

Abstract
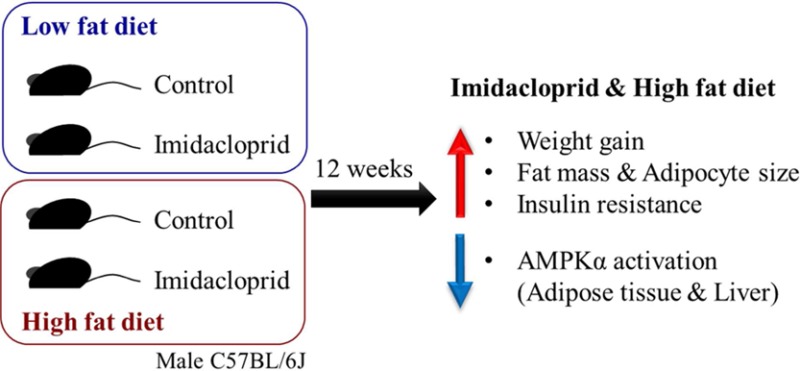
Imidacloprid, a neonicotinoid insecticide widely used in agriculture worldwide, has been reported to promote adipogenesis and cause insulin resistance in vitro. The purpose of the current study was to determine the effects of imidacloprid and its interaction with dietary fat in the development of adiposity and insulin resistance using male C57BL/6J mice. Imidacloprid (0.06, 0.6, or 6 mg/kg bw/day) was mixed in a low-fat (4% w/w) or high-fat (20% w/w) diet and given to mice ad libitum for 12 weeks. Imidacloprid significantly promoted high fat diet-induced body weight gain and adiposity. In addition, imidacloprid treatment with the high fat diet resulted in impaired glucose metabolism. Consistently, there were significant effects of imidacloprid on genes regulating lipid and glucose metabolisms, including the AMP-activated protein kinase-α (AMPKα) pathway in white adipose tissue and liver. These results suggest that imidacloprid may potentiate high fat diet-induced adiposity and insulin resistance in male C57BL/6J mice.
Keywords: imidacloprid, neonicotinoids, adiposity, insulin resistance, AMPKα
Introduction
Since its introduction to the market in 1991, imidacloprid (N-{1-[(6-chloro-3-pyridyl)methyl]-4,5-dihydroimidazol-2-yl}nitramide) is the most successful neonicotinoid insecticide containing the 2-(N-nitroimidazolidin) pharmacophore and the (6-chloropyrid-3-yl) methyl residue.1 Imidacloprid has been widely used on various types of grains, vegetables, fruits, and turfs to control agricultural insect pests as well as on domesticated animals to control ectoparasitic arthropods (e.g., K9 Advantix II and Advantage).2−4 In 2009, the market sales for imidacloprid in the United States alone was estimated to be U.S. $1.1 billion.2 This extensive use of imidacloprid in agriculture, including seed dressing, in recent years might further add to its presence in soil5,6 and water7 as well as detection in various kinds of fresh and processed fruits and vegetables.8 These possibilities suggest that the potential for human exposure to imidacloprid, in the general public as well as agricultural workers, would be relatively high. Imidacloprid use was restricted by the European Commission in 2013 along with two other neonicotinoids due to its potential risk for the collapse of bee populations.9 The selective potency of imidacloprid to insects versus mammals has been well characterized and attributed to the higher affinity at the insect nicotinic acetylcholine receptors (nAChRs).10−14 This selective action of imidacloprid and its systemic property make imidacloprid a preferable choice of insecticide in various field situations.2 In addition, imidacloprid is relatively more water-soluble when compared to other classes of insecticides (e.g., organochlorine, organophosphorus, and pyrethroid insecticides) and penetrates human skin slowly. Imidacloprid is also known to be quite persistent, with an approximately 39 day photolysis half-life at the soil surface (a range of 26.5–229 days) and an aerobic half-life of ∼3 years.15 Moreover, it is known that a plant metabolite, desnitro-imidacloprid, has been determined to be more toxic to mammals than the parent compound.16,17
Epidemiological studies suggest a link between exposure to persistent organic pollutants, including insecticides, and the epidemic of obesity and diabetes.18−22 Recently, one animal study reported that exposure to contaminated salmon oil containing persistent organic pollutants along with a high fat diet resulted in insulin resistance, represented by hyperinsulinemia, glucose intolerance, and hypertriglyceridemia, as well as hepatic steatosis, compared to control fed high fat diet alone in rats.23 Another study reported that exposure to dichlorodiphenyldichloroethylene (DDE) has a biphasic effect on fasting blood glucose in high fat diet fed male mice.24 Previously, a single study reported that 20 mg imidacloprid/kg body weight/day through oral gavage decreased body weight accompanied by significant elevation of serum glucose, glutamic oxaloacetic transaminase (GOT), glutamic pyruvic transaminase (GPT), and blood urea nitrogen in rats, whereas 10 mg imidacloprid/kg body weight/day did not show such toxic effects.25 Another study concluded that imidacloprid (5 and 10 mg/kg bw/day) has immunosuppressive effects, which might result from the direct cytotoxic effects of imidacloprid against T-cells.4 Moreover, there are recent reports that imidacloprid may have adverse effects on development.26,27 Along with these papers, our previously published studies reported that several pesticides, including imidacloprid, promote adipogenesis in 3T3-L1 adipocytes and induce insulin resistance in C2C12 myotubes.28−32 It is not known, however, if imidacloprid exposure alone or in combination with other factors of obesity and insulin resistance, such as high fat diet, will exacerbate obesity and insulin resistance symptoms. Thus, the current study was conducted to determine if exposure to imidacloprid aggravated high fat diet-induced metabolic disorders characterized by adiposity, dyslipidemia, hyperglycemia, and insulin resistance in male C57BL/6J mice.
Materials and Methods
Materials
Imidacloprid (>98%) was purchased from Chem Service Inc. (West Chester, PA, USA). Three-week-old male C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Semipurified powdered diets (TD 94048, 4% fat w/w, low fat diet; and TD 07518, 20% fat w/w, high fat diet) were based on Harlan Laboratories (Madison, WI, USA) diets. Food ingredients were obtained from Bio-Serv (Flemington, NJ, USA). Glucose and total cholesterol assay kits were purchased from Genzyme Diagnostics (Charlottetown, PE, Canada). Mouse leptin assay kit was purchased from R&D Systems (Minneapolis, MN, USA). Nonesterified fatty acid assay kit was from Wako Life Sciences, Inc. (Mountain View, CA, USA). Serum insulin level was analyzed with mouse insulin ELISA kit from ALPCO (Salem, NH, USA). The amounts of triglyceride were quantified using Infinity Triglycerides Reagent from Thermo Scientific (Waltham, MA, USA), and other chemicals were purchased from Fisher Scientific (Pittsburgh, PA, USA). Radioimmunoprecipitation assay (RIPA) buffer supplemented with 1% protease inhibitor was purchased from Boston Bioproducts Inc. (Ashland, MA, USA). Rabbit antibodies to AMPKα, phosphorylated AMPKα (pAMPKα), acetyl-CoA carboxylase (ACC), phosphorylated ACC (pACC), and horseradish peroxidase-conjugated goat anti-rabbit IgG were purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit antibodies to Sirtuin 1 (SIRT1), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and β-actin were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Ca2+/calmodulin-dependent protein kinase kinase β (CAMKKβ) rabbit antibody was purchased Abcam Inc. (Cambridge, MA, USA). Human recombinant insulin (Novolin, 100 U/mL) was purchased from Novo Nordisk Pharmaceuticals Industries Inc. (Seattle, WA, USA). Dextrose (50%) and bacteriostatic 0.9% sodium chloride injection were obtained from Hospira, Inc. (Lake Forest, IL, USA).
Animals and Diet
All animal work was conducted in compliance with the Institutional Animal Care and Use Committee at the University of Massachusetts under protocol no. 2013-0014. Mice were housed in a temperature- and humidity-controlled room with a 12 h light/dark cycle. The compositions of diets are shown in Table 1. Diet and water were given to mice ad libitum throughout the experiment and were provided fresh twice a week. Body weight and food intake were monitored weekly. After a 2 week adaptation, the mice were fed a control diet (4% fat w/w) and subjected to baseline tests for insulin tolerance and glucose tolerance. All animals were then divided into two dietary groups (low fat and high fat diets); each dietary treatment group contained four imidacloprid treatment doses (0, 0.06, 0.6, and 6 mg/kg bw/day). The treatment lasted for 12 weeks. Doses of imidacloprid were determined on the basis of previous publications; the highest imidacloprid dose was based on a no-observed-adverse-effect-level (NOAEL) of imidacloprid at 5.7 mg/kg bw/day, and the lowest dose of imidacloprid was based on estimated average daily intake of imidacloprid of 60 μg/kg body weight/day.33−35 Imidacloprid contents in low fat diets were 0, 0.516, 5.16, and 51.6 mg imidacloprid/kg diet. Imidacloprid contents in high fat diets were 0, 0.744, 7.44, and 74.4 mg imidacloprid/kg diet. The average consumptions of imidacloprid at the end of the study were 0, 0.07, 0.7, and 7 mg/kg bw/day in low fat dietary groups and 0, 0.08, 0.8, and 7 mg/kg bw/day in high fat dietary groups. Imidacloprid intakes between low fat diet and high fat diet were not statistically different.
