

Abstract
Inspired by the discovery of the antimalarial drug artemisinin from a traditional Chinese medicine (TCM), a natural product library of 44 lindenane-type sesquiterpenoids was assessed for activities against the Dd2 chloroquine-resistant strain of the malaria parasite Plasmodium falciparum. These compounds were mainly isolated from plants of the Chloranthus genus, many species of which are named “Sikuaiwa” in TCM and have long been used to treat malaria. The compounds consisted of 41 sesquiterpenoid dimers and three monomers, including the twelve new dimers 1–12 isolated from C. fortunei. The results showed that 16 dimers exhibited potent antiplasmodial activities (<100 nM); in particular, compounds 1, 14 and 19 exhibited low nanomolar activities with IC50 values ranging from 1 to 7 nM, which is comparable to the potency of artemisinin, and selectivity index values toward mammalian cells greater than 500. A comprehensive structure-activity relationship (SAR) study clearly indicated that three functional groups are essential and two motifs can be modified.
Keywords: Antimalarial activity, Structure-activity relationship, Natural products, Lindenane-type sesquiterpenoids
Graphical Abstract
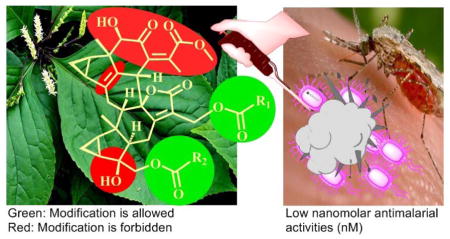
Malaria is a global parasitic infectious disease caused by Plasmodium parasites, which are transmitted by the bite of female anopheles mosquitoes. The disease is widespread in tropical and subtropical regions, with the majority of deaths occurring in Africa and caused by P. falciparum, resulting in losses of US $12 billion a year.1 Approximately 3.2 billion people remain at risk of contracting malaria, leading to an estimated 214 million malaria cases, and 438,000 deaths in 2014 alone.2 Drug resistance also poses a growing problem for malaria treatment, including the gold standard artemisinin combination therapies.3 Therefore, the development of new antimalarial agents to expand the repertoire of drugs suitable for use in combination therapies remains a priority to retain effective malaria control and elimination.
The Chloranthaceae family has three genera with a total of 16 species, including Chloranthus (thirteen sp.), Sarcandra (two sp.), and Hedyosmum (one sp.), which are mainly distributed in the southern area of China.4 Our ongoing studies on plants of the former two genera in the Chloranthaceae family have led to the isolation of a large number of sesquiterpenoids and sesquiterpenoid dimers with diverse bioactivities.5
Natural products have played a very important role in antimalarial drug development, and have led to the well-known antimalarial drugs quinine and artemisinin.6 The potent antimalarial artemisinin was developed from a traditional Chinese medicine, the aerial parts of Artemisia annua dubbed “Qinghao”, which was used to treat malaria as early as 340 CE, as documented in the ancient Chinese pharmacopeia “Principal Prescription Emergency” in China’s Eastern Jin Dynasty7 Interestingly, many Chloranthus species, called “Sikuaiwa” in TCM, were also used for the same purpose.8 A small library of structurally related compounds isolated from five Chloranthus species and one Sarcandra specie was thus subjected to antiplasmodial bioassay against the chloroquine-resistant Dd2 strain of P. falciparum. These compounds included three lindenane-type sesquiterpenoid monomers and 41 dimers (Figures 1–3). Compounds 1–12 are new dimeric sesquiterpenoids isolated from the twigs of C. fortunei and reported in this study. The antiplasmodial screening revealed that several of the sesquiterpenoid dimers exhibited very potent antiplasmodial activities, and three compounds had potent activities similar to that of artemisinin. These results open the way to further studies on this new class of promising antiplasmodial compounds that may act through a new mechanism of action because of their unique structural architectures. We herein present the isolation and structure elucidation of the new compounds, as well as the antiplasmodial activities and structure-activity relationships of all 44 compounds.
Figure 1.

The tested compounds 1–6, and 13–27.
Figure 3.

The tested compounds 29–44.
RESULTS AND DISCUSSION
Compound 1 had a molecular formula of C41H46O14 as determined by the sodiated (+)-HRESIMS ion at m/z 785.2781 [M + Na]+ (calcd 785.2780) and 13C NMR data. Analysis of the NMR data (Tables 1 and 4) suggested that 1 is a lindenane-type sesquiterpenoid dimer with the distinct features of two 1,2-substituted cyclopropane rings, one α,β-unsaturated-γ-lactone, and a persubstituted double bond. Comprehensive analysis of the 1D and 2D NMR data further revealed that its structure is similar to that of chlorajaponol G9 except for the presence of a γ-formylsenecioate motif instead of the angeloate moiety of the latter. This conclusion was verified by HMBC correlations of H-4″ (δH 9.67)/C-2″ and C-5″ within the motif that was attached to C-15′ by the key HMBC correlation from H2-15′ to C-1″ (δC 165.5) (Figure 4A). Additionally, a methyl succinoate group was attached to C-13′ as indicated by the key HMBC correlation of H2-13′/C-9″ (δC 172.1). The relative configuration of 1 was assigned primarily by its ROESY data (Figure 4B), in which the correlations of H-1/H-2α, H-1/H-3, H-1/H-9, H-1′/H-2′α, H-2′α/H-3′, H-3′/H-15′, and H-5′/H-15′ indicated that they are spatially close and assigned arbitrarily as α-oriented. Accordingly, the ROESY correlations of H-2β/H3-14, H3-14/H-6, H-2′β/H3-14′, and H3-14′/H-9′ revealed that they were β-oriented. The Δ2″ double bond was assigned E by the ROESY correlation of H2-4″/H-2″. Thus, the structure of fortunilide A (1) was depicted as shown.
Table 1.
1H NMR Data of Compounds 1–4 (500 MHz, CDCl3)
| No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
|
| ||||
| (mult., J in Hz) | (mult., J in Hz) | (mult., J in Hz) | (mult., J in Hz) | |
| 1 | 2.05, ddd (8.3, 5.7, 4.2) | 2.05, ddd (8.1, 5.7, 4.3) | 2.08, ddd (8.3, 5.8, 4.2) | 2.02, ddd (8.5, 6.2, 4.3) |
| 2α | 0.98, m | 0.98, m | 1.02, m | 0.95, m |
| 2β | 0.27, m | 0.29, m | 0.33, m | 1.26, m |
| 3 | 1.83, m | 1.82, m | 1.87, m | 1.57, m |
| 6 | 3.93, d (3.7) | 3.89, d (3.9) | 3.93, d (3.9) | |
| 9 | 3.95, s | 3.92, s | 3.97, s | 3.78, s |
| 13 | 1.87, s | 1.90, s | 1.94, s | 1.78, s |
| 14 | 1.00, s | 0.98, s | 1.02, s | 1.01, s |
| 15 | 2.75, m 2.60, m |
2.78, m 2.55, ddd (16.4, 6.2, 3.9) |
2.81, d (16.2) 2.58, ddd (16.2, 6.2, 3.9) |
3.03, dd (14.2, 7.1) 1.61, m |
| 1′ | 1.60, m | 1.57, dt (8.4, 3.8) | 1.60, dt (8.8, 5.7) | 1.84, m |
| 2′α | 0.74, m | 0.68, m | 0.71, m | 0.60, m |
| 2′β | 1.32, m | 1.23, m | 1.26, m | 1.18, m |
| 3′ | 1.45, m | 1.49, ddd (9.0, 7.3, 3.8) | 1.52, m | 1.25, m |
| 5′ | 1.81, m | 1.82, m | 1.87, dd (13.7, 6.1) | 1.66, dd (13.2, 6.5) |
| 6′α | 2.52, dd (18.4, 5.8) | 2.28, dd (18.5, 6.1) | 2.28, dd (18.4, 6.1) | 2.29, dd (17.6, 6.5) |
| 6′β | 2.77, m | 2.72, m | 2.72, dd (18.4, 13.7) | 2.89, dd (17.6, 13.2) |
| 9′ | 1.83, m | 1.89, m | 1.94, m | 2.56, dd (10.2, 7.1) |
| 13′ | 4.97, d (13.4) 4.82, d (13.4) |
4.37, d (13.6) 4.30, d (13.6) |
4.41, d (13.6) 4.34, d (13.6) |
4.44, d (13.8) 4.37, d (13.8) |
| 14′ | 0.88, s | 0.85, s | 0.88, s | 0.98, s |
| 15′ | 4.30, d (11.8) 3.83, d (11.8) |
4.07, d (11.5) 3.81, d (11.5) |
4.83, d (11.5) 4.10, d (11.5) |
4.03, d (11.4) 3.97, d (11.4) |
| 2″ | 6.65, q (1.5) | 5.89, s | 5.73, m | 5.89, d (1.4) |
| 4″ | 9.67, s | 4.62, s | 1.94, s | 4.66, m |
| 5″ | 2.19, d (1.5) | 2.13, s | 2.19, s | 2.16, s |
| 7″ | 2.67, m | 2.67, m | ||
| 8″ | 2.73, m | 2.73, m | ||
| 12-OMe | 3.68, s | 3.75, s | 3.65, s | 3.77, s |
| 6″-OMe | 3.66, s | |||
| 9″-OMe | 3.67, s | 3.69, s | ||
| 4-OOH | 8.64, s | |||
Table 4.
