

Abstract
High-risk human papillomaviruses (HPVs) encode oncoproteins which manipulate gene expression patterns in the host keratinocytes to facilitate viral replication, regulate viral transcription, and promote immune evasion and persistence. In some cases, oncoprotein-induced changes in host cell behavior can cause progression to cancer, but a complete picture of the functions of the viral oncoproteins in the productive HPV life cycle remains elusive. E7 is the HPV-encoded factor most responsible for maintaining cell cycle competence in differentiating keratinocytes. Through interactions with dozens of host factors, E7 has an enormous impact on host gene expression patterns. In this review, we will examine the role of E7 specifically as a regulator of transcription. We will discuss mechanisms of regulation of cell cycle-related genes by E7 as well as genes involved in immune regulation, growth factor signaling, DNA damage responses, microRNAs, and others pathways. We will also discuss some unanswered questions about how transcriptional regulation by E7 impacts the biology of HPV in both benign and malignant conditions.
Keywords: E7, pRb family, histone deacetylase, transcription factor, innate immunity, cell cycle control
1. Introduction
Human papillomaviruses (HPVs) are small DNA viruses that cause benign and malignant lesions of mucosal and cutaneous stratified squamous epithelia, including uterine cervix, vagina, vulva, penis, and increasingly the oropharynx (reviewed in (zur Hausen 2009; Moody and Laimins 2010; Forman, de Martel et al. 2012)). Genital HPVs are the most common sexually transmitted infectious agents and are extremely widespread (Weinstock, Berman et al. 2004; Forman, de Martel et al. 2012). There are over 200 HPV types, which can be broadly classified into “high-risk” (cancer-associated) types and “low-risk” (non-cancer-associated) types (Klingelhutz and Roman 2012). HPV16 is the high-risk HPV responsible for the majority of cervical cancers (Lorincz, Reid et al. 1992; Lombard, Vincent-Salomon et al. 1998; Forman, de Martel et al. 2012), and so is highly represented in the HPV literature, including this review. Other high-risk types include HPV18 and 31, while HPV6 and 11 are prototypical low-risk types.
High-risk HPV infections usually last 1–2 years (Scott, Nakagawa et al. 2001; Moscicki, Schiffman et al. 2012), which means that HPV can replicate in host tissues for months in the face of constant cellular turnover and immune surveillance. Its ability to do so depends largely on its potential to regulate both viral and host gene expression (Frazer 2009; Bodily and Laimins 2011). Regulation of viral gene transcription is linked to differentiation of host keratinocytes (Moody and Laimins 2010; Bodily and Laimins 2011). Consequently, capsid proteins are only detectible upon terminal differentiation of the host cell, restricting high levels of viral antigens to tissue strata expressing lower levels of major histocompatibility complex (MHC) type I and other immune surveillance molecules (Gielen, Schmitt et al. 1988; Nardelli, Zaritskaya et al. 2002; Caberg, Hubert et al. 2008). In addition to its differentiation-dependent life cycle organization, HPV actively suppresses both innate and adaptive immune responses. Uninfected keratinocytes constitutively express low levels of a variety of cytokines (Wang, Amerio et al. 1999), but HPV-positive keratinocytes show significantly reduced expression of many inflammatory mediators (Arany and Tyring 1996; Fichorova and Anderson 1999; Alcocer-Gonzalez, Berumen et al. 2006). HPV also inhibits the differentiation and function of Langerhans cells, interferon (IFN) responses, MHC-I expression, and other immune mediators, through the regulation of host gene transcription patterns (Spinillo, Tenti et al. 1993; Bielenberg, McCarty et al. 1999; Connor, Ferrer et al. 1999; Georgopoulos, Proffitt et al. 2000; Delvenne, Hubert et al. 2001; Scott, Nakagawa et al. 2001; Matthews, Leong et al. 2003; Guess and McCance 2005; Hubert, Caberg et al. 2005; Herman, Hubert et al. 2006; Li, Ou et al. 2006; Caberg, Hubert et al. 2008; Caberg, Hubert et al. 2009; Tirone, Peghini et al. 2009; Leong, Doorbar et al. 2010; Li, Deng et al. 2010; Deng, Li et al. 2011; Heller, Weisser et al. 2011; Hong, Mehta et al. 2011; Karim, Meyers et al. 2011; Reiser, Hurst et al. 2011; Abd Warif, Stoitzner et al. 2015). HPV also has a significant impact on the growth factor environment of the host cell, thereby creating a tissue environment within which viral infection can persist (Bequet-Romero and Lopez-Ocejo 2000; Nakamura, Bodily et al. 2009; Walker, Smiley et al. 2011; Dimaio and Petti 2013).
The HPV oncoprotein E7 is essential for the normal virus life cycle and throughout the cancer development pathway from benign precursor lesions to invasive carcinomas (Thomas, Hubert et al. 1999; Flores, Allen-Hoffmann et al. 2000; Munger, Baldwin et al. 2004; Jabbar, Abrams et al. 2009; Bodily, Mehta et al. 2011). E7 acts primarily as a transcriptional regulator. The best-recognized mechanism of gene regulation by E7 is through the retinoblastoma (pRb)/E2F system. pRb family proteins, also called “pocket proteins,” function in part by binding to E2F transcription factors and recruiting transcriptional inhibitory complexes to promoters (Brehm, Miska et al. 1998; Cobrinik 2005; Sadasivam and DeCaprio 2013). E7 binds to and targets pRb family members for degradation thereby disrupting inhibitory complexes and facilitating the expression of DNA synthesis machinery in otherwise post-mitotic, differentiated keratinocytes (Watt 1998; Nees, Geoghegan et al. 2000; Munger, Baldwin et al. 2004; McLaughlin-Drubin and Munger 2009; Roman and Munger 2013). However, elimination of pRb family members cannot entirely account for the impact of E7 on host cells (Balsitis, Sage et al. 2003; Balsitis, Dick et al. 2006; Shin, Sage et al. 2012), indicating that there are other functionally important mechanisms by which E7 acts to support the viral life cycle and cancer development.
In addition to the pRb/E2F system, E7 suppresses many genes regulated through the NFκB and IFN pathways (Barnard and McMillan 1999; Chang and Laimins 2000; O’Brien and Saveria Campo 2002; Spitkovsky, Hehner et al. 2002; De Andrea, Mondini et al. 2007; Li, Zhan et al. 2009; Heller, Weisser et al. 2011; Hong, Mehta et al. 2011; Reiser, Hurst et al. 2011; Vandermark, Deluca et al. 2012; Zhou, Chen et al. 2013), thereby enabling the evasion of innate and adaptive immunity. The IFN response is particularly important in suppressing HPV replication and persistence (Barnard and McMillan 1999; Nees, Geoghegan et al. 2001; Herdman, Pett et al. 2006; Hong, Mehta et al. 2011; Reiser, Hurst et al. 2011). Transforming growth factor (TGF)-β signaling, which arrests keratinocyte proliferation, inhibits viral gene expression, and promotes immunity, is also counteracted by E7 (Pietenpol, Stein et al. 1990; Woodworth, Notario et al. 1990; Moses 1992; Creek, Geslani et al. 1995; Ozbun and Meyers 1996; Nees, Geoghegan et al. 2000; Murvai, Borbely et al. 2004; Habig, Smola et al. 2006). HIF1, KLF4, p73, p53, and AP-1 are among the many other reported E7 targets (Morosov, Phelps et al. 1994; Massimi and Banks 1997; Brooks, Sullivan et al. 2002; Eichten, Westfall et al. 2002; Nakamura, Bodily et al. 2009; Bodily, Mehta et al. 2011; Gunasekharan, Li et al. 2016). In addition to these host factors, we have found that E7 plays a role in the regulation of the HPV16 late promoter (Bodily, Hennigan et al. 2013). Dozens of interactions have been reported between E7 and cellular factors, including transcriptional regulatory proteins such as histone deacteylases (HDACs), DNA methyltransferases (DNMTs), histone acetyl transferases (HATs), general and promoter-specific transcription factors, and others (Roman and Munger 2013). How these interactions work together to promote virus production or carcinogenesis, whether in cell culture or animal models of cancer, is not yet known.
In this review, we will discuss E7 specifically as a regulator of gene expression. E7 possesses some activities and interactions not directly related to transcription (Roman and Munger 2013), but because the theme of transcriptional regulation can unify the major functions of E7, that will be our focus. Although we will review the interaction of E7 with the pRb/E2F system, we will touch only briefly on the role of E7 in transformation or immortalization, as these topics have frequently been reviewed by others (Wise-Draper and Wells 2008; McLaughlin-Drubin and Munger 2009; Ghittoni, Accardi et al. 2010; Moody and Laimins 2010; Klingelhutz and Roman 2012). We will also not examine the effects of E7 on large arrays of genes that are downstream of many signaling pathways regulated by E7. Instead we will discuss what is known of the mechanisms used by E7 to regulate viral and host gene expression and focus on genes that are more or less direct targets. Our goal is to better understand how E7 impacts the transcriptional process.
1.1. A tour of E7
HPV16 E7, which is the subject of most of the papers reviewed here, has 98 amino acids and no known enzymatic activities or DNA binding domain (Figure 1)(Munger, Baldwin et al. 2004). Sequences in the N terminus similar to domains of adenovirus E1A and polyomavirus T antigens are designated conserved region 1 (CR1) and CR2 (Phelps, Yee et al. 1988). A C terminal zinc finger-like domain is also similar to other viral oncoproteins and is designated CR3 (Phelps, Yee et al. 1988). These structural similarities are reflected in functional similarities in transformation and transactivation between the viral oncoproteins (Phelps, Yee et al. 1988; Berk 2005).
Figure 1.
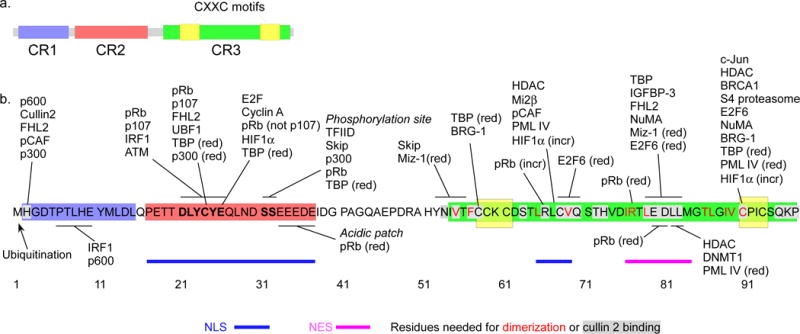
a. The CR1 (blue), CR2 (red) and CR3 (green) domains of E7, along with the zinc-coordinating CXXC motifs (yellow). b. Sites of interaction in E7 with known transcriptional regulators. Numbers represent amino acid positions. Factors that are not involved in transcription or whose binding sites in E7 have not been mapped by mutation are not shown. In some cases binding is reduced (indicated “red”) or increased (indicated “incr”) by mutation of a specific residue. The location of the NLSs (blue bars) and NES (pink bar) are indicated (Knapp, McManus et al. 2009; Eberhard, Onder et al. 2013). Residues colored red are important for dimerization and residues shaded grey are important for cullin 2 binding (Huh, Zhou et al. 2007; Todorovic, Hung et al. 2012).
The E7 N terminus is intrinsically disordered and flexible, perhaps accounting for its promiscuous binding abilities (Heck, Yee et al. 1992; Calcada, Felli et al. 2013; Lee, Kim et al. 2016). The N terminus includes both CR1 and CR2. The CR2 region contains the LXCXE motif and the primary phosphorylation site. The LXCXE motif is similar to motifs found in other viral and host factors that bind to pocket proteins, and it is essential for E7-pocket protein interaction (Munger, Baldwin et al. 2004; Dick and Rubin 2013). Although sometimes called the “pRb binding domain,” the LXCXE sequence is also important for binding other factors in addition to pocket proteins (Roman and Munger 2013). Although the LXCXE motif is critical for binding pRb, sequences in the CR1 region are needed for pRb degradation (Helt and Galloway 2001; Munger, Baldwin et al. 2004). Among several factors that bind to the CR1 region is the cullin 2 ubiquitin ligase complex which is thought to be important for the degradation of pRb (Huh, Zhou et al. 2007), although this is controversial (Todorovic, Hung et al. 2012). The roles of CR1 and CR2 in E7 function will be discussed extensively below.
Similar to other viral oncoproteins such as HPV E6 and human adenovirus E1A and E1B, HPV E7 is a phosphoprotein, and its phosphorylation is thought to be important for its function and regulation (Barbosa, Edmonds et al. 1990; Helt and Galloway 2003; Ching, Dobner et al. 2012; Boon, Tomaic et al. 2015). The primary site of phosphorylation is a pair of serines at positions 31 and 32 in CR2 which are phosphorylated by casein kinase II (CKII)(Firzlaff, Galloway et al. 1989; Barbosa, Edmonds et al. 1990; Chien, Parker et al. 2000). Whether CKII is the only kinase that phosphorylates these residues in vivo is not known. Serine 71 in the C terminus is also phosphorylated by an unknown cellular kinase (Massimi and Banks 2000), but we will refer to the S31–32 residues as the “phosphorylation site.” The phosphorylation level of E7 of high-risk HPV types is greater than that of low-risk HPV types, and mutation of the phosphorylation site has either significant or modest effects on pocket protein binding, degradation, cell transformation, and progeny virus production, depending on the model system and HPV type (Edmonds and Vousden 1989; Banks, Edmonds et al. 1990; Barbosa, Edmonds et al. 1990; Chesters, Vousden et al. 1990; Firzlaff, Luscher et al. 1991; Phelps, Munger et al. 1992; Demers, Espling et al. 1996; Jones, Thompson et al. 1997; Thomas, Hubert et al. 1999; Chien, Parker et al. 2000; Southern, Lewis et al. 2004; Tugizov, Berline et al. 2005; Genovese, Banerjee et al. 2008; Bodily, Mehta et al. 2011). In the case of HPV16 E7, phosphorylation by CKII may be cell cycle dependent (Massimi and Banks 2000). Phosphorylation increases the binding affinity of E7 to general transcription factors (GTFs) such as TATA box-binding protein (TBP) and TBP-associated factor 110 (TAF-110)(McDougall 1994; Mazzarelli, Atkins et al. 1995; Massimi, Pim et al. 1996; Maldonado, Cabrejos et al. 2002). HPV16 E7 phosphorylation is required for association of E7 with p300 and p53 (Massimi and Banks 1997; Rey, Lee et al. 2000; Bernat, Avvakumov et al. 2003). Mutation of the phosphorylation site does not disrupt episomal maintenance of HPV16 (Bodily, Mehta et al. 2011) or HPV31 (Thomas, Hubert et al. 1999) but does reduce the yield of infectious virus particles and prevents induction of cyclin A during differentiation (Bodily, Mehta et al. 2011). A patch of acidic amino acids immediately downstream of the phosphorylation site contributes to binding to pRb via ionic interactions with basic residues in pRb (Dick and Dyson 2002) and is necessary for phosphorylation of E7 by CKII (Phelps, Munger et al. 1992). Phosphorylation of S31–32 essentially increases the size of the acidic patch, suggesting that these residues may function as a unit.
The C terminal domain or CR3 of E7 is more structured than the N terminus. It consists of an atypical zinc finger-like fold and is responsible for E7 dimerization via a broad hydrophobic surface (Phelps, Yee et al. 1988; McIntyre, Frattini et al. 1993; Clemens, Brent et al. 1995; Liu, Clements et al. 2006). Although the functional role of dimerization is not clear and it is not required for transformation, dimerization is important for maintaining the tertiary structure of the CR3 domain (Liu, Clements et al. 2006; Todorovic, Massimi et al. 2011). Zinc is coordinated by two conserved CXXC motifs in the CR3 domain. There is a secondary binding site for pRb in the C terminus, and the CR3 domain alone can disrupt pRb/E2F complexes, although at reduced levels compared to full length E7 (Liu, Clements et al. 2006). The CR3 domain has transcriptional activation potential analogous to the CR3 domain of E1A (Rawls, Pusztai et al. 1990; Berk 2005), and binding of E7 to a range of transcriptional regulatory proteins can be disrupted by mutations in the CR3 domain (Roman and Munger 2013). However, some CR3 mutants used over the years for functional studies may alter the overall structure of the zinc finger rather than affect specific protein-protein interactions (McIntyre, Frattini et al. 1993; Clemens, Brent et al. 1995; Liu, Clements et al. 2006), so it is unclear how many of the reported interactions with the C terminus are physiological. A new series of surface mutations based on the C terminal crystal structure has been partially characterized and should yield additional insight (Todorovic, Massimi et al. 2011).
E7 has several other interesting features. Endogenous E7 is predominantly nuclear with some cytoplasmic localization and evidence of shuttling (Guccione, Massimi et al. 2002; Dreier, Scheiden et al. 2011; Laurson and Raj 2011), consistent with both nuclear and cytoplasmic binding partners (Roman and Munger 2013). There is a nuclear localization sequence (NLS) in the N terminus and one in the C terminus of E7 (Knapp, McManus et al. 2009; Eberhard, Onder et al. 2013). There is also a nuclear export sequence (NES) in the C terminus (Knapp, McManus et al. 2009). Nuclear localization requires an intact zinc finger but not the phosphorylation site or the LXCXE motif (Knapp, McManus et al. 2009; Eberhard, Onder et al. 2013). Localization may depend on cell cycle stage or cellular confluency (Dreier, Scheiden et al. 2011; Laurson and Raj 2011). E7 contains free cysteine residues that can exist in different oxidation states and affect protein structure (Chemes, Camporeale et al. 2014). Given that stratified epithelia form a redox gradient during differentiation (Conway, Alam et al. 2009), these cysteines may have regulatory consequences. E7 can be ubiquitinated at the N terminal amine group and subjected to proteolysis (Reinstein, Scheffner et al. 2000; Munger, Basile et al. 2001; Wang, Sampath et al. 2001), but E7 turnover is not needed for pRb degradation (Gonzalez, Stremlau et al. 2001).
1.2. Biological activities of E7
E7 plays a role early in the viral life cycle. HPV16 (Bodily, Mehta et al. 2011) and HPV31 (Thomas, Hubert et al. 1999) genomes containing translational termination mutations in E7 cannot immortalize keratinocytes or maintain themselves episomally. E7 is not needed for transient genome replication (Thomas, Hubert et al. 1999), so failure of E7 mutants to be maintained episomally may be a consequence of the inability of the viral genome to immortalize the host cell. This idea is supported by the finding that HPV16 is maintained episomally at normal copy number in the absence of E7 if the host keratinocytes are already immortalized (Flores, Allen-Hoffmann et al. 2000; Collins, Nakahara et al. 2005). The LXCXE motif of E7 is indispensable for early viral activities, including immortalization, in the context of the complete genome in primary keratinocytes (Longworth and Laimins 2004; Bodily, Mehta et al. 2011).
E7 also acts later on in the life cycle. HPV18 E7 is not necessary for immortalization or episomal replication in keratinocytes (McLaughlin-Drubin, Bromberg-White et al. 2005), indicating that 18E7 plays a different role in immortalization and maintenance as compared to 16E7 or 31E7. But both 16E7 and 18E7 are necessary for genome amplification and virion morphogenesis under differentiating conditions (Flores, Allen-Hoffmann et al. 2000; Collins, Nakahara et al. 2005; McLaughlin-Drubin, Bromberg-White et al. 2005). E7 can uncouple host cell differentiation from cell cycle arrest (Munger, Basile et al. 2001), something that also happens when pRb is knocked out in the skin (Ruiz, Santos et al. 2004). In cervical lesions, there is a small overlap between cells expressing MCM7 (which is a replication marker used as a surrogate for E7) and E4, suggesting that E7 levels decline just as the viral late promoter is induced (Isaacson Wechsler, Wang et al. 2012). Mutations of the HPV16 phosphorylation site and CR3 primarily impact the differentiation-dependent phase of the life cycle to reduce viral titers, although per-particle infectivity is not altered (Bodily, Mehta et al. 2011). HPV genomes lacking E7 are unable to induce DNA synthesis or markers of active cell cycling in suprabasal epithelial layers (Flores, Allen-Hoffmann et al. 2000; Isaacson Wechsler, Wang et al. 2012), suggesting that induction of the host DNA replication machinery may be a critical function of E7 in differentiating cells. However, genomes with mutations in pRb binding or degradation domains are still able to make late gene products in previously immortalized cells, even though they are reduced for the ability to induce suprabasal DNA synthesis, indicating that ability to induce DNA synthesis is genetically separable from late gene expression (Collins, Nakahara et al. 2005). E7 can delay differentiation in a manner that depends on both CR1 and CR2, but inducing suprabasal DNA synthesis depends only on CR2 (Noya, Chien et al. 2002; Collins, Nakahara et al. 2005), indicating that delaying differentiation and inducing DNA synthesis are also genetically separable. Using conditional nuclear localization, it was found that E7 can induce DNA synthesis in suprabasal layers of organotypic cultures, even in cells that were differentiated and quiescent before the activation of E7 (Banerjee, Genovese et al. 2006). E7 is therefore able to reestablish S phase de novo in differentiated cells, but whether E7 in real infections forces differentiated cells to re-enter S phase or instead prevents cell cycle withdrawal in the first place is not known. It is possible E7 targets different pocket proteins at different stages of differentiation (Collins, Nakahara et al. 2005)(see below), but this also remains to be fully understood.