Table 1. Composition of Diet.
| ingredient | low fat diet amount (g/kg) | high fat diet amount (g/kg) |
|---|---|---|
| casein | 140 | 169.1 |
| l-cystine | 1.8 | 2.2 |
| sucrose | 100 | 100 |
| cornstarch | 465.692 | 288.5 |
| maltodextrin | 155 | 132 |
| cellulose | 50 | 50 |
| soybean oil | 40 | 200 |
| mineral mix, AIN-93M-MX (TD 94049) | 35 | 42.8 |
| vitamin mix, AIN-93-VX (TD 94047) | 10 | 12.4 |
| choline bitartrate | 2.5 | 3 |
| tert-butylhydroquinone | 0.008 | 0.04 |
| total | 1000 | 1000 |
At the end of the study, mice were fasted for 4 h and sacrificed by CO2 asphyxiation. Blood was collected by cardiac puncture, and serum was separated by centrifugation at 2000g for 20 min. The organs (kidney, heart, liver, spleen, pancreas, and adipose tissues including epididymal, mesenteric, retroperitoneal, and subcutaneous adipose tissues) were weighed following sacrifice. One-eighth of the epididymal adipose tissue was kept in 10% buffered formalin and used for adipocyte size determination. The other parts of the epididymal adipose tissue and all other organs including gastrocnemius were snap frozen with liquid nitrogen and stored at −80 °C until analysis.
Serum Parameters
The levels of total cholesterol, leptin, nonesterified fatty acids (NEFA), triglyceride (TG), and insulin in serum were determined with commercial kits following the manufacturers’ instructions.
Insulin Tolerance Test (ITT)
ITT was performed during adaptation and treatment at weeks 5 and 9 as previously described.36 All mice were fasted for 4 h. Blood glucose from the tail vein was measured at 0, 15, 30, 60, and 120 min post-intraperitoneal injection of insulin solution (0.75 U/kg body weight), with a blood glucose meter, Advocate Redi-Code+ (Advocate Meters Inc., Dorado, Puerto Rico). The areas under the curve (AUCs) were calculated with Sigma Plot 11.0 (Systat Software, Inc., San Jose, CA, USA).
Glucose Tolerance Test (GTT)
GTT was performed during adaptation and treatment at weeks 6 and 11 as described previously.36 All mice were fasted for 6 h before the first measurement of blood glucose from the tail vein, and blood glucose was measured with a blood glucose meter (0 min). Then, a 20% glucose solution (2 g glucose/kg of body weight) was administered intraperitoneally, and blood glucose levels were further monitored at 15, 30, 60, and 120 min. Blood insulin level was also measured at 0, 30, 60, and 120 min with a mouse insulin ELISA kit from ALPCO. AUCs were calculated with Sigma Plot. The homeostasis model assessment-insulin resistance (HOMA-IR) score was calculated with a HOMA-IR calculator (University of Oxford).37
Adipocyte Size Measurement
Epididymal adipose tissue was fixed in 10% phosphate-buffered formalin and processed for paraffin sectioning.38 Tissue sections (4 μm) were cut and stained with hematoxylin and eosin (HE) staining. Pictures were taken with an Olympus CK2 inverted microscope (Olympus, Tokyo, Japan) (100× magnification) and microscope eyepiece camera (AmScope, Irvine, CA, USA). Adipocyte size was measured as previously described with ImageJ software with minor modification.39 The mean area of 50 cells from each sample was measured with ImageJ software. Briefly, pictures of the calibration slide (AmScope) were taken under the same settings as the tissue sections and were used to set the scale in ImageJ, and then the areas of the adipocytes were measured with ImageJ.
mRNA Expression Analysis
Mouse epididymal adipose tissue, liver, and gastrocnemius muscle were homogenized in Trizol reagent to extract total RNA under RNase-free conditions. Total RNA was reverse-transcribed with a high-capacity reverse transcription kit (Applied Biosystems, Carlsbad, CA, USA). mRNA expression levels of fatty acid translocase (FAT/CD36), sterol regulatory element-binding protein 1c (SREBP1c), tumor necrosis factor α (TNFα), phosphoenolpyruvate carboxykinase (PEPCK), peroxisome proliferator-activated receptor alpha (PPARα), glucose transporter type 4 (GLUT4), pyruvate dehydrogenase kinase 4 (PDK4), carnitine palmitoyltransferase 1B (CPT1b), and 18S rRNA (18S rRNA) were analyzed. Real-time polymerase chain reaction (PCR) was completed using a StepOne Plus real-time PCR instrument (Applied Biosystems) and Taqman probe-based gene expression analysis (Applied Biosystems). Respective integrated sequences used were NM_001159555.1 (CD36), NM_011480.3 (SREBP1c), NM_001278601.1 (TNFα), NM_028994.2 (PEPCK), NM_001113418.1 (PPARα), NM_009204.2 (GLUT4), NM_013743.2 (PDK4), NM_009948.2 (CPT1b), and NR_003278.3 (18S rRNA).
Immunoblotting
Mouse tissues were prepared for immunoblotting as previously described.32 Protein quantities were determined using the assay kit (Bio-Rad Co., Hercules, CA, USA). β-Actin was used as an internal control. The secondary antibody was goat anti-rabbit IgG conjugated with horseradish peroxidase. Detections were performed on an image Station 4000MM (Carestream Health, New Haven, CT, USA) with a Clarity Western ECL Substrate Kit (Bio-Rad Co.). Images were quantified with ImageJ software.40
Statistical Analyses
Data were analyzed by PROC MIXED of the SAS software (version 9.3, SAS Institute Inc., Cary, NC, USA). For the result on body weight (Figure 1A), two-way repeated measures analysis of variance (ANOVA) and the slice option in the least square (LS) means statement were used to determine the differences at each time point. For all other results, two-way ANOVA along with LS means statement was used. The Tukey–Kramer method was applied for the multiple comparisons among the groups. P values <0.05 are reported as statistically significant. If there were significant interactions between diet and imidacloprid, letters are used in the figures to present the differences between each group. When there were no interactions between diet and imidacloprid, brackets are used in the figures to represent overall differences between imidacloprid treatments and control groups.
Figure 1.

Effects of imidacloprid on body weight, body weight gain, and food intake: (A) body weight monitored weekly; (B) body weight gain for 12 weeks; (C) total food intake. Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. Numbers are the mean ± SE (n = 5–8 for panels A and B; n = 3–4 for panel C). Means with different letters are significantly different at P < 0.05.
Results
Imidacloprid Promoted Weight Gain in High Fat Diet Fed Mice
To understand the effect of imidacloprid on dietary fat induced changes, we chose C57BL/6J male mice fed either a low fat (4%) or a high fat (20%) diet. High fat diets (∼40–70% of kcal) are known to induce obesity in this strain.41 There were overall effects of diet or imidacloprid on body weight (Figure 1A). Significant three-way interaction (imidacloprid × diet × time) was also observed. In high fat diet fed mice, all imidacloprid groups showed a significant increase in body weight compared to the control group from week 7 onward and maintained the trend throughout the experiment (P < 0.001 for all weeks from 7 to 12) (Figure 1A). No differences of body weight were found in the low fat diet fed mice among all groups throughout the experiment (Figure 1A). Overall, both dietary fat and imidacloprid significantly affected body weight gain, and there was a significant interaction between dietary fat and imidacloprid (Figure 1B). In high fat diet fed mice, imidacloprid treatment groups (0.06, 0.6, and 6 mg/kg bw/day) had significantly greater weight gain compared to the high fat control group (P = 0.0057, 0.0149, and 0.0038, respectively) (Figure 1B).
Food intake as determined by calorie consumption is shown in Figure 1C. Both imidacloprid and dietary fat significantly affected the calorie intake alone but without any interaction between them (Figure 1C). Mice fed the high fat diet had greater calorie intake compared to mice fed the low fat diet (P = 0.0080). There were no significant differences of calorie intake between the control and three imidacloprid treatment groups. However, there was a significant difference of calorie intake between the 0.06 and 6 mg/kg bw/day imidacloprid treatment groups (P = 0.0223) (Figure 1C).
Influence of Imidacloprid on Tissue and Organ Weights
Organ weights (liver, pancreas, heart, kidneys, and spleen) as well as adipose tissue weights (epididymal, subcutaneous, mesenteric, retroperitoneal, and total adipose tissue) measured as percent of body weights are shown in Table 2. There was a significant diet effect, but there were no imidacloprid and diet × imidacloprid interaction effects on liver, heart, and spleen weights. Both imidacloprid and diet significantly affected the kidney weight with a significant interaction. In high fat diet fed mice, imidacloprid treatment groups (0.06 and 6 mg/kg bw/day) had reduced kidney weight compared to the control group (P = 0.0297 and 0.0006, respectively). There were overall effects of diet and imidacloprid on all adipose tissue weights with interaction except for mesenteric adipose tissue, for which only a diet effect was observed. In high fat diet fed mice, animals treated with imidacloprid (0.6 and 6 mg/kg bw/day) had significantly greater epididymal (P = 0.0337 and 0.0002, respectively) and retroperitoneal (P = 0.0078 and 0.0008, respectively) adipose tissue weights compared to the control group.