13C NMR Data of Compounds 1–12 at 125 MHz
| No. | 1a | 2a | 3a | 4a | 5a | 6b | 7a | 8a | 9b | 10b | 11a | 12a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 25.7 | 26.1 | 26.1 | 26.0 | 26.0 | 27.3 | 26.1 | 28.2 | 30.4 | 34.3 | 28.5 | 28.6 |
| 2 | 16.0 | 16.1 | 16.1 | 8.3 | 8.3 | 9.0 | 16.0 | 14.3 | 10.0 | 11.7 | 15.8 | 15.8 |
| 3 | 24.8 | 24.9 | 25.0 | 27.5 | 27.9 | 28.4 | 24.9 | 23.9 | 28.4 | 25.9 | 26.6 | 26.7 |
| 4 | 142.4 | 142.6 | 142.6 | 90.8 | 90.7 | 91.3 | 141.3 | 138.0 | 89.9 | 146.1 | 151.3 | 151.3 |
| 5 | 131.5 | 132.2 | 132.2 | 158.7 | 158.9 | 159.5 | 132.8 | 155.8 | 161.0 | 43.8 | 135.4 | 135.4 |
| 6 | 40.7 | 41.1 | 41.1 | 127.1 | 127.0 | 129.2 | 41.9 | 115.7 | 126.8 | 44.6 | 151.4 | 151.4 |
| 7 | 131.2 | 131.1 | 131.0 | 143.2 | 143.3 | 132.6 | 134.8 | 124.8 | 154.6 | 153.3 | 129.7 | 129.7 |
| 8 | 200.2 | 200.2 | 200.0 | 198.8 | 198.4 | 200.6 | 204.1 | 104.7 | 105.2 | 108.0 | 197.0 | 197.1 |
| 9 | 80.5 | 80.1 | 80.1 | 77.9 | 77.8 | 78.7 | 80.6 | 75.9 | 78.8 | 80.0 | 83.2 | 83.2 |
| 10 | 51.3 | 51.1 | 51.2 | 50.3 | 50.2 | 51.5 | 50.4 | 49.3 | 50.5 | 47.4 | 57.6 | 57.6 |
| 11 | 148.1 | 147.5 | 147.6 | 129.0 | 128.8 | 141.5 | 144.2 | 153.8 | 125.3 | 133.2 | 65.3 | 65.4 |
| 12 | 170.5 | 171.8 | 171.2 | 170.3 | 170.5 | 172.3 | 169.4 | 171.8 | 173.2 | 173.2 | 172.0 | 172.0 |
| 13 | 20.8 | 20.5 | 20.5 | 21.4 | 21.4 | 21.0 | 19.2 | 10.4 | 10.6 | 10.7 | 18.9 | 19.1 |
| 14 | 15.2 | 15.5 | 15.6 | 15.5 | 15.5 | 16.1 | 15.5 | 14.5 | 14.3 | 22.7 | 14.9 | 14.9 |
| 15 | 25.4 | 25.4 | 25.4 | 36.9 | 36.8 | 36.8 | 25.4 | 121.0 | 36.1 | 112.5 | 29.3 | 29.2 |
| 1′ | 25.6 | 25.7 | 25.7 | 27.9 | 27,5 | 27.4 | 26.2 | 25.6 | 28.2 | 26.6 | 26.6 | 26.6 |
| 2′ | 12.0 | 12.0 | 12.0 | 10.2 | 10.2 | 16.9 | 11.8 | 11.2 | 11.0 | 11.4 | 10.3 | 10.4 |
| 3′ | 28.0 | 28.4 | 28.6 | 29.3 | 29.0 | 24.1 | 27.2 | 29.2 | 30.4 | 29.8 | 29.8 | 29.7 |
| 4′ | 77.3 | 77.3 | 77.5 | 77.3 | 77.1 | 49.4 | 77.3 | 77.3 | 78.1 | 78.4 | 79.0 | 78.8 |
| 5′ | 61.0 | 59.9 | 59.8 | 54.6 | 54.0 | 54.5 | 61.8 | 55.8 | 53.1 | 57.1 | 55.7 | 56.5 |
| 6′ | 23.4 | 22.3 | 22.2 | 22.0 | 22.8 | 26.2 | 25.3 | 21.7 | 22.5 | 24.9 | 27.8 | 27.8 |
| 7′ | 171.9 | 168.5 | 168.5 | 166.3 | 170.3 | 170.0 | 174.4 | 171.6 | 173.9 | 173.2 | 59.1 | 59.1 |
| 8′ | 93.3 | 93.5 | 93.7 | 87.7 | 87.6 | 88.8 | 93.3 | 85.9 | 87.5 | 92.0 | 95.6 | 95.5 |
| 9′ | 55.9 | 55.1 | 55.1 | 52.7 | 52.8 | 52.4 | 55.0 | 55.5 | 52.1 | 54.0 | 52.2 | 52.1 |
| 10′ | 44.9 | 44.8 | 44.9 | 45.1 | 45.0 | 45.7 | 44.7 | 47.8 | 46.0 | 46.5 | 43.0 | 43.1 |
| 11′ | 123.6 | 127.5 | 127.4 | 128.6 | 125.3 | 129.5 | 123.2 | 123.6 | 124.7 | 128.4 | 145.9 | 146.1 |
| 12′ | 171.4 | 172.4 | 172.5 | 172.9 | 171.6 | 174.9 | 171.9 | 171.8 | 173.4 | 173.9 | 168.4 | 168.4 |
| 13′ | 55.8 | 54.8 | 54.9 | 55.1 | 55.8 | 54.5 | 55.2 | 55.3 | 56.6 | 54.6 | 123.1 | 129.7 |
| 14′ | 26.6 | 26.4 | 26.4 | 24.4 | 24.3 | 23.1 | 26.2 | 25.3 | 24.5 | 25.8 | 24.0 | 24.0 |
| 15′ | 72.4 | 70.5 | 70.1 | 69.8 | 69.1 | 64.1 | 72.2 | 68.3 | 69.3 | 72.4 | 69.5 | 70.4 |
| 1″ | 165.5 | 166.1 | 166.8 | 166.0 | 166.3 | 166.2 | 167.0 | 168.4 | 168.3 | 167.9 | 166.0 | |
| 2″ | 135.4 | 114.9 | 115.4 | 115.2 | 114.5 | 113.0 | 113.4 | 114.4 | 114.1 | 128.2 | 114.3 | |
| 3″ | 151.2 | 153.4 | 158.8 | 153.9 | 160.9 | 153.9 | 159.1 | 159.9 | 160.1 | 138.7 | 154.2 | |
| 4″ | 195.1 | 67.4 | 27.6 | 67.9 | 67.1 | 66.2 | 67.2 | 67.2 | 67.3 | 14.7 | 67.3 | |
| 5″ | 11.0 | 15.9 | 20.6 | 16.1 | 15.8 | 15.9 | 16.0 | 16.0 | 15.8 | 12.3 | 16.0 | |
| 6″ | 173.2 | 171.8 | 171.9 | 173.3 | 172.2 | 174.8 | 174.8 | 171.7 | ||||
| 7″ | 28.7 | 28.8 | 28.9 | 28.6 | 29.5 | 28.5 | 173.9 | 28.9 | ||||
| 8″ | 28.7 | 29.0 | 29.1 | 28.7 | 29.2 | 28.5 | 29.5 | 29.0 | ||||
| 9″ | 172.1 | 173.0 | 173.0 | 172.1 | 172.9 | 171.9 | 29.5 | 172.9 | ||||
| 12-OMe | 52.8 | 52.9 | 52.9 | 53.0 | 53.1 | 53.1 | 53.0 | 52.2 | 52.1 | |||
| 6″-OMe | 52.2 | 52.3 | 53.0 | 52.6 | ||||||||
| 9″-OMe | 52.1 | 52.2 | 52.1 |
Data were measured in CDCl3 and
data were measured in CD3OD.
Figure 4.

(A) HMBC and (B) ROESY correlations of 1.
The molecular formula of compound 2 was established as C41H48O14, by the sodium adduct (+)-HRESIMS ion at m/z 787.2964 [M + Na]+ (calcd 787.2936) and the 13C NMR data. The NMR data (Tables 1 and 4) of 2 showed many similarities to those of shizukaol F,10 suggesting that they are structural analogues. Comprehensive analysis of the spectroscopic data of 2 revealed that it shared the same dimeric sesquiterpenoid core with shizukaol F and the structural differences were the substituent groups at C-13′ and C-15′. Compound 2 was assigned as a methanolysis product of shizukaol F with the cleavage between C-13′ and C-9″ in the 18-membered macrocyclic trilactone of the latter. This conclusion was confirmed by the HMBC correlations (Figure S13, Supporting Information) from H-4″ (δH 4.62) to C-2″ (δC 114.8) and C-6″ (δC 171.8), from both H-7″ (δH 2.67) and H-8″ (δH 2.73) to C-6″ and C-9″ (δC 173.0), and from OCH3 (δH 3.67) to C-9″. The relative configuration of 2 was established by ROESY data (Figure S14, Supporting Information), in particular, the Δ2″ double bond was assigned as E-geometry by the key ROESY interaction between H-4″ and H-2″. Hence, the structure of 2, fortunilide B, was unequivocally characterized.