E7 and the other major viral oncoprotein E6 are the only two viral genes which are consistently retained in all cervical cancers (Munger, Baldwin et al. 2004). E7’s ability to transform cells has long been known, and mutations in CR1, CR2, and C3 have all been found to affect transformation, depending on the model system (Edmonds and Vousden 1989; Phelps, Munger et al. 1992; Schmitt, Harry et al. 1994; Todorovic, Massimi et al. 2011). Moreover, E7 expressed alone as a transgene in the skins of mice disrupts differentiation and can cause cutaneous tumors, although these tumors are mostly benign and occur late in life (Herber, Liem et al. 1996; Gulliver, Herber et al. 1997). Interestingly, in order to cause tumors in the cervical epithelium, estrogen is required as a co-carcinogen (Riley, Duensing et al. 2003). Continuous expression of E7 is required for maintenance of tumors in transgenic mice (Jabbar, Abrams et al. 2009). Binding to and inducing pRb degradation is largely responsible for E7’s oncogenic activities in the mouse skin but other targets are important in the mouse cervix (Gulliver, Herber et al. 1997; Balsitis, Sage et al. 2003; Balsitis, Dick et al. 2005; Balsitis, Dick et al. 2006).
Because HPV E6 and E7 are always expressed together in nature (Munger, Baldwin et al. 2004), it can be difficult to distinguish the separate contributions of each oncoprotein in physiological systems. Moreover, the genes regulated by each protein when expressed alone may be different from those regulated by the two working together. Over the years, many genome-wide studies have investigated the host gene expression patterns in cells containing E7, either alone or in combination with E6, in a variety of model systems (Chang and Laimins 2000; Nees, Geoghegan et al. 2000; Nees, Geoghegan et al. 2001; Garner-Hamrick, Fostel et al. 2004; Johung, Goodwin et al. 2007; Lu, Wright et al. 2007; Cortes-Malagon, Bonilla-Delgado et al. 2013; Gyongyosi, Szalmas et al. 2014; Zhou, Zhang et al. 2016). Cell cycle-related genes have consistently been found to be activated by E7, including not only genes activated at G1, but also G2/M phase genes (Nees, Geoghegan et al. 2000; Garner-Hamrick, Fostel et al. 2004; Johung, Goodwin et al. 2007; Zhou, Zhang et al. 2016). In addition, differentiation-related genes (Nees, Geoghegan et al. 2000; Johung, Goodwin et al. 2007; Gyongyosi, Szalmas et al. 2014), innate immune response genes (Chang and Laimins 2000; Nees, Geoghegan et al. 2001; Cortes-Malagon, Bonilla-Delgado et al. 2013; Zhou, Zhang et al. 2016), DNA damage and stress response genes (Zhou, Zhang et al. 2016), and growth factors (Nees, Geoghegan et al. 2000) have appeared repeatedly. In studies that take differentiation into account, E7 has been found to have a more profound effect in differentiating cells than in undifferentiated cells (Nees, Geoghegan et al. 2001; Vazquez-Ortiz, Garcia et al. 2007).
2. Transcriptional regulation
The primary role of E7 is as a regulator of transcription. The molecular interactions that result in the activation or inhibition of the transcriptional process are complex and have been extensively reviewed by others (Li, Carey et al. 2007; Lenhard, Sandelin et al. 2012; Allen and Taatjes 2015; Zhang, Cooper et al. 2015), so we will give only a brief outline here. The pattern of modifications on histone proteins is thought to comprise a complex, interactive “histone code,” which serves both to communicate information to transcriptional regulatory proteins and to physically alter the structure of the chromatin in order to increase or decrease promoter activity (Zhang, Cooper et al. 2015). Enzymes that modify histones and other chromatin components are designated “writer” proteins, and include HATs, methyltransferases, ubiquitin ligases, and others. Proteins that recognize these modifications are referred to as “reader” proteins. Reader proteins bind to the modified histone moieties and recruit other proteins to the growing transcriptional complex. Finally, “eraser” enzymes remove modifying groups from the histones (Zhang, Cooper et al. 2015). Erasers include HDACs, demethylases, deubiquitinases, and so forth (Li, Carey et al. 2007).
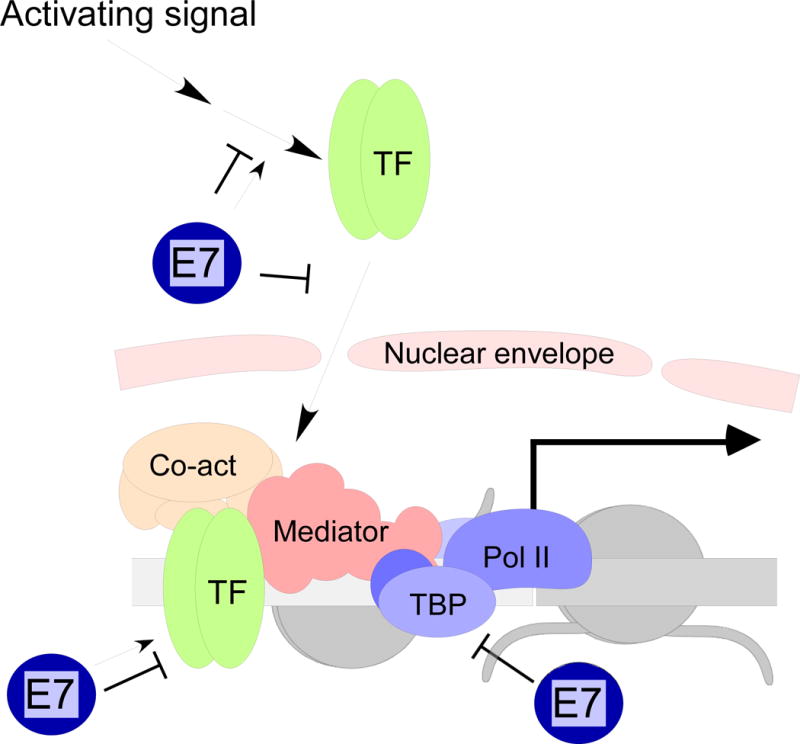
The current model of transcriptional activation begins with chromatin in a closed or inactive conformation, possessing a characteristic pattern of histone modifications (Figure 2). Inactive chromatin is not accessible to most transcriptional activators or to the general transcription machinery, which includes the GTFs (TFIID, TFIIH, etc.) and RNA polymerase II (Pol II). In addition to repressive modifications on the histones, the DNA itself may undergo inactivating modifications, primarily through CpG methylation. However, some transcription factor binding sites are thought to remain accessible even in the context of inactive chromatin. “Pioneer” transcription factors binding to these accessible sites recruit chromatin remodeling enzymes which modify the chromatin to promote a more open conformation. The best known enzymes that promote chromatin accessibility are the HATs, which acetylate histone tails. Transcriptionally active chromatin is characterized by high levels of histone acetylation, which is thought to reduce interactions between nucleosomes to promote chromatin accessibility, as well as providing recognition sites for reader proteins (Zhang, Cooper et al. 2015). As more modifications occur, reader proteins are recruited, which interact to promote binding of additional transcriptional effectors (Zhang, Cooper et al. 2015). As transcription factors accumulate on the promoter, the Mediator complex is recruited. Mediator is a large, multi-subunit complex that serves as a bridge between promoter-specific transcription factors and the GTFs (Allen and Taatjes 2015). Facilitated by Mediator, the GTFs are assembled. They, in turn, recruit Pol II and promote transcriptional initiation (Allen and Taatjes 2015). Transcription can also be regulated at steps following transcriptional initiation, through polymerase pausing, transcript elongation, and termination (Lenasi and Barboric 2010; Jonkers and Lis 2015; Proudfoot 2016), but the effect of E7 on these later processes is not known.
Figure 2.
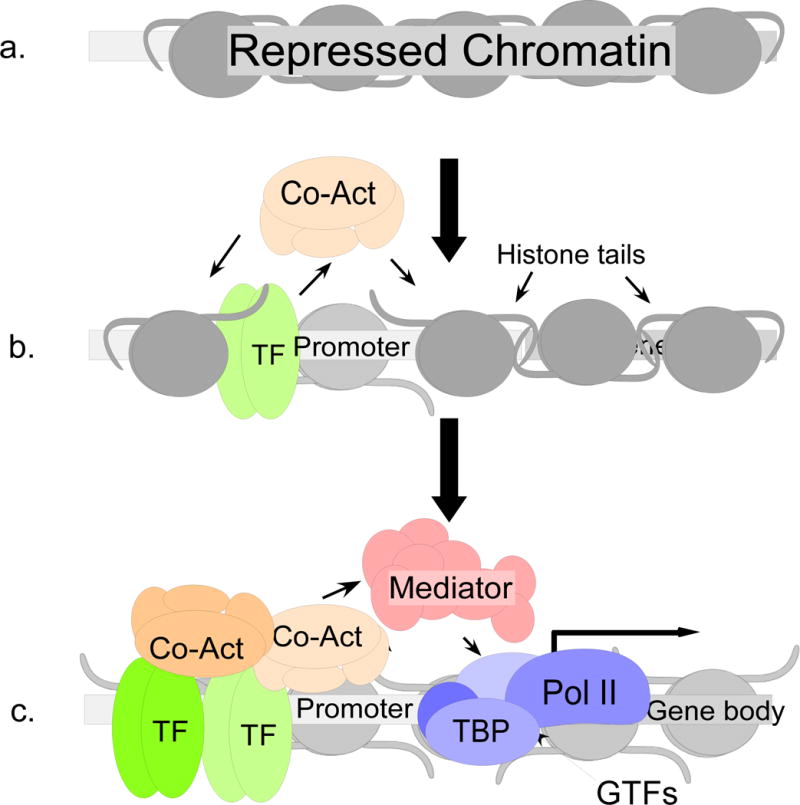
A simplified depiction of promoter activation. a. Repressed or closed chromatin prevents the binding of transcriptional regulators to the promoter region, in part because of the tight inter-nucleosomal interactions between histone tails. b. Binding of pioneer transcription factors results in the recruitment of histone and chromatin modifying enzymes, or co-activators (co-act), which modify histone tails and chromatin structure to make it more accessible for binding of other protein complexes. c. In active chromatin, additional transcription factors and co-activator proteins are recruited, including the Mediator complex, which recruits the GTFs and Pol II close to the transcriptional start site to form a pre-initiation complex. Through the action of the GTFs, the pre-initiation complex transitions to active transcription.
3. Effects of E7 on chromatin structure
E7 can associate with and alter the activities of enzymes that regulate chromatin structure.
HDACs
Because they remove acetyl groups from histones, HDACs generally repress transcription and associate with many transcriptionally repressive complexes (Frolov and Dyson 2004; Sadasivam and DeCaprio 2013; Zhang, Cooper et al. 2015). HDAC1 and -2 are components of nucleosome remodeling and deacetylase (NURD) complexes which also contain the chromatin remodeling ATPase Mi2β (Denslow and Wade 2007). E7 can bind to Mi2β and thus associate with HDACs in cells and in vitro (Brehm, Nielsen et al. 1999; Longworth and Laimins 2004). It is not known whether E7 can associate with HDACs other than in the context of NURD. Mutations in the CR3 region that disrupt the zinc finger domain fail to bind to HDAC complexes (Brehm, Nielsen et al. 1999; Longworth and Laimins 2004). Because HDACs immunoprecipitated with E7 are still active, E7 does not inhibit HDAC activity through binding (Brehm, Nielsen et al. 1999), but several instances of transcriptional regulation by E7 have been attributed to the ability of E7 to bind to HDACs. For example, in cells containing HPV31, levels of E2F2, a cell cycle promoting E2F family member, are increased in both differentiated and undifferentiated conditions (Longworth, Wilson et al. 2005). The ability to upregulate E2F2 is abrogated by mutations of either the LXCXE motif or the HDAC binding site in the C terminus (Longworth, Wilson et al. 2005). Importantly, HPV31 E7 is able to reduce the levels of HDAC1 and -3 found at the E2F2 promoter (Figure 3a), suggesting that the ability to displace HDACs from the promoter may be a significant mechanism of transcriptional activation (Longworth, Wilson et al. 2005). E7 binding to HDACs can repress transcription as well as activate it. E7 from both high and low-risk HPVs can interact with interferon response factor 1 (IRF1) via the CR1 and LXCXE motifs and can then recruit HDACs via the CR3 domain to suppress IRF1 transcriptional activity (Park, Kim et al. 2000; Um, Rhyu et al. 2002)(Figure 3b). Other examples of regulation of transcription by E7 through manipulating HDAC binding or recruitment will be discussed below.
Figure 3.
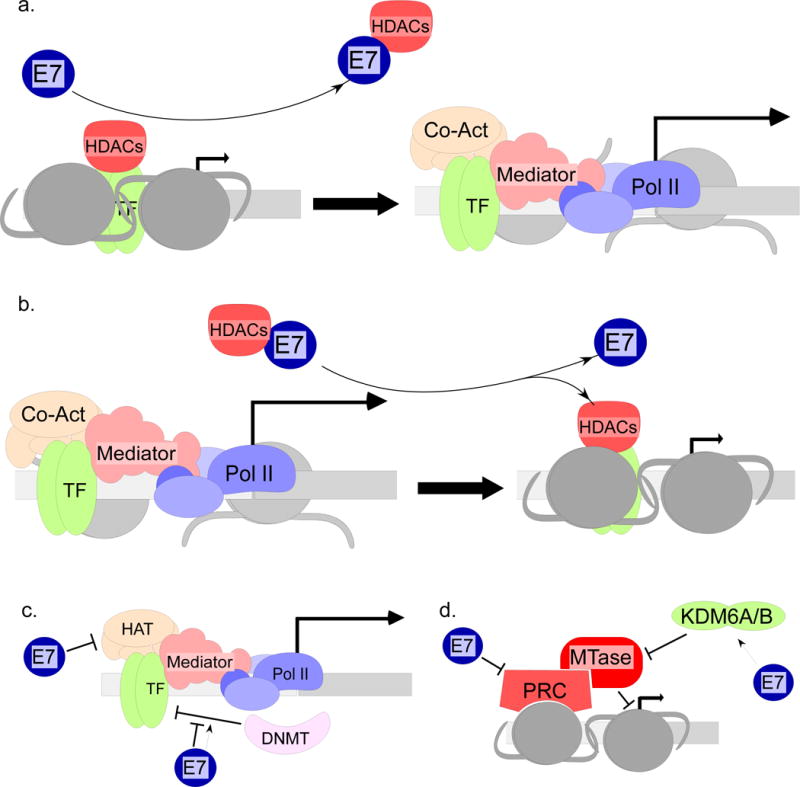
Regulation of chromatin structure by E7. a. E7 can bind to HDACs and displace them from promoters, such as the E2F2 promoter (Longworth, Wilson et al. 2005). E7 can also displace HDACs from binding to transcription factors, such as HIF1α (Bodily, Mehta et al. 2011). b. Alternatively, E7 can recruit HDACs to promoters in a transcription factor-specific manner, as in the case of MHC-I and TLR9 (Park, Kim et al. 2000; Um, Rhyu et al. 2002; Hasan, Zannetti et al. 2013). c. E7 can bind to HAT co-activators and inhibit their activity. pCAF, p300/CBP, Skip, and SRC are examples (Prathapam, Kuhne et al. 2001; Huang and McCance 2002; Avvakumov, Torchia et al. 2003; Bernat, Avvakumov et al. 2003; Baldwin, Huh et al. 2006; Fera and Marmorstein 2012; Jansma, Martinez-Yamout et al. 2014). Binding of E7 to HATs results in the inhibition of the interleukin 8 (IL8) gene (Huang and McCance 2002) and possibly contributes to the inhibition of p53-mediated transcription (Bischof, Nacerddine et al. 2005; Fera and Marmorstein 2012). E7 can also upregulate transcriptional co-activator Sirt1 (Langsfeld, Bodily et al. 2015). E7 can either promote or prevent promoter DNA methylation by DNMTs (Laurson, Khan et al. 2010; Chalertpet, Pakdeechaidan et al. 2015; Cicchini, Westrich et al. 2016; Yin, Wang et al. 2016). d. PRCs inhibit transcription by recruiting histone methyltransferases (MTases) to promoters. E7 suppresses the expression of PRC components and upregulates the demethylase enzymes KDM6A and KDM6B (Hyland, McDade et al. 2011; McLaughlin-Drubin, Crum et al. 2011).
The HDAC binding function of E7 is important in the context of the viral life cycle. The L67R and C91G mutations, which abrogate HDAC binding, abolish episomal maintenance of HPV31 genomes (Longworth and Laimins 2004). In HPV16, the L67R and C91S mutants are able to immortalize and be maintained episomally but show reduced late gene expression and are totally defective for the production of infectious virus particles (Bodily, Mehta et al. 2011). It should be noted that the role of E7 binding to HDAC complexes has been inferred because of the phenotypes of the L67R and C91G mutations. These mutants, although genuinely defective for binding HDACs, may also have disruptions in the overall structure of the C terminus, and thus of other interactions mediated by the CR3 region (McIntyre, Frattini et al. 1993; Clemens, Brent et al. 1995; Liu, Clements et al. 2006). Further work in this area will be required to confirm that the interaction between E7 and HDACs is relevant to the phenotypes of these mutants.
HATs
E7 interacts with several chromatin remodeling complexes (Figure 3c). CBP and p300 are highly homologous HATs. CBP was first characterized as CREB target (Chrivia, Kwok et al. 1993) and p300 as E1A binding partner (Stein, Corrigan et al. 1990; Eckner, Ewen et al. 1994). CBP/p300 are involved in the regulation of many genes (Avantaggiati, Ogryzko et al. 1997; Gu, Shi et al. 1997; Lill, Grossman et al. 1997; Perkins, Felzien et al. 1997; Snowden and Perkins 1998; Ito, Lai et al. 2001; Huang and McCance 2002; Gray, Zhang et al. 2005). E7 binds to p300/CBP in a manner dependent on CR1, the LXCXE motif, and the phosphorylation site (Bernat, Avvakumov et al. 2003; Fera and Marmorstein 2012; Jansma, Martinez-Yamout et al. 2014). p300/CBP-associated factor (pCAF) is another HAT which is bound by E7 via its CR1 and the C terminus (Huang and McCance 2002; Avvakumov, Torchia et al. 2003). The significance of HAT/E7 interaction is not clear, but E7 can reduce their activities (Huang and McCance 2002; Avvakumov, Torchia et al. 2003), and the IL8 promoter is suppressed through this mechanism (Huang and McCance 2002). E7 binding to p300 can also increase acetylation of pRb, reducing its inhibitory activity on E2F-mediated transcription (Jansma, Martinez-Yamout et al. 2014).
Polycomb repressive complexes (PRCs)
PRCs are important for stem cell and fate determination, including the differentiation of keratinocytes (McLaughlin-Drubin and Munger 2013). PRC2 contains the methyltransferase EZH1 which trimethylates histone H3 on lysine 27 (H3K27Me3); this mark, in turn, can recruit additional repressive complexes (McLaughlin-Drubin and Munger 2013; Zhang, Cooper et al. 2015). E7 upregulates KDM6A and KDM6B, which are demethylases for H2K27Me3, and E7 downregulates the expression or activity of PRC complex components (Figure 3d)(Hyland, McDade et al. 2011; McLaughlin-Drubin, Park et al. 2013). As a consequence of KDM6A and KDM6B upregulation, overall cellular levels of H3K27Me3 are dramatically reduced (Hyland, McDade et al. 2011; McLaughlin-Drubin and Munger 2013; McLaughlin-Drubin, Park et al. 2013). HOX genes and other genes regulated by PRC complexes are consequently de-repressed in E6/E7-expressing cells (Hyland, McDade et al. 2011). One important target of KDM6B is the cyclin dependent kinase (CDK) inhibitor p16. Because excess CDK4/6 activity is toxic to cells expressing E7, upregulation of p16 via KDM6B activity is necessary for cell survival (McLaughlin-Drubin, Park et al. 2013).