Table 2. Organ Weights (Percent of Body Weight)a.
| low
fat |
high
fat |
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| imidacloprid
dose |
imidacloprid
dose |
P value |
|||||||||
| control | 0.06 mg/kg | 0.6 mg/kg | 6 mg/kg | control | 0.06 mg/kg | 0.6 mg/kg | 6 mg/kg | dietary fat | imidacloprid | interaction | |
| liver | 3.90 ± 0.12 | 3.86 ± 0.15 | 3.78 ± 0.07 | 3.92 ± 0.16 | 3.51 ± 0.18 | 3.57 ± 0.08 | 3.34 ± 0.21 | 3.58 ± 0.17 | 0.002 | ns | ns |
| heart | 0.44 ± 00.02 | 0.50 ± 0.05 | 0.48 ± 0.02 | 0.48 ± 0.02 | 0.47 ± 0.03 | 0.41 ± 0.03 | 0.41 ± 0.03 | 0.38 ± 0.03 | 0.011 | ns | ns |
| spleen | 0.26 ± 0.03 | 0.20 ± 0.01 | 0.23 ± 0.02 | 0.19 ± 0.02 | 0.30 ± 0.02 | 0.27 ± 0.02 | 0.30 ± 0.01 | 0.35 ± 0.05 | 0.0002 | ns | ns |
| kidney | 1.06 ± 0.05ab | 1.09 ± 0.05ab | 1.15 ± 0.04a | 1.10 ± 0.04ab | 1.12 ± 0.07a | 0.88 ± 0.04bc | 0.96 ± 0.03abc | 0.80 ± 0.05c | <0.0001 | 0.027 | 0.003 |
| pancreas | 0.43 ± 0.04 | 0.45 ± 0.02 | 0.48 ± 0.04 | 0.47 ± 0.03 | 0.54 ± 0.05 | 0.38 ± 0.02 | 0.42 ± 0.02 | 0.39 ± 0.04 | ns | ns | 0.037 |
| adipose tissue | |||||||||||
| epididymal | 2.19 ± 0.20d | 2.29 ± 0.42cd | 1.64 ± 0.17d | 2.00 ± 0.25d | 3.57 ± 0.42bc | 5.0 ± 0.31ab | 5.03 ± 0.27a | 5.82 ± 0.40a | <0.0001 | 0.011 | 0.001 |
| subcutaneous | 1.36 ± 0.24cd | 1.43 ± 0.37cd | 0.94 ± 0.14d | 1.43 ± 0.30cd | 2.65 ± 0.45bc | 3.79 ± 0.37ab | 3.59 ± 0.28a | 4.91 ± 0.56a | <0.0001 | 0.014 | 0.026 |
| retroperitoneal | 0.59 ± 0.08c | 0.57 ± 0.13c | 0.39 ± 0.08c | 0.51 ± 0.09c | 1.09 ± 0.12b | 1.50 ± 0.09ab | 1.64 ± 0.09a | 1.75 ± 0.14a | <0.0001 | 0.038 | 0.001 |
| mesenteric | 1.33 ± 0.10 | 1.61 ± 0.20 | 1.23 ± 0.15 | 1.23 ± 0.11 | 2.02 ± 0.23 | 2.60 ± 0.19 | 2.64 ± 0.17 | 2.80 ± 0.26 | <0.0001 | ns | ns |
| total | 5.47 ± 0.58bc | 5.90 ± 1.10bc | 4.20 ± 0.52c | 5.17 ± 0.74bc | 9.34 ± 1.12b | 12.9 ± 0.88a | 12.9 ± 0.68a | 15.3 ± 1.29a | <0.0001 | 0.001 | <0.001 |
Mice were treated with three doses of imidacloprid (0.06, 0.6, and 6 mg/kg bw/day). Values represent means ± SE (n = 5–8). Means with different letters within the same row are significantly different at P < 0.05. Abbreviations: ns, not significant.
Histological analysis revealed that there were significant effects of diet and imidacloprid with diet × imidacloprid interaction on epididymal adipocyte size (Figure 2). Imidacloprid (0.06, 0.6, and 6 mg/kg bw/day) treatment significantly increased adipocyte size in high fat diet fed mice (P = 0.0081, 0.0002, and 0.0001, respectively), but not in low fat diet fed mice (Figure 2A,B).
Figure 2.
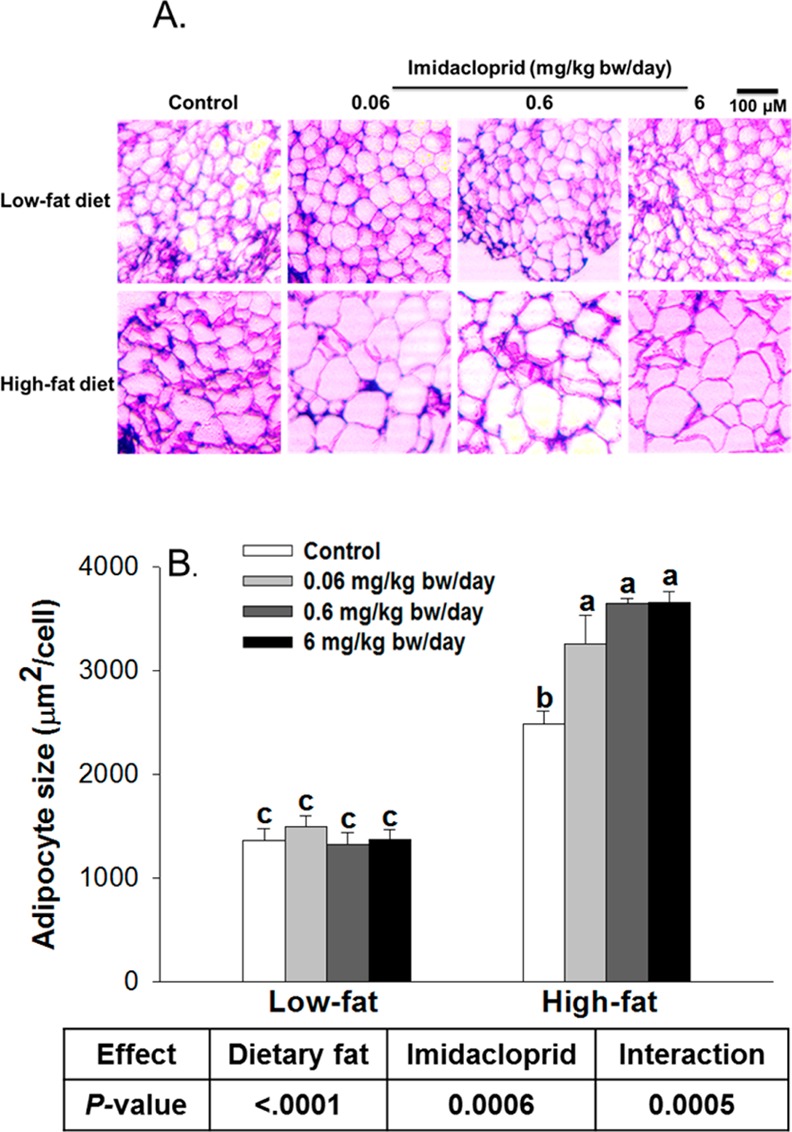
Effects of imidacloprid on adipocyte size: (A) representative pictures of epididymal adipose tissues after H&E staining (100× magnification); (B) adipocyte size. Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. The mean area of 50 cells from each sample was measured with ImageJ. Data are expressed as the mean ± SE (n = 3). Means with different letters are significantly different at P < 0.05.
Influence of Imidacloprid on Serum Markers
The serum levels of glucose, cholesterol, leptin, NEFA, TG, and insulin are shown in Table 3. There were overall treatment effects from both diet and imidacloprid (without interaction) on blood glucose and insulin levels. Mice fed imidacloprid (0.06 and 6 mg/kg bw/day) had significantly higher levels of blood glucose compared to control groups (P = 0.0096 and 0.0144, respectively). Mice fed imidacloprid (6 mg/kg bw/day) also had significantly higher blood insulin levels compared to control groups (P = 0.0325). There were overall treatment effects from both diet and imidacloprid with their interaction on leptin level. Compared to the high fat control group, animals fed the high fat diet with (0.06, 0.6, and 6 mg/kg bw/day) imidacloprid had higher blood leptin levels (P < 0.0001, 0.0011, and <0.0001, respectively). There were overall treatment effects from imidacloprid, but not diet and their interaction, on serum TG levels. Mice fed imidacloprid (6 mg/kg bw/day) had significantly higher serum TG level compared to control groups (P = 0.0099). There were significant effects of diet, but not to imidacloprid or to their interaction, on cholesterol levels. There were no significant effects of both diet and imidacloprid on serum NEFA levels.
Table 3. Serum Parametersa.
| low
fat |
high
fat |
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| imidacloprid
dose |
imidacloprid
dose |
P value |
|||||||||
| control | 0.06 mg/kg | 0.6 mg/kg | 6 mg/kg | control | 0.06 mg/kg | 0.6 mg/kg | 6 mg/kg | dietary fat | imidacloprid | interaction | |
| glucose, mg/dL | 162.8 ± 7.9 | 183.1 ± 24.1 | 169.7 ± 4.6 | 169.3 ± 17.7 | 178.2 ± 17.9 | 234.7 ± 19.1 | 193.7 ± 18.7 | 229.9 ± 15.4 | 0.0312 | 0.0034 | ns |
| insulin, ng/mL | 0.77 ± 0.11 | 0.97 ± 0.25 | 0.81 ± 0.07 | 0.97 ± 0.21 | 2.05 ± 0.34 | 3.10 ± 0.33 | 2.51 ± 0.37 | 3.36 ± 0.58 | <0.0001 | 0.0312 | ns |
| leptin, ng/mL | 4.9 ± 1.0b | 4.2 ± 1.4b | 3.4 ± 0.7b | 4.4 ± 1.2b | 14.4 ± 3.7b | 59.6 ± 3.2a | 47.9 ± 6.1a | 66.4 ± 11.5a | <0.0001 | <0.0001 | <0.0001 |
| TG, mg/dL | 77.2 ± 6.3 | 94.2 ± 6.1 | 76.3 ± 12.2 | 91.2 ± 8.1 | 63.6 ± 5.1 | 82.4 ± 6.0 | 85.0 ± 2.3 | 94.5 ± 6.8 | ns | 0.0104 | ns |
| cholesterol, mg/dL | 161.5 ± 8.7 | 163.7 ± 18.9 | 117.6 ± 11.3 | 150.5 ± 9.5 | 171 ± 15 | 213.4 ± 21 | 182.6 ± 16 | 219.9 ± 21.9 | 0.0003 | ns | ns |
| NEFA, mequiv/L | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.2 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | ns | ns | ns |
Mice were treated with three doses of imidacloprid (0.06, 0.6, and 6 mg/kg bw/day). Values represent means ± SE (n = 5–8). Means with different letters within the same row are significantly different at P < 0.05. Abbreviations: ns, not significant; TG, triglyceride; NEFA, nonesterified fatty acid.