Compound 3 had the molecular formula C36H42O10 based on its 13C NMR data and sodium adduct (+)-HRESIMS ion at m/z 657.2672 [M + Na]+ (calcd 657.2670). Analysis of its NMR data (Tables 1 and 4) revealed that its structure is closely related to that of shizukaol K11 with the differences being the substituents at C-13′ and C-15′. The presence of a senecioate moiety was identified by HMBC correlations within this motif, and its attachment to C-15′ was indicated by the key HMBC cross-peak from H2-15′ (δH 4.83 and 4.10) to C-1″ (δC 166.8) (Figure S22, Supporting Information). A hydroxy group was located at C-13′ by the chemical shifts of H-13′ (δH 4.41 and 4.34, each 1H, d, J = 13.6 Hz) and C-13′ (δC 54.9). A ROESY experiment (Figure S23, Supporting Information) showed that the relative configuration of 3 was identical to that of shizukaol K. The structure of compound 3, fortunilide C, was thus established as shown.
The molecular formula, C41H48O16, of compound 4 was determined by its sodium adduct (+)-HRESIMS ion at m/z 819.2851 [M + Na]+ (calcd 819.2835). Detailed analysis of its NMR data (Tables 1 and 4) revealed it to be a methanolysis product of spicachlorantin D12 with the cleavage between C-13′ and C-9″ in the 18-membered macrocyclic trilactone of the latter. This conclusion was confirmed by the HMBC correlations (Figure S31, Supporting Information) from H-4″ (δH 4.66) to C-2″ (δC 115.2) and C-6″ (δC 171.9), from both H-7″ (δH 2.67) and H-8′ (δH 2.73) to C-6″ and C-9″ (δC 173.0), and from OCH3 (δH 3.69) to C-9″. In addition, a characteristic singlet signal at δH 8.64 (OOH) and a quaternary carbon resonance at δC 90.8 indicated the presence of a hydroperoxy group at C-4.12 The relative configuration of 4 was identical to that of spicachlorantin D as indicated by its ROESY data (Figure S32, Supporting Information). The structure of 4, fortunilide D, was thereby determined as shown.
Compound 5 had the same molecular formula C41H48O16 as 4, as determined by the (+)-HRESIMS ion at m/z 819.2842 [M + Na]+ (calcd 819.2835) and 13C NMR data. Analysis of the NMR data (Tables 2 and 4) revealed that 4 and 5 were isomers, with differing substituents at C-13′ and C-15′. A methyl succinoate and a γ-hydroxysenecioate moieties were attached to C-13′ and C-15′ of 5 as shown by the key HMBC correlations of H2-13′/C-9″ and H2-15′/C-1″ (Figure S40, Supporting Information), respectively. The Δ2″ double bond was assigned as E by the ROESY interaction between H2-4″ and H-2″ (Figure S41, Supporting Information). Thus, the structure of fortunilide E (5) was unequivocally characterized as shown.
Table 2.
1H NMR Data of Compounds 5–8 at 500 MHz
| No. | 5a | 6b | 7a | 8a |
|---|---|---|---|---|
|
| ||||
| (mult., J in Hz) | (mult., J in Hz) | (mult., J in Hz) | (mult., J in Hz) | |
| 1 | 1.98, m | 1.98, ddd (8.3, 6.3, 4.3) | 1.99, m | 2.09, m |
| 2α | 0.95, m | 0.94, ddd (8.3, 8.3, 5.7) | 0.97, m | 1.07, m |
| 2β | 1.24, m | 1.00, ddd (5.7, 4.3, 4.0) | 0.33, m | 0.63, m |
| 3 | 1.55, m | 1.86, ddd (8.3, 6.3, 4.0) | 1.80, m | 2.21, m |
| 6 | 4.65, br d | |||
| 9 | 3.85, s | 3.94, s | 3.77, s | 3.85, s |
| 13 | 1.78, s | 1.69, s | 2.05, s | 1.75, s |
| 14 | 1.00, s | 1.05, s | 1.05, s | 0.81, s |
| 15 | 3.06, dd (14.2, 7.1) 1.65, dd (14.2, 10.2) |
3.20, dd (14.3, 7.2) 1.70, dd (14.3, 10.1) |
2.73, m 2.50, m |
6.16, d (4.6) |
| 1′ | 1.87, m | 1.50, ddd (8.9, 7.2, 4.0) | 1.58, m | 1.88, m |
| 2′α | 0.60, m | 0.69, m | 0.72, m | 0.66, m |
| 2′β | 1.22, m | 0.61, m | 1.32, m | 1.20, m |
| 3′ | 1.55, m | 1.32, m | 1.37, m | 1.71, m |
| 4′ | 1.38, m | |||
| 5′ | 1.76, m | 1.78 ddd (13.2, 11.0, 6.8) | 1.76, m | 2.26, dd (12.9, 6.1) |
| 6′α | 2.34, dd (17.7, 6.9) | 2.34, dd (17.6, 6.8) | 2.57, m | 2.37, dd (18.5, 6.1) |
| 6′β | 2.82, dd (17.7, 13.2) | 2.66, dd (17.6, 13.2) | 2.83, m | 2.82, dd (18.5, 12.9) |
| 9′ | 2.58, dd (10.2, 7.1) | 2.63, dd (10.1, 7.2) | 1.76, m | 2.65, m |
| 13′ | 5.06, dd (13.7, 2.0) 4.78, d (13.7) |
4.29, d (13.3) 4.24, d (13.3) |
4.98, d (12.1) 4.50, d (12.1) |
5.12, dd (13.2, 1.4) 4.81, d (13.2) |
| 14′ | 0.96, s | 0.85, s | 0.77, s | 0.98, s |
| 15′ | 4.02, s | 3.57, dd (10.5, 4.2) 3.35, dd (10.5, 6.2) |
4.49, d (11.4) 3.65, d (11.4) |
4.09, d (11.2) 3.87, d (11.2) |
| 2″ | 5.90, dd (2.8, 1.5) | 6.01, d (1.7) | 5.98, m | |
| 4″ | 4.16, m | 4.84, d (16.7) 4.59, d (16.7) |
4.14, d (16.6) 4.09, d (16.6) |
|
| 5″ | 2.19, d (1.5) | 2.19, s | 2.05, s | |
| 7″ | 2.64, m | 2.58, m | 2.50, m | |
| 8″ | 2.70, m | 2.84, m | 2.70, m | |
| 12-OMe | 3.77, s | 3.75, s | 3.77, s | |
| 6″-OMe | 3.67, s | 3.68, s | ||
| 4-OOH | 8.53, s | |||
Data were measured in CDCl3 and
data were measured in CD3OD.
Compound 6 had the molecular formula C31H36O10, based on its (+)-HRESIMS ion at m/z 591.2211 [M + Na]+ (calcd 591.2201) and 13C NMR data. Comparison of its NMR data (Tables 2 and 4) with those of spicachlorantin J13 revealed that their structures are closely related and both of them possessed a hydroperoxy moiety at C-4. The only variation was the presence of a hydroxymethyl group at C-4′ in 6 instead of the exocyclic Δ4′(15′) double bond in the latter, as supported by the HMBC correlations from H2-15′ (δH 3.57 and 3.35) to C-3′, C-4′, and C-5′ (Figure S49, Supporting Information). The structure of 6, fortunilide F, was finally confirmed by the HMBC and NOESY spectra (Figure S50, Supporting Information).
Compound 7 had the molecular formula C40H44O13 as determined by the mass of the sodiated molecular ion at m/z 755.2683 [M + Na]+ (calcd 755.2674) in the (+)-HRESIMS and the 13C NMR data. The NMR data (Tables 2 and 4) of 7 showed strong similarities to those of henriol C14 except for the changes regarding the location of CH3-5″, which was attached to C-3″ in the lactone bridge of 7 instead of C-2″ in that of henriol C, as deduced by the key HMBC correlation from H-5″ to C-4″. The chemical shift of H2-4″ (δH 4.71) suggested an E-geometry15 for the Δ2″double bond in the macrolide motif. The structure of 7, fortunilide G, was further verified by HMBC and ROESY spectra (Figures S58 and S59, Supporting Information).
Compound 8, named fortunilide H, was assigned the molecular formula C40H44O14 on the basis of its (+)-HRESIMS ion at m/z 771.2625 (calcd 771.2623) and its 13C NMR data. Analysis of the NMR data (Tables 2 and 4) showed many similarities with those of chloramultiol F,16 with a large conjugated system of two additional double bonds and one α,β-unsaturated-γ-lactone. The main differences were the presence of methyl succinoate and γ-hydroxysenecioate residues at C-13′ and C-15′, respectively, instead of the 18-membered macrocyclic trilactone ring of chloramultiol F. The locations of these groups were assigned by the HMBC correlations of H2-13′/C-9″ and H2-15′/C-1″ (Figure S67, Supporting Information). In addition, a hydroxy group was placed at C-8 by the chemical shift (δC 104.7) instead of the methoxy group in chloramultiol F, which was assigned as α-oriented by the pyridine-induced solvent shifts for H-5′ (Δδ = –0.63).17 The Δ2″ double bond was assigned as E-geometry by the key ROESY correlation of H2-4″/C-2″ (Figure S68, Supporting Information). The structure of 8 was further verified by 2D NMR spectra.
Compound 9 had the molecular formula C40H46O16, as determined by its (+)-HRESIMS ion peak at m/z 805.2659 [M + Na]+ (calcd 805.2678) and 13C NMR data. The NMR data (Tables 3 and 4) of 9 showed many similarities to those of 8, suggestive of structural analogues. In particular, an oxygenated quaternary carbon signal (δC 89.9, C-4) and a methylene carbon signal (δC 36.1, C-15) were observed for 9 in place of the carbon signals of an Δ4(15) double bond for 8, indicating the presence of a hydroperoxy moiety at C-4. Two groups of a methyl succinoate and a γ-hydroxysenecioate were assigned to C-13′ and C-15′ respectively by the HMBC correlations of H2-13′/C-9″ and H2-15′/C-1″ (Figure S76, Supporting Information). 8-OH was assigned as α-oriented by the pyridine-induced solvent shifts for H-5′ (Δδ = –0.74).17 The structure of fortunilide I (9) finally confirmed by HMBC and ROESY data (Figure S77, Supporting Information), was elucidated as shown.
