.1021/mp400681n. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Lin H, Lin S, et al. Cell-penetrating peptide TAT-mediated delivery of acidic FGF to retina and protection against ischemia-reperfusion injury in rats. J Cell Mol Med. 2010;14(7):1998–2005. doi: 10.1111/j.1582-4934.2009.00786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leavesley DI, Schwartz MA, Rosenfeld M, Cheresh DA. Integrin beta 1- and beta 3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J Cell Biol. 1993;121(1):163–170. doi: 10.1083/jcb.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedlander M, Theesfeld CL, Sugita M, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci U S A. 1996;93(18):9764–9769. doi: 10.1073/pnas.93.18.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luna J, Tobe T, Mousa SA, Reilly TM, Campochiaro PA. Antagonists of integrin alpha v beta 3 inhibit retinal neovascularization in a murine model. Lab Invest. 1996;75(4):563–573. [PubMed] [Google Scholar]
- 18.Schraa AJ, Kok RJ, Moorlag HE, et al. Targeting of RGD-modified proteins to tumor vasculature: a pharmacokinetic and cellular distribution study. Int J Cancer. 2002;102(5):469–475. doi: 10.1002/ijc.10727. [DOI] [PubMed] [Google Scholar]
- 19.Singh SR, Grossniklaus HE, Kang SJ, Edelhauser HF, Ambati BK, Kompella UB. Intravenous transferrin, RGD peptide and dual-targeted nanoparticles enhance anti-VEGF intraceptor gene delivery to laser-induced CNV. Gene Ther. 2009;16(5):645–659. doi: 10.1038/gt.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. doi: 10.2147/IJN.S46962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1–2):1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 22.Toropainen E, Ranta VP, Talvitie A, Suhonen P, Urtti A. Culture model of human corneal epithelium for prediction of ocular drug absorption. Invest Ophthalmol Vis Sci. 2001;42(12):2942–2948. [PubMed] [Google Scholar]
- 23.Edelman JL, Castro MR. Quantitative image analysis of laser-induced choroidal neovascularization in rat. Exp Eye Res. 2000;71(5):523–533. doi: 10.1006/exer.2000.0907. [DOI] [PubMed] [Google Scholar]
- 24.Cressman S, Dobson I, Lee JB, Tam YY, Cullis PR. Synthesis of a labeled RGD-lipid, its incorporation into liposomal nanoparticles, and their trafficking in cultured endothelial cells. Bioconjug Chem. 2009;20(7):1404–1411. doi: 10.1021/bc900041f. [DOI] [PubMed] [Google Scholar]
- 25.Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H. Dual-ligand modification of PEGylated liposomes shows better cell selectivity and efficient gene delivery. J Control Release. 2011;153(2):141–148. doi: 10.1016/j.jconrel.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98(15):8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Araki-Sasaki K, Ohashi Y, Sasabe T, et al. An SV40-immortalized human corneal epithelial cell line and its characterization. Invest Ophthalmol Vis Sci. 1995;36(3):614–621. [PubMed] [Google Scholar]
- 28.Giannola LI, de Caro V, Giandalia G, Siragusa MG, Cordone L. Ocular gelling microspheres: in vitro precorneal retention time and drug permeation through reconstituted corneal epithelium. J Ocul Pharmacol Ther. 2008;24(2):186–196. doi: 10.1089/jop.2007.0113. [DOI] [PubMed] [Google Scholar]
- 29.Reichl S, Dohring S, Bednarz J, Muller-Goymann CC. Human cornea construct HCC-an alternative for in vitro permeation studies? A comparison with human donor corneas. Eur J Pharm Biopharm. 2005;60(2):305–308. doi: 10.1016/j.ejpb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamizuru H, Kimura H, Yasukawa T, Tabata Y, Honda Y, Ogura Y. Monoclonal antibody-mediated drug targeting to choroidal neovascularization in the rat. Invest Ophthalmol Vis Sci. 2001;42(11):2664–2672. [PubMed] [Google Scholar]
- 32.Lee EK, Kim YJ, Kim JY, Song HB, Yu HG. Melissa officinalis extract inhibits laser-induced choroidal neovascularization in a rat model. PLoS One. 2014;9(10):e110109. doi: 10.1371/journal.pone.0110109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura H, Yasukawa T, Tabata Y, Ogura Y. Drug targeting to choroidal neovascularization. Adv Drug Deliv Rev. 2001;52(1):79–91. doi: 10.1016/s0169-409x(01)00190-9. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt-Erfurth U, Hasan T, Gragoudas E, Michaud N, Flotte TJ, Birngruber R. Vascular targeting in photodynamic occlusion of subretinal vessels. Ophthalmology. 1994;101(12):1953–1961. doi: 10.1016/s0161-6420(13)31079-3. [DOI] [PubMed] [Google Scholar]
- 35.Hughes PM, Olejnik O, Chang-Lin JE, Wilson CG. Topical and systemic drug delivery to the posterior segments. Adv Drug Deliv Rev. 2005;57(14):2010–2032. doi: 10.1016/j.addr.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Schopf LR, Popov AM, Enlow EM, et al. Topical ocular drug delivery to the back of the eye by mucus-penetrating particles. Transl Vis Sci Technol. 2015;4(3):11. doi: 10.1167/tvst.4.3.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmed I, Patton TF. Importance of the noncorneal absorption route in topical ophthalmic drug delivery. Invest Ophthalmol Vis Sci. 1985;26(4):584–587. [PubMed] [Google Scholar]
- 38.Bloomfield SE, Miyata T, Dunn MW, Bueser N, Stenzel KH, Rubin AL. Soluble gentamicin ophthalmic inserts as a drug delivery system. Arch Ophthalmol. 1978;96(5):885–887. doi: 10.1001/archopht.1978.03910050487020. [DOI] [PubMed] [Google Scholar]
- 39.Amrite AC, Kompella UB. Size-dependent disposition of nanoparticles and microparticles following subconjunctival administration. J Pharm Pharmacol. 2005;57(12):1555–1563. doi: 10.1211/jpp.57.12.0005. [DOI] [PubMed] [Google Scholar]
- 40.Xu P, Gullotti E, Tong L, et al. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Mol Pharm. 2009;6(1):190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun X, Li F, Wang Y, Liang W. Cellular uptake and elimination of lipophilic drug delivered by nanocarriers. Pharmazie. 2010;65(10):737–742. [PubMed] [Google Scholar]
- 42.Lin T-T, Gao D-Y, Liu Y-C, et al. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J Control Release. 2016;221:62–70. doi: 10.1016/j.jconrel.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Paka GD, Ramassamy C. Optimization of curcumin loaded-PEG-PLGA nanoparticles by GSH functionalization. Investigation of the internalization pathway in neuronal cells. Mol Pharm. 2016;14(1):93–106. doi: 10.1021/acs.molpharmaceut.6b00738. [DOI] [PubMed] [Google Scholar]