Effects of Imidacloprid on Glucose Homeostasis
To determine the effect of imidacloprid and diet on glucose homeostasis, we measured serum glucose levels during ITT and GTT and measured serum insulin level during GTT, which are presented in Figure 3 and Figure 1S. Before treatment, there were no differences between any of the groups for ITT, GTT, and insulin level (Figure 3A,D,G).
Figure 3.

Effects of imidacloprid on insulin tolerance test (ITT) (A, B, C), glucose tolerance test (GTT) (D, E, F), and insulin level determination with serum from GTT (G, H, I) as the area under the curve (AUC). Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. ITT was measured during the adaptation period, weeks 5 and 9. GTT was measured during the adaptation period, weeks 6 and 11. Blood was collected from the tail vein, and glucose levels were determined at 0 min; then the insulin (ITT) or glucose solution (GTT) was administered by intraperitoneal injection, and the glucose level was further measured at 15, 30, 60, and 120 min. During GTT, the blood was collected from the tail vein at 0, 30, 60, and 120 min for determination of insulin level. Numbers represent means ± SE (n = 5–8). Means with different letters are significantly different at P < 0.05.
As shown in Figure 3B, there were significant diet and imidacloprid effects without interaction on insulin tolerance after 5 weeks of treatment. In particular, mice that received the highest dose of imidacloprid (6 mg/kg bw/day) had significantly higher AUC compared to control groups (P = 0.0018) in week 5 (Figure 3B). After 9 weeks of treatment, there were significant effects of diet and imidacloprid with interaction (Figure 3C). High fat diet imidacloprid treated mice at 6 mg/kg bw/day had significantly elevated AUC compared to the high fat control (P = 0.0006) (Figure 3C).
We further performed GTT at 6 and 11 weeks. As shown in Figure 3E, only diet was found to have a significant effect on impaired glucose tolerance at week 6. At week 11, both diet and imidacloprid effects were observed without interaction (Figure 3F). At the highest dose of imidacloprid (6 mg/kg bw/day), treated mice showed more severe glucose intolerance compared to control groups (P = 0.0331) (Figure 3F).
The serum samples collected from the GTT tests were further utilized for insulin level measurements. There were significant diet and imidacloprid effects with interaction on serum insulin level at both weeks 6 and 11 (Figure 3H,I). High fat diet with imidacloprid treated animals at 0.6 and 6 mg/kg bw/day had significantly higher AUC of insulin levels compared to high fat controls (P <0.0001 for both 0.6 and 6 mg/kg bw/day at week 6, P = 0.0007 and 0.0211 at week 11, respectively) (Figure 3H,I). Similarly, there were significant diet and imidacloprid effects on HOMA-IR scores with significant interaction at weeks 6, 11, and 12 (Figure 4). High fat diet with imidacloprid (0.6 and 6 mg/kg bw/day) treated mice had higher HOMA-IR scores compared with the high fat control (Figure 4).
Figure 4.

Effects of imidacloprid on HOMA-IR score. Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. HOMA-IR score was calculated during the adaptation period, weeks 6, 11, and 12, with a HOMA-IR calculator.37 Numbers represent means ± SE (n = 5–8). Means with different letters are significantly different at P < 0.05.
Effects of Imidacloprid on mRNA Expression Levels of Genes Regulating Lipid Metabolism and AMPKα Pathway in Epididymal Adipose Tissue
Because imidacloprid and high fat diet significantly affected the lipid and glucose metabolism in mice, we first investigated their effects on mRNA expression levels of genes regulating lipid metabolism in white adipose tissue. CD36, one of the fatty acid transporter proteins in adipocytes, facilitates the cellular entry of lipoprotein derived fatty acids, thus promoting the storage of triglycerides in adipose tissue.42 There were significant diet and imidacloprid effects without interaction on CD36 mRNA expression (Figure 5A). Compared to control groups, mice fed with imidacloprid (6 mg/kg bw/day) had significantly higher expression of CD36 (P = 0.011) (Figure 5A).
Figure 5.

Effects of imidacloprid on molecular target genes involved in lipid metabolism and inflammation in white adipose tissue: (A) FAT/CD36, fatty acid translocase; (B) SREBP1c, sterol regulatory element binding protein 1c; (C) TNFα, tumor necrosis factor α; (D) representative pictures; (E) CaMKKβ, Ca2+/calmodulin-dependent protein kinase kinase β; (F) pAMPKα/AMPKα, phosphorylated AMP-activated protein kinase-α/AMP-activated protein kinase-α; (G) pACC/ACC, phosphorylated acetyl-CoA carboxylase/acetyl-CoA carboxylase. Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. Numbers represent means ± SE (n = 4–6). Means with different letters are significantly different at P < 0.05.
SREBP1c is highly expressed in adipose tissue and is a well-known master regulator of the lipogenic pathway.43,44 There was significant imidacloprid effect, without diet effects or interaction, on SREBP1c mRNA expression in the white adipose tissue (Figure 5B). Mice fed 6 mg/kg bw/day imidacloprid had a significant increase in SREBP1 expression compared to controls (P = 0.0035) (Figure 5B).
TNFα, a pro-inflammatory cytokine, is highly expressed in the adipose tissue of obese mice.45 TNFα is regarded as an important link between obesity and insulin resistance.46 There were significant effects of diet and imidacloprid as well as their interaction on TNFα expression (Figure 5C). Mice fed a high fat diet and imidacloprid at 6 mg/kg bw/day had an increased TNFα mRNA expression level compared to high fat control (P = 0.0029) (Figure 5C).
AMPK is known as a master regulator of energy metabolism, and activation of AMPK leads to the stimulation of fatty acid oxidation and inhibition of lipogenesis.47 Previously we reported that imidacloprid may potentiate adipogenesis via an AMPK mediated mechanism in 3T3-L1 adipocytes.28 To determine if oral administration of imidacloprid exerts its effect via a similar mechanism, we have detected AMPK and its downstream target (acetyl Co-A carboxylase, ACC) as well as one of its upstream regulators, CaMKKβ,48,49 from epididymal adipose tissue (Figure 5D–G). There were significant imidacloprid and imidacloprid × diet interaction effects without diet effect on the expression of CaMKKβ (Figure 5E). High fat diet with imidacloprid (6 mg/kg bw/day) had significantly reduced expression of CaMKKβ compared to controls (P = 0.0002) (Figure 5E). There were significant diet and imidacloprid effects with interaction on the ratio of expressions of pAMPKα/AMPKα (Figure 5F). High fat diet and imidacloprid (6 mg/kg bw/day) fed mice had a significant decrease in the ratio of pAMPKα/AMPKα compared to high fat control (P = 0.0028) (Figure 5F). There was only an imidacloprid effect, without diet effect, on the ratio of phosphorylated ACC/ACC (pACC/ACC) (Figure 5G). Mice treated with imidacloprid at 0.06 and 6 mg/kg bw/day had significantly lower pACC/ACC ratios compared to controls (P = 0.015 and 0.0403, respectively) (Figure 5G). These results suggest that oral administration of imidacloprid might promote adipogenesis via a CaMKKβ-AMPKα-dependent pathway in white adipose tissue of mice.
Effects of Imidacloprid on mRNA Expression of Genes Regulating Lipid and Glucose Metabolism as well as AMPKα Pathway in Liver
Liver is an important organ in regulating glucose and fatty acid metabolisms; thus, we further investigated the effects of imidacloprid and high fat diet on two genes, PEPCK and PPARα, which are the rate-limiting enzyme for gluconeogenesis50 and a key regulator of fatty acid oxidation in liver, respectively.51 There were significant diet and imidacloprid effects with their interaction on the expression of PEPCK (Figure 6A). In mice fed the high fat diet, imidacloprid (0.06 and 6 mg/kg bw/day) treatment groups showed significantly higher mRNA expression of PEPCK, compared to control (P = 0.0166 and 0.0336) (Figure 6A). There were significant diet and imidacloprid effects without interaction on PPARα mRNA expression (Figure 6B). Mice fed imidacloprid at 0.6 and 6 mg/kg bw/day had significantly reduced mRNA expression of PPARα compared to controls (P = 0.0159 and 0.023, respectively) (Figure 6B).
Figure 6.