DNA methylation
E7 can influence DNA methylation, which is another form of epigenetic regulation (Figure 3c). E7 decreases E-cadherin levels in keratinocytes containing complete HPV16 genomes by increasing the levels of DNMT1, which methylates the E-cadherin promoter and represses the gene (Laurson, Khan et al. 2010). E7 also suppresses expression of CXCL14, an angiogenesis inhibitor, by promoting increased methylation of the CXCL14 promoter (Cicchini, Westrich et al. 2016). By contrast, STK31, a kinase involved in proliferation, invasiveness, and survival, is upregulated by E7 in a manner dependent on demethylation of the promoter DNA (Yin, Wang et al. 2016). E7 can bind to DNMT1 via the CR3 region and stimulate its activity and association with the cyclin A1 promoter (Burgers, Blanchon et al. 2007; Chalertpet, Pakdeechaidan et al. 2015). However, the overall effect of E7-mediated regulation of DNA methylation and the mechanisms by which it is accomplished are not fully understood.
4. The pRb/E2F system
The example of HDACs shows that E7 can operate by disrupting the interaction between transcription factors and their inhibitors. Another example is the activation of E2F-regulated genes by disruption of their repressor, pRb (Munger, Baldwin et al. 2004). The E2F family consists of both activating (E2F1, 2, 3) and inhibitory (E2F4, 5, 6, 7, 8) transcription factors (Frolov and Dyson 2004; Macaluso, Montanari et al. 2006). Activating E2Fs primarily function by binding to the promoters of genes involved in cell cycle regulation and increasing their transcription (Frolov and Dyson 2004). pRb binds to E2Fs, inhibiting their transcriptional function. Through the activity of mitogen-activated cyclin dependent kinases (CDKs), pRb is phosphorylated, resulting in the release of E2Fs and the activation of genes needed for entry into S phase (Frolov and Dyson 2004; Macaluso, Montanari et al. 2006).
E7 binds to and targets pRb for degradation via the proteasome, promoting cell cycle entry (Figure 4a). The LXCXE motif in CR2 is the primary binding site for pRb (Munger, Werness et al. 1989; Barbosa, Edmonds et al. 1990; Huang, Patrick et al. 1993; Chien, Parker et al. 2000; Dong, Caldeira et al. 2001; Helt and Galloway 2001), although CR3 has an independent, lower affinity pRb binding site (Patrick, Oliff et al. 1994; Todorovic, Hung et al. 2012). The CR1 domain is important for pRb degradation (Jones, Thompson et al. 1997; Huh, Zhou et al. 2007). As a consequence of E7 targeting pRb for degradation, E2F-mediated cell-cycle progression can occur in the absence of mitogenic signals, including in differentiating keratinocytes (Munger, Baldwin et al. 2004; Moody and Laimins 2010). This classic regulatory scheme is well known, but some important nuances are needed to understand how cell cycle-promoting genes are regulated by E7.
Figure 4.
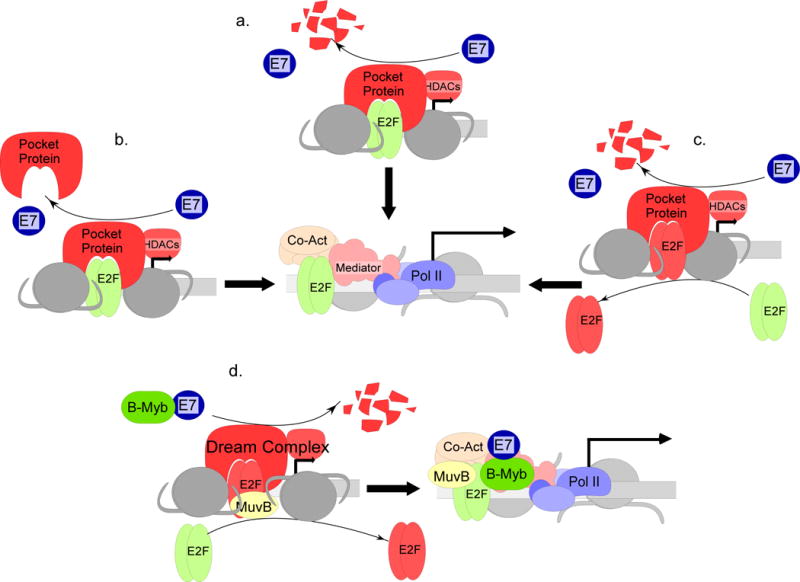
Regulation of pocket proteins by E7. a. E7 can bind to pocket proteins and target them for degradation, thus facilitating transcription by activating E2Fs (green)(Munger, Werness et al. 1989; Barbosa, Edmonds et al. 1990; Huang, Patrick et al. 1993; Chien, Parker et al. 2000; Dong, Caldeira et al. 2001; Helt and Galloway 2001). b. E7 can also displace pocket protein binding to E2Fs without targeting them for degradation (Chellappan, Kraus et al. 1992; Gonzalez, Stremlau et al. 2001; Collins, Nakahara et al. 2005). c. Inhibitory E2Fs (red), in combination with pocket proteins, can repress E2F dependent promoters. In addition to targeting pocket proteins for degradation, E7 can cause an exchange of activating E2Fs for inhibitory E2F complexes, upregulating gene expression (Longworth, Wilson et al. 2005; McLaughlin-Drubin, Huh et al. 2008). d. DREAM complexes are repressive complexes associated with pocket proteins, inhibitory E2Fs, and MuvB. E7 can cause the disruption of DREAM complexes and recruitment of B-Myb to associate with MuvB and activate mitotic gene expression (Nor Rashid, Yusof et al. 2011; Sadasivam and DeCaprio 2013; Fischer, Quaas et al. 2014; Pang, Toh et al. 2014).
4.1. pRb degradation is not the whole story
Similar to E1A, E7 can disrupt the E2F/pRb complex without necessarily targeting pRb for degradation (Figure 4b)(Chellappan, Kraus et al. 1992; Gonzalez, Stremlau et al. 2001). CR2 and CR3 domains of E7 are both needed to displace E2F from pRb (Huang, Patrick et al. 1993; Helt and Galloway 2001). The mechanisms of E2F/pRb complex disruption are not entirely clear. Although E2F binds to a site on pRb distinct from the CR2 binding site (Huang, Patrick et al. 1993), the CR3 binding domain in pRb overlaps the E2F binding site (Patrick, Oliff et al. 1994), suggesting that simple competition may occur. The dissociation of E2F from pRb may also be due to the overexpression of MDM2 in E7-containing cells (Thomas and Laimins 1998; Seavey, Holubar et al. 1999; Eichten, Westfall et al. 2002; Liu, Disbrow et al. 2007), which binds to E2F, displacing pRb and promoting S phase entry (Martin, Trouche et al. 1995; Xiao, Chen et al. 1995). How much of the biological effect of E7 on the pRb/E2F system is mediated through degradation vs. complex disruption is also not clear. We have observed that levels of pRb family members in immortalized keratinocyte cell lines containing HPV16 episomes do not differ substantially from levels in uninfected keratinocytes, in contrast to cells in which E7 is expressed from a retroviral vector ((Bodily, Mehta et al. 2011); W.S. and J.B., unpublished observations). This finding may suggest that reducing pRb levels through degradation may not be critical in cells containing intact viral episomes. A CR1 mutant that cannot degrade pRb can still induce suprabasal DNA synthesis in differentiating keratinocytes although to a lower level than wild type (Collins, Nakahara et al. 2005), suggesting that although pRb degradation contributes to induction of S phase it is not strictly necessary.
E7 can also bind E2F via the E7 C terminus and activate E2F-driven promoter activity in a pocket protein-independent manner (Hwang, Lee et al. 2002). On the other hand, E2F6 is an inhibitory E2F family member that does not bind to pocket proteins but instead is associated with PRCs (Frolov and Dyson 2004; Cobrinik 2005; McLaughlin-Drubin and Munger 2013) and thus would not be inhibited by E7’s ability to displace or degrade pocket proteins. However, both high and low-risk E7s can bind to E2F6 via the C terminus and prevent its ability to repress transcription (McLaughlin-Drubin, Huh et al. 2008).
4.2. Not just pRb
The pocket protein family includes p107 and p130 in addition to pRb (Macaluso, Montanari et al. 2006). The three pocket proteins sometimes share redundant functions and sometimes they do not, although p130 and p107 tend to be more similar to each other than to pRb (Dick and Rubin 2013). pRb prefers to bind the activating E2F1–3, and p107/p130 bind the inhibitory E2F4–5 (Macaluso, Montanari et al. 2006). Pocket proteins can be recruited to the DNA in promoter-selective ways, suggesting that they have promoter-selective functions (Cobrinik 2005). Although pRb is one of the most common genes mutated in cancers, p107 and p130 are not, for reasons that remain unclear (Chinnam and Goodrich 2011). Ablation of p107 and p130, alone or in combination, does not predispose to cancer, but p107 and p130 can help suppress cancer in the context of pRb deficiency (Rizzolio, Esposito et al. 2010). pRb is present in both proliferating and non-proliferating cells, whereas p107 is found in proliferating, and p130 mostly in non-proliferating cells (Cobrinik 2005; Macaluso, Montanari et al. 2006). In the skin, pRb is concentrated in the basal layers, and p130 is found in more differentiated layers (Genovese, Banerjee et al. 2008).
E7 can target p107 and p130 for degradation through the CR1 and CR2 domains (Helt and Galloway 2001; Zhang, Chen et al. 2006; Genovese, Banerjee et al. 2008; Barrow-Laing, Chen et al. 2010). Interestingly, point mutations within the LXCXE motif can affect binding to one pocket protein but not others (Jewers, Hildebrandt et al. 1992; Demers, Espling et al. 1996; Chien, Parker et al. 2000), suggesting that the mode of interaction may differ somewhat between family members. Efficient binding and degradation of p130 is also influenced by the E7 phosphorylation site (Genovese, Banerjee et al. 2008). p130 is more consistently eliminated from the differentiating layers of the epithelium by E7 than pRb is, and elimination of p130 appears to be more important than pRb for inducing DNA synthesis in the suprabasal layers (Collins, Nakahara et al. 2005). Importantly, p130 is degraded by low-risk HPVs as well as high-risk, which is unique among pRb family members (Zhang, Chen et al. 2006; Barrow-Laing, Chen et al. 2010). HPV11 E7 can induce S phase in differentiating epithelial cultures in an LXCXE-dependent manner (Banerjee, Genovese et al. 2006; Genovese, Banerjee et al. 2008), suggesting that p130 rather than pRb may be the more important target for promoting S phase in the differentiating epithelium.
4.3. pRb/E2F complexes can actively inhibit transcription
When pocket proteins bind to E2F family members, they bind to the activation domain, thus preventing the recruitment of positive transcriptional cofactors (Frolov and Dyson 2004; Cobrinik 2005). However, pocket proteins also interact with chromatin modifying complexes, such as HDACs, which actively and even permanently repress target promoters bound by inhibitory E2F family members (Magnaghi-Jaulin, Groisman et al. 1998; Frolov and Dyson 2004; Cobrinik 2005; Macaluso, Montanari et al. 2006). Because of the existence of inhibitory E2Fs which recruit pocket proteins and promote transcriptional repression, E2F binding sites may be either activating or inhibitory, or both (Sadasivam and DeCaprio 2013). For example, in keratinocytes the important E2F binding site in the Cdc25A promoter is inhibitory, since mutation increases promoter activity (Nguyen, Westbrook et al. 2002). Expression of E7 results in degradation of pocket proteins, increasing the activity of the wild type promoter to levels similar to the E2F binding site mutant (Nguyen, Westbrook et al. 2002). Thus E7 does not simply release E2Fs to activate promoters. Instead, E7 can counteract the active inhibition exerted by inhibitory E2F/pocket protein complexes by replacing the inhibitory E2F with an activating E2F, thus promoting transcription (Figure 4c). It is possible that the ability of E7 to associate with HDACs may play a role in displacing HDAC-associated pocket protein repressive complexes in favor of activating E2F complexes (Longworth, Wilson et al. 2005), but the molecular interactions involved are not fully understood.
4.4. The DREAM complex
E2F/pRb is important for regulation of genes at the G1/S transition, but some genes are specifically regulated later in the cell cycle, such as at G2/M. The dimerization partner, pRb-like, E2F and multi-vulval class B (MuvB)(DREAM) complex is an example of a pocket protein-associated transcriptional repressor (reviewed in (Sadasivam and DeCaprio 2013)). The DREAM complex contains the multi-subunit protein MuvB, which associates with different additional factors throughout the cell cycle (Sadasivam, Duan et al. 2012; Sadasivam and DeCaprio 2013). Under non-proliferative conditions such as quiescence or senescence, p130 and possibly p107 bind to MuvB along with repressive E2Fs such as E2F4 and E2F5, forming the DREAM complex and inhibiting expression of G2/M phase genes (Muller and Engeland 2010; Sadasivam and DeCaprio 2013). Phosphorylation of the pocket protein by mitogen driven-CDKs disrupts the DREAM complex, facilitating recruitment of B-Myb and/or FOXM1, which then cooperate with MuvB to activate gene expression (Sadasivam, Duan et al. 2012; Sadasivam and DeCaprio 2013). Because B-Myb is an E2F responsive gene, it is induced at the G1/S transition and then serves to activate genes in G2/M (Sadasivam, Duan et al. 2012; Sadasivam and DeCaprio 2013). Following mitosis, B-Myb and FOXM1 are degraded, allowing the reconstitution of the DREAM complex and promoter inhibition (Sadasivam and DeCaprio 2013). DREAM complexes bind close to the transcription start sites of many promoters at E2F binding sites or cycle-dependent element (CDE) - cell cycle genes homology region (CHR) elements (Muller and Engeland 2010; Sadasivam and DeCaprio 2013; Fischer, Quaas et al. 2014). CDE-CHR elements are characteristic of genes that are expressed late in the cell cycle (Perkins 2002; Muller and Engeland 2010; Sadasivam and DeCaprio 2013).
It is not clear whether pRb is functionally distinct from the DREAM complex or whether DREAM regulates genes other than cell cycle genes (Sadasivam and DeCaprio 2013), but given the role of pocket proteins in their activity, DREAM complexes are found at reduced levels in cells containing E7, along with increased levels of B-Myb/MuvB complexes (Nor Rashid, Yusof et al. 2011; Sadasivam and DeCaprio 2013). Both high and low-risk HPVs can disrupt DREAM complexes to similar extents and the LXCXE motif is necessary (Figure 4d)(Nor Rashid, Yusof et al. 2013; Fischer, Quaas et al. 2014). In the case of the E2F2 promoter, E7 causes a loss of p130/E2F4/HDAC complexes from the promoter region in a manner dependent on the C terminal HDAC binding region and the LXCXE motif, resulting in transcriptional upregulation (Longworth, Wilson et al. 2005). E7 also promotes the formation of B-Myb/MuvB complexes. B-Myb was the first E7-responsive cellular gene to be identified (Lam, Morris et al. 1994). The E2F site in the B-Myb promoter is repressive in cells that lack E7, but in the presence of E7, the E2F site becomes transcriptionally activating (Lam, Morris et al. 1994). E7 upregulates B-Myb by disrupting p107/E2F complexes at the B-Myb promoter using the CR2 and possibly the CR1 domains (Lam, Morris et al. 1994; Pang, Toh et al. 2014). E7 also interacts with and can cooperate with B-Myb to activate genes regulated by B-Myb-MuvB complexes (Figure 4d)(Pang, Toh et al. 2014). The CR2 domain of E7 is needed for binding to the B-Myb-MuvB complex, and pocket protein degradation is necessary but not sufficient to activate transcription (Pang, Toh et al. 2014).
4.5. E2F responsive genes
Through its regulation of the E2F/pocket protein system, E7 can regulate a large set of promoters (Nees, Geoghegan et al. 2000; Johung, Goodwin et al. 2007; Gyongyosi, Szalmas et al. 2014; Zhou, Zhang et al. 2016). Here are two examples for the purposes of illustration.
The first example of transactivation by E7 to be discovered was the adenovirus E2 (AdE2) promoter (Phelps, Yee et al. 1988; Phelps, Bagchi et al. 1991). Activation of a promoter in a different virus is obviously not physiological, but it highlights the similarity between E7 and the promoter’s physiological activator, E1A (Berk 2005). The AdE2 promoter drives expression of the adenovirus DNA replication genes, and so it is regulated by E2F in a manner similar to cellular genes that regulate DNA replication. E7 transactivation of the AdE2 promoter is similar to E1A 12S in requiring the CR2 domain, specifically the LXCXE motif (Phelps, Yee et al. 1988; Edmonds and Vousden 1989; Watanabe, Kanda et al. 1990; Phelps, Bagchi et al. 1991; Phelps, Munger et al. 1992). The ability of E7s from different HPV types to transactivate does not depend simply on the affinity of binding to pRb (Schmitt, Harry et al. 1994). Activation also requires the C terminus (Rawls, Pusztai et al. 1990; Phelps, Munger et al. 1992; Mavromatis, Jones et al. 1997), although the specific interaction partners that mediate this activity are not known. Dimerization of E7 can influence AdE2 transactivation (Todorovic, Massimi et al. 2011), but the CR1 domain is not necessary (Watanabe, Kanda et al. 1990; Phelps, Munger et al. 1992). The phosphorylation domain has either a modest or no contribution, depending on the study (Watanabe, Kanda et al. 1990; Firzlaff, Luscher et al. 1991; Heck, Yee et al. 1992; Phelps, Munger et al. 1992), whereas deletion of the acidic patch reduces transactivation (Phelps, Bagchi et al. 1991).
As mentioned above, Cdc25A is upregulated by E7 at the promoter level through both LXCXE and C terminal domains (Nguyen, Westbrook et al. 2002). Upregulation requires the E2F binding site, and in cells expressing E7, Cdc25A is no longer regulated in a cell-cycle dependent manner, even if DNA synthesis is inhibited (Nguyen, Westbrook et al. 2002). E7 also upregulates cyclin E, cyclin A, and p73 by increasing the activity of E2F1 (Caldeira, de Villiers et al. 2000; Brooks, Sullivan et al. 2002).
5. Transcription factors regulated by E7
E2Fs are the best studied example of how E7 can regulate transcription through a specific family of transcription factors, but the E7-mediated gene regulation includes more than the pocket protein/E2F system. In transgenic mouse models, pRb is responsible for essentially all the effects of E7 in the cutaneous epidermis (Balsitis, Sage et al. 2003; Balsitis, Dick et al. 2005); however, disrupting pRb-E7 interaction has very little effect on the phenotype of E7 in the murine cervix (Balsitis, Dick et al. 2006). In fact, knocking out all three pocket proteins cannot mimic the effect of E7 in causing cervical lesions (Shin, Sage et al. 2012), indicating that other proteins are relevant targets of E7. The basis of these tissue-specific effects is not clear, nor is it clear what effect these differences may have on the replication of the complete HPV genome. What is clear is that E7 has physiologically important targets in addition to pocket proteins. Here we will discuss other transcription factors regulated by E7 and highlight the various mechanisms involved (Figure 5).
Figure 5.
Impact of E7 on transcription factor function. E7 can activate some transcription factors by stimulating upstream signaling pathways. STAT5 (Hong and Laimins 2013) and HIF1 (Wang, Zhan et al. 2014) are two examples. E7 can also inhibit upstream signaling pathways, reducing the function of other transcription factors, including STAT1, NFκB, SMAD2/3, and IRF1 (Perea, Lopezocejo et al. 1997; Mi, Borger et al. 2000; Nees, Geoghegan et al. 2001; Spitkovsky, Hehner et al. 2002; Murvai, Borbely et al. 2004; Hypes, Pirisi et al. 2009; Li, Zhan et al. 2009; Vandermark, Deluca et al. 2012; Hasan, Zannetti et al. 2013; Zhou, Chen et al. 2013; Zhou, Chen et al. 2013; Lau, Gray et al. 2015). E7 can interfere with nuclear localization of STAT1 and NFκB (Perea, Lopezocejo et al. 1997; Nees, Geoghegan et al. 2001; Spitkovsky, Hehner et al. 2002; Li, Zhan et al. 2009; Zhou, Chen et al. 2013; Zhou, Chen et al. 2013). E7 can directly bind to and stimulate transcription factors, including E2F1, B-Myb, c-Myc, and c-Jun (Antinore, Birrer et al. 1996; Hwang, Lee et al. 2002; Wang, Chang et al. 2007; Pang, Toh et al. 2014). Alternatively, E7 can bind and inhibit transcription factors including E2F6, SMAD2/3, Miz1, IRF1, and TBP (Massimi, Pim et al. 1996; Massimi, Pim et al. 1997; Park, Kim et al. 2000; Maldonado, Cabrejos et al. 2002; Habig, Smola et al. 2006; McLaughlin-Drubin, Huh et al. 2008; Morandell, Kaiser et al. 2012). E7 can also induce the expression of transcription factors such as B-Myb and c-Fos (Lam, Morris et al. 1994; Morosov, Phelps et al. 1994; Pang, Toh et al. 2014), and inhibit the expression of transcription factors such as STAT1 and IRF1 (Hong, Mehta et al. 2011; Zhou, Chen et al. 2013).