Table 3.
1H NMR Data of 9–12 at 500 MHz
| No. | 9a | 10a | 11b | 12b |
|---|---|---|---|---|
|
| ||||
| (mult., J in Hz) | (mult., J in Hz) | δH (mult, J in Hz) | δH (mult, J in Hz) | |
| 1 | 1.91, ddd (8.7, 6.4, 4.7) | 1.59, ddd (8.1, 5.8, 4.0) | 2.18, dt (8.6, 4.9) | 2.18, dt (8.6, 4.9) |
| 2α | 0.93, ddd (8.7, 5.7, 5.7) | 0.58, m | 1.17, m | 1.18, m |
| 2β | 1.12, m | 0.80, ddd (8.7, 8.1, 5.1) | 0.62, m | 0.62, m |
| 3 | 1.82, ddd (8.5, 6.4, 3.8) | 2.13, ddd (8.7, 5.8, 3.4) | 1.96, ddd (2, 5.5, 3.1) | 1.97, ddd (8.2, 5.4, 3.1) |
| 5 | 2.20, m | |||
| 6 | 2.86, m | |||
| 9 | 3.78, s | 3.97, s | 3.94, s | 3.99, s |
| 13 | 1.56, s | 1.84, s | 1.35, s | 1.38, s |
| 14 | 0.77, s | 1.07, s | 1.16, s | 1.16, s |
| 15 | 3.22, dd (14.0, 6.9) 1.73, dd (14.0, 10.6) |
5.82, s | 2.30, dd (18.0, 7.1) 2.96, dd (18.0, 9.5) |
2.29, dd (18.0, 7.2) 2.95, dd (18.0, 9.5) |
| 1′ | 1.73, m | 1.93, m | 1.66, m | 1.65, m |
| 2′α | 0.64, ddd (8.9, 5.3, 5.3) | 0.63, m | 0.64, m | 0.64, m |
| 2′β | 1.22, m | 1.20, m | 1.20, m | 1.20, m |
| 3′ | 1.73, m | 1.38, ddd (8.9, 7.3, 3.5) | 1.66, m | 1.66, m |
| 5′ | 2.36, dd (12.7, 6.8) | 2.49, dd (13.5, 5.6) | 1.65, m | 1.58, dd (14.6, 2.9) |
| 6′α | 2.46, dd (17.9, 6.8) | 2.37, dd (18.7, 5.6) | 2.45, d (13.0) | 2.54, dd (14.6, 14.1) |
| 6′β | 2.91, dd (17.9, 12.7) | 2.87, dd (18.7, 13.5) | 1.69, m | 1.73, dd (14.1, 2.9) |
| 9′ | 2.71, dd (10.6, 6.8) | 2.21, m | 2.64, dd (9.5 7.1) | 2.64, dd (9.5 7.2) |
| 13′ | 4.87, d (13.3) 4.84, dd (13.3, 1.4) |
4.37, d (13.2) 4.31, dd (13.2, 1.3) |
5.52, brs 6.20, brs |
5.51, brs 6.19, brs |
| 14′ | 1.02, s | 0.91, s | 0.94, s | 0.94, s |
| 15′ | 4.04, d (11.0) 4.01, d (11.0) |
4.41, d (11.2) 3.90, d (11.2) |
4.23, d (11.1) 4.18, d (11.1) |
4.30, d (11.1) 4.08, d (11.1) |
| 2″ | 6.07, m | 5.99, q (1.5) | 6.86, m | 5.91, q (1.5) |
| 4″ | 4.10, m | 4.08, m | 1.81, brs | 4.62, m |
| 5″ | 2.12, d (1.3) | 2.07, (br s) | 1.82, brs | 2.13, d (1.5) |
| 7″ | 2.57, m | 2.68, m | ||
| 8″ | 2.67, m | 2.75, m | ||
| 12-OMe | 3.44, s | 3.43, s | ||
| 6″-OMe | 3.69, s | |||
| 9″-OMe | 3.69, s | |||
Data were measured in CD3OD and
data were measured in CDCl3.
Compound 10, named fortunilide J, was obtained as a white amorphous powder. A molecular formula of C35H40O11 was assigned for 10 with 16 DBEs on the basis of its (−)-HRESIMS ion at m/z 635.2475 [M − H]− (calcd 635.2498) as well as its 13C NMR data. Analysis of the NMR data (Tables 3 and 4) suggested that 10 is a lindenane-type sesquiterpenoid dimer with the distinct features of two 1,2-substituted propane rings, two α,β-unsaturated-γ-lactone groups, a trisubstituted double bond and a γ-hydroxysenecioate residue. Comprehensive investigation of the spectroscopic data including 2D NMR revealed that it is structurally related to henriol B,14 and the differences were the presence of a Δ4(15) double bond instead of the Δ5 double bond of the latter, and the γ-hydroxysenecioate residue in place of the senecioate residue of henriol B. The different position of the double bond was confirmed by the key HMBC correlations (Figure 5A) from H-15 (δH 5.82) to C-3 and C-8′ (δC 92.0), and from H-6 to C-4 (δC 146.1). In addition, the γ-hydroxysenecioate group was attached to C-15′ by the HMBC correlations of H2-15′/C-1″ (δC 168.3). The relative configuration of 10 was verified mainly by its ROESY data (Figure 5B), in which ROESY correlations of H-1/H-2α, H-1/H-3, H-1/H-9, H-1′/H-2′α, H-2′α/H-3′, H-3′/H-15′, and H-5′/H-15′ showed that these protons were co-facial and were assigned arbitrarily as α-oriented. In consequence, the ROESY correlations of H-2β/H3-14, H3-14/H-5, H3-14/H-6, H-2′β/H3-14′, and H3-14′/H-9′ indicated that they were β-oriented The Δ2″ double bond was assigned as E by the ROESY correlation of H2-4″/H-2″. The 8-OH was assigned as α by the significant pyridine-induced solvent shift17 for H-5′ (Δδ = −0.81).
Figure 5.
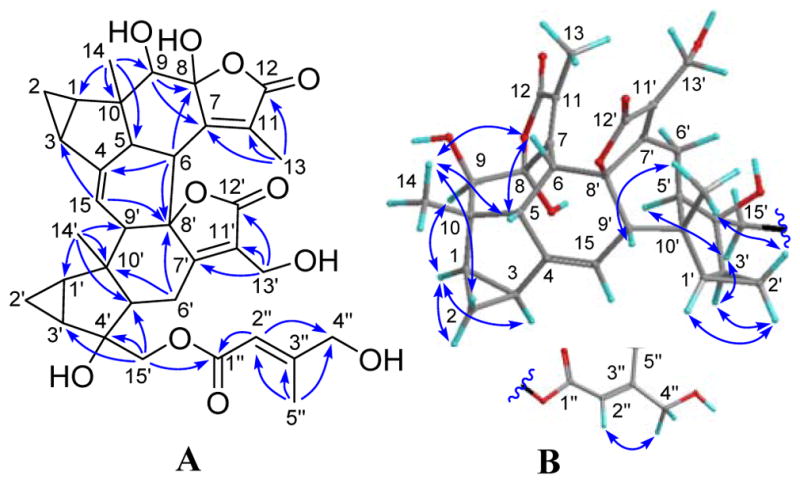
(A) HMBC and (B) ROESY correlations of 10.
Compound 11 was obtained as a colorless gum and was assigned the molecular formula C36H40O9 by the (−)-HRESIMS ion peak at m/z 615.2588 [M − H]− (calcd for 615.2600), requiring 17 double bond equivalents (DBE). The IR spectrum revealed the presence of hydroxy (3455 cm-1), carbonyl (1761 cm-1), and olefinic (1656 cm-1) functionalities. Initial analysis of the NMR data (Tables 3 and 4) of 11 suggested it was a lindenane-type sesquiterpenoid dimer possessing the rare sarcanolide A skeleton.18 Comprehensive analysis of the 1D and 2D NMR spectra of 11 further revealed that the only structural difference between it and sarcanolide A was the existence of Δ4 double bond in place of the 4,5-diol of the latter. This was verified by the multiple HMBC correlations (Figure 6) of H-15/C-4 (δC 151.3) and C-5 (δC 135.4), and H-14/C-5. The unique β-directed 3-methylenedihydrofuran-2(3H)-one motif was deduced by the key HMBC correlations from H-13′ (δH 5.52 and 6.20) to C-7′, C-11′ (δC 145.9), and C-12′ (δC 168.4). The tigloyl unit was assigned to C-15′ by the key HMBC correlation from H-15′ to C-1″ (δC 167.9). The relative configuration of 1 was elucidated by its ROESY data (Figure 6), which is similar to that of sarcanolide A, except for that CH3-13 was assigned as α-configured on the basis of the strong correlation between H3-13 and H-6′α. The structure of compound 11 (fortunilide K) was hence characterized as shown.
Figure 6.