Effects of imidacloprid on molecular target genes regulating lipid and glucose metabolism as well as the AMPKα pathway in liver: (A) PEPCK, phosphoenolpyruvate carboxykinase; (B) PPARα, peroxisome proliferator-activated receptor-α; (C) representative pictures; (D) CaMKKβ, Ca2+/calmodulin-dependent protein kinase kinase β; (E) pAMPKα/AMPKα, phosphorylated AMP-activated protein kinase-α/AMP-activated protein kinase-α; (F) pACC/ACC, phosphorylated acetyl-CoA carboxylase/acetyl-CoA carboxylase; (G) SIRT1, sirtuin 1; (H) PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α. Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. Numbers represent means ± SE (n = 4–6). Means with different letters are significantly different at P < 0.05.
We further investigated the effect of imidacloprid on protein markers important for lipid and glucose homeostasis in the liver. PGC-1α is highly expressed in the liver and is known as a master regulator of fatty acid oxidation.52,53 Two metabolic sensors, AMPK and SIRT1, have been reported to directly increase PGC-1α activity.54 CaMKKβ is known to be the upstream regulator of AMPK.48 There were significant effects of imidacloprid and imidacloprid × diet interaction without significant effect of diet on CaMKKβ expression (Figure 6D). High fat diet with 6 mg/kg bw/day imidacloprid fed mice had a decrease in expression of CaMKKβ compared to high fat control (P = 0.0049) (Figure 6D). Significant effects of diet and imidacloprid with their interaction were observed for the pAMPKα/AMPKα and pACC/ACC ratios (Figure 6E,F). High fat imidacloprid (6 mg/kg bw/day) fed mice exhibited decreased ratios of pAMPKα/AMPKα and pACC/ACC compared to high fat control (P < 0.0001 and 0.0475, respectively) (Figure 6E,F). There were effects of both diet and imidacloprid on SIRT1 and PGC-1α expression without interaction (Figure 6G,H). Mice treated with imidacloprid (0.6 and 6 mg/kg bw/day) had a significant decrease in expression of SIRT1 (P = 0.0026 and 0.0121) and PGC-1α compared to controls (P = 0.0356 and 0.011, respectively) (Figure 6G,H). These results suggest that imidacloprid might contribute to decreased fatty acid oxidation in the liver.
Effects of Imidacloprid on mRNA Expression of Genes Regulating Lipid and Glucose Metabolism in Skeletal Muscle
Muscle is responsible for 70–90% of glucose disposal in our body,55 and insulin resistance in skeletal muscle is the primary defect in type 2 diabetes.56 In muscle, GLUT4 is the main glucose transporter responsible for glucose uptake in response to insulin stimulation.57 It has been reported that mice lacking or overexpressing GLUT4 have a decrease or increase in whole-body insulin sensitivity, respectively.58 In the current study, there were significant effects of diet and imidacloprid without interaction on muscle GLUT4 mRNA expression (Figure 7A). Mice fed imidacloprid at 0.06 and 6 mg/kg bw/day had significantly lower mRNA expression of GLUT4 compared to controls (P = 0.0003 and 0.0012, respectively) (Figure 7A).
Figure 7.

Effects of imidacloprid on markers regulating lipid and glucose metabolism in gastrocnemius muscle: (A) GLUT4, glucose transporter type 4; (B) PDK4, pyruvate dehydrogenase kinase 4; (C) CPT1, carnitine palmitoyltransferase 1. Mice were fed a low fat diet or high fat diet supplemented with (0, 0.06, 0.6, and 6 mg/kg bw/day) imidacloprid for 12 weeks. Numbers represent means ± SE (n = 4–6). Means with different letters are significantly different at P < 0.05.
PDK4, a kinase enzyme highly expressed in skeletal muscle, inactivates the enzyme pyruvate dehydrogenase.59 As a result, the pyruvate formed from glycolysis cannot be oxidized, which leads to hyperglycemia due to the decrease in blood glucose oxidation.60 A previous study also reported that PDK4 is overexpressed in skeletal muscle of people with type 2 diabetes, resulting in impaired glucose disposal.61 In the current mouse study, there were effects of diet and imidacloprid on PDK4 mRNA expression without interaction (Figure 7B). Mice fed imidacloprid at 6 mg/kg bw/day had a higher expression level of PDK4 compared to controls (P = 0.0015) (Figure 7B).
CPT1 is a mitochondrial transmembrane enzyme, recognized as rate limiting for mitochondrial fatty acid β-oxidation.62 In the current study, there was a significant diet effect on CPT1 mRNA expression without imidacloprid effect and interaction. Taken together, these results suggested that imidacloprid and high fat diet might affect the expression of genes important for lipid and glucose metabolism and contribute to adiposity and insulin resistance in the current study.
Discussion
In this study, we demonstrated that dietary exposure to relatively low doses of imidacloprid (at or lower than NOAEL) exacerbated high fat diet induced weight gain and insulin resistance in C57BL/6J male mice during a 12 week treatment. As such, it is the first mouse study demonstrating the role of imidacloprid on the development of high fat diet induced obesity and type 2 diabetes. The current results also suggest that imidacloprid may elicit its effects by post-translational regulation of AMPK via CaMKKβ and/or SIRT1 in adipocytes and the liver. This finding is consistent with our previous reports that imidacloprid inhibits the phosphorylation of AMPKα in 3T3-L1 adipocytes.28
Several studies have reported an interaction between organophosphorous insecticides (diazinon and parathion) and high fat diet, including their contribution to increased weight gain and insulin resistance.63−67 Ruzzin et al.23 reported that persistent organic pollutants (POPs) in crude fish oil exacerbated high fat diet induced increased visceral adipose tissue and insulin resistance compared to refined salmon oil (without POPs) in male Sprague–Dawley rats. In the Ruzzin study, POPs with high fat diet were reported to induce increased liver SREBP1c expression. Similarly, imidacloprid exposure increased the expression of SREBP1c in adipose tissue in the current study. In addition, our current results showed that imidacloprid with high fat diet increased TNFα (a pro-inflammatory cytokine) in adipose tissue of high fat diet fed mice, which may have significance in insulin resistance.
A previous mouse study reported that exposure to DDE initially facilitated high fat diet induced hyperglycemia at weeks 4 and 8, which returned to normal at 12–13 weeks.24 However, Ruzzin et al.23 reported that POPs with high fat diet increased HOMA-IR score and reduced insulin-stimulated glucose uptake in skeletal muscle and adipose tissue. This study is consistent with the current results that imidacloprid with high fat diet increased the blood glucose and insulin levels along with impaired insulin resistance. Moreover, we observed that animals fed imidacloprid with high fat diet potentiated gluconeogenesis in the liver by targeting PEPCK and reduced glucose disposal from the skeletal muscle by decreasing GLUT4 expression and glucose oxidation, both of which may have contributed to overall impairment of glucose homeostasis. It is important to point out, however, that our results are limited because we did not measure insulin-stimulated GLUT4 translocation in skeletal muscle. Nevertheless, it was previously reported that imidacloprid exposure might induce insulin resistance by reducing insulin-stimulated glucose uptake and phosphorylation of protein kinase B (AKT) in adipocytes, hepatocytes, and myotubes.29 Thus, it is likely that imidacloprid exacerbated high fat diet induced insulin resistance by targeting the insulin signaling pathway. Alternatively, imidacloprid might cause oxidative stress and inflammation as previously suggested.68−72 Inflammation, including the significance of pro-inflammatory cytokine TNFα, and oxidative stress were reported to be correlated with the development of obesity and insulin resistance.46,73−75 Inflammation and oxidative stress are known to activate JNK (c-Jun N-terminal kinase),76,77 which is linked to insulin resistance.78 Thus, further studies of imidacloprid’s role in oxidative stress and inflammation, including the role of TNFα and their contributions to obesity and insulin resistance, are needed to understand how imidacloprid elicits altered metabolism.
Mice fed the high fat diet showed physiological and metabolic changes compared to low fat diet fed mice as expected;79−82 including increased body weight, calorie intake, serum insulin level, HOMA-IR score, expression of epididymal adipose tissue CD36, TNFα, adipocyte size, and impaired insulin tolerance as well as glucose tolerance. However, several studies reported inconsistent results of lipogenesis in mice consuming a high fat diet: increased,83 no change84 or decreased lipogenesis after high fat diet feeding.85,86 Our current results show no significant different expression levels of SREBP1, a key lipogenic gene, in adipose tissue between high fat diet and low fat diet fed mice.
Imidacloprid is the most widely used neonicotinoid insecticide,2 and compared to highly lipophilic organochlorine and organophosphorus insecticides, imidacloprid is relatively water-soluble.15 Imidacloprid is considered to be moderately toxic, with an oral LD50 in rats of 420 mg/kg body weight, and the typical symptoms of poisoning include fatigue, cramps, and muscle weakness.87 However, the dose we used in the current study was based on a NOAEL of 5.7 mg imidacloprid/kg body weight/day or lower; thus, we did not expect any of those acute toxic symptoms in these animals.88 A previous mouse study showed that oral gavage of >5 mg/kg bw/day imidacloprid for 28 days resulted in immunosuppressive effects; however, no changes in body weight were observed for all doses of imidacloprid treatment in that study.4 Another study reported that oral administration of 20 mg/kg bw/day imidacloprid decreased the body weight in rats; however, this was not observed in 10 mg/kg bw/day imidacloprid treated rats.25 Thus, the significance of the current study is the interaction between high fat diet and imidacloprid, which would have significance for understanding potential health implications of imidacloprid.