5.1. General transcription factors: TFIID
The GTFs form the heart of the transcription initiation machinery. They are recruited by promoter-binding transcription factors and Mediator, and then provide a binding surface for the recruitment, positioning, and initiation of RNA polymerase II (reviewed in (Gupta, Sari-Ak et al. 2016)). TBP is the component of the GTF TFIID that binds to the TATA box which positions the Pol II complex on the promoter (Gupta, Sari-Ak et al. 2016). HPV16 E7, like E1A, can form a complex with TBP dependent on the CR3 domain (Massimi, Pim et al. 1997). Interestingly, TBP binding is enhanced by the negative charge resulting from phosphorylation of S31–S32 and is reduced by mutation of those residues to alanine (Massimi, Pim et al. 1996; Massimi, Pim et al. 1997). Interaction of E7 with TBP disrupts the DNA binding activity of TBP (Maldonado, Cabrejos et al. 2002), suggesting that the effect of the E7-TBP interaction would be inhibitory to transcription. Although binding to TBP correlates with transforming activity (Massimi, Pim et al. 1997), the role of TBP binding by E7 in the context of viral infection is not clear. HPV16 E7, like E1A, can also bind to another component of TFIID, TAF110 (Mazzarelli, Atkins et al. 1995), although the consequences of this interaction are not known. E1A can also interact directly with the Mediator complex and with the transcriptional elongation machinery, recruiting them to promoters to drive transcription (Vijayalingam and Chinnadurai 2013). Whether E7 has a similar activity is not known.
5.2. Interferon signaling: IRFs and STAT1
IFNs are antiviral cytokines that are critical components of the innate immune response (reviewed in (Vilaysane and Muruve 2009; Schneider, Chevillotte et al. 2014)). IFN treatment can prevent infection of human keratinocytes with HPV particles (Warren, Griffin et al. 2014), prevent immortalization (Khan, Tolleson et al. 1993), trigger growth arrest and reduce viral episome levels (Chang, Pena et al. 2002), reduce HPV gene expression, and promote viral integration (Herdman, Pett et al. 2006; Pett, Herdman et al. 2006; Lace, Anson et al. 2015). As a consequence, HPV has many mechanisms for downregulating or evading IFN responses (Beglin, Melar-New et al. 2009; Stanley 2009).
Although a detailed discussion of IFN signaling is beyond the scope of this review, two transcription factor families are important for regulation of IFN responses. First, the interferon response factors (IRFs) are responsible for driving expression of the IFN genes themselves in response to pathogen-associated pattern recognition signaling (Tamura, Yanai et al. 2008). Following binding of IFNs to their receptors, the signal transducer and activator of transcription (STAT) family of transcription factors are activated to promote expression of interferon stimulated genes (ISGs), which mediate antiviral responses (Ivashkiv and Donlin 2014; Schneider, Chevillotte et al. 2014). Of the STAT family members, STAT1 is especially important in mediating the antiviral effects of IFN both on its own through heterodimer formation and by forming part of the interferon stimulated gene factor 3 (ISGF3) transcription factor complex (Ivashkiv and Donlin 2014).
We have already mentioned IRF1 in our discussion of HDACs above. IRF1 is upregulated and activated by many cytokines and innate immune sensors (Taniguchi, Ogasawara et al. 2001; Tamura, Yanai et al. 2008). IRF1 target genes include TAP1, MHC-I, and other critical immune mediators, and IRF1 can act as a genetic tumor suppressor (Taniguchi, Ogasawara et al. 2001; Tamura, Yanai et al. 2008). E7 can bind IRF1 and recruit HDACs to IRF1 dependent promoters, including MHC-I and TAP1 (Georgopoulos, Proffitt et al. 2000; Park, Kim et al. 2000; Um, Rhyu et al. 2002; Li, Ou et al. 2006; Bottley, Watherston et al. 2008; Manning, Indrova et al. 2008; Li, Zhan et al. 2009; Deng, Li et al. 2011; Heller, Weisser et al. 2011). E7 also inhibits IRF1 DNA binding activity (Perea, Massimi et al. 2000) and suppresses the upregulation of IRF1 levels in response to IFNγ (Zhou, Chen et al. 2013). E6 and E7 both prevent upregulation of the IRF-dependent IFNβ promoter by infection with other viruses or by IFNγ (Perea, Lopezocejo et al. 1997), but whether this is through IRF1 or other IRFs is not clear. In addition to inhibiting IRF1-mediated transactivation directly, E7 can interfere with signals that activate IRFs. For example, E7 and E1A can each inhibit the cytoplasmic DNA sensor STING, thus preventing upregulation of IFNs in response to DNA stimulation (Lau, Gray et al. 2015). Although not tested for E7, the CR3 domain and LXCXE motif of E1A are important for this effect, but pRb is not (Lau, Gray et al. 2015). Suppression of IRF1 by E7 facilitates evasion of T cell-mediated keratinocyte death, and restoration of IRF1 expression can restore sensitivity to T cell killing (Zhou, Chen et al. 2013).
STAT1 is a central effector of signaling downstream of the IFN receptors by regulating expression of genes in response to all subgroups of IFNs, and so STAT1-regulated genes are important targets of host gene regulation by HPV (Chang and Laimins 2000; Nees, Geoghegan et al. 2001). HPV31 E7 can suppress STAT1 at the transcriptional level, resulting in reduced IFN-mediated gene expression (Hong, Mehta et al. 2011). HPV16 E7 inhibits IFN-induced phosphorylation and nuclear translocation of STAT1 and downstream expression of ISGs (Perea, Lopezocejo et al. 1997; Nees, Geoghegan et al. 2001; Zhou, Chen et al. 2013; Zhou, Chen et al. 2013). In addition to targeting STAT1 itself, HPV18 E7 can bind to the IRF9 subunit of the ISGF3 complex and prevent translocation to the nucleus (Barnard and McMillan 1999; Antonsson, Payne et al. 2006).
Interestingly, E7 can also selectively activate STAT1 dependent promoters. For example, IL-18 binding protein (IL18BP) is an anti-inflammatory factor that is upregulated by IFNγ (Richards, Doble et al. 2014). Because it is an ISG, we might expect IL18BP levels to be lower in E7 expressing cells, but cells expressing E7 have higher levels IL18BP in response to IFNγ than controls (Richards, Doble et al. 2014). Activation by E7 requires STAT1 phosphorylation and the STAT binding element in the IL18BP promoter (Richards, Doble et al. 2014), indicating that E7 is able to activate STAT1-dependent transcription rather than just inhibit it. The mechanism responsible for this selectivity is not clear, but both low and high-risk E7s can do it, and deletions in the C terminus inhibit the stimulatory activity (Richards, Doble et al. 2014).
5.3. Inflammatory signaling: NFκB
Nuclear factor kappa-B (NFκB) is another transcription factor critical for regulating immunity. Pathogen recognition receptors, cytokine receptors, and many other stimuli trigger the degradation of the NFκB inhibitor, IκB. IκB degradation allows NFκB to translocate to the nucleus and increase expression of inflammatory cytokines, pro-survival factors, and IFNs to promote immune responses (reviewed in (Pahl 1999; Gilmore 2006)). The interface between NFκB and HPV is complex and poorly understood (Da Costa, Bastos et al. 2016), but E7 suppresses many genes regulated through the NFκB pathway, including cytokines, IFN, MHC-I, and others (Spitkovsky, Hehner et al. 2002; Li, Zhan et al. 2009; Vandermark, Deluca et al. 2012). The primary mechanism appears to be preventing NFkB nuclear translocation by inhibiting the degradation of IκB and/or preventing NFkB acetylation (Spitkovsky, Hehner et al. 2002; Li, Zhan et al. 2009; Richards, Wasson et al. 2015). Failure of NFkB to enter the nucleus would explain how E7 can prevent both DNA binding and transactivation ability (Perea, Massimi et al. 2000). On the other hand E7 can inhibit toll-like receptor 9 (TLR9) expression by inducing the formation of an inhibitory complex on the TLR9 promoter (Hasan, Zannetti et al. 2013). This complex contains NFkB, but rather than being activating, the complex contains HDACs and histone demethylase to repress TLR9 expression (Hasan, Zannetti et al. 2013). Again, how selectivity is achieved is not clear.
5.4. DNA damage: STAT5
STAT proteins are involved in innate immune responses, but some family members also mediate growth factor and DNA damage signaling (Quesnelle, Boehm et al. 2007). An example of a growth factor-stimulated STAT is STAT5. DNA damage signaling is critical for the amplification of viral genomes and completion of the viral life cycle (Moody and Laimins 2009; Gautam and Moody 2016). Cells containing HPV31 genomes have high levels of total and phosphorylated (activated) STAT5 even in the absence of exogenous growth factors (Hong and Laimins 2013). E7 is responsible for STAT5 upregulation, perhaps through activating Akt (Hong and Laimins 2013). STAT5 induced by E7 drives the expression of factors needed for the DNA damage response, including TopBP1 (Hong and Laimins 2013; Hong, Cheng et al. 2015). In the absence of STAT5 and TopBP1, E7-induced DNA damage signaling is attenuated, resulting in reduced viral genome amplification and late gene expression (Hong and Laimins 2013; Hong, Cheng et al. 2015).
5.5. Genome stress: p53
p53 is a tumor suppressor and transcription factor that drives the expression of genes regulating cell cycle arrest, DNA repair, apoptosis, senescence, and metabolism in response to genotoxic, oncogenic, and other cellular stresses (reviewed in (Yee and Vousden 2005; Riley, Sontag et al. 2008; Zilfou and Lowe 2009)). Levels of p53 are tightly controlled by the ubiquitin ligase MDM2, which is also a p53 target gene (Bode and Dong 2004). MDM2 normally targets p53 for degradation, keeping steady state p53 protein levels low. However, in response to DNA damage or other signals, p53 undergoes phosphorylation which disrupts the MDM2-p53 interaction, resulting in increased p53 levels and activity (Xu 2003; Bode and Dong 2004). p53 transcriptional activity is also regulated extensively by other post-translational modifications, including acetylation by pCAF and CBP/p300, which promotes p53 DNA binding and transcriptional function (Avantaggiati, Ogryzko et al. 1997; Gu, Shi et al. 1997; Lill, Grossman et al. 1997; Sakaguchi, Herrera et al. 1998; Liu, Scolnick et al. 1999; Barlev, Liu et al. 2001; Ito, Lai et al. 2001; Xu 2003; Bode and Dong 2004). In response to p53 activation, cells can arrest the cell cycle, upregulate DNA repair, or undergo apoptosis (Riley, Sontag et al. 2008).
In the context of oncogenic HPV infection, uncontrolled DNA synthesis and DNA damage caused by E7 results in p53 stabilization and inhibition of cell cycling and viral DNA synthesis (Lepik, Ilves et al. 1998; Korzeniewski, Spardy et al. 2011). However, HPV has evolved various mechanisms to regulate this response. The most important and well-known mechanism is expression of the E6 oncoprotein, which recruits ubiquitin ligase enzyme, E6AP, to target p53 for degradation (Werness, Levine et al. 1990; Patel, Huang et al. 1999; Thomas and Chiang 2005). In addition, many studies have shown that E7 can also inhibit p53-mediated transcriptional programs, including p53-induced cell cycle arrest (Demers, Foster et al. 1994; Massimi and Banks 1997; Morozov, Shiyanov et al. 1997; Patel, Huang et al. 1999; Munger, Basile et al. 2001; Eichten, Westfall et al. 2002), suggesting a functional cooperation between E7 and E6.
The specific molecular mechanisms by which E7 interferes with the p53 pathway remain unclear. On the one hand, cells expressing E7 have high levels of p53 with increased half-life (Demers, Halbert et al. 1994; Jones, Thompson et al. 1997; Massimi and Banks 1997; Massimi, Pim et al. 1997; Stoppler, Stoppler et al. 1998; Thomas and Laimins 1998; Jones, Thompson et al. 1999; Patel, Huang et al. 1999; Seavey, Holubar et al. 1999; Flores, Allen-Hoffmann et al. 2000; Eichten, Westfall et al. 2002; Balsitis, Dick et al. 2005; Bischof, Nacerddine et al. 2005; Liu, Disbrow et al. 2007; Kho, Wang et al. 2013). The ability to stabilize p53 depends on the CR1 domain and the phosphorylation site and LXCXE motifs in the CR2 domain (Jones, Thompson et al. 1997). The mechanisms of p53 stabilization are not clear. It could be that p53 stabilization results from oncogene stress induced by E7 (Korzeniewski, Spardy et al. 2011), but other mechanisms are possible. Despite high p53 levels, MDM2 levels are also high in cells expressing E7 (Thomas and Laimins 1998; Seavey, Holubar et al. 1999; Eichten, Westfall et al. 2002; Liu, Disbrow et al. 2007), suggesting that the MDM2 may not be functioning to properly target p53 for degradation. Indeed, the association of p53 and MDM2 is inhibited in cervical cell culture (Barnard and McMillan 1999; Hengstermann, Linares et al. 2001), although this effect may depend on the cell type examined (Eichten, Westfall et al. 2002).
On the other hand, despite high p53 protein levels in E7-expressing cells, p53 activity is reduced. The stabilized p53 in E7 expressing cells lacks normal endogenous and reporter-driven transcriptional activity and it fails to activate key p53 target genes that are known to regulate cell cycle events (W.S. and J.B., unpublished observations)(Massimi and Banks 1997; Jones, Thompson et al. 1999; Patel, Huang et al. 1999; Eichten, Westfall et al. 2002; Bischof, Nacerddine et al. 2005). However, MDM2, which is a p53 target gene as well as inhibitor (Momand, Zambetti et al. 1992; Oliner, Pietenpol et al. 1993), is increased in E7-expressing cells (Thomas and Laimins 1998; Seavey, Holubar et al. 1999; Eichten, Westfall et al. 2002; Kho, Wang et al. 2013). Whether this is due to the increased p53 levels or other mechanisms is unclear. Inhibition of p53 reporter activity by E7 does not require pRb binding but does require the CKII phosphorylation site (Massimi and Banks 1997), which differs from the E7 domains required for p53 stabilization. The adenovirus E1A protein and simian virus 40 large T antigen, homologs of E7, have also been shown to inhibit p53 transcriptional activity (Mietz, Unger et al. 1992; Steegenga, van Laar et al. 1996; Somasundaram and El-Deiry 1997).
Like the stabilization of p53, the mechanism responsible for the disruption of p53-mediated transactivation by E7 remains unknown. Subcellular localization of p53 is not altered by E7 since p53-MDM2 complexes remain localized to the nucleus (Eichten, Westfall et al. 2002). Additionally, neither phosphorylation nor the wild-type conformation of p53 is altered in E7-expressing cells (Eichten, Westfall et al. 2002). One study showed that E7 could form a complex with p53 in a TBP-dependent manner (Massimi and Banks 1997), although the consequences of this association are not clear. Post-translational modification of p53 has the potential to inhibit p53 activity (Stewart, Tang et al. 2001; Xu 2003). E7 can bind and inhibit p300, whose HAT activity is important for p53 transactivation and turnover (Grossman, Perez et al. 1998; Bernat, Avvakumov et al. 2003; Xu 2003; Fera and Marmorstein 2012). E7 and p53 bind the same domain in p300, and E7 can disrupt association between p300/CBP and p53 (Bischof, Nacerddine et al. 2005; Fera and Marmorstein 2012). Accordingly, E7 prevents acetylation of p53, reducing its activity (Bischof, Nacerddine et al. 2005). Whether altering p53 post-translational modifications is sufficient to explain all of the effects of E7 on p53 activity remains to be clarified.
5.6. Proliferation and differentiation: c-Myc
c-Myc is a transcription factor that plays a critical role in cellular differentiation, growth regulation, and apoptosis (reviewed in (Eisenman 2001; Watt, Frye et al. 2008; Gandarillas 2012)). c-Myc can promote keratinocyte differentiation by mobilizing stem cells into the transit amplifying compartment, where they show increased proliferation but are committed to differentiate (Watt, Frye et al. 2008; Gandarillas 2012). c-Myc can either activate or repress cellular genes via several mechanisms, notably the recruitment of chromatin remodeling proteins, DNA methyltransferases, and transcriptional elongation factors (Batsche, Lipp et al. 1994; Dang, O’;Donnell et al. 2006; Rahl, Lin et al. 2010). High levels of c-Myc have been reported in cervical cancer (Nesbit, Tersak et al. 1999). c-Myc heterodimerizes with Max and binds to E-box elements in promoters (Ansieau, Strobl et al. 2001; Palomero, Lim et al. 2006). HPV16 E7 interacts with c-Myc to enhance DNA binding to and transcriptional activation of the hTert promoter (Wang, Chang et al. 2007). The zinc finger domain of E7 is required for c-Myc interaction (Wang, Chang et al. 2007). Additionally, the c-Myc promoter itself can be activated by E2F to induce unscheduled entry into S-phase (Hiebert, Lipp et al. 1989; Thalmeier, Synovzik et al. 1989; Leone, Sears et al. 2001).
E7 can act as a transcription repressor by binding to and inhibiting the transcriptional activity of Myc-interacting zinc-finger protein 1 (Miz1)(Morandell, Kaiser et al. 2012). Miz1 is an anti-proliferative transcription factor that inhibits the positive transcriptional activity of c-Myc and is required for transcriptional activation of the CDK inhibitor p21 in response to DNA damage (Dulic, Kaufmann et al. 1994; Herold, Wanzel et al. 2002; Wang, Chang et al. 2007; Barrilleaux, Burow et al. 2014). HPV16 E7, but not HPV11 E7, binds to the p21 promoter and inhibits Miz-1-dependent transcriptional regulation of p21 gene after DNA damage, thus overriding the Miz-1 mediated G1-cell cycle arrest (Morandell, Kaiser et al. 2012).
5.7. TGFβ signaling: SMADs
TGFβ is a critical regulator of immune responses, cell growth, epithelial-stromal interactions, and differentiation (Massague 2012; Pickup, Novitskiy et al. 2013; Travis and Sheppard 2014). TGFβ signaling is initiated when TGFβ binds to the TGFβ receptor complex, activating the receptor-associated kinase to phosphorylate SMAD2 and SMAD3. SMAD2/3 form a complex with SMAD4 and translocate to the nucleus to promote gene expression (Massague 2012). TGFβ is produced by many cells and stimulates in a variety of biological effects that are both cell type and context dependent (reviewed in (Li and Flavell 2008; Gigante, Gesualdo et al. 2012; Massague 2012). In early stages of tumorigenesis TGFβ acts as a tumor suppressor, whereas it promotes tumor growth during later stages of cancer development (Wakefield and Sporn 1990; Creek, Geslani et al. 1995; Pickup, Novitskiy et al. 2013; Polanska and Orimo 2013). High levels of TGFβ are found in cervical cancers (Li, Huang et al. 2009) and might be responsible for epithelial-to-mesenchymal transition during cervical malignancy (Helleman, Smid et al. 2010).
TGFβ signaling can arrest keratinocyte proliferation and inhibit HPV gene expression; consequently, TGFβ signaling is a frequent target of HPV oncogenes including E7 (Pietenpol, Stein et al. 1990; Woodworth, Notario et al. 1990; Moses 1992; Creek, Geslani et al. 1995; Ozbun and Meyers 1996; Nees, Geoghegan et al. 2000; Murvai, Borbely et al. 2004; Habig, Smola et al. 2006). E7 can inhibit growth suppression by blocking TGFβ expression and signaling, so that TGFβ treatment of HPV-containing cells stimulates rather than arrests growth (Pietenpol, Stein et al. 1990; Moses 1992; Woodworth, Chung et al. 1996). The ability of E7 to abrogate TGFβ-induced growth arrest depends on the LXCXE motif and the CR1 domain (Demers, Espling et al. 1996), and E7 can reverse TGFβ-induced downregulation of c-Myc in a CR2 dependent manner (Pietenpol, Stein et al. 1990; Moses 1992). Two possible mechanisms by which E7 interferes with TGFβ have been reported. First, E7 interacts with SMAD2, SMAD3, and SMAD4 resulting in disruption of the SMAD complex and inhibition of TGF-β-driven gene expression (Habig, Smola et al. 2006). Second, E7 can inhibit the TGFβ2 promoter in a manner dependent on the CR2 and CR3 domains (Murvai, Borbely et al. 2004). E7 also may contribute to the inhibition of the TGFβ receptor type I gene, although the precise mechanism is unknown (Mi, Borger et al. 2000; Hypes, Pirisi et al. 2009).