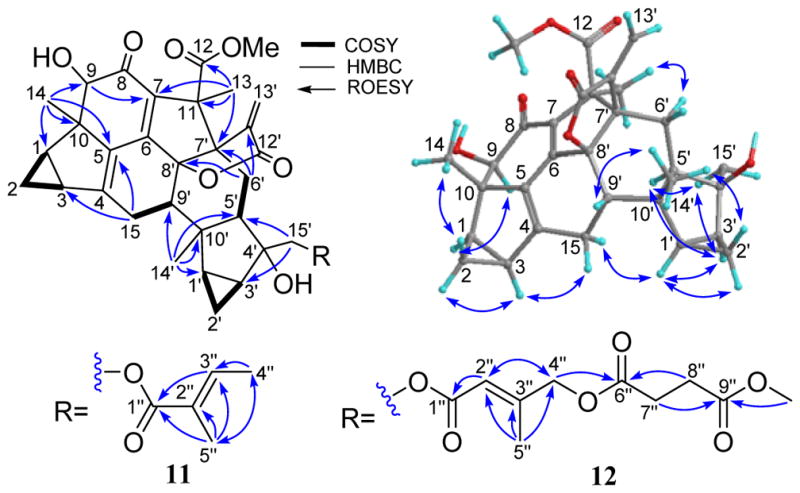
Selected HMBC, COSY, and ROESY correlations of 11 and 12.
Fortunilide L (12) had the molecular formula C41H46O13 as determined by its (+)-HRESIMS ion at m/z 769.2830 [M + Na]+ (calcd 769.2831), requiring 19 DBEs. Detailed analysis of NMR data (Tables 3 and 4) of 12 showed that it shared a common dimeric sesquiterpenoid core with 11, and the only structural difference was in the substituent at C-15′. HMBC correlations (Figure 6) within the substituent confirmed it to be an (E)-4-((4-methoxy-4-oxobutanoyl)oxy)-2-methylbut-2-enoyloxy group, and it was assigned to C-15′ by the HMBC correlations from H-15′ to C-1″. The E-configuration of the Δ2″ double bond was assigned based on the ROESY cross-peak between H2-4″ and H-2″ (Figure S105, Supporting Information). The structure of 12 was thus established as depicted.
The 15 known lindenane-type sesquiterpenoid dimers, sarglabolide I,19 sarglabolide J,19 shizukaol F,10 shizukaol K,11 shizukaol I,10 shizukaol C,20 shizukaol M,11 chlorajaponilide C,21 chlorahololides D,5j spicachlorantin D,12 chloramultilide C,22 shizukanolide F,23 chloranthalactone C,24 and isoshizukanolide,25 and sarcandrolide J5a were also isolated and identified by spectroscopic data and analogy with the reported data in literature.
Antiplasmodial Activity and Structure-Activity Relationships
A total of 44 structurally related compounds obtained from our previous studies were assessed for potential antiplasmodial activity. Twelve new compounds fortunilides A-L (1–12), as well as 10 known analogues sarglabolide I (13), sarglabolide J (14), shizukaol K (15), shizukaol I (16), shizukaol C (17), schizukaol M (18), chlorahololide D (20), shizukanolide F (42), chloranthalactone C (43), and isoshizukanolide (44) were isolated currently from C. fortunei. Chlorahololide A (33),15b chlorahololide C (34),17b and chlorahololide E (38)17b were obtained from C. holosteigius. Chloramultilide A (35),26 chloramultilide D (37),22 chloramultilide C (39),22 and chloramultilide B (41)22 came from C. multisachys. Chlorajaponilide C (19),21 shizukaol N (21),11 shizukaol E (25),10 shizukaol D (26),20 shizukaol F (28),10 shizukaol G (29),10 shizukaol B (30),20 spicachlorantin D (31),27 shizukaol A (32),28 and spicachlorantin B (40)29 were isolated from the plants C. serratus and C. spicatus. Sarcandrolide B (22),5b sarcandrolide A (23),5b sarcandrolide J (24),5a sarcandrolide E (27),5b and sarcandrolide D (36)5b were isolated from S. glabra.
The antiplasmodial activities of all the compounds were tested using a SYBR-Green assay against P. falciparum strain Dd2 (chloroquine-resistant) with artemisinin as the positive control as described previously.30,31 Sixteen compounds exhibited IC50 values below 100 nM, eight compounds showed mid to high nanomolar IC50 values (100–860 nM), eight compounds were in the micromolar range, and the others were inactive at 25 μM, which was the highest concentration tested (Table 5). Fifteen of the compounds that exhibited IC50 values below 100 nM were tested for mammalian cytotoxicity toward normal embryonic lung tissue (WI-38 cell line). Nine of the 15 compounds presented a selectivity index (SI) value of ≥ 100 (Table 6). Compounds 1, 14, and 19 had IC50 values of 5.2 ± 0.6, and 7.2 ± 1.3, and 1.1 ± 0.2 nM, respectively, and thus have similar potencies to the positive control artemisinin (IC50 = 4.0 ± 4.2 nM). Moreover, these compounds showed a SI value of 1,700, 561 and 4,900 respectively (Table 6) supporting their selective activity against the malaria parasite.
Table 5.
P. falciparum growth inhibition for compounds 1–44
| Comps | IC50 ± SD (nM) | Comps | IC50 ± SD (nM) |
|---|---|---|---|
| 1 | 5.2 ± 0.6 | 23 | 320 ± 130 |
| 2 | 19 ± 8 | 24 | 11,400 ± 1,600 |
| 3 | 211 ± 56 | 25 | 1,800 ± 400 |
| 4 | 30 ± 8 | 26 | 580 ± 90 |
| 5 | 43 ± 3 | 27 | IA |
| 6 | 5,300 ± 2,000 | 28 | 11 ± 1 |
| 7 | 46 ± 3 | 29 | 13 ± 1 |
| 8 | 198 ± 22 | 30 | 27 ± 3 |
| 9 | 94 ± 30 | 31 | 474 ± 12 |
| 10 | 9,900 ± 2,700 | 32 | 1,500± 300 |
| 11 | 4,700 ± 500 | 33 | IA |
| 12 | 99 ± 18 | 34 | IA |
| 13 | 4,600 ± 200 | 35 | IA |
| 14 | 7.2 ± 1.3 | 36 | IA |
| 15 | 860 ± 89 | 37 | IA |
| 16 | 111 ± 12 | 38 | IA |
| 17 | 21 ± 9 | 39 | IA |
| 18 | 96 ± 37 | 40 | IA |
| 19 | 1.1 ± 0.2 | 41 | 7,100 ± 1,000 |
| 20 | 13 ± 3 | 42 | IA |
| 21 | 100 ± 10 | 43 | IA |
| 22 | 265 ± 5 | 44 | IA |
IA represents inactive (IC50 > 25 mM).
Artemisinin (IC50 = 4.0 ± 4.2 nM) was used as the positive control.
Table 6.
Mammalian cytotoxic activity (WI-38 cell line) of 15 most active natural compounds (IC50 ≤ 100 nM)
| Compound | WI-38 IC50(μM) | P. falciparum IC50(μM) | SI |
|---|---|---|---|
| 1 | 8.84 | 0.0052 | 1700 |
| 2 | 3.09 | 0.019 | 163 |
| 4 | 0.53 | 0.030 | 18 |
| 5 | > 100 | 0.043 | Not cytotoxic |
| 7 | 1.24 | 0.046 | 27 |
| 12 | 15.5 | 0.099 | 157 |
| 14 | 4.04 | 0.0072 | 561 |
| 17 | 0.77 | 0.021 | 37 |
| 18 | 4.45 | 0.096 | 46 |
| 19 | 5.39 | 0.0011 | 4900 |
| 20 | 0.16 | 0.013 | 12 |
| 21 | 10.04 | 0.100 | 100 |
| 28 | 0.23 | 0.011 | 21 |
| 29 | 1.74 | 0.013 | 134 |
| 30 | 16.7 | 0.027 | 619 |
| Adriamycina | 0.08 | - | - |
adriamycin was used as the positive control
To assess the structure-activity relationships of this new class of antiplasmodial compounds, we first compared the structures of the most active compounds, 1, 2, 7, 14, 17–21, and 28–30 with those inactive and/or less potent compounds, 10, 13, 24, 25, 27, 32–44 (Figures 1–3) in Table 5. All the active compounds against P. falciparum are lindenane-type sesquiterpenoid dimers, and the three lindenane-type sesquiterpenoid monomers tested (42–44) were inactive. The most active dimers, e.g. compounds 1, 2, 14, 17, 19, 20, and 28–30, featured the common motifs of a conjugated system of methyl (Z)-5-hydroxy-4-oxopent-2-enoate, a Δ4 double bond, and a hydroxy group at C-4′ as marked in red. All the compounds without methyl (Z)-5-hydroxy-4-oxopent-2-enoate and/or Δ4 double bond motifs showed marginal activities or were inactive. Compounds 20 and 27 showed very similar structures except for the facts that 20 (13 ± 3 nM) had a Δ4 double bond, while 27 (> 25 μM) possessed a 4-OH and Δ5 double bond, suggesting that a Δ4 double bond is necessary for the activity, and this was supported by the very similar observations between compounds 30 (27 ± 3 nM) and 35 (>25 μM). Moreover, a similar observation between compounds 19 and 5 was observed where the absence of the Δ4 double bond in compound 5 reduced the antiplasmodial activity about 4-fold; however, the toxicity toward mammalian cells was also tremendously reduced since no toxicity was observed for compound 5 at 100 μM (Table 6). Interestingly, the absence of the Δ4 double bond in compound 4 reduced the antiplasmodial activity by less than 1-fold as compared with the structurally similar counterpart compound 2; however, the toxicity toward mammalian cells was also increased by 6-fold (Table 6). The absence of the 4′-OH renders the compounds less active as compared with structurally similar counterparts, e.g. the compound pairs 21 (100 ± 10 nM)/26 (without 4′-OH, 580 ± 90 nM), and 13 (4.6 ± 0.2 μM)/24 (without 4′-OH, 11.4 ± 1.6 μM), and this was consistent with the fact that compounds 25 and 32 without 4′-OH groups also showed P. falciparum growth inhibition in the micromolar range. The rare dimeric compounds 11 and 12 without the (Z)-configured Δ7(11) double bond showed attenuated antiplasmodial activities with IC50 values of 4.7 ± 0.5 μM and 99 ± 18 nM, respectively, as compared with their structurally similar analogues 2, 17, and 20. In addition, when the methyl (Z)-5-hydroxy-4-oxopent-2-enoate motif of compound 28 (11 ± 1 nM) changed to the E-geometry of 7 (48 ± 12 nM), the antiplasmodial activity was reduced about 4-fold, suggesting that the motif with a Z-geometry seemed more favorable for the antiplasmodial activity. However, both compounds showed SI values below 30 where the E-geometry also decreased the mammalian cytotoxicity by 5-fold (Table 6). Furthermore, compound 8 (198 ± 22 nM) had a very similar structure with that of the most potent antiplasmodial compound 19 (1.1 ± 0.2 nM) except for the absence of the Δ4 double bond (replaced by Δ4(15) and Δ5 double bonds) and the methyl (Z)-5-hydroxy-4-oxopent-2-enoate motif (ring closed to a five-membered α,β-unsaturated semiketal lactone), suggesting that these two groups are very important for the antiplasmodial activity observed with compound 19.