It is known that imidacloprid is quickly absorbed from the gastrointestinal tract and distributed in almost all organs and tissues.33 The oral absorption of imidacloprid was estimated as 92–99%.33 Although imidacloprid intake between low fat diet and high fat diet in the current study were not statistically different, we cannot exclude the possibility that high fat and low fat diet might have different effects on the bioavailability of imidacloprid, thus leading to the differences observed in the current study. Once absorbed, imidacloprid is degraded into a variety of metabolites: 6-chloronicotinic acid, imidazolidine 4- and 5-hydroxy compounds, olefinic imidacloprid, desnitro-imidacloprid, and the nitrosoimine compound.33 These metabolites are known to be excreted mainly in the urine as glutathione and glycine conjugates of mercaptonicotinic acid and hippuric acid.33 Imidacloprid also photodegrades rapidly in water, with a half-life of <3 h. However, imidacloprid has been found to be persistent in the environment with a photolysis half-life of 39 days at the soil surface (a range of 26.5–229 days) when absorbed by the soil.15
Despite intensive investigations and establishment of preventive and therapeutic strategies for obesity and type 2 diabetes, these diseases have increased significantly in the past few decades.89,90 Sedentary lifestyle and excessive caloric intake can only partially explain the dramatic increase of metabolic diseases worldwide. Recently, air pollution and environmental contaminants such as bisphenol A, as well as other POPs, have been suggested to contribute to the development of obesity and insulin resistance.23,91−93 Along with the current results, these reports provide compelling evidence that environmental contaminants may contribute to the epidemic of obesity, insulin resistance, and eventually type 2 diabetes.
To conclude, our present finding is the first report that imidacloprid exposure aggravates high fat diet induced obesity and insulin resistance in C57BL/6J male mice. The current results are significant in substantiating a potential link between insecticide exposure, particularly imidacloprid, and dietary fat and adiposity as well as insulin resistance. However, the underlying mechanisms by which imidacloprid promotes high fat diet induced obesity and insulin resistance still need to be further explored.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jafc.6b04322.
Figure 1S. Effects of imidacloprid on insulin tolerance test (ITT) (A, B, C), glucose tolerance test (GTT) (D, E, F), and insulin level determination (G, H, I) (PDF)
The China Scholarship Council supported Q.S. This project is supported by NIH R21ES023128.
The authors declare no competing financial interest.
Supplementary Material
References
- Jeschke P.; Nauen R. Neonicotinoids—from zero to hero in insecticide chemistry. Pest Manage. Sci. 2008, 64, 1084–1098. 10.1002/ps.1631. [DOI] [PubMed] [Google Scholar]
- Jeschke P.; Nauen R.; Schindler M.; Elbert A. Overview of the status and global strategy for neonicotinoids. J. Agric. Food Chem. 2011, 59, 2897–2908. 10.1021/jf101303g. [DOI] [PubMed] [Google Scholar]
- Herms D. A.; McCullough D.; Smitley D. R.; Sadof C.; Williamson R. C.; Nixon P. L.. Insecticide Options for Protecting Ash Trees from Emerald Ash Borer, 2009; pp 1–20, www.emeraldashborer.info.
- Badgujar P. C.; Jain S. K.; Singh A.; Punia J. S.; Gupta R. P.; Chandratre G. A. Immunotoxic effects of imidacloprid following 28 days of oral exposure in BALB/c mice. Environ. Toxicol. Pharmacol. 2013, 35, 408–418. 10.1016/j.etap.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Wettstein F. E.; Kasteel R.; Garcia Delgado M. F.; Hanke I.; Huntscha S.; Balmer M. E.; Poiger T.; Bucheli T. D. Leaching of the neonicotinoids thiamethoxam and imidacloprid from sugar beet seed dressings to subsurface tile drains. J. Agric. Food Chem. 2016, 64, 6407–6415. 10.1021/acs.jafc.6b02619. [DOI] [PubMed] [Google Scholar]
- Sarkar M. A.; Roy S.; Kole R. K.; Chowdhury A. Persistence and metabolism of imidacloprid in different soils of West Bengal. Pest Manage. Sci. 2001, 57, 598–602. 10.1002/ps.328.abs. [DOI] [PubMed] [Google Scholar]
- Sánchez-Bayo F.; Goka K.; Hayasaka D. Contamination of the aquatic environment with neonicotinoids and its implication for ecosystems. Front. Environ. Sci. 2016, 4, 00071. 10.3389/fenvs.2016.00071. [DOI] [Google Scholar]
- Gervais J. A.; Luukinen B.; Buhl K.; Stone D.. Imidacloprid General Fact Sheet; National Pesticide Information Center, Oregon State University Extension Services, 2010; http://npic.orst.edu/factsheets/imidacloprid. [Google Scholar]
- The European Commision. Decision by the European Commission to restrict the use of imidacloprid, thiamethoxam and clothianidin (Regulation EU No. 485/2013), 2016; http://eurlex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:139:0012:0026:EN:PDFS.
- Liu M. Y.; Casida J. E. High affinity binding of [3H]imidacloprid in the insect acetylcholine receptor. Pestic. Biochem. Physiol. 1993, 46, 40–46. 10.1006/pest.1993.1034. [DOI] [Google Scholar]
- Tomizawa M.; Casida J. E. Minor structural changes in nicotinoid insecticides confer differential subtype selectivity for mammalian nicotinic acetylcholine receptors. Br. J. Pharmacol. 1999, 127, 115–122. 10.1038/sj.bjp.0702526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa M.; Lee D. L.; Casida J. E. Neonicotinoid insecticides: molecular features conferring selectivity for insect versus mammalian nicotinic receptors. J. Agric. Food Chem. 2000, 48, 6016–6024. 10.1021/jf000873c. [DOI] [PubMed] [Google Scholar]
- Matsuda K.; Buckingham S. D.; Kleier D.; Rauh J. J.; Grauso M.; Sattelle D. B. Neonicotinoids: insecticides acting on insect nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 2001, 22, 573–580. 10.1016/S0165-6147(00)01820-4. [DOI] [PubMed] [Google Scholar]
- Tomizawa M.; Casida J. E. Selective toxicity of neonicotinoids attributable to specificity of insect and mammalian nicotinic receptors. Annu. Rev. Entomol. 2003, 48, 339–364. 10.1146/annurev.ento.48.091801.112731. [DOI] [PubMed] [Google Scholar]
- Fossen M.Environmental Fate of Imidacloprid; California Department of Pesticide Regulation, 2006; pp 1–16
- Lee Chao S.; Casida J. E. Interaction of imidacloprid metabolites and analogs with the nicotinic acetylcholine receptor of mouse brain in relation to toxicity. Pestic. Biochem. Physiol. 1997, 58, 77–88. 10.1006/pest.1997.2284. [DOI] [Google Scholar]
- Thurman E. M.; Ferrer I.; Zavitsanos P.; Zweigenbaum J. A. Identification of imidacloprid metabolites in onion (Allium cepa L.) using high-resolution mass spectrometry and accurate mass tools. Rapid Commun. Mass Spectrom. 2013, 27, 1891–1903. 10.1002/rcm.6637. [DOI] [PubMed] [Google Scholar]
- Lee D.-H.; Steffes M. W.; Sjödin A.; Jones R. S.; Needham L. L.; Jacobs D. R. Jr Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS One 2011, 6, e15977. 10.1371/journal.pone.0015977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezg R.; Mornagui B.; El-Fazaa S.; Gharbi N. Organophosphorus pesticides as food chain contaminants and type 2 diabetes: a review. Trends Food Sci. Technol. 2010, 21, 345–357. 10.1016/j.tifs.2010.04.006. [DOI] [Google Scholar]
- Son H.-K.; Kim S.-A.; Kang J.-H.; Chang Y.-S.; Park S.-K.; Lee S.-K.; Jacobs D.; Lee D.-H. Strong associations between low-dose organochlorine pesticides and type 2 diabetes in Korea. Environ. Int. 2010, 36, 410–414. 10.1016/j.envint.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Montgomery M.; Kamel F.; Saldana T.; Alavanja M.; Sandler D. Incident diabetes and pesticide exposure among licensed pesticide applicators: Agricultural Health Study, 1993–2003. Am. J. Epidemiol. 2008, 167, 1235–1246. 10.1093/aje/kwn028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada L. D.; Lara P. C.; Álvarez-León E. E.; Losada A.; Zumbado M. L.; Limiñana-Cañal J. M.; Apolinario R.; Serra-Majem L.; Luzardo O. P. Serum levels of insulin-like growth factor-I in relation to organochlorine pesticides exposure. Growth Horm. IGF Res. 2007, 17, 506–511. 10.1016/j.ghir.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Ruzzin J.; Petersen R.; Meugnier E.; Madsen L.; Lock E.-J.; Lillefosse H.; Ma T.; Pesenti S.; Sonne S. B.; Marstrand T. T.; Malde M. K.; Du Z.-Y.; Chavey C.; Fajas L.; Lundebye A.-K.; Brand C. L.; Vidal H.; Kristiansen K.; Frøyland L. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ. Health Perspect. 2010, 118, 465–471. 10.1289/ehp.0901321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell G. E.; Mulligan C.; Meek E.; Chambers J. E. Effect of chronic p,p′-dichlorodiphenyldichloroethylene (DDE) exposure on high fat diet-induced alterations in glucose and lipid metabolism in male C57BL/6H mice. Toxicology 2015, 328, 112–122. 10.1016/j.tox.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj S.; Srivastava M. K.; Kapoor U.; Srivastava L. P. A 90 days oral toxicity of imidacloprid in female rats: morphological, biochemical and histopathological evaluations. Food Chem. Toxicol. 2010, 48, 1185–1190. 10.1016/j.fct.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Wang C. J.; Wang G.; Wang X. Y.; Liu M.; Chuai M.; Lee K. K.; He X. S.; Lu D. X.; Yang X. Imidacloprid exposure suppresses neural crest cells generation during early chick embryo development. J. Agric. Food Chem. 2016, 64, 4705–4715. 10.1021/acs.jafc.6b01478. [DOI] [PubMed] [Google Scholar]
- Gao L.-r.; Li S.; Zhang J.; Liang C.; Chen E.-n.; Zhang S.-y.; Chuai M.; Bao Y.; Wang G.; Yang X. Excess imidacloprid exposure causes the heart tube malformation of chick embryos. J. Agric. Food Chem. 2016, 64, 9078–9088. 10.1021/acs.jafc.6b03381. [DOI] [PubMed] [Google Scholar]
- Park Y.; Kim Y.; Kim J.; Yoon K. S.; Clark J.; Lee J.; Park Y. Imidacloprid, a neonicotinoid insecticide, potentiates adipogenesis in 3T3-L1 adipocytes. J. Agric. Food Chem. 2013, 61, 255–259. 10.1021/jf3039814. [DOI] [PubMed] [Google Scholar]
- Kim J.; Park Y.; Yoon K. S.; Clark J. M.; Park Y. Imidacloprid, a neonicotinoid insecticide, induces insulin resistance. J. Toxicol. Sci. 2013, 38, 655–660. 10.2131/jts.38.655. [DOI] [PubMed] [Google Scholar]
- Kim J.; Park Y.; Yoon K. S.; Clark J. M.; Park Y. Permethrin alters adipogenesis in 3T3-L1 adipocytes and causes insulin resistance in C2C12 myotubes. J. Biochem. Mol. Toxicol. 2014, 28, 418–424. 10.1002/jbt.21580. [DOI] [PubMed] [Google Scholar]
- Kim J.; Sun Q.; Yue Y.; Yoon K. S.; Whang K. Y.; Marshall Clark J.; Park Y. 4,4′-Dichlorodiphenyltrichloroethane (DDT) and 4,4′-dichlorodiphenyldichloroethylene (DDE) promote adipogenesis in 3T3-L1 adipocyte cell culture. Pestic. Biochem. Physiol. 2016, 131, 40–45. 10.1016/j.pestbp.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Qi W.; Yang J. J.; Yoon K. S.; Clark J. M.; Park Y. Fipronil promotes adipogenesis via AMPKα-mediated pathway in 3T3-L1 adipocytes. Food Chem. Toxicol. 2016, 92, 217–223. 10.1016/j.fct.2016.04.011. [DOI] [PubMed] [Google Scholar]
- California Environmental Protection Agency. Imidacloprid risk characterization document dietary and drinking water exposure, 2016; http://www.cdpr.ca.gov/docs/risk/rcd/imidacloprid.pdf.