5.8. Ski interacting protein (Skip)
Skip was first identified as the binding partner of viral and cellular Ski isoforms (Dahl, Wani et al. 1998) and can act as a transcriptional co-activator at many promoters, including AdE2 and p21 (Prathapam, Kuhne et al. 2001). Skip interacts with pocket proteins and inhibits their transcriptional repression activity (Prathapam, Kuhne et al. 2002), and interacts with SMAD2/3 to induce TGFβ-dependent transcription (Leong, Subramaniam et al. 2001). High-risk E7 can interact with Skip. This interaction depends on the S31–32 phosphorylation site and sequences in the CR3 region but not the presence of pRb (Prathapam, Kuhne et al. 2001). E7 binding to Skip can prevent Ski-Skip dependent transcriptional activation (Prathapam, Kuhne et al. 2001). The genes regulated by the E7-Skip interaction in the context of HPV infection or cancer are not yet known.
5.9. Hypoxic response: HIF1
Hypoxia inducible factor 1 (HIF1) is an important transcription factor that induces angiogenesis and metabolic adaptation in both benign and malignant lesions (Zhou, Schmid et al. 2006; Semenza 2009). Under hypoxic (low oxygen) conditions, cells with episomal HPV genomes have increased levels of, HIF1 and HIF1 targets, including carbonic anhydrase IX and vascular endothelial growth factor (VEGF)(Nakamura, Bodily et al. 2009; Bodily, Mehta et al. 2011; Walker, Smiley et al. 2011). The HIF1α subunit is associated with HDACs, which reduces the activity of HIF1 dependent promoters. However, E7 can disrupt the association of HDACs with HIF1, thus increasing the level of HIF1 dependent transcription (Bodily, Mehta et al. 2011). E7 is also able to stabilize HIF1α protein levels (Nakamura, Bodily et al. 2009) and bind to HIF1α through the CR2 domain (Bodily, Mehta et al. 2011). How E7 binding to HIF1α relates to protein stabilization and HDAC displacement is not clear. E7 upregulates ribonucleotide reductase M2 subunit (RRM2) through the E2F site in the promoter, and RRM2 enhances the expression of HIF1α and VEGF through ERK activation and contributes to angiogenesis (Wang, Zhan et al. 2014).
5.10. Other transcription factors regulated by E7
E7 can regulate aspects of the AP-1 system. AP-1 is a family of transcription factors composed of dimers of the Jun and Fos family proteins (reviewed in (Zenz, Eferl et al. 2008)). In the skin, AP-1 regulates both differentiation and inflammatory processes (Zenz, Eferl et al. 2008). High-risk E7 enhances c-Fos gene expression in a manner dependent on the CR1 and CR2 region of E7 and a cAMP response element in the promoter (Morosov, Phelps et al. 1994). E7 binds (via the C terminus) to AP-1 proteins including c-Jun, JunB, JunD and c-Fos and activates c-Jun (Antinore, Birrer et al. 1996). On the other hand, E7 can also inhibit the activity of AP-1, perhaps contributing to suppression of differentiation-related genes by E7 (Gyongyosi, Szalmas et al. 2014).
E7 can separate the proliferation arrest function of C/EBP from the differentiation-inducing function, enabling cells to continue to proliferate even while undergoing differentiation (Gyongyosi, Szalmas et al. 2014). The ability of E7 to act on C/EBP requires the phosphorylation site, but not the CR1 or LXCXE domains (Muller, Alunni-Fabbroni et al. 1999). E7 can upregulate or stimulate the activity of the pro-proliferative transcription factors FoxM1b (Jaiswal, John et al. 2015) and MPP2 (Luscher-Firzlaff, Westendorf et al. 1999) and can downregulate the activity of the steroid receptor co-activator (SRC), blocking its transcriptional and HAT activity by sequestering it in the cytoplasm (Baldwin, Huh et al. 2006).
6. Other promoters regulated by E7
We have discussed how E7 can regulate transcription by interacting with transcriptional co-activators, co-repressors, inhibitory complexes, and transcription factors. Here we discuss other promoters that are regulated by mechanisms not yet fully understood.
E7 can reduce CCL20 expression and secretion (Guess and McCance 2005; Herman, Hubert et al. 2006; Caberg, Hubert et al. 2008; Caberg, Hubert et al. 2009). Interestingly, TGFβ, CCL20, and E-cadherin are all inhibited by E7, and each is critical for the recruitment, differentiation, and retention of Langerhans cells in the skin (Guess and McCance 2005; Mayumi, Watanabe et al. 2013; Travis and Sheppard 2014). Levels of these critical antigen presenting cells are markedly reduced in HPV-containing tissues (Spinillo, Tenti et al. 1993; Connor, Ferrer et al. 1999; Delvenne, Hubert et al. 2001; Scott, Nakagawa et al. 2001; Matthews, Leong et al. 2003; Hubert, Caberg et al. 2005; Herman, Hubert et al. 2006; Caberg, Hubert et al. 2008; Caberg, Hubert et al. 2009; Leong, Doorbar et al. 2010). When E7 is expressed in the murine epidermis, Langerhans cells are functionally impaired in both migration and antigen uptake, with fewer dendritic processes (Abd Warif, Stoitzner et al. 2015). It is possible that regulation of these transcriptional targets by E7 may be important for immune evasion by HPV.
The LXCXE motif in E7 contributes to hTert promoter activity in cells expressing E6 and E7 (Liu, Roberts et al. 2008). The E2F site in the hTert promoter is inhibitory, suggesting that E7 acts through abrogating the effect of inhibitory pocket protein/E2F complexes (Liu, Roberts et al. 2008).
Sirt1 is an NAD-dependent deacetylase that is involved in the regulation of the DNA damage response. In cooperation with E6, E7 increases Sirt1 levels through the C terminus and LXCXE motifs (Allison, Jiang et al. 2009; Langsfeld, Bodily et al. 2015). Sirt1 binds to the viral genome and recruits homologous recombination factors which are important for episomal maintenance, amplification, and late gene expression (Langsfeld, Bodily et al. 2015; Gautam and Moody 2016). Sirt1 levels may also drive changes in cellular gene expression but this is as yet unknown.
E7 can induce patterns of gene expression typical of the epithelial-mesenchymal transition, a frequent precursor to cancer metastasis, in which fibroblast-like genes are expressed in epithelial cells (Hellner, Mar et al. 2009; Hanahan and Weinberg 2011). E7 can also upregulate matrix metalloproteinases, which are also involved in cancer metastasis, but which may be important for the activation of growth factors and other molecules stored in the extracellular matrix (Smola-Hess, Pahne et al. 2005; Page-McCaw, Ewald et al. 2007; Cardeal, Boccardo et al. 2012; Kaewprag, Umnajvijit et al. 2013; Zhu, Ye et al. 2015). The impact of these factors on the viral life cycle is unclear.
Although most of the literature focuses on the effect of E7 on cellular gene expression, E7 also increases HPV16 late promoter reporter activity (Bodily, Hennigan et al. 2013). E2, the DNA binding transcription factor encoded by HPV16, has no effect on E7 activation of the late promoter reporter and E7 has no effect on E2 inhibiting the early promoter (Bodily, Hennigan et al. 2013). LXCXE and C terminal mutants each reduce activation (Bodily, Hennigan et al. 2013). The mechanism of activation remains unclear, but factors that increase cell cycling, such as CDK1/2 and activating E2Fs, actually inhibit the promoter (Bodily, Hennigan et al. 2013), so it is unlikely that E7 activates the late promoter through promoting E2F activity.
7. MicroRNAs
Finally, in recent years, many microRNAs (miRNAs) have been found to be regulated by E7. Table 1 shows a summary of these findings, including miRNAs that have been shown to be regulated directly by E7. Other miRNAs have been shown to be regulated by HPV but the specific contribution of E7 (vs. the other viral oncogenes) is not yet known (Martinez, Gardiner et al. 2008; Wang, Wang et al. 2009; Bonnet, Tatari et al. 2010; McKenna, McDade et al. 2010; Jung, Phillips et al. 2014; Leung, Deng et al. 2014; Liu, Gao et al. 2014; McKenna, Patel et al. 2014; Zheng, Liu et al. 2015; Liu, Zhang et al. 2016). Only a few of these miRNAs are yet known to have a clear impact on the viral life cycle or on cancer development.
Table 1.
MicroRNAs shown to be regulated by E7 NT=not tested
| miR | Direction of regulation | Mechanism of regulation | miR target | Consequence | Reference | |||
|---|---|---|---|---|---|---|---|---|
| 15b | ↑ | E2F activation? | Cyclin E1 | Arrest in G0/G1 | (Myklebust, Bruland et al. 2011) | |||
| 20a | ↑ | NT | NT | Less proliferation, migration, invasion | (Hu, Ge et al. 2016; Liu, Zhang et al. 2016) | |||
| 21 | ↑ | NT; estrogen involved | PTEN | Increased proliferation and migration; more GF signaling? | (Kong, Wang et al. 2015; Gomez- Gomez, Organista-Nava et al. 2016) | |||
| 27b | ↑ | Increased processing via DGCR8 | PPARγ, PLK2 | Invasion, proliferation and transformation, reduced apoptosis. | (Zhang, Liu et al. 2015; Liu, Zhang et al. 2016) | |||
| 34a | ↑ | NT; p53 target? | Myc, CDK4, Cdc25a | NT | (Chiantore, Mangino et al. 2016) | |||
| 143 | ↓ | NT; estrogen involved | Bcl2 | Reduced apoptosis? | (Gomez- Gomez, Organista-Nava et al. 2016) | |||
| 145 | ↓ | NT | HPV E1/E2 | More amplification, late gene expression | (Gunasekhara n and Laimins 2013; Gunasekharan, Li et al. 2016) | |||
| 203 | ↓ | Transcription via inhibition of MAPK/PKC | P63 | Increased CARM1, p21, Bax; genome maintenance and amplification | (Melar-New and Laimins 2010) | |||
| 205 | ↓ | Loss of pRb | NT | Increased proliferation | (McKenna, Patel et al. 2014) | |||
| MicroRNAs regulated by E7 but not otherwise characterized | ||||||||
| 18a, 19a, 93, 106b, 509-5p | ↑ | NT | NT | NT | (Chiantore, Mangino et al. 2016; Liu, Zhang et al. 2016) | |||
miR-145 is suppressed by E7 during differentiation in keratinocytes containing HPV31 genomes (Gunasekharan and Laimins 2013). miR-145 targets a region in the HPV E1 open reading frame and reduces genome amplification and late transcripts (Gunasekharan and Laimins 2013). miR-145 is thus an example of a miR regulating viral transcripts. miR-145 also targets the transcription factor KLF4 which binds to the viral regulatory region and promotes viral amplification and late gene expression (Gunasekharan, Li et al. 2016). This may be a mechanism by which E7 could contribute to viral gene expression. miR-375 also targets HPV transcripts (Jung, Phillips et al. 2014), although its regulation by E7 specifically has not been tested.
miR-203 can increase differentiation of keratinocytes and can downregulate p63; E7 inhibits miR-203 expression at the transcriptional level, so that p63 and its targets are maintained at higher levels during differentiation, facilitating viral genome replication (Melar-New and Laimins 2010). Finally, miR-21 (upregulated) and miR-143 (downregulated) are targets of E7 in the estrogen-E7 transgenic mouse model, where they suppress PTEN and Bcl-2, respectively (Gomez-Gomez, Organista-Nava et al. 2016).
8. Summary and additional questions
We have seen that E7 can regulate gene expression through a wide array of mechanisms, ranging from direct interaction with the core transcriptional machinery to the regulation of signaling pathways to promote or inhibit transcription factor activation. Clearly the binding of E7 to cellular factors is flexible to the point of promiscuity, which is one of the features that make E7 such a powerful regulator of cell behavior. One of the most important questions resulting from these studies is how (or whether) E7 can mediate such a wide array of effects in real HPV infections. Even though some of the interactions between E7 and host proteins are likely to be catalytic (such as promoting degradation of pocket proteins) rather than stoichiometric, it is not clear that there is enough E7 present in an infected cell to engage in all these interactions at the same time. It is possible that some of these reported effects are artifacts of overexpression or of expression of E7 in the absence of the other viral gene products. Alternatively, it is possible that some interactions only occur under specific conditions, such as upon activation of a signaling pathway or during differentiation. It is also possible that this diverse array of effects may ultimately be traceable to only a few critical regulatory nodes. For example, the degradation of pocket proteins unleashes a wide variety of downstream events, even though E7 itself may be engaging in only one activity. Much additional study will be needed to work out these possibilities.
There is evidence that E7 may have tissue-specific effects, as seen most clearly in the role of E7 in cutaneous vs. cervical carcinogenesis in the mouse (Herber, Liem et al. 1996; Gulliver, Herber et al. 1997; Riley, Duensing et al. 2003). Are such tissue-specific differences relevant for human infection, tissue tropism, or carcinogenesis (for example in the cervix vs. oropharynx)? E7 can activate or suppress whole pathways, such as the IRF or STAT pathways, but it can also mediate the opposite effect on specific genes (for example IL18BP) that are regulated by those pathways. What can explain the selectivity of gene expression by E7? Some interactions between E7 and cellular factors have the potential to bring about enormous changes in host gene expression – for example the interaction with p300/CBP – but these interactions have not, as yet, been linked to very many specific regulatory changes. What are the downstream consequences of the interaction between E7 and these critical transcriptional regulators?
Because of their link to cancer, the HPV oncogenes have usually been viewed through the lens of cell transformation, cell proliferation, and tumorigenesis, using cervical cancer cell lines as models. However, because of the focus on carcinogenic transformation, the contributions that the viral oncogenes have evolved to make in the productive HPV life cycle are not fully understood. Although HPVs are medically important because of their ability to cause cancers, progression toward malignancy is detrimental to HPV from an evolutionary perspective because it is associated with failure to produce progeny virus (Pett and Coleman 2007; Doorbar, Quint et al. 2012). Therefore, high-risk HPV should not benefit from inducing cellular changes that result in frank malignancy or metastasis. Yet evolution has retained these activities in the viral oncoproteins. Low-risk HPV types use broadly similar life cycle strategies, promote unscheduled cell proliferation, replicate to high levels, and are evolutionarily successful to a degree comparable to the high-risk types (Doorbar, Quint et al. 2012), and yet they are not associated with cancer and their oncogenes lack much of the tumorigenic potential of the high-risk homologs (Klingelhutz and Roman 2012). These observations suggest an important evolutionary question: Why do high-risk HPV oncogenes have cancer-promoting activities, while low-risk HPV oncogenes, under similar conditions, do not promote cancer development? What is it about the high-risk HPV life cycles that requires oncogenic HPV activities that is not necessary for the low-risk HPVs?
Examining the functions of viral oncogenes from the perspective of the evolutionary needs of the virus rather than cancer per se may provide additional insight into how the viral oncogenes impact host cell biology. For example, all small DNA viruses target pRb, p53, and the PI3K/Akt/mTOR pathway (Moore and Chang 2010). These pathways regulate cell cycling and proliferation, and therefore control the availability of cellular resources for replicating viral genomes. However, each of these host pathways also has critical effects on innate and adaptive immunity (Tang, Amagai et al. 1993; Takaoka, Hayakawa et al. 2003; Munoz-Fontela, Macip et al. 2008; Youlyouz-Marfak, Gachard et al. 2008; Zaheer, Koetzler et al. 2009; Moore and Chang 2010; Lupberger, Duong et al. 2013; Mayumi, Watanabe et al. 2013; Kalinowski, Ueki et al. 2014; Lulli, Carbone et al. 2016; Miciak and Bunz 2016; Zheng, Stamminger et al. 2016). It is possible that the immune effects of these oncogenic pathways may be more important in the context of productive virus infections than simply making nucleotides and enzymes available for replicating genomes. How viruses exploit the relative contributions of the oncogenic and immune activities of tumor suppressor genes in their productive life cycles is not clearly known. Fundamentally, in order to understand the functions that E7 evolved to fulfill, we need to better understand which of all these interactions are important for the productive viral life cycle. Better appreciating the role of E7 in the normal viral life cycle will enable us to better understand the context from which HPV-induced carcinogenesis arises.
Highlights.
Human papillomavirus E7 both positively and negatively regulates transcription of host cell genes.
E7 regulates factors that control chromatin structure, including histone modifying enzymes and DNA methylation machinery.
E7 modifies the functions of many transcription factor systems, including pRb/E2F, IRFs, SMADs, HIF1, p53, and others.
Transcriptional regulation by E7 impacts host cell proliferation, innate and adaptive immunity, stress responses, differentiation, and DNA damage responses.