Interestingly, the presence of two ester chains R1 and R2 at C-13′ and C-15′, respectively, dramatically improves the antiplasmodial activities as observed for the low nanomolar antiplasmodial compounds 1, 14, and 19. When the R1 ester group was absent, for example compounds 2, 17, 18, and 21, the antiplasmodial potencies were reduced considerably. In addition, when the two ester chains formed an 18-membered macrocyclic trilactone ring, e.g. compounds 7, 28–30, the antiplasmodial potencies were slightly reduced as compared with the ring-opened analogues. For the compounds possessing the 18-membered macrocyclic trilactone rings, the antiplasmodial activities of compounds with an E-acyloxy-3-methylbut-2-enoate (28 and 41) were better than those with an E-acyloxy-2-methylbut-2-enoate (30 and 39); however, those with those with an E-acyloxy-2-methylbut-2-enoate showed lower mammalian cytotoxicity and better SI values. The presence of a 7′′α-OH at the 18-membered macrocyclic trilactone ring (29, 13 ± 1 nM) also increased the potency as compared with compound 30 (27 ± 3 nM), however, also increased mammalian cytotoxicity by ~10-fold (Table 6). It is evident that chain lengths, geometry of the double bonds, and oxidation patterns of the R1 and R2 motifs influence the P. falciparum growth inhibition activities significantly, indicating that the R1 and R2 motifs can be modified to improve the antiplasmodial activities of this compound class.
Compounds 4, 5, 9, and 31 with a hydroperoxy group at C-4 (4-OOH) and Δ5 double bond showed remarkable antiplasmodial activities with IC50 values ranging from the low nanomolar to the low micromolar range (Table 5). These potencies are much stronger than those predicted by the previous SAR results, with inactive analogues 27, 33–35 bearing a 4-OH and Δ5 double bond (Figures 1 and 3), suggesting that the 4-OOH is the crucial moiety for this antiplasmodial compound subclass that likely operates by a different mechanism and/or has a different SAR than the other active dimers. On the other hand, the absence of the 4′-OH group and the two acyl groups at C-13′ and C-15′ reduced antiplasmodial potency dramatically. Thus compound 6 (5.3 ± 2 μM) is much less active than compound 4 and 5, indicating that the 4′-OH and the two ester chains R1 and R2 at C-13′ and C-15′ are also important motifs for the antiplasmodial activities for this compound subclass.
Taken together, the above analysis has outlined a clear SAR for the tested compounds as follows (Figure 7). (1) All the active compounds are the Diels-Alder adducts of lindenane-type sesquiterpenoid dimers, while the monomers were inactive. (2) The most potent dimers feature common motifs of a Δ4 double bond, a 4′-OH, and a conjugated system of methyl (Z)-5-hydroxy-4-oxopent-2-enoate. (3) The presence of two ester chains R1 and R2 at C-13′ and C-15′, respectively, dramatically affects the antiplasmodial activities, suggesting that these groups can be modified to improve the antiplasmodial potency of this class of compounds. (4) For the 4-OOH compound subclass, the 4-OOH moiety is the crucial motif for the antiplasmodial activity, which likely operates by a different mechanism and/or has a different SAR than the other active dimers.
Figure 7.
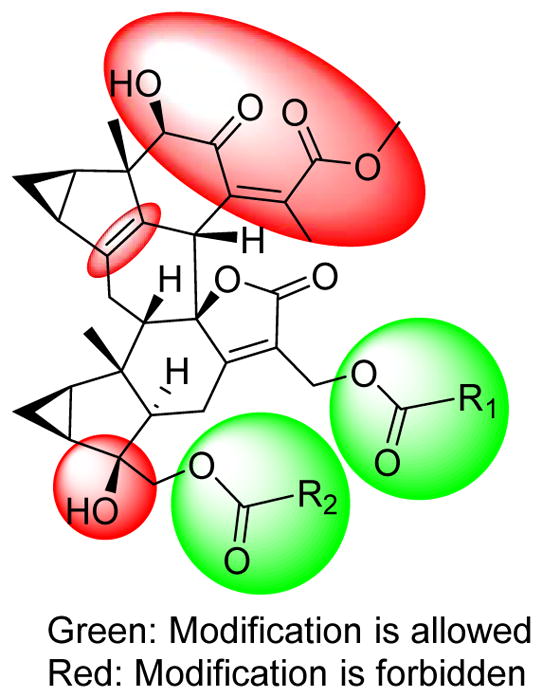
Brief SAR of antiplasmodial compounds.
In conclusion, a new class of potent antiplasmodial agents against the chloroquine-resistant strain of P. falciparum with low mammalian cytotoxicity was discovered from the plants of Chloranthus genus known as “sikuaiwa”, which have long been used in traditional Chinese medicine to treat malaria. Among the active compounds, three of them presented low nanomolar IC50 values antiplasmodial similar to artemisinin and a selectivity index value of ≥ 500. A SAR study of this new class has also been performed and clearly indicated that two motifs in this class can been modified to enhance the antiplasmodial potency. Therefore, lindenane sesquiterpnoid dimers are new class of promising antiplasmodial agents, and likely act through a new mode-of-action because of their unique scaffold that is different from all currently known antiplasmodial agents. Further investigation of this new antiplasmodial class is thus warranted and is currently underway.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were recorded on an Autopol VI polarimeter. The UV data were obtained by using a Shimadzu UV-2550 spectrophotometer. The IR spectra were acquired on a Thermo IS5 spectrometer with KBr disks. The NMR spectra were run on a Bruker AM-500 spectrometer with TMS as internal standard. The ESIMS and HRESIMS were obtained on a Brucker Daltonics Esquire 3000 plus and a Waters-Micromass Q-TQF Ultima Global mass spectrometer, respectively. Semipreparative HPLC was performed on a Waters 1525 binary pump system with a Waters 2489 detector (210 nm) using a YMC-Pack ODS-A (250×10 mm, S-5 μm). Silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd), C18 reversed-phase (RP-18) silica gel (20–45 μM, Fuji Silysia Chemical LTD), CHP20P MCI gel (75–150 μm, Mitsubishi Chemical Corporation), D101-macroporous absorption resin (Shanghai Hualing Resin Co., Ltd), and Sephadex LH-20 gel (Amersham Biosciences) were used for column chromatography (CC). Precoated silica gel GF254 plates (Qingdao Haiyang Chemical Co., Ltd.) were used for TLC monitors. All the solvents used for CC were of analytical grade (Shanghai Chemical Reagents Co., Ltd.), and the solvents used for HPLC were of HPLC grade (J & K Scientific Ltd.).
Plant Material
Twigs of C. fortunei were collected in June of 2013 in Guilin city, Guangxi Province, China, and were authenticated by Professor Shao-Qing Tang of Guangxi Normal University. A voucher specimen has been deposited in Shanghai Institute of Materia Medica, Chinese Academy of Sciences (accession no: CHF-2011-1Y).