- Marshall I B. S.Health investigation level for endosulfan in soil. In Proceedings of the Fifth National Workshop on the Assessment of Site Contamination; Environment Protection and Heritage Council: Adelaide, Australia, 2003; pp 1–10.
- Craig M. S.; Gupta R. C.; Candery T. D.; Britton D. A. Human exposure to imidacloprid from dogs treated with Advantage(R). Toxicol. Mech. Methods 2005, 15, 287–291. 10.1080/15376520590968842. [DOI] [PubMed] [Google Scholar]
- Ayala J. E.; Samuel V. T.; Morton G. J.; Obici S.; Croniger C. M.; Shulman G. I.; Wasserman D. H.; McGuinness O. P. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Models Mech. 2010, 3, 525–534. 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar F. H.; Mojiminiyi O. A.; Al-Rumaih H. M.; Diejomaoh M. F. Computational methods are significant determinants of the associations and definitions of insulin resistance using the homeostasis model assessment in women of reproductive age. Clin. Chem. 2011, 57, 279–285. 10.1373/clinchem.2010.152025. [DOI] [PubMed] [Google Scholar]
- Berry R.; Church C.; Gericke M. T.; Jeffery E.; Colman L.; Rodeheffer M. S. Methods in enzymology (MIE): methods of adipose tissue biology. Chapter 7: Imaging of adipose tissue. Methods Enzymol. 2014, 537, 47–73. 10.1016/B978-0-12-411619-1.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlee S. D.; Lentz S. I.; Mori H.; MacDougald O. A. Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol. 2014, 537, 93–122. 10.1016/B978-0-12-411619-1.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Jackson Laboratory. Diet-induced obesity models, 2016; https://www.jax.org/jax-mice-and-services/find-and-order-jax-mice/surgical-and-preconditioning-services/diet-induced-obesity-models.
- Vroegrijk I. O.; van Klinken J. B.; van Diepen J. A.; van den Berg S. A.; Febbraio M.; Steinbusch L. K.; Glatz J. F.; Havekes L. M.; Voshol P. J.; Rensen P. C.; van Dijk K. W.; van Harmelen V. CD36 is important for adipocyte recruitment and affects lipolysis. Obesity 2013, 21, 2037–2045. 10.1002/oby.20354. [DOI] [PubMed] [Google Scholar]
- Eberle D.; Hegarty B.; Bossard P.; Ferre P.; Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Horton J. D.; Shimomura I.; Ikemoto S.; Bashmakov Y.; Hammer R. E. Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J. Biol. Chem. 2003, 278, 36652–36660. 10.1074/jbc.M306540200. [DOI] [PubMed] [Google Scholar]
- Tzanavari T.; Giannogonas P.; Karalis K. P. TNF-alpha and obesity. Curr. Dir. Autoimmun. 2010, 11, 145–156. 10.1159/000289203. [DOI] [PubMed] [Google Scholar]
- Nieto-Vazquez I.; Fernandez-Veledo S.; Kramer D. K.; Vila-Bedmar R.; Garcia-Guerra L.; Lorenzo M. Insulin resistance associated to obesity: the link TNF-alpha. Arch. Physiol. Biochem. 2008, 114, 183–194. 10.1080/13813450802181047. [DOI] [PubMed] [Google Scholar]
- Viollet B.; Foretz M.; Guigas B.; Horman S.; Dentin R.; Bertrand L.; Hue L.; Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A.; Dickerson K.; Heath R.; Hong S. P.; Momcilovic M.; Johnstone S. R.; Carlson M.; Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Zhang B. B.; Zhou G.; Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Shoji I.; Deng L.; Hotta H. Molecular mechanism of hepatitis C virus-induced glucose metabolic disorders. Front. Microbiol. 2012, 2, 1–5. 10.3389/fmicb.2011.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. 10.1016/j.molmet.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck B. N.; Gropler M. C.; Chen Z.; Leone T. C.; Croce M. A.; Harris T. E.; Lawrence J. C. Jr; Kelly D. P. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 2006, 4, 199–210. 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Rodgers J. T.; Lerin C.; Gerhart-Hines Z.; Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2008, 582, 46–53. 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C.; Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J.; Rustad P. I.; Kolnes A. J.; Lai Y.-C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physiol. 2011, 2, 1–11. 10.3389/fphys.2011.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo R. A.; Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnen A. M.; Leguisamo N. M.; Pinto G. H.; Markoski M. M.; De Angelis K.; Machado U. F.; Schaan B. The beneficial effects of exercise in rodents are preserved after detraining: a phenomenon unrelated to GLUT4 expression. Cardiovasc. Diabetol. 2010, 9, 1–8. 10.1186/1475-2840-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho E.; Kotani K.; Peroni O. D.; Kahn B. B. Adipose-specific overexpression of GLUT4 reverses insulin resistance and diabetes in mice lacking GLUT4 selectively in muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E551–E561. 10.1152/ajpendo.00116.2005. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Hulver M. W.; McMillan R. P.; Cline M. A.; Gilbert E. R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 1–9. 10.1186/1743-7075-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holness M. J.; Sugden M. C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- Wynn R. M.; Kato M.; Chuang J. L.; Tso S. C.; Li J.; Chuang D. T. Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. J. Biol. Chem. 2008, 283, 25305–25315. 10.1074/jbc.M802249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens F. B.; Constantin-Teodosiu D.; Greenhaff P. L. New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. J. Physiol. 2007, 581, 431–444. 10.1113/jphysiol.2006.125799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roegge C. S.; Timofeeva O. A.; Seidler F. J.; Slotkin T. A.; Levin E. D. Developmental diazinon neurotoxicity in rats: later effects on emotional response. Brain Res. Bull. 2008, 75, 166–172. 10.1016/j.brainresbull.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassiter T. L.; Ryde I. T.; Mackillop E. A.; Brown K. K.; Levin E. D.; Seidler F. J.; Slotkin T. A. Exposure of neonatal rats to parathion elicits sex-selective reprogramming of metabolism and alters the response to a high-fat diet in adulthood. Environ. Health Perspect. 2008, 116, 1456–1462. 10.1289/ehp.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassiter T. L.; Ryde I. T.; Levin E. D.; Seidler F. J.; Slotkin T. A. Neonatal exposure to parathion alters lipid metabolism in adulthood: Interactions with dietary fat intake and implications for neurodevelopmental deficits. Brain Res. Bull. 2010, 81, 85–91. 10.1016/j.brainresbull.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adigun A. A.; Wrench N.; Levin E. D.; Seidler F. J.; Slotkin T. A. Neonatal parathion exposure and interactions with a high-fat diet in adulthood: adenylyl cyclase-mediated cell signaling in heart, liver and cerebellum. Brain Res. Bull. 2010, 81, 605–612. 10.1016/j.brainresbull.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin T. A. Does early-life exposure to organophosphate insecticides lead to prediabetes and obesity?. Reprod. Toxicol. 2011, 31, 297–301. 10.1016/j.reprotox.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzguner V.; Erdogan S. Chronic exposure to imidacloprid induces inflammation and oxidative stress in the liver and central nervous system of rats. Pestic. Biochem. Physiol. 2012, 104, 58–64. 10.1016/j.pestbp.2012.06.011. [DOI] [Google Scholar]
- Kapoor U.; Srivastava M. K.; Bhardwaj S.; Srivastava L. P. Effect of imidacloprid on antioxidant enzymes and lipid peroxidation in female rats to derive its No Observed Effect Level (NOEL). J. Toxicol. Sci. 2010, 35, 577–581. 10.2131/jts.35.577. [DOI] [PubMed] [Google Scholar]