Acknowledgments
Thanks to Gaurav Raikhy, Brittany Woodby, and Matthew Scott for careful reading of the manuscript and helpful discussions. The authors are supported by the National Institute of Allergy and Infectious Diseases (R01AI118904), National Institute of General Medical Sciences (P30GM110703), and the Feist-Weiller Cancer Center. We apologize to any of our colleagues whose work we were not able to cite due to space requirements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abd Warif NM, Stoitzner P, et al. Langerhans cell homeostasis and activation is altered in hyperplastic human papillomavirus type 16 E7 expressing epidermis. PLoS One. 2015;10(5):e0127155. doi: 10.1371/journal.pone.0127155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcocer-Gonzalez JM, Berumen J, et al. In vivo expression of immunosuppressive cytokines in human papillomavirus-transformed cervical cancer cells. Viral Immunol. 2006;19(3):481–491. doi: 10.1089/vim.2006.19.481. [DOI] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ, et al. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16(3):155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison SJ, Jiang M, et al. Oncogenic viral protein HPV E7 up-regulates the SIRT1 longevity protein in human cervical cancer cells. Aging (Albany NY) 2009;1(3):316–327. doi: 10.18632/aging.100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansieau S, Strobl LJ, et al. Activation of the Notch-regulated transcription factor CBF1/RBP-Jkappa through the 13SE1A oncoprotein. Genes Dev. 2001;15(4):380–385. doi: 10.1101/gad.189301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinore MJ, Birrer MJ, et al. The human papillomavirus type 16 E7 gene product interacts with and trans-activates the AP1 family of transcription factors. Embo J. 1996;15(8):1950–1960. [PMC free article] [PubMed] [Google Scholar]
- Antonsson A, Payne E, et al. The human papillomavirus type 16 E7 protein binds human interferon regulatory factor-9 via a novel PEST domain required for transformation. J Interferon Cytokine Res. 2006;26(7):455–461. doi: 10.1089/jir.2006.26.455. [DOI] [PubMed] [Google Scholar]
- Arany I, Tyring SK, et al. Status of local cellular immunity in interferon-responsive and -nonresponsive human papillomavirus-associated lesions. Sex Transm Dis. 1996;23(6):475–480. doi: 10.1097/00007435-199611000-00007. [DOI] [PubMed] [Google Scholar]
- Avantaggiati ML, Ogryzko V, et al. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89(7):1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Avvakumov N, Torchia J, et al. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene. 2003;22(25):3833–3841. doi: 10.1038/sj.onc.1206562. [DOI] [PubMed] [Google Scholar]
- Baldwin A, Huh KW, et al. Human papillomavirus E7 oncoprotein dysregulates steroid receptor coactivator 1 localization and function. J Virol. 2006;80(13):6669–6677. doi: 10.1128/JVI.02497-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis S, Dick F, et al. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006;66(19):9393–9400. doi: 10.1158/0008-5472.CAN-06-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis S, Dick F, et al. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J Virol. 2005;79(17):11392–11402. doi: 10.1128/JVI.79.17.11392-11402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis SJ, Sage J, et al. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol Cell Biol. 2003;23(24):9094–9103. doi: 10.1128/MCB.23.24.9094-9103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee NS, Genovese NJ, et al. Conditionally activated E7 proteins of high-risk and low-risk human papillomaviruses induce S phase in postmitotic, differentiated human keratinocytes. J Virol. 2006;80(13):6517–6524. doi: 10.1128/JVI.02499-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks L, Edmonds C, et al. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene. 1990;5(9):1383–1389. [PubMed] [Google Scholar]
- Barbosa MS, Edmonds C, et al. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. Embo J. 1990;9(1):153–160. doi: 10.1002/j.1460-2075.1990.tb08091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev NA, Liu L, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8(6):1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Barnard P, McMillan NA, et al. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology. 1999;259(2):305–313. doi: 10.1006/viro.1999.9771. [DOI] [PubMed] [Google Scholar]
- Barrilleaux BL, Burow D, et al. Miz-1 activates gene expression via a novel consensus DNA binding motif. PLoS One. 2014;9(7):e101151. doi: 10.1371/journal.pone.0101151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow-Laing L, Chen W, et al. Low- and high-risk human papillomavirus E7 proteins regulate p130 differently. Virology. 2010;400(2):233–239. doi: 10.1016/j.virol.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsche E, Lipp M, et al. Transcriptional repression and activation in the same cell type of the human c-MYC promoter by the retinoblastoma gene protein: antagonisation of both effects by SV40 T antigen. Oncogene. 1994;9(8):2235–2243. [PubMed] [Google Scholar]
- Beglin M, Melar-New M, et al. Human papillomaviruses and the interferon response. J Interferon Cytokine Res. 2009;29(9):629–635. doi: 10.1089/jir.2009.0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bequet-Romero M, Lopez-Ocejo O. Angiogenesis modulators expression in culture cell lines positives for HPV-16 oncoproteins. Biochem Biophys Res Commun. 2000;277(1):55–61. doi: 10.1006/bbrc.2000.3628. [DOI] [PubMed] [Google Scholar]
- Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24(52):7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- Bernat A, Avvakumov N, et al. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22(39):7871–7881. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- Bielenberg DR, McCarty MF, et al. Expression of interferon-beta is associated with growth arrest of murine and human epidermal cells. J Invest Dermatol. 1999;112(5):802–809. doi: 10.1046/j.1523-1747.1999.00566.x. [DOI] [PubMed] [Google Scholar]
- Bischof O, Nacerddine K, et al. Human Papillomavirus Oncoprotein E7 Targets the Promyelocytic Leukemia Protein and Circumvents Cellular Senescence via the Rb and p53 Tumor Suppressor Pathways. Mol Cell Biol. 2005;25(3):1013–1024. doi: 10.1128/MCB.25.3.1013-1024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bode AM, Dong Z, et al. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- Bodily J, Laimins LA, et al. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 2011;19(1):33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodily JM, Hennigan C, et al. Regulation of the human papillomavirus type 16 late promoter by E7 and the cell cycle. Virology. 2013;443(1):11–19. doi: 10.1016/j.virol.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodily JM, Mehta KP, et al. The E7 Open Reading Frame Acts in cis and in trans To Mediate Differentiation-Dependent Activities in the Human Papillomavirus Type 16 Life Cycle. J Virol. 2011;85(17):8852–8862. doi: 10.1128/JVI.00664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodily JM, Mehta KP, et al. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011;71(3):1187–1195. doi: 10.1158/0008-5472.CAN-10-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet E, Tatari M, et al. Module network inference from a cancer gene expression data set identifies microRNA regulated modules. PLoS One. 2010;5(4):e10162. doi: 10.1371/journal.pone.0010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon SS, Tomaic V, et al. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J Virol. 2015;89(3):1579–1586. doi: 10.1128/JVI.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottley G, Watherston OG, et al. High-risk human papillomavirus E7 expression reduces cell-surface MHC class I molecules and increases susceptibility to natural killer cells. Oncogene. 2008;27(12):1794–1799. doi: 10.1038/sj.onc.1210798. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391(6667):597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Brehm A, Nielsen SJ, et al. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. Embo J. 1999;18(9):2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks LA, Sullivan A, et al. E7 proteins from oncogenic human papillomavirus types transactivate p73: role in cervical intraepithelial neoplasia. Br J Cancer. 2002;86(2):263–268. doi: 10.1038/sj.bjc.6600033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers WA, Blanchon L, et al. Viral oncoproteins target the DNA methyltransferases. Oncogene. 2007;26(11):1650–1655. doi: 10.1038/sj.onc.1209950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberg JH, Hubert P, et al. Increased migration of Langerhans cells in response to HPV16 E6 and E7 oncogene silencing: role of CCL20. Cancer Immunol Immunother. 2008 doi: 10.1007/s00262-008-0522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberg JH, Hubert P, et al. Increased migration of Langerhans cells in response to HPV16 E6 and E7 oncogene silencing: role of CCL20. Cancer Immunol Immunother. 2009;58(1):39–47. doi: 10.1007/s00262-008-0522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberg JH, Hubert PM, et al. Silencing of E7 oncogene restores functional E-cadherin expression in human papillomavirus 16-transformed keratinocytes. Carcinogenesis. 2008;29(7):1441–1447. doi: 10.1093/carcin/bgn145. [DOI] [PubMed] [Google Scholar]
- Calcada EO, Felli IC, et al. The heterogeneous structural behavior of E7 from HPV16 revealed by NMR spectroscopy. Chembiochem. 2013;14(14):1876–1882. doi: 10.1002/cbic.201300172. [DOI] [PubMed] [Google Scholar]
- Caldeira S, de Villiers EM, et al. Human papillomavirus E7 proteins stimulate proliferation independently of their ability to associate with retinoblastoma protein. Oncogene. 2000;19(6):821–826. doi: 10.1038/sj.onc.1203375. [DOI] [PubMed] [Google Scholar]
- Cardeal LB, Boccardo E, et al. HPV16 oncoproteins induce MMPs/RECK-TIMP-2 imbalance in primary keratinocytes: possible implications in cervical carcinogenesis. PLoS One. 2012;7(3):e33585. doi: 10.1371/journal.pone.0033585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalertpet K, Pakdeechaidan W, et al. Human papillomavirus type 16 E7 oncoprotein mediates CCNA1 promoter methylation. Cancer Sci. 2015;106(10):1333–1340. doi: 10.1111/cas.12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YE, Laimins LA, et al. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol. 2000;74(9):4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YE, Pena L, et al. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J Virol. 2002;76(17):8864–8874. doi: 10.1128/JVI.76.17.8864-8874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappan S, Kraus VB, et al. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A. 1992;89(10):4549–4553. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemes LB, Camporeale G, et al. Cysteine-rich positions outside the structural zinc motif of human papillomavirus E7 provide conformational modulation and suggest functional redox roles. Biochemistry. 2014;53(10):1680–1696. doi: 10.1021/bi401562e. [DOI] [PubMed] [Google Scholar]
- Chesters PM, Vousden KH, et al. Analysis of human papillomavirus type 16 open reading frame E7 immortalizing function in rat embryo fibroblast cells. J Gen Virol. 1990;71(Pt 2):449–453. doi: 10.1099/0022-1317-71-2-449. [DOI] [PubMed] [Google Scholar]
- Chiantore MV, Mangino G, et al. Human papillomavirus E6 and E7 oncoproteins affect the expression of cancer-related microRNAs: additional evidence in HPV-induced tumorigenesis. J Cancer Res Clin Oncol. 2016;142(8):1751–1763. doi: 10.1007/s00432-016-2189-1. [DOI] [PubMed] [Google Scholar]
- Chien WM, Parker JN, et al. Casein kinase II phosphorylation of the human papillomavirus-18 E7 protein is critical for promoting S-phase entry. Cell Growth Differ. 2000;11(8):425–435. [PubMed] [Google Scholar]
- Ching W, Dobner T, et al. The human adenovirus type 5 E1B 55-kilodalton protein is phosphorylated by protein kinase CK2. J Virol. 2012;86(5):2400–2415. doi: 10.1128/JVI.06066-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnam M, Goodrich DW, et al. RB1, development, and cancer. Curr Top Dev Biol. 2011;94:129–169. doi: 10.1016/B978-0-12-380916-2.00005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365(6449):855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cicchini L, Westrich JA, et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. MBio. 2016;7(3) doi: 10.1128/mBio.00270-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens KE, Brent R, et al. Dimerization of the human papillomavirus E7 oncoprotein in vivo. Virology. 1995;214(1):289–293. doi: 10.1006/viro.1995.9926. [DOI] [PubMed] [Google Scholar]
- Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24(17):2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- Collins AS, Nakahara T, et al. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J Virol. 2005;79(23):14769–14780. doi: 10.1128/JVI.79.23.14769-14780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JP, Ferrer K, et al. Evaluation of Langerhans’ cells in the cervical epithelium of women with cervical intraepithelial neoplasia. Gynecol Oncol. 1999;75(1):130–135. doi: 10.1006/gyno.1999.5559. [DOI] [PubMed] [Google Scholar]
- Conway MJ, Alam S, et al. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J Virol. 2009;83(20):10515–10526. doi: 10.1128/JVI.00731-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Malagon EM, Bonilla-Delgado J, et al. Gene expression profile regulated by the HPV16 E7 oncoprotein and estradiol in cervical tissue. Virology. 2013;447(1–2):155–165. doi: 10.1016/j.virol.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creek KE, Geslani G, et al. Progressive loss of sensitivity to growth control by retinoic acid and transforming growth factor-beta at late stages of human papillomavirus type 16-initiated transformation of human keratinocytes. Adv Exp Med Biol. 1995;375:117–135. doi: 10.1007/978-1-4899-0949-7_11. [DOI] [PubMed] [Google Scholar]
- Da Costa R, Bastos MM, et al. The NFkappaB Signaling Pathway in Papillomavirus-induced Lesions: Friend or Foe? Anticancer Res. 2016;36(5):2073–2083. [PubMed] [Google Scholar]
- Dahl R, Wani B, et al. The Ski oncoprotein interacts with Skip, the human homolog of Drosophila Bx42. Oncogene. 1998;16(12):1579–1586. doi: 10.1038/sj.onc.1201687. [DOI] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, et al. The c-Myc target gene network. Semin Cancer Biol. 2006;16(4):253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- De Andrea M, Mondini M, et al. Alpha- and betapapillomavirus E6/E7 genes differentially modulate pro-inflammatory gene expression. Virus Res. 2007;124(1–2):220–225. doi: 10.1016/j.virusres.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Delvenne P, Hubert P, et al. The organotypic culture of HPV-transformed keratinocytes: an effective in vitro model for the development of new immunotherapeutic approaches for mucosal (pre)neoplastic lesions. Vaccine. 2001;19(17–19):2557–2564. doi: 10.1016/s0264-410x(00)00489-8. [DOI] [PubMed] [Google Scholar]
- Demers GW, Espling E, et al. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70(10):6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers GW, Foster SA, et al. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci U S A. 1994;91(10):4382–4386. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers GW, Halbert CL, et al. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology. 1994;198(1):169–174. doi: 10.1006/viro.1994.1019. [DOI] [PubMed] [Google Scholar]
- Deng XM, Li W, et al. RNA interference of human papillomavirus type 16 E7 increases HLA class I antigen expression in HaCaT-E7 cells. Int J Gynecol Cancer. 2011;21(1):28–34. doi: 10.1097/IGC.0b013e3181ffcca1. [DOI] [PubMed] [Google Scholar]
- Denslow SA, Wade PA, et al. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26(37):5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- Dick FA, Dyson NJ, et al. Three regions of the pRB pocket domain affect its inactivation by human papillomavirus E7 proteins. J Virol. 2002;76(12):6224–6234. doi: 10.1128/JVI.76.12.6224-6234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Rubin SM, et al. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaio D, Petti LM, et al. The E5 proteins. Virology. 2013;445(1–2):99–114. doi: 10.1016/j.virol.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong WL, Caldeira S, et al. Determination of the binding affinity of different human papillomavirus E7 proteins for the tumour suppressor pRb by a plate-binding assay. J Virol Methods. 2001;98(1):91–98. doi: 10.1016/s0166-0934(01)00361-5. [DOI] [PubMed] [Google Scholar]
- Doorbar J, Quint W, et al. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(Suppl 5):F55–70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- Dreier K, Scheiden R, et al. Subcellular localization of the human papillomavirus 16 E7 oncoprotein in CaSki cells and its detection in cervical adenocarcinoma and adenocarcinoma in situ. Virology. 2011;409(1):54–68. doi: 10.1016/j.virol.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V, Kaufmann WK, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76(6):1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- Eberhard J, Onder Z, et al. Nuclear import of high risk HPV16 E7 oncoprotein is mediated by its zinc-binding domain via hydrophobic interactions with Nup62. Virology. 2013;446(1–2):334–345. doi: 10.1016/j.virol.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R, Ewen ME, et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8(8):869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- Edmonds C, Vousden KH, et al. A point mutational analysis of human papillomavirus type 16 E7 protein. J Virol. 1989;63(6):2650–2656. doi: 10.1128/jvi.63.6.2650-2656.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichten A, Westfall M, et al. Stabilization and functional impairment of the tumor suppressor p53 by the human papillomavirus type 16 E7 oncoprotein. Virology. 2002;295(1):74–85. doi: 10.1006/viro.2002.1375. [DOI] [PubMed] [Google Scholar]
- Eisenman RN. Deconstructing myc. Genes Dev. 2001;15(16):2023–2030. doi: 10.1101/gad928101. [DOI] [PubMed] [Google Scholar]
- Fera D, Marmorstein R. Different regions of the HPV-E7 and Ad-E1A viral oncoproteins bind competitively but through distinct mechanisms to the CH1 transactivation domain of p300. Biochemistry. 2012;51(47):9524–9534. doi: 10.1021/bi3011863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichorova RN, Anderson DJ, et al. Differential expression of immunobiological mediators by immortalized human cervical and vaginal epithelial cells. Biol Reprod. 1999;60(2):508–514. doi: 10.1095/biolreprod60.2.508. [DOI] [PubMed] [Google Scholar]
- Firzlaff JM, Galloway DA, et al. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989;1(1):44–53. [PubMed] [Google Scholar]
- Firzlaff JM, Luscher B, et al. Negative charge at the casein kinase II phosphorylation site is important for transformation but not for Rb protein binding by the E7 protein of human papillomavirus type 16. Proc Natl Acad Sci U S A. 1991;88(12):5187–5191. doi: 10.1073/pnas.88.12.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Quaas M, et al. Polo-like kinase 4 transcription is activated via CRE and NRF1 elements, repressed by DREAM through CDE/CHR sites and deregulated by HPV E7 protein. Nucleic Acids Res. 2014;42(1):163–180. doi: 10.1093/nar/gkt849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, et al. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74(14):6622–6631. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman D, de Martel C, et al. Global burden of human papillomavirus and related diseases. Vaccine. 2012;30(Suppl 5):F12–23. doi: 10.1016/j.vaccine.2012.07.055. [DOI] [PubMed] [Google Scholar]
- Frazer IH. Interaction of human papillomaviruses with the host immune system: a well evolved relationship. Virology. 2009;384(2):410–414. doi: 10.1016/j.virol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Frolov MV, Dyson NJ, et al. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117(Pt 11):2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- Gandarillas A. The mysterious human epidermal cell cycle, or an oncogene-induced differentiation checkpoint. Cell Cycle. 2012;11(24):4507–4516. doi: 10.4161/cc.22529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner-Hamrick PA, Fostel JM, et al. Global effects of human papillomavirus type 18 E6/E7 in an organotypic keratinocyte culture system. J Virol. 2004;78(17):9041–9050. doi: 10.1128/JVI.78.17.9041-9050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam D, Moody CA, et al. Impact of the DNA Damage Response on Human Papillomavirus Chromatin. PLoS Pathog. 2016;12(6):e1005613. doi: 10.1371/journal.ppat.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese NJ, Banerjee NS, et al. Casein kinase II motif-dependent phosphorylation of human papillomavirus E7 protein promotes p130 degradation and S-phase induction in differentiated human keratinocytes. J Virol. 2008;82(10):4862–4873. doi: 10.1128/JVI.01202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulos NT, Proffitt JL, et al. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene. 2000;19(42):4930–4935. doi: 10.1038/sj.onc.1203860. [DOI] [PubMed] [Google Scholar]
- Ghittoni R, Accardi R, et al. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes. 2010;40(1):1–13. doi: 10.1007/s11262-009-0412-8. [DOI] [PubMed] [Google Scholar]
- Gielen V, Schmitt D, et al. HLA class I antigen (heavy and light chain) expression by Langerhans cells and keratinocytes of the normal human epidermis: ultrastructural quantitation using immunogold labelling procedure. Arch Dermatol Res. 1988;280(3):131–136. doi: 10.1007/BF00456841. [DOI] [PubMed] [Google Scholar]
- Gigante M, Gesualdo L, et al. TGF-beta: a master switch in tumor immunity. Curr Pharm Des. 2012;18(27):4126–4134. doi: 10.2174/138161212802430378. [DOI] [PubMed] [Google Scholar]
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- Gomez-Gomez Y, Organista-Nava J, et al. The expression of miR-21 and miR-143 is deregulated by the HPV16 E7 oncoprotein and 17beta-estradiol. Int J Oncol. 2016;49(2):549–558. doi: 10.3892/ijo.2016.3575. [DOI] [PubMed] [Google Scholar]
- Gonzalez SL, Stremlau M, et al. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75(16):7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MJ, Zhang J, et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24(19):3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Perez M, et al. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2(4):405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- Gu W, Shi XL, et al. Synergistic activation of transcription by CBP and p53. Nature. 1997;387(6635):819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- Guccione E, Massimi P, et al. Comparative analysis of the intracellular location of the high- and low-risk human papillomavirus oncoproteins. Virology. 2002;293(1):20–25. doi: 10.1006/viro.2001.1290. [DOI] [PubMed] [Google Scholar]
- Guess JC, McCance DJ, et al. Decreased Migration of Langerhans Precursor-Like Cells in Response to Human Keratinocytes Expressing Human Papillomavirus Type 16 E6/E7 Is Related to Reduced Macrophage Inflammatory Protein-3{alpha} Production. J Virol. 2005;79(23):14852–14862. doi: 10.1128/JVI.79.23.14852-14862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulliver GA, Herber RL, et al. Both conserved region 1 (CR1) and CR2 of the human papillomavirus type 16 E7 oncogene are required for induction of epidermal hyperplasia and tumor formation in transgenic mice. J Virol. 1997;71(8):5905–5914. doi: 10.1128/jvi.71.8.5905-5914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekharan V, Laimins LA, et al. Human Papillomaviruses Modulate MicroRNA 145 Expression To Directly Control Genome Amplification. J Virol. 2013;87(10):6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekharan VK, Li Y, et al. Post-Transcriptional Regulation of KLF4 by High-Risk Human Papillomaviruses Is Necessary for the Differentiation-Dependent Viral Life Cycle. PLoS Pathog. 2016;12(7):e1005747. doi: 10.1371/journal.ppat.1005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Sari-Ak D, et al. Zooming in on Transcription Preinitiation. J Mol Biol. 2016;428(12):2581–2591. doi: 10.1016/j.jmb.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyongyosi E, Szalmas A, et al. Transcriptional regulation of genes involved in keratinocyte differentiation by human papillomavirus 16 oncoproteins. Arch Virol. 2014 doi: 10.1007/s00705-014-2305-y. [DOI] [PubMed] [Google Scholar]