Extraction and Isolation
Dried powder of C. fortunei (5 kg) was extracted with 95% EtOH at room temperature to give a crude extract (520 g), which was then partitioned between EtOAc and H2O. The EtOAc soluble fraction (230 g) was subjected to CC (D101-macroporous absorption resin) eluted with 30%, 50%, 80% and 95% MeOH in H2O to give four fractions 1–4, respectively. Fraction 2 (120 g) was separated by an MCI gel column (MeOH/H2O, 4:6 to 9:1) to afford three fractions A–C, and fraction 3 (100 g) was treated similarly to afford two fractions D and E. Fraction A (15g) was chromatographed over a silica gel column and eluted with petroleum ether-acetone (from 50:1 to 1:5) in gradient to afford seven subfractions A1–A7. Fraction A2 (600 mg) was purified by semipreparative HPLC (50% CH3CN in H2O, 3 mL/min) to yield shizukanolide F (4.7 mg), chloranthalactone C (5.7 mg), and isoshizukanolide (3.7 mg). Fraction A6 (1.3 g) was chromatographed over a column of Sephadex LH-20 to yield three major parts, and each of them was purified by semipreparative HPLC (65% CH3OH in H2O, 3 mL/min) to give shizukaol F (10 mg), shizukaol K (8.3 mg), chlorajaponilide C (11 mg), and chlorahololide D (6.5 mg). Fraction A7 (6.1g) was further separated on a column of reversed phase C18 silica gel (30–80% MeOH in H2O) to yield three major components (A7a–A7c). Component A7a (3.5 g) was fractioned on a silica gel column (CHCl3-MeOH, 500:1 to 50:1) to give four major parts A7a1–A7a4. Fraction A7a1 (80 mg) was purified by semipreparative HPLC (55% CH3CN in H2O, 3 mL/min) to yield compounds 11 (9.5 mg). In the same ways, fraction A7a2 (400 mg) gave 1 (2.2 mg) and shizukaol I (3.7 mg); fraction A7a3 (1.5 g) yielded compounds 2 (44 mg), 4 (4.7 mg), 5 (43 mg), 7 (22 mg), and spicachlorantin D (3 mg). Fraction A7a4 (900 mg) was chromatographed over a column of Sephadex LH-20 to yield three major parts, and each of them was purified by semipreparative HPLC (40% CH3CN in H2O, 3 mL/min) to give 8 (3.0 mg), 10 (8.3 mg), sarglabolide I (5.8 mg), chloramultilide C (3.3 mg), and sarcandrolide J (6.8 mg). By the similar separating procedures used in fraction A, fraction D (52 g) was successively subjected to silica gel column (petroleum ether-acetone: 10:1 to 1:5), Sephadex LH-20 (eluted with EtOH), reversed phase C18 silica gel column (MeOH, 30–80%), silica gel column (CHCl3-MeOH, 100:1 to 10:1), and finally purified by semipreparative HPLC to yield 3 (49 mg), 6 (23 mg), 9 (4.0 mg), 12 (28 mg), shizukaol C (23 mg), shizukaol M (13 mg), and sarglabolide J (23 mg).
Fortunilide A (1): white, amorphous powder; [α]24D −14.8 (c 0.2, MeOH); UV(MeOH) λmax (log ε) 222 (4.40) nm; IR (KBr) νmax 3446, 2926, 1733, 1632, 1438, 1384, 1223,1159, 1037, 991 cm-1; 1H NMR data (CDCl3), see Table 1 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 785.3 [M + Na]+; (−)-ESIMS m/z 761.9 [M − H]−; (+)-HRESIMS m/z 785.2781 [M + Na++ (calcd for C41H46O14Na, 785.2780).
Fortunilide B (2): white, amorphous powder; [α]24D −115.1 (c 1.0, MeOH); UV(MeOH) λmax (log ε) 219 (4.49) nm; IR (KBr) νmax 3458, 2952, 1736, 1604, 1438, 1376, 1225,1157, 1086, 991 cm-1; 1H NMR data (CDCl3), see Table 1 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 787.2 [M + Na]+; (−)-ESIMS m/z 763.2 [M − H]−; (+)-HRESIMS m/z 787.2964 [M + Na]+ (calcd for C41H48O14Na, 787.2936).
Fortunilide C (3): white, amorphous powder; [α]24D −152.6 (c 1.0, MeOH); UV(MeOH) λmax (log ε) 219 (4.50) nm; IR (KBr) νmax 3465, 2949, 1739, 1601, 1437, 1383, 1228, 1144, 991 cm-1; 1H NMR data (CDCl3), see Table 1 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 657.2 [M + Na]+; (−)-ESIMS m/z 633.3 [M − H]−; (+)-HRESIMS m/z 657.2672 [M + Na]+ (calcd for C36H42O10Na, 657.2670).
Fortunilide D (4): white, amorphous powder; [α]24D −79.5 (c 0.5, MeOH); UV(MeOH) λmax (log ε) 219 (4.47) nm; IR (KBr) νmax 3454, 2952, 1735, 1438, 1277, 1223, 1153, 976 cm-1; 1H NMR data (CDCl3), see Table 1 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 819.2 [M + Na]+; (−)-ESIMS m/z 795.3 [M − H]−; (+)-HRESIMS m/z 819.2851 [M + Na]+ (calcd for C41H48O16Na, 819.2835).
Fortunilide E (5): white, amorphous powder; [α]24D −94.2 (c 0.9, MeOH); UV(MeOH) λmax (log ε) 219 (4.44) nm; IR (KBr) νmax 3442, 2955, 1736, 1439, 1280, 1216, 1151, 976 cm-1; 1H NMR data (CDCl3), see Table 2 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 778.9 [M − H2O + H]+; (+)-HRESIMS m/z 819.2840 [M + Na]+ (calcd for C41H48O16Na, 819.2835).
Fortunilide F (6): white, amorphous powder [α]24D −130.2 (c 0.8, MeOH); UV(MeOH) λmax (log ε) 227 (4.24) nm; IR (KBr) νmax 3436, 2925, 2878, 1735, 1677, 1434, 1383, 1109, 971 cm-1; 1H NMR data (CD3OD), see Table 2 and 13C NMR (CD3OD), see Table 4; (+)-ESIMS m/z 569.3 [M + H]+, 1159.6 [2 M + Na]+; (+)-HRESIMS m/z 591.2211 [M + Na]+ (calcd for C31H36O10Na, 591.2201).
Fortunilide G (7): white, amorphous powder; [α]24D −49.8 (c 0.7, MeOH); UV (MeOH) λmax (log ε) 213 (4.37) nm; IR (KBr) νmax 3371, 2941, 2855, 1751, 1658, 1438, 1223, 1158, 998, 871 cm-1; 1H NMR data (CDCl3), see Table 2 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 755.3 [M + Na]+; (−)-ESIMS m/z 731.7 [M − H]−; (+)-HRESIMS m/z 755.2683 [M + Na]+ (calcd for C40H44O13Na, 755.2674).
Fortunilide H (8): white, amorphous powder; [α]24D −45.1 (c 0.8, MeOH); UV(MeOH) λmax (log ε) 215 (4.46) nm; IR (KBr) νmax 3432, 2923, 1761, 1655, 1387, 1223, 1153, 1075 cm-1; 1H NMR data (CDCl3), see Table 2 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 749.3 [M + H]+; (+)-HRESIMS m/z 771.2625 [M + Na]+ (calcd for C40H44O14Na, 771.2623).
Fortunilide I (9): white, amorphous powder; [α]24D +6.0 (c 0.4, MeOH); UV(MeOH) λmax (log ε) 221 (4.42) nm; IR (KBr) νmax 3433, 2930, 1751, 1441, 1220, 1153, 1081, 1012, 970 cm-1; 1H NMR data (CD3OD), see Table 3 and 13C NMR (CD3OD), see Table 4; (+)-ESIMS m/z 800.3 [M − H2O + H]+; (+)-HRESIMS m/z 805.2659 [M + Na]+ (calcd for C40H46O16Na, 805.2678).
Fortunilide J (10): white, amorphous powder; [α]24D +107.9 (c 0.3, MeOH); UV(MeOH) λmax (log ε) 208 (4.56) nm; IR (KBr) νmax 3432, 2925, 1730, 1660, 1448, 1384, 1226, 1145, 959 cm-1; 1H NMR data (CD3OD), see Table 3 and 13C NMR (CD3OD), see Table 4; (−)-ESIMS m/z 635.6 [M − H]−; (−)-HRESIMS m/z 635.2475 [M − H]− (calcd for C35H39O11, 635.2498).
Fortunilide K (11): colorless gum; [α]24D −47.2 (c 1.0, MeOH); UV(MeOH) λmax (log ε) 215 (4.37) nm; IR (KBr) νmax 3455, 2926, 1761, 1656, 1444, 1377, 1252, 1133, 1092, 951 cm-1; 1H NMR data (CDCl3), see Table 3 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 617.4 [M + H]+; (−)-ESIMS m/z 615.7 [M − H]−; (−)-HRESIMS m/z 615.2588 [M − H]− (calcd for C36H39O9, 615.2600).
Fortunilide L (12): colorless gum; [α]24D −26.9 (c 0.9, MeOH); UV(MeOH) λmax (log ε) 215 (4.53) nm; IR (KBr) νmax 3465, 2952, 1737, 1617, 1438, 1384, 1242, 1041, 968 cm-1; 1H NMR data (CDCl3), see Table 3 and 13C NMR (CDCl3), see Table 4; (+)-ESIMS m/z 747.5 [M + H]+, m/z 769.3 [M + Na]+; (−)-ESIMS m/z 745.8 [M − H]−; (+)-HRESIMS m/z 769.2830 [M + Na]+ (calcd for C41H46O13Na, 769.2831).
Antiplasmodial Evaluation of Compounds 1–44
P. falciparum in vitro growth inhibition assay
Dose-dependent growth inhibition against P. falciparum strain Dd2 (chloraquine-resistant) was measured in a 72 h growth assay in the presence of inhibitor as described previously.30–31 Artemisinin was used as a positive control. Parasite growth was normalized to untreated controls in the presence of DMSO. Ring stage parasite cultures (100 μL per well, with 1% hematocrit and 1% parasitaemia) were grown for 72 h in the presence of increasing concentrations of the inhibitor in a 5.05% CO2, 4.93% O2 and 90.2% N2 gas mixture at 37 °C. After 72 h in culture, parasite viability was determined by DNA quantitation using SYBR Green I as described previously. The half-maximum inhibitory concentration (IC50) values were calculated with Kaleida Graph using nonlinear regression curve fitting, and the reported values represent averages of at least three independent experiments performed in triplicates with standard deviations.