- Aydin B. Effects of thiacloprid, deltamethrin and their combination on oxidative stress in lymphoid organs, polymorphonuclear leukocytes and plasma of rats. Pestic. Biochem. Physiol. 2011, 100, 165–171. 10.1016/j.pestbp.2011.03.006. [DOI] [Google Scholar]
- El-Gendy K. S.; Aly N. M.; Mahmoud F. H.; Kenawy A.; El-Sebae A. K. H. The role of vitamin C as antioxidant in protection of oxidative stress induced by imidacloprid. Food Chem. Toxicol. 2010, 48, 215–221. 10.1016/j.fct.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Duzguner V.; Erdogan S. Acute oxidant and inflammatory effects of imidacloprid on the mammalian central nervous system and liver in rats. Pestic. Biochem. Physiol. 2010, 97, 13–18. 10.1016/j.pestbp.2009.11.008. [DOI] [Google Scholar]
- Youn J. Y.; Siu K. L.; Lob H. E.; Itani H.; Harrison D. G.; Cai H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes 2014, 63, 2344–2355. 10.2337/db13-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. L.; Maddux B. A.; Goldfine I. D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signaling 2005, 7, 1040–1052. 10.1089/ars.2005.7.1040. [DOI] [PubMed] [Google Scholar]
- Greenberg A. S.; Obin M. S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [DOI] [PubMed] [Google Scholar]
- Wicovsky A.; Muller N.; Daryab N.; Marienfeld R.; Kneitz C.; Kavuri S.; Leverkus M.; Baumann B.; Wajant H. Sustained JNK activation in response to tumor necrosis factor is mediated by caspases in a cell type-specific manner. J. Biol. Chem. 2007, 282, 2174–2183. 10.1074/jbc.M606167200. [DOI] [PubMed] [Google Scholar]
- Kaneto H.; Nakatani Y.; Kawamori D.; Miyatsuka T.; Matsuoka T.-a. Involvement of oxidative stress and the JNK pathway in glucose toxicity. Rev. Diabet. Stud. 2004, 1, 165–174. 10.1900/RDS.2004.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J.; Tuncman G.; Chang L.; Gorgun C. Z.; Uysal K. T.; Maeda K.; Karin M.; Hotamisligil G. S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Winzell M. S.; Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53 (Suppl. 3), S215–S219. 10.2337/diabetes.53.suppl_3.S215. [DOI] [PubMed] [Google Scholar]
- Borst S. E.; Conover C. F. High-fat diet induces increased tissue expression of TNF-alpha. Life Sci. 2005, 77, 2156–2165. 10.1016/j.lfs.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Aliou Y.; Liao M.-C.; Zhao X.-P.; Chang S.-Y.; Chenier I.; Ingelfinger J. R.; Zhang S.-L. Post-weaning high-fat diet accelerates kidney injury, but not hypertension programmed by maternal diabetes. Pediatr. Res. 2016, 79, 416–424. 10.1038/pr.2015.236. [DOI] [PubMed] [Google Scholar]
- Hong Q.; Xia C.; Xiangying H.; Quan Y. Capsinoids suppress fat accumulation via lipid metabolism. Mol. Med. Rep. 2015, 11, 1669–1674. 10.3892/mmr.2014.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavens K. F.; Easton R. M.; Shulman G. I.; Previs S. F.; Birnbaum M. J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009, 10, 405–418. 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterveer M. H.; van Dijk T. H.; Tietge U. J.; Boer T.; Havinga R.; Stellaard F.; Groen A. K.; Kuipers F.; Reijngoud D. J. High fat feeding induces hepatic fatty acid elongation in mice. PLoS One 2009, 4, e6066. 10.1371/journal.pone.0006066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. N.; Bassilian S.; Ajie H. O.; Schoeller D. A.; Edmond J.; Bergner E. A.; Byerley L. O. In vivo measurement of fatty acids and cholesterol synthesis using D2O and mass isotopomer analysis. Am. J. Physiol. 1994, 266, 699–708. [DOI] [PubMed] [Google Scholar]
- Brunengraber D. Z.; McCabe B. J.; Kasumov T.; Alexander J. C.; Chandramouli V.; Previs S. F. Influence of diet on the modeling of adipose tissue triglycerides during growth. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E917–E925. 10.1152/ajpendo.00128.2003. [DOI] [PubMed] [Google Scholar]
- Doull J.; Klassen C. D.; Amdur M. O.. Cassarett and Doull’s Toxicology. The Basic Science of Poisons, 4th ed.; Pergamon Press: Elmsford, NY, USA, 1991. [Google Scholar]
- Marshall I.; Begg S.. Health Investigation Level for Imidacloprid in Soil; Proceedings of the Fifth National Workshop on the Assessment of Site Contamination, 2003; pp 211–214; http://www.scew.gov.au/system/files/resources/27b740ab-880f-d7f4-d10e-bab0fa964e48/files/asc-wkshoppaper-14-pest-marshall-hil-imidacloprid-200301.pdf (accessed 2016).
- Ng M.; Fleming T.; Robinson M.; Thomson B.; Graetz N.; Margono C.; Mullany E. C.; Biryukov S.; Abbafati C.; Abera S. F.; Abraham J. P.; Abu-Rmeileh N. M. E.; Achoki T.; AlBuhairan F. S.; Alemu Z. A.; Alfonso R.; Ali M. K.; Ali R.; Guzman N. A.; Ammar W.; Anwari P.; Banerjee A.; Barquera S.; Basu S.; Bennett D. A.; Bhutta Z.; Blore J.; Cabral N.; Nonato I. C.; Chang J.-C.; Chowdhury R.; Courville K. J.; Criqui M. H.; Cundiff D. K.; Dabhadkar K. C.; Dandona L.; Davis A.; Dayama A.; Dharmaratne S. D.; Ding E. L.; Durrani A. M.; Esteghamati A.; Farzadfar F.; Fay D. F. J.; Feigin V. L.; Flaxman A.; Forouzanfar M. H.; Goto A.; Green M. A.; Gupta R.; Hafezi-Nejad N.; Hankey G. J.; Harewood H. C.; Havmoeller R.; Hay S.; Hernandez L.; Husseini A.; Idrisov B. T.; Ikeda N.; Islami F.; Jahangir E.; Jassal S. K.; Jee S. H.; Jeffreys M.; Jonas J. B.; Kabagambe E. K.; Khalifa S. E. A. H.; Kengne A. P.; Khader Y. S.; Khang Y.-H.; Kim D.; Kimokoti R. W.; Kinge J. M.; Kokubo Y.; Kosen S.; Kwan G.; Lai T.; Leinsalu M.; Li Y.; Liang X.; Liu S.; Logroscino G.; Lotufo P. A.; Lu Y.; Ma J.; Mainoo N. K.; Mensah G. A.; Merriman T. R.; Mokdad A. H.; Moschandreas J.; Naghavi M.; Naheed A.; Nand D.; Narayan K. M. V.; Nelson E. L.; Neuhouser M. L.; Nisar M. I.; Ohkubo T.; Oti S. O.; Pedroza A.; Prabhakaran D.; Roy N.; Sampson U.; Seo H.; Sepanlou S. G.; Shibuya K.; Shiri R.; Shiue I.; Singh G. M.; Singh J. A.; Skirbekk V.; Stapelberg N. J. C.; Sturua L.; Sykes B. L.; Tobias M.; Tran B. X.; Trasande L.; Toyoshima H.; van de Vijver S.; Vasankari T. J.; Veerman J. L.; Velasquez-Melendez G.; Vlassov V. V.; Vollset S. E.; Vos T.; Wang C.; Wang X.; Weiderpass E.; Werdecker A.; Wright J. L.; Yang Y. C.; Yatsuya H.; Yoon J.; Yoon S.-J.; Zhao Y.; Zhou M.; Zhu S.; Lopez A. D.; Murray C. J. L.; Gakidou E. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study. Lancet 2013, 384, 766–781. 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E. S.; Giles W. H.; Mokdad A. H. Increasing prevalence of the metabolic syndrome among u.s. Adults. Diabetes Care 2004, 27, 2444–2449. 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- Alonso-Magdalena P.; Morimoto S.; Ripoll C.; Fuentes E.; Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. 10.1289/ehp.8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelishadi R.; Mirghaffari N.; Poursafa P.; Gidding S. S. Lifestyle and environmental factors associated with inflammation, oxidative stress and insulin resistance in children. Atherosclerosis 2009, 203, 311–319. 10.1016/j.atherosclerosis.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Yue P.; Deiuliis J. A.; Lumeng C. N.; Kampfrath T.; Mikolaj M. B.; Cai Y.; Ostrowski M. C.; Lu B.; Parthasarathy S.; Brook R. D.; Moffatt-Bruce S. D.; Chen L. C.; Rajagopalan S. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009, 119, 538–546. 10.1161/CIRCULATIONAHA.108.799015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.