- Habig M, Smola H, et al. E7 proteins from high- and low-risk human papillomaviruses bind to TGF-beta-regulated Smad proteins and inhibit their transcriptional activity. Arch Virol. 2006;151(10):1961–1972. doi: 10.1007/s00705-006-0768-1. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, et al. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hasan UA, Zannetti C, et al. The Human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J Exp Med. 2013 doi: 10.1084/jem.20122394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck DV, Yee CL, et al. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc Natl Acad Sci U S A. 1992;89(10):4442–4446. doi: 10.1073/pnas.89.10.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleman J, Smid M, et al. Pathway analysis of gene lists associated with platinum-based chemotherapy resistance in ovarian cancer: the big picture. Gynecol Oncol. 2010;117(2):170–176. doi: 10.1016/j.ygyno.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Heller C, Weisser T, et al. Identification of key amino acid residues that determine the ability of high risk HPV16-E7 to dysregulate major histocompatibility complex class I expression. J Biol Chem. 2011;286(13):10983–10997. doi: 10.1074/jbc.M110.199190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellner K, Mar J, et al. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology. 2009;391(1):57–63. doi: 10.1016/j.virol.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helt AM, Galloway DA, et al. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75(15):6737–6747. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helt AM, Galloway DA, et al. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003;24(2):159–169. doi: 10.1093/carcin/24.2.159. [DOI] [PubMed] [Google Scholar]
- Hengstermann A, Linares LK, et al. Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc Natl Acad Sci U S A. 2001;98(3):1218–1223. doi: 10.1073/pnas.031470698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber R, Liem A, et al. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70(3):1873–1881. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdman MT, Pett MR, et al. Interferon-beta treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis. 2006;27(11):2341–2353. doi: 10.1093/carcin/bgl172. [DOI] [PubMed] [Google Scholar]
- Herman L, Hubert P, et al. MIP3 alpha stimulates the migration of Langerhans cells in models of human papillomavirus (HPV)-associated (pre)neoplastic epithelium. Cancer Immunol Immunother. 2006 doi: 10.1007/s00262-006-0255-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Wanzel M, et al. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell. 2002;10(3):509–521. doi: 10.1016/s1097-2765(02)00633-0. [DOI] [PubMed] [Google Scholar]
- Hiebert SW, Lipp M, et al. E1A-dependent trans-activation of the human MYC promoter is mediated by the E2F factor. Proc Natl Acad Sci U S A. 1989;86(10):3594–3598. doi: 10.1073/pnas.86.10.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Cheng S, et al. STAT-5 Regulates Transcription of the Topoisomerase IIbeta-Binding Protein 1 (TopBP1) Gene To Activate the ATR Pathway and Promote Human Papillomavirus Replication. MBio. 2015;6(6):e02006–02015. doi: 10.1128/mBio.02006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Laimins LA, et al. The JAK-STAT Transcriptional Regulator, STAT-5, Activates the ATM DNA Damage Pathway to Induce HPV 31 Genome Amplification upon Epithelial Differentiation. PLoS Pathog. 2013;9(4):e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Mehta KP, et al. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol. 2011;85(18):9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Ge W, et al. HPV 16 E7 inhibits OSCC cell proliferation, invasion, and metastasis by upregulating the expression of miR-20a. Tumour Biol. 2016;37(7):9433–9440. doi: 10.1007/s13277-016-4817-4. [DOI] [PubMed] [Google Scholar]
- Huang PS, Patrick DR, et al. Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol Cell Biol. 1993;13(2):953–960. doi: 10.1128/mcb.13.2.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, McCance DJ, et al. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol. 2002;76(17):8710–8721. doi: 10.1128/JVI.76.17.8710-8721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert P, Caberg JH, et al. E-cadherin-dependent adhesion of dendritic and Langerhans cells to keratinocytes is defective in cervical human papillomavirus-associated (pre)neoplastic lesions. J Pathol. 2005;206(3):346–355. doi: 10.1002/path.1771. [DOI] [PubMed] [Google Scholar]
- Huh K, Zhou X, et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81(18):9737–9747. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SG, Lee D, et al. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J Biol Chem. 2002;277(4):2923–2930. doi: 10.1074/jbc.M109113200. [DOI] [PubMed] [Google Scholar]
- Hyland PL, McDade SS, et al. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J Virol. 2011;85(21):10999–11006. doi: 10.1128/JVI.00160-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hypes MK, Pirisi L, et al. Mechanisms of decreased expression of transforming growth factor-beta receptor type I at late stages of HPV16-mediated transformation. Cancer Lett. 2009;282(2):177–186. doi: 10.1016/j.canlet.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson Wechsler E, Wang Q, et al. Reconstruction of human papillomavirus type 16-mediated early-stage neoplasia implicates E6/E7 deregulation and the loss of contact inhibition in neoplastic progression. J Virol. 2012;86(11):6358–6364. doi: 10.1128/JVI.07069-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Lai CH, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. Embo J. 2001;20(6):1331–1340. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT, et al. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbar SF, Abrams L, et al. Persistence of high-grade cervical dysplasia and cervical cancer requires the continuous expression of the human papillomavirus type 16 E7 oncogene. Cancer Res. 2009;69(10):4407–4414. doi: 10.1158/0008-5472.CAN-09-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal N, John R, et al. Oncogenic Human Papillomavirus 16E7 modulates SUMOylation of FoxM1b. Int J Biochem Cell Biol. 2015;58C:28–36. doi: 10.1016/j.biocel.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Jansma AL, Martinez-Yamout MA, et al. The high-risk HPV16 E7 oncoprotein mediates interaction between the transcriptional coactivator CBP and the retinoblastoma protein pRb. J Mol Biol. 2014;426(24):4030–4048. doi: 10.1016/j.jmb.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewers RJ, Hildebrandt P, et al. Regions of human papillomavirus type 16 E7 oncoprotein required for immortalization of human keratinocytes. J Virol. 1992;66(3):1329–1335. doi: 10.1128/jvi.66.3.1329-1335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johung K, Goodwin EC, et al. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J Virol. 2007;81(5):2102–2116. doi: 10.1128/JVI.02348-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Thompson DA, et al. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239(1):97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- Jones DL, Thompson DA, et al. Expression of the HPV E7 oncoprotein mimics but does not evoke a p53- dependent cellular DNA damage response pathway. Virology. 1999;258(2):406–414. doi: 10.1006/viro.1999.9733. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Lis JT, et al. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16(3):167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HM, Phillips BL, et al. miR-375 activates p21 and suppresses telomerase activity by coordinately regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3zeta. Mol Cancer. 2014;13:80. doi: 10.1186/1476-4598-13-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaewprag J, Umnajvijit W, et al. HPV16 oncoproteins promote cervical cancer invasiveness by upregulating specific matrix metalloproteinases. PLoS One. 2013;8(8):e71611. doi: 10.1371/journal.pone.0071611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski A, Ueki I, et al. EGFR activation suppresses respiratory virus-induced IRF1-dependent CXCL10 production. Am J Physiol Lung Cell Mol Physiol. 2014;307(2):L186–196. doi: 10.1152/ajplung.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim R, Meyers C, et al. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS One. 2011;6(3):e17848. doi: 10.1371/journal.pone.0017848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Tolleson WH, et al. Inhibition of growth, transformation, and expression of human papillomavirus type 16 E7 in human keratinocytes by alpha interferons. J Virol. 1993;67(6):3396–3403. doi: 10.1128/jvi.67.6.3396-3403.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho EY, Wang HK, et al. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc Natl Acad Sci U S A. 2013;110(19):7542–7549. doi: 10.1073/pnas.1304855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhutz AJ, Roman A, et al. Cellular transformation by human papillomaviruses: lessons learned by comparing high- and low-risk viruses. Virology. 2012;424(2):77–98. doi: 10.1016/j.virol.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp AA, McManus PM, et al. Identification of the nuclear localization and export signals of high risk HPV16 E7 oncoprotein. Virology. 2009;383(1):60–68. doi: 10.1016/j.virol.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Wang W, et al. Regulator role of HPV E7 protein on miR-21 expression in cervical carcinoma cells and its functional implication. Int J Clin Exp Pathol. 2015;8(12):15808–15813. [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski N, Spardy N, et al. Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Lett. 2011;305(2):113–122. doi: 10.1016/j.canlet.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace MJ, Anson JR, et al. Interferon treatment of human keratinocytes harboring extrachromosomal, persistent HPV-16 plasmid genomes induces de novo viral integration. Carcinogenesis. 2015;36(1):151–159. doi: 10.1093/carcin/bgu236. [DOI] [PubMed] [Google Scholar]
- Lam EW, Morris JD, et al. HPV16 E7 oncoprotein deregulates B-myb expression: correlation with targeting of p107/E2F complexes. Embo J. 1994;13(4):871–878. doi: 10.1002/j.1460-2075.1994.tb06330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langsfeld ES, Bodily JM, et al. The Deacetylase Sirtuin 1 Regulates Human Papillomavirus Replication by Modulating Histone Acetylation and Recruitment of DNA Damage Factors NBS1 and Rad51 to Viral Genomes. PLoS Pathog. 2015;11(9):e1005181. doi: 10.1371/journal.ppat.1005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L, Gray EE, et al. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350(6260):568–571. doi: 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- Laurson J, Khan S, et al. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis. 2010;31(5):918–926. doi: 10.1093/carcin/bgq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurson J, Raj K. Localisation of human papillomavirus 16 e7 oncoprotein changes with cell confluence. PLoS One. 2011;6(6):e21501. doi: 10.1371/journal.pone.0021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kim DH, et al. Structural investigation on the intrinsically disordered N-terminal region of HPV16 E7 protein. BMB Rep. 2016;49(8):431–436. doi: 10.5483/BMBRep.2016.49.8.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenasi T, Barboric M. P-TEFb stimulates transcription elongation and pre-mRNA splicing through multilateral mechanisms. RNA Biol. 2010;7(2):145–150. doi: 10.4161/rna.7.2.11057. [DOI] [PubMed] [Google Scholar]
- Lenhard B, Sandelin A, et al. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet. 2012;13(4):233–245. doi: 10.1038/nrg3163. [DOI] [PubMed] [Google Scholar]
- Leone G, Sears R, et al. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol Cell. 2001;8(1):105–113. doi: 10.1016/s1097-2765(01)00275-1. [DOI] [PubMed] [Google Scholar]
- Leong CM, Doorbar J, et al. Deregulation of E-cadherin by human papillomavirus is not confined to high-risk, cancer-causing types. Br J Dermatol. 2010;163(6):1253–1263. doi: 10.1111/j.1365-2133.2010.09968.x. [DOI] [PubMed] [Google Scholar]
- Leong GM, Subramaniam N, et al. Ski-interacting protein interacts with Smad proteins to augment transforming growth factor-beta-dependent transcription. J Biol Chem. 2001;276(21):18243–18248. doi: 10.1074/jbc.M010815200. [DOI] [PubMed] [Google Scholar]
- Lepik D, Ilves I, et al. p53 protein is a suppressor of papillomavirus DNA replication. J Virol. 1998;72(8):6822–6831. doi: 10.1128/jvi.72.8.6822-6831.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CO, Deng W, et al. miR-135a leads to cervical cancer cell transformation through regulation of beta-catenin via a SIAH1-dependent ubiquitin proteosomal pathway. Carcinogenesis. 2014;35(9):1931–1940. doi: 10.1093/carcin/bgu032. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, et al. The role of chromatin during transcription. Cell. 2007;128(4):707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Li H, Ou X, et al. HPV16E7 mediates HADC chromatin repression and downregulation of MHC class I genes in HPV16 tumorigenic cells through interaction with an MHC class I promoter. Biochem Biophys Res Commun. 2006;349(4):1315–1321. doi: 10.1016/j.bbrc.2006.08.182. [DOI] [PubMed] [Google Scholar]
- Li H, Zhan T, et al. Repression of MHC class I transcription by HPV16E7 through interaction with a putative RXRbeta motif and NF-kappaB cytoplasmic sequestration. Biochem Biophys Res Commun. 2009;388(2):383–388. doi: 10.1016/j.bbrc.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA, et al. TGF-beta: a master of all T cell trades. Cell. 2008;134(3):392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Huang W, et al. Expression of CD34, alpha-smooth muscle actin and transforming growth factor-beta1 in squamous intraepithelial lesions and squamous cell carcinoma of the cervix. J Int Med Res. 2009;37(2):446–454. doi: 10.1177/147323000903700220. [DOI] [PubMed] [Google Scholar]
- Li W, Deng XM, et al. Down-regulation of HLA class I antigen in human papillomavirus type 16 E7 expressing HaCaT cells: correlate with TAP-1 expression. Int J Gynecol Cancer. 2010;20(2):227–232. doi: 10.1111/IGC.0b013e3181cceec5. [DOI] [PubMed] [Google Scholar]
- Lill NL, Grossman SR, et al. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387(6635):823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhang S, et al. MicroRNA-27b up-regulated by human papillomavirus 16 E7 promotes proliferation and suppresses apoptosis by targeting polo-like kinase2 in cervical cancer. Oncotarget. 2016;7(15):19666–19679. doi: 10.18632/oncotarget.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19(2):1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Gao G, et al. Activation of miR-9 by human papillomavirus in cervical cancer. Oncotarget. 2014;5(22):11620–11630. doi: 10.18632/oncotarget.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Clements A, et al. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J Biol Chem. 2006;281(1):578–586. doi: 10.1074/jbc.M508455200. [DOI] [PubMed] [Google Scholar]
- Liu X, Disbrow GL, et al. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. J Virol. 2007;81(22):12689–12695. doi: 10.1128/JVI.00669-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Roberts J, et al. HPV E7 contributes to the telomerase activity of immortalized and tumorigenic cells and augments E6-induced hTERT promoter function. Virology. 2008;375(2):611–623. doi: 10.1016/j.virol.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard I, Vincent-Salomon A, et al. Human papillomavirus genotype as a major determinant of the course of cervical cancer. J Clin Oncol. 1998;16(8):2613–2619. doi: 10.1200/JCO.1998.16.8.2613. [DOI] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA, et al. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol. 2004;78(7):3533–3541. doi: 10.1128/JVI.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Wilson R, et al. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. Embo J. 2005;24(10):1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz AT, Reid R, et al. Human papillomavirus infection of the cervix: relative risk associations of 15 common anogenital types. Obstet Gynecol. 1992;79(3):328–337. doi: 10.1097/00006250-199203000-00002. [DOI] [PubMed] [Google Scholar]
- Lu ZH, Wright JD, et al. Hypoxia-inducible factor-1 facilitates cervical cancer progression in human papillomavirus type 16 transgenic mice. Am J Pathol. 2007;171(2):667–681. doi: 10.2353/ajpath.2007.061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulli D, Carbone ML, et al. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7(30):47777–47793. doi: 10.18632/oncotarget.10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J, Duong FH, et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology. 2013;58(4):1225–1235. doi: 10.1002/hep.26404. [DOI] [PubMed] [Google Scholar]
- Luscher-Firzlaff JM, Westendorf JM, et al. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: enhancement of transformation and transactivation. Oncogene. 1999;18(41):5620–5630. doi: 10.1038/sj.onc.1202967. [DOI] [PubMed] [Google Scholar]
- Macaluso M, Montanari M, et al. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene. 2006;25(38):5263–5267. doi: 10.1038/sj.onc.1209680. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391(6667):601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Maldonado E, Cabrejos ME, et al. Human papillomavirus-16 E7 protein inhibits the DNA interaction of the TATA binding transcription factor. J Cell Biochem. 2002;85(4):663–669. doi: 10.1002/jcb.10172. [DOI] [PubMed] [Google Scholar]
- Manning J, Indrova M, et al. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology. 2008;123(2):218–227. doi: 10.1111/j.1365-2567.2007.02689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Trouche D, et al. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375(6533):691–694. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- Martinez I, Gardiner AS, et al. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene. 2008;27(18):2575–2582. doi: 10.1038/sj.onc.1210919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimi P, Banks L. Repression of p53 transcriptional activity by the HPV E7 proteins. Virology. 1997;227(1):255–259. doi: 10.1006/viro.1996.8315. [DOI] [PubMed] [Google Scholar]
- Massimi P, Banks L. Differential phosphorylation of the HPV-16 E7 oncoprotein during the cell cycle. Virology. 2000;276(2):388–394. doi: 10.1006/viro.2000.0514. [DOI] [PubMed] [Google Scholar]
- Massimi P, Pim D, et al. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J Gen Virol. 1997;78(Pt 10):2607–2613. doi: 10.1099/0022-1317-78-10-2607. [DOI] [PubMed] [Google Scholar]
- Massimi P, Pim D, et al. HPV-16 E7 and adenovirus E1a complex formation with TATA box binding protein is enhanced by casein kinase II phosphorylation. Oncogene. 1996;12(11):2325–2330. [PubMed] [Google Scholar]
- Matthews K, Leong CM, et al. Depletion of Langerhans cells in human papillomavirus type 16-infected skin is associated with E6-mediated down regulation of E-cadherin. J Virol. 2003;77(15):8378–8385. doi: 10.1128/JVI.77.15.8378-8385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromatis KO, Jones DL, et al. The carboxyl-terminal zinc-binding domain of the human papillomavirus E7 protein can be functionally replaced by the homologous sequences of the E6 protein. Virus Res. 1997;52(1):109–118. doi: 10.1016/s0168-1702(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Mayumi N, Watanabe E, et al. E-cadherin interactions are required for Langerhans cell differentiation. Eur J Immunol. 2013;43(1):270–280. doi: 10.1002/eji.201242654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarelli JM, Atkins GB, et al. The viral oncoproteins Ad5 E1A, HPV16 E7 and SV40 TAg bind a common region of the TBP-associated factor-110. Oncogene. 1995;11(9):1859–1864. [PubMed] [Google Scholar]
- McDougall JK. Immortalization and transformation of human cells by human papillomavirus. Curr Top Microbiol Immunol. 1994;186:101–119. doi: 10.1007/978-3-642-78487-3_6. [DOI] [PubMed] [Google Scholar]
- McIntyre MC, Frattini MG, et al. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J Virol. 1993;67(6):3142–3150. doi: 10.1128/jvi.67.6.3142-3150.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna DJ, McDade SS, et al. MicroRNA 203 expression in keratinocytes is dependent on regulation of p53 levels by E6. J Virol. 2010;84(20):10644–10652. doi: 10.1128/JVI.00703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna DJ, Patel D, et al. miR-24 and miR-205 expression is dependent on HPV onco-protein expression in keratinocytes. Virology. 2014;448:210–216. doi: 10.1016/j.virol.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Bromberg-White JL, et al. The role of the human papillomavirus type 18 E7 oncoprotein during the complete viral life cycle. Virology. 2005;338(1):61–68. doi: 10.1016/j.virol.2005.04.036. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Crum CP, et al. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A. 2011;108(5):2130–2135. doi: 10.1073/pnas.1009933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Huh KW, et al. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J Virol. 2008;82(17):8695–8705. doi: 10.1128/JVI.00579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Munger K, et al. The human papillomavirus E7 oncoprotein. Virology. 2009;384(2):335–344. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Munger K, et al. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143(2):195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Munger K, et al. Biochemical and functional interactions of human papillomavirus proteins with polycomb group proteins. Viruses. 2013;5(5):1231–1249. doi: 10.3390/v5051231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Park D, et al. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc Natl Acad Sci U S A. 2013;110(40):16175–16180. doi: 10.1073/pnas.1310432110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melar-New M, Laimins LA, et al. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;84(10):5212–5221. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Y, Borger DR, et al. Loss of transforming growth factor-beta (TGF-beta) receptor type I mediates TGF-beta resistance in human papillomavirus type 16-transformed human keratinocytes at late stages of in vitro progression. Virology. 2000;270(2):408–416. doi: 10.1006/viro.2000.0283. [DOI] [PubMed] [Google Scholar]
- Miciak J, Bunz F. Long story short: p53 mediates innate immunity. Biochim Biophys Acta. 2016;1865(2):220–227. doi: 10.1016/j.bbcan.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietz JA, Unger T, et al. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J. 1992;11(13):5013–5020. doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Moody CA, Laimins LA, et al. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5(10):e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA, et al. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- Moore PS, Chang Y, et al. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10(12):878–889. doi: 10.1038/nrc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandell D, Kaiser A, et al. The human papillomavirus type 16 E7 oncoprotein targets Myc-interacting zinc-finger protein-1. Virology. 2012;422(2):242–253. doi: 10.1016/j.virol.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morosov A, Phelps WC, et al. Activation of the c-fos gene by the HPV16 oncoproteins depends upon the cAMP-response element at −60. J Biol Chem. 1994;269(28):18434–18440. [PubMed] [Google Scholar]
- Morozov A, Shiyanov P, et al. Accumulation of human papillomavirus type 16 E7 protein bypasses G1 arrest induced by serum deprivation and by the cell cycle inhibitor p21. J Virol. 1997;71(5):3451–3457. doi: 10.1128/jvi.71.5.3451-3457.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscicki AB, Schiffman M, et al. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine. 2012;30(Suppl 5):F24–33. doi: 10.1016/j.vaccine.2012.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses HL. TGF-beta regulation of epithelial cell proliferation. Mol Reprod Dev. 1992;32(2):179–184. doi: 10.1002/mrd.1080320215. [DOI] [PubMed] [Google Scholar]