Mammalian Cytotoxicity Evaluation of Selected Compounds with Antimalarial Activity
WI-38 cell line in vitro growth inhibition assay
Compounds were evaluated for their cytotoxicity against normal cell line WI-38 (normal embryonic lung tissue). Briefly, 10,000 cells per well were plated in a clear-bottom 96 well plate. Cells were allowed to adhere and then the media was replaced with 100 μL of media containing varying amounts of the test inhibitor and incubated for 24 h. After the incubation time was completed, 10 μL of resazurin sodium salt (Sigma) at 0.125 mg/mL was added to each well and incubated for 2 h. Cell viability was determined by measuring the fluorescence at 585 nm after excitation at 540 nm. The IC50 values were calculated as described above, and the reported values represent averages of at least two independent.
Supplementary Material
Figure 2.

The tested compounds 7–12, and 28.
Acknowledgments
This project was supported by the National Natural Science Foundation (No. 21532007, 81321092) and the Foundation (2012CB721105) from the Ministry of Science and Technology of the People’s Republic of China. We thank Prof. S. Q. Tang of Guangxi Normal University for the identification of the plant of C. fortunei. The work of DGIK and MBC was supported by the National Center for Complementary and Integrative Health under award 1 R01 AT008088, and this support is gratefully acknowledged.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. IR, ESIMS, HRESIMS, 1D and 2D NMR spectra of compounds 1–12, and the purities of compounds 1–44 are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Greenwood BM, Bojang K, Whitty CJM, Targett GAT. Malaria. The Lancet. 2005;365:1487–1498. doi: 10.1016/S0140-6736(05)66420-3. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. World malarial report. http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/
- 3.(a) Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, von Seidlein L. Nat Rev Microbio. 2010;8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]; (b) Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ. New Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) White NJ. J Clin Invest. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cowman AF, Morry MJ, Biggs BA, Cross G, Foote SJ. P Natl Acad Sci. 1988;85:9109–9113. doi: 10.1073/pnas.85.23.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YQ, Cheng DZ, Wu GF, Cheng PS, Zhu PZ. Zhongguo Zhiwu Zhi. Vol. 20. Science Press; Beijing: 1982. pp. 80–96. [Google Scholar]
- 5.(a) Ni G, Zhang H, Liu HC, Yang SP, Geng MY, Yue JM. Tetrahedron. 2013;69:564–569. [Google Scholar]; (b) He XF, Yin S, Ji YC, Su ZS, Geng MY, Yue JM. J Nat Prod. 2009;73:45–50. doi: 10.1021/np9006469. [DOI] [PubMed] [Google Scholar]; (c) Zhang S, Su ZS, Yang SP, Yue JM. J Asian Nat Prod Res. 2010;12:522–528. doi: 10.1080/10286020.2010.492599. [DOI] [PubMed] [Google Scholar]; (d) Yuan T, Zhu RX, Yang SP, Zhang H, Zhang CR, Yue JM. Org Lett. 2012;14:3198–3201. doi: 10.1021/ol301306e. [DOI] [PubMed] [Google Scholar]; (e) Yuan T, Zhang CR, Yang SP, Yin S, Wu WB, Dong L, Yue JM. J Nat Prod. 2008;71:2021–2025. doi: 10.1021/np800543f. [DOI] [PubMed] [Google Scholar]; (f) Zhang S, Yang SP, Yuan T, Lin BD, Wu Y, Yue JM. Tetrahedron Lett. 2010;51:764–766. [Google Scholar]; (g) Yang SP, Zhang CR, Chen HD, Liao SG, Yue JM. Chin J Chem. 2007;25:1892–1895. [Google Scholar]; (h) Yang SP, Yue JM. Tetrahedron Lett. 2006;47:1129–1132. [Google Scholar]; (i) Yang SP, Gao ZB, Wu Y, Hu GY, Yue JM. Tetrahedron. 2008;64:2027–2034. [Google Scholar]; (j) Yang SP, Gao ZB, Wang FD, Liao SG, Chen HD, Zhang CR, Hu GY, Yue JM. Org Lett. 2007;9:903–906. doi: 10.1021/ol0700759. [DOI] [PubMed] [Google Scholar]
- 6.(a) Schwikkard S, van Heerden FR. Nat Prod Rep. 2002;19:675–692. doi: 10.1039/b008980j. [DOI] [PubMed] [Google Scholar]; (b) Kaur K, Jain M, Kaur T, Jain R. Bioorg Med Chem. 2009;17:3229–3256. doi: 10.1016/j.bmc.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ge H. Principal Prescription Emergency. AD 340;2:111. [Google Scholar]; (b) Klayman DL. Science. 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 8.Yunnan Zhongcaoyao. Yunnan People Press; Kunming: 1971. pp. 80–96. [Google Scholar]
- 9.Wang QH, Kuang HX, Yang BY, Xia YG, Wang JS, Kong LY. J Nat Prod. 2010;74:16–20. doi: 10.1021/np100504m. [DOI] [PubMed] [Google Scholar]
- 10.Kawabata J, Fukushi E, Mizutani J. Phytochemistry. 1995;39:121–125. [Google Scholar]
- 11.Wang XC, Zhang YN, Wang LL, Ma SP, Liu JH, Hu LH. J Nat Prod. 2008;71:674–677. doi: 10.1021/np7007544. [DOI] [PubMed] [Google Scholar]
- 12.Kim SY, Kashiwada Y, Kawazoe K, Murakami K, Sun HD, Li SL, Takaishi Y. Tetrahedron Lett. 2009;50:6032–6035. [Google Scholar]
- 13.Kim SY, Kashiwada Y, Kawazoe K, Murakami K, Sun HD, Li SL, Takaishi Y. Chem Pharm Bull. 2011;59:1281–1284. doi: 10.1248/cpb.59.1281. [DOI] [PubMed] [Google Scholar]
- 14.Li CJ, Zhang DM, Luo YM, Yu SS, Li Y, Lu Y. Phytochemistry. 2008;69:2867–2874. doi: 10.1016/j.phytochem.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 15.(a) Tully L, Carson M, McMurry T. Tetrahedron lett. 1987;28:5925–5928. [Google Scholar]; (b) Yang SP, Gao ZB, Wang FD, Liao SG, Chen HD, Zhang CR, Hu GY, Yue JM. Org Lett. 2007;9:903–906. doi: 10.1021/ol0700759. [DOI] [PubMed] [Google Scholar]
- 16.Ran XH, Teng F, Chen CX, Wei G, Hao XJ, Liu HY. J Nat Prod. 2010;73:972–975. doi: 10.1021/np900764n. [DOI] [PubMed] [Google Scholar]
- 17.(a) Demarco PV, Farkas E, Doddrell D, Mylari BL, Wenkert E. J Am Chem Soc. 1968;90:5480–5486. [Google Scholar]; (b) Yang SP, Gao ZB, Wu Y, Hu GY, Yue JM. Tetrahedron. 2008;64:2027–2034. [Google Scholar]
- 18.He XF, Zhang S, Zhu RX, Yang SP, Yuan T, Yue JM. Tetrahedron. 2011;67:3170–3174. [Google Scholar]
- 19.Wang P, Luo J, Zhang YM, Kong LY. Tetrahedron. 2015;71:5362–5370. [Google Scholar]
- 20.Kawabata J, Mizutani J. Phytochemistry. 1992;31:1293–1296. [Google Scholar]
- 21.Fang PL, Cao YL, Yan H, Pan LL, Liu SC, Gong NB, Lü Y, Chen CX, Zhong HM, Guo Y. J Nat Prod. 2011;74:1408–1413. doi: 10.1021/np200087d. [DOI] [PubMed] [Google Scholar]
- 22.Xu YJ, Tang CP, Ke CQ, Zhang JB, Weiss HC, Gesing ER, Ye Y. J Nat Prod. 2007;70:1987–1990. doi: 10.1021/np070433g. [DOI] [PubMed] [Google Scholar]
- 23.Kawabata J, Mizutani J. Agri Biol Chem. 1989;53:203–207. [Google Scholar]
- 24.Wang XC, Wu WQ, Ma SP, Liu JH, Hu LH. Chin J Nat Med. 2008;6:404–407. [Google Scholar]
- 25.Kawabata J, Tahara S, Mizutani J. Agri Biol Chem. 1981;45:1447–1453. [Google Scholar]
- 26.Yang SP, Yue JM. Tetrahedron Lett. 2006;47:1129–1132. [Google Scholar]
- 27.Kim SY, Kashiwada Y, Kawazoe K, Murakami K, Sun HD, Li SL, Takaishi Y. Tetrahedron Lett. 2009;50:6032–6035. [Google Scholar]
- 28.Kawabata J, Fukushi Y, Tahara S, Mizutani J. Phytochemistry. 1990;29:2332–2334. [Google Scholar]
- 29.Kim SY, Kashiwada Y, Kawazoe K, Murakami K, Sun HD, Li SL, Takaishi Y. Phytochemistry Lett. 2009;2:110–113. [Google Scholar]
- 30.Liu J, He XF, ang GH, Merino EF, Yang SP, Zhu RX, Gan LS, Zhang H, Cassera MB, Wang HY. J Org Chem. 2013;79:599–607. doi: 10.1021/jo402340h. [DOI] [PubMed] [Google Scholar]
- 31.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.