- Muller C, Alunni-Fabbroni M, et al. Separation of C/EBPalpha-mediated proliferation arrest and differentiation pathways. Proc Natl Acad Sci U S A. 1999;96(13):7276–7281. doi: 10.1073/pnas.96.13.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller GA, Engeland K, et al. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. FEBS J. 2010;277(4):877–893. doi: 10.1111/j.1742-4658.2009.07508.x. [DOI] [PubMed] [Google Scholar]
- Munger K, Baldwin A, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Basile JR, et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20(54):7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Munger K, Werness BA, et al. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Embo J. 1989;8(13):4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Fontela C, Macip S, et al. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008;205(8):1929–1938. doi: 10.1084/jem.20080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murvai M, Borbely AA, et al. Effect of human papillomavirus type 16 E6 and E7 oncogenes on the activity of the transforming growth factor-beta2 (TGF-beta2) promoter. Arch Virol. 2004;149(12):2379–2392. doi: 10.1007/s00705-004-0376-x. [DOI] [PubMed] [Google Scholar]
- Myklebust MP, Bruland O, et al. MicroRNA-15b is induced with E2F-controlled genes in HPV-related cancer. Br J Cancer. 2011;105(11):1719–1725. doi: 10.1038/bjc.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Bodily JM, et al. Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology. 2009;387(2):442–448. doi: 10.1016/j.virol.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardelli B, Zaritskaya L, et al. Regulatory effect of IFN-kappa, a novel type I IFN, on cytokine production by cells of the innate immune system. J Immunol. 2002;169(9):4822–4830. doi: 10.4049/jimmunol.169.9.4822. [DOI] [PubMed] [Google Scholar]
- Nees M, Geoghegan JM, et al. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol. 2001;75(9):4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nees M, Geoghegan JM, et al. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Res. 2000;60(15):4289–4298. [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, et al. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18(19):3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Westbrook TF, et al. Human papillomavirus type 16 E7 maintains elevated levels of the cdc25A tyrosine phosphatase during deregulation of cell cycle arrest. J Virol. 2002;76(2):619–632. doi: 10.1128/JVI.76.2.619-632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nor Rashid N, Yusof R, et al. Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J Gen Virol. 2011;92(Pt 11):2620–2627. doi: 10.1099/vir.0.035352-0. [DOI] [PubMed] [Google Scholar]
- Nor Rashid N, Yusof R, et al. Disruption of pocket protein dream complexes by E7 proteins of different types of human papillomaviruses. Acta Virol. 2013;57(4):447–451. doi: 10.4149/av_2013_04_447. [DOI] [PubMed] [Google Scholar]
- Noya F, Chien WM, et al. The promoter of the human proliferating cell nuclear antigen gene is not sufficient for cell cycle-dependent regulation in organotypic cultures of keratinocytes. J Biol Chem. 2002;277(19):17271–17280. doi: 10.1074/jbc.M112441200. [DOI] [PubMed] [Google Scholar]
- O’Brien PM, Saveria Campo M, et al. Evasion of host immunity directed by papillomavirus-encoded proteins. Virus Res. 2002;88(1–2):103–117. doi: 10.1016/s0168-1702(02)00123-5. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362(6423):857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- Ozbun MA, Meyers C, et al. Transforming growth factor beta1 induces differentiation in human papillomavirus-positive keratinocytes. J Virol. 1996;70(8):5437–5446. doi: 10.1128/jvi.70.8.5437-5446.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, et al. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Palomero T, Lim WK, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103(48):18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang CL, Toh SY, et al. A functional interaction of E7 with B-Myb-MuvB complex promotes acute cooperative transcriptional activation of both S- and M-phase genes. (129 c) Oncogene. 2014;33(31):4039–4049. doi: 10.1038/onc.2013.426. [DOI] [PubMed] [Google Scholar]
- Park JS, Kim EJ, et al. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000;275(10):6764–6769. doi: 10.1074/jbc.275.10.6764. [DOI] [PubMed] [Google Scholar]
- Patel D, Huang SM, et al. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. Embo J. 1999;18(18):5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick DR, Oliff A, et al. Identification of a novel retinoblastoma gene product binding site on human papillomavirus type 16 E7 protein. J Biol Chem. 1994;269(9):6842–6850. [PubMed] [Google Scholar]
- Perea S, Lopezocejo O, et al. Human papillomavirus type-16 (HPV-16) major transforming proteins functionally interact with interferon signaling mechanisms. Int J Oncol. 1997;11(1):169–173. doi: 10.3892/ijo.11.1.169. [DOI] [PubMed] [Google Scholar]
- Perea SE, Massimi P, et al. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int J Mol Med. 2000;5(6):661–666. doi: 10.3892/ijmm.5.6.661. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Not just a CDK inhibitor: regulation of transcription by p21(WAF1/CIP1/SDI1) Cell Cycle. 2002;1(1):39–41. [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, et al. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275(5299):523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Pett M, Coleman N. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J Pathol. 2007;212(4):356–367. doi: 10.1002/path.2192. [DOI] [PubMed] [Google Scholar]
- Pett MR, Herdman MT, et al. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc Natl Acad Sci U S A. 2006;103(10):3822–3827. doi: 10.1073/pnas.0600078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps WC, Bagchi S, et al. Analysis of trans activation by human papillomavirus type 16 E7 and adenovirus 12S E1A suggests a common mechanism. J Virol. 1991;65(12):6922–6930. doi: 10.1128/jvi.65.12.6922-6930.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps WC, Munger K, et al. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J Virol. 1992;66(4):2418–2427. doi: 10.1128/jvi.66.4.2418-2427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps WC, Yee CL, et al. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell. 1988;53(4):539–547. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- Pickup M, Novitskiy S, et al. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietenpol JA, Stein RW, et al. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990;61(5):777–785. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- Polanska UM, Orimo A, et al. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. J Cell Physiol. 2013;228(8):1651–1657. doi: 10.1002/jcp.24347. [DOI] [PubMed] [Google Scholar]
- Prathapam T, Kuhne C, et al. The HPV-16 E7 oncoprotein binds Skip and suppresses its transcriptional activity. Oncogene. 2001;20(52):7677–7685. doi: 10.1038/sj.onc.1204960. [DOI] [PubMed] [Google Scholar]
- Prathapam T, Kuhne C, et al. Skip interacts with the retinoblastoma tumor suppressor and inhibits its transcriptional repression activity. Nucleic Acids Res. 2002;30(23):5261–5268. doi: 10.1093/nar/gkf658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prathapam T, Kuhne C, et al. Ski interacts with the evolutionarily conserved SNW domain of Skip. Nucleic Acids Res. 2001;29(17):3469–3476. doi: 10.1093/nar/29.17.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science. 2016;352(6291) doi: 10.1126/science.aad9926. aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnelle KM, Boehm AL, et al. STAT-mediated EGFR signaling in cancer. J Cell Biochem. 2007;102(2):311–319. doi: 10.1002/jcb.21475. [DOI] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141(3):432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JA, Pusztai R, et al. Chemical synthesis of human papillomavirus type 16 E7 oncoprotein: autonomous protein domains for induction of cellular DNA synthesis and for trans activation. J Virol. 1990;64(12):6121–6129. doi: 10.1128/jvi.64.12.6121-6129.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinstein E, Scheffner M, et al. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000;19(51):5944–5950. doi: 10.1038/sj.onc.1203989. [DOI] [PubMed] [Google Scholar]
- Reiser J, Hurst J, et al. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol. 2011;85(21):11372–11380. doi: 10.1128/JVI.05279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey O, Lee S, et al. The E7 oncoprotein of human papillomavirus type 16 interacts with F-actin in vitro and in vivo. Virology. 2000;268(2):372–381. doi: 10.1006/viro.1999.0175. [DOI] [PubMed] [Google Scholar]
- Richards KH, Doble R, et al. Human papillomavirus E7 oncoprotein increases production of the anti-inflammatory interleukin-18 binding protein in keratinocytes. J Virol. 2014;88(8):4173–4179. doi: 10.1128/JVI.02546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KH, Wasson CW, et al. The human papillomavirus (HPV) E7 protein antagonises an Imiquimod-induced inflammatory pathway in primary human keratinocytes. Sci Rep. 2015;5:12922. doi: 10.1038/srep12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RR, Duensing S, et al. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63(16):4862–4871. [PubMed] [Google Scholar]
- Riley T, Sontag E, et al. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Rizzolio F, Esposito L, et al. RB gene family: genome-wide ChIP approaches could open undiscovered roads. J Cell Biochem. 2010;109(5):839–843. doi: 10.1002/jcb.22448. [DOI] [PubMed] [Google Scholar]
- Roman A, Munger K. The papillomavirus E7 proteins. Virology. 2013;445(1–2):138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Santos M, et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004;131(11):2737–2748. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- Sadasivam S, DeCaprio JA, et al. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer. 2013;13(8):585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, Duan S, et al. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26(5):474–489. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12(18):2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Harry JB, et al. Comparison of the properties of the E6 and E7 genes of low- and high-risk cutaneous papillomaviruses reveals strongly transforming and high Rb-binding activity for the E7 protein of the low-risk human papillomavirus type 1. J Virol. 1994;68(11):7051–7059. doi: 10.1128/jvi.68.11.7051-7059.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, et al. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M, Nakagawa M, et al. Cell-mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol. 2001;8(2):209–220. doi: 10.1128/CDLI.8.2.209-220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seavey SE, Holubar M, et al. The E7 oncoprotein of human papillomavirus type 16 stabilizes p53 through a mechanism independent of p19(ARF) J Virol. 1999;73(9):7590–7598. doi: 10.1128/jvi.73.9.7590-7598.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19(1):12–16. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Shin MK, Sage J, et al. Inactivating all three Rb family pocket proteins is insufficient to initiate cervical cancer. Cancer Res. 2012;72(20):5418–5427. doi: 10.1158/0008-5472.CAN-12-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola-Hess S, Pahne J, et al. Expression of membrane type 1 matrix metalloproteinase in papillomavirus-positive cells: role of the human papillomavirus (HPV) 16 and HPV8 E7 gene products. J Gen Virol. 2005;86(Pt 5):1291–1296. doi: 10.1099/vir.0.80551-0. [DOI] [PubMed] [Google Scholar]
- Snowden AW, Perkins ND, et al. Cell cycle regulation of the transcriptional coactivators p300 and CREB binding protein. Biochem Pharmacol. 1998;55(12):1947–1954. doi: 10.1016/s0006-2952(98)00020-3. [DOI] [PubMed] [Google Scholar]
- Somasundaram K, El-Deiry WS, et al. Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene. 1997;14(9):1047–1057. doi: 10.1038/sj.onc.1201002. [DOI] [PubMed] [Google Scholar]
- Southern SA, Lewis MH, et al. Induction of tetrasomy by human papillomavirus type 16 E7 protein is independent of pRb binding and disruption of differentiation. Br J Cancer. 2004;90(10):1949–1954. doi: 10.1038/sj.bjc.6601827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinillo A, Tenti P, et al. Langerhans’ cell counts and cervical intraepithelial neoplasia in women with human immunodeficiency virus infection. Gynecol Oncol. 1993;48(2):210–213. doi: 10.1006/gyno.1993.1035. [DOI] [PubMed] [Google Scholar]
- Spitkovsky D, Hehner SP, et al. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa B kinase complex. J Biol Chem. 2002;277(28):25576–25582. doi: 10.1074/jbc.M201884200. [DOI] [PubMed] [Google Scholar]
- Stanley MA. Immune responses to human papilloma viruses. Indian J Med Res. 2009;130(3):266–276. [PubMed] [Google Scholar]
- Steegenga WT, van Laar T, et al. Adenovirus E1A proteins inhibit activation of transcription by p53. Mol Cell Biol. 1996;16(5):2101–2109. doi: 10.1128/mcb.16.5.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RW, Corrigan M, et al. Analysis of E1A-mediated growth regulation functions: binding of the 300-kilodalton cellular product correlates with E1A enhancer repression function and DNA synthesis-inducing activity. J Virol. 1990;64(9):4421–4427. doi: 10.1128/jvi.64.9.4421-4427.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart ZA, Tang LJ, et al. Increased p53 phosphorylation after microtubule disruption is mediated in a microtubule inhibitor- and cell-specific manner. Oncogene. 2001;20(1):113–124. doi: 10.1038/sj.onc.1204060. [DOI] [PubMed] [Google Scholar]
- Stoppler H, Stoppler MC, et al. The E7 protein of human papillomavirus type 16 sensitizes primary human keratinocytes to apoptosis. Oncogene. 1998;17(10):1207–1214. doi: 10.1038/sj.onc.1202053. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Hayakawa S, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- Tamura T, Yanai H, et al. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- Tang A, Amagai M, et al. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature. 1993;361(6407):82–85. doi: 10.1038/361082a0. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, et al. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- Thalmeier K, Synovzik H, et al. Nuclear factor E2F mediates basic transcription and trans-activation by E1a of the human MYC promoter. Genes Dev. 1989;3(4):527–536. doi: 10.1101/gad.3.4.527. [DOI] [PubMed] [Google Scholar]
- Thomas JT, Hubert WG, et al. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc Natl Acad Sci U S A. 1999;96(15):8449–8454. doi: 10.1073/pnas.96.15.8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JT, Laimins LA, et al. Human papillomavirus oncoproteins E6 and E7 independently abrogate the mitotic spindle checkpoint. J Virol. 1998;72(2):1131–1137. doi: 10.1128/jvi.72.2.1131-1137.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM, et al. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17(2):251–264. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Tirone NR, Peghini BC, et al. Local expression of interferon-alpha and interferon receptors in cervical intraepithelial neoplasia. Cancer Immunol Immunother. 2009;58(12):2003–2010. doi: 10.1007/s00262-009-0707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic B, Hung K, et al. Conserved region 3 of human papillomavirus 16 E7 contributes to deregulation of the retinoblastoma tumor suppressor. J Virol. 2012;86(24):13313–13323. doi: 10.1128/JVI.01637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic B, Massimi P, et al. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J Virol. 2011;85(19):10048–10057. doi: 10.1128/JVI.00643-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis MA, Sheppard D, et al. TGF-beta activation and function in immunity. Annu Rev Immunol. 2014;32:51–82. doi: 10.1146/annurev-immunol-032713-120257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugizov S, Berline J, et al. Inhibition of human papillomavirus type 16 E7 phosphorylation by the S100 MRP-8/14 protein complex. J Virol. 2005;79(2):1099–1112. doi: 10.1128/JVI.79.2.1099-1112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SJ, Rhyu JW, et al. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002;179(2):205–212. doi: 10.1016/s0304-3835(01)00871-0. [DOI] [PubMed] [Google Scholar]
- Vandermark ER, Deluca KA, et al. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology. 2012;425(1):53–60. doi: 10.1016/j.virol.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Ortiz G, Garcia JA, et al. Differentially expressed genes between high-risk human papillomavirus types in human cervical cancer cells. Int J Gynecol Cancer. 2007;17(2):484–491. doi: 10.1111/j.1525-1438.2007.00831.x. [DOI] [PubMed] [Google Scholar]
- Vijayalingam S, Chinnadurai G. Adenovirus L-E1A activates transcription through mediator complex-dependent recruitment of the super elongation complex. J Virol. 2013;87(6):3425–3434. doi: 10.1128/JVI.03046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilaysane A, Muruve DA, et al. The innate immune response to DNA. Semin Immunol. 2009;21(4):208–214. doi: 10.1016/j.smim.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Wakefield LM, Sporn MB, et al. Suppression of carcinogenesis: a role for TGF-beta and related molecules in prevention of cancer. Immunol Ser. 1990;51:217–243. [PubMed] [Google Scholar]
- Walker J, Smiley LC, et al. Expression of human papillomavirus type 16 E7 is sufficient to significantly increase expression of angiogenic factors but is not sufficient to induce endothelial cell migration. Virology. 2011;410(2):283–290. doi: 10.1016/j.virol.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Amerio P, et al. Role of cytokines in epidermal Langerhans cell migration. J Leukoc Biol. 1999;66(1):33–39. doi: 10.1002/jlb.66.1.33. [DOI] [PubMed] [Google Scholar]
- Wang J, Sampath A, et al. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene. 2001;20(34):4740–4749. doi: 10.1038/sj.onc.1204655. [DOI] [PubMed] [Google Scholar]
- Wang N, Zhan T, et al. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br J Cancer. 2014;110(4):1034–1044. doi: 10.1038/bjc.2013.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang HK, et al. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. Rna. 2009;15(4):637–647. doi: 10.1261/rna.1442309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YW, Chang HS, et al. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int J Biochem Cell Biol. 2007;39(2):402–412. doi: 10.1016/j.biocel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Warren CJ, Griffin LM, et al. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS One. 2014;9(5):e96579. doi: 10.1371/journal.pone.0096579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Kanda T, et al. Mutational analysis of human papillomavirus type 16 E7 functions. J Virol. 1990;64(1):207–214. doi: 10.1128/jvi.64.1.207-214.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM. Epidermal stem cells: markers, patterning and the control of stem cell fate. Philos Trans R Soc Lond B Biol Sci. 1998;353(1370):831–837. doi: 10.1098/rstb.1998.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM, Frye M, et al. MYC in mammalian epidermis: how can an oncogene stimulate differentiation? Nat Rev Cancer. 2008;8(3):234–242. doi: 10.1038/nrc2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock H, Berman S, et al. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health. 2004;36(1):6–10. doi: 10.1363/psrh.36.6.04. [DOI] [PubMed] [Google Scholar]
- Werness BA, Levine AJ, et al. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248(4951):76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Wise-Draper TM, Wells SI, et al. Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci. 2008;13:1003–1017. doi: 10.2741/2739. [DOI] [PubMed] [Google Scholar]
- Woodworth CD, Chung J, et al. Transforming growth factor beta 1 supports autonomous growth of human papillomavirus-immortalized cervical keratinocytes under conditions promoting squamous differentiation. Cell Growth Differ. 1996;7(6):811–820. [PubMed] [Google Scholar]
- Woodworth CD, Notario V, et al. Transforming growth factors beta 1 and 2 transcriptionally regulate human papillomavirus (HPV) type 16 early gene expression in HPV-immortalized human genital epithelial cells. J Virol. 1990;64(10):4767–4775. doi: 10.1128/jvi.64.10.4767-4775.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao ZX, Chen J, et al. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature. 1995;375(6533):694–698. doi: 10.1038/375694a0. [DOI] [PubMed] [Google Scholar]
- Xu Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003;10(4):400–403. doi: 10.1038/sj.cdd.4401182. [DOI] [PubMed] [Google Scholar]
- Yee KS, Vousden KH, et al. Complicating the complexity of p53. Carcinogenesis. 2005;26(8):1317–1322. doi: 10.1093/carcin/bgi122. [DOI] [PubMed] [Google Scholar]
- Yin FF, Wang N, et al. Serine/threonine kinases 31(STK31) may be a novel cellular target gene for the HPV16 oncogene E7 with potential as a DNA hypomethylation biomarker in cervical cancer. Virol J. 2016;13(1):60. doi: 10.1186/s12985-016-0515-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youlyouz-Marfak I, Gachard N, et al. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ. 2008;15(2):376–385. doi: 10.1038/sj.cdd.4402270. [DOI] [PubMed] [Google Scholar]
- Zaheer RS, Koetzler R, et al. Selective transcriptional down-regulation of human rhinovirus-induced production of CXCL10 from airway epithelial cells via the MEK1 pathway. J Immunol. 2009;182(8):4854–4864. doi: 10.4049/jimmunol.0802401. [DOI] [PubMed] [Google Scholar]
- Zenz R, Eferl R, et al. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther. 2008;10(1):201. doi: 10.1186/ar2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chen W, et al. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. 2006;103(2):437–442. doi: 10.1073/pnas.0510012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Liu F, et al. Elevation of miR-27b by HPV16 E7 inhibits PPARgamma expression and promotes proliferation and invasion in cervical carcinoma cells. Int J Oncol. 2015;47(5):1759–1766. doi: 10.3892/ijo.2015.3162. [DOI] [PubMed] [Google Scholar]
- Zhang T, Cooper S, et al. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16(11):1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Liu Z, et al. miR-31 functions as an oncogene in cervical cancer. Arch Gynecol Obstet. 2015;292(5):1083–1089. doi: 10.1007/s00404-015-3713-2. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Stamminger T, et al. E2F/Rb Family Proteins Mediate Interferon Induced Repression of Adenovirus Immediate Early Transcription to Promote Persistent Viral Infection. PLoS Pathog. 2016;12(1):e1005415. doi: 10.1371/journal.ppat.1005415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Chen J, et al. Human papillomavirus 16-encoded E7 protein inhibits IFN-gamma-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J Gen Virol. 2013;94(Pt 11):2504–2514. doi: 10.1099/vir.0.054486-0. [DOI] [PubMed] [Google Scholar]
- Zhou F, Chen J, et al. Human papillomavirus 16-encoded E7 protein inhibits IFN-gamma-mediated MHC Class I antigen presentation and CTL-induced lysis through blocking IRF-1 expression in mouse keratinocytes. J Gen Virol. 2013 doi: 10.1099/vir.0.054486-0. [DOI] [PubMed] [Google Scholar]
- Zhou J, Schmid T, et al. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237(1):10–21. doi: 10.1016/j.canlet.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang Q, et al. Role of WDHD1 in Human Papillomavirus-Mediated Oncogenesis Identified by Transcriptional Profiling of E7-Expressing Cells. J Virol. 2016;90(13):6071–6084. doi: 10.1128/JVI.00513-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Ye M, et al. E6/E7 oncoproteins of high risk HPV-16 upregulate MT1-MMP, MMP-2 and MMP-9 and promote the migration of cervical cancer cells. Int J Clin Exp Pathol. 2015;8(5):4981–4989. [PMC free article] [PubMed] [Google Scholar]
- Zilfou JT, Lowe SW, et al. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1(5):a001883. doi: 10.1101/cshperspect.a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology. 2009;384(2):260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]