

ABSTRACT
The diarrheal pathogen Clostridium difficile consists of at least six distinct evolutionary lineages. The RT017 lineage is anomalous, as strains only express toxin B, compared to strains from other lineages that produce toxins A and B and, occasionally, binary toxin. Historically, RT017 initially was reported in Asia but now has been reported worldwide. We used whole-genome sequencing and phylogenetic analysis to investigate the patterns of global spread and population structure of 277 RT017 isolates from animal and human origins from six continents, isolated between 1990 and 2013. We reveal two distinct evenly split sublineages (SL1 and SL2) of C. difficile RT017 that contain multiple independent clonal expansions. All 24 animal isolates were contained within SL1 along with human isolates, suggesting potential transmission between animals and humans. Genetic analyses revealed an overrepresentation of antibiotic resistance genes. Phylogeographic analyses show a North American origin for RT017, as has been found for the recently emerged epidemic RT027 lineage. Despite having only one toxin, RT017 strains have evolved in parallel from at least two independent sources and can readily transmit between continents.
KEYWORDS: Clostridium difficile, sequencing, SNPs, ribotype 017, evolution, phylogenetics, antibiotic resistance, phylogeny
INTRODUCTION
Clostridium difficile is a spore-forming obligate anaerobe that continues to be the leading cause of health care-associated infections in the developed world (1, 2). There are six main lineages that broadly split into PCR ribotypes (RTs) associated with RT027, RT023, RT017, RT078, a grouping of diverse RTs, and the recently identified novel lineage containing RT131 (3). The global emergence of the RT027 strain was responsible for multiple outbreaks and increased disease severity in Canada and the United States in 2001 (4). This strain has since spread to South America (5–7), China (8), Japan (9), Hong Kong (10), South Korea (11, 12), Taiwan (13), Singapore (14), Australia (15, 16), Saudi Arabia (17), Israel (18), New Zealand (19), and throughout Europe (5, 20–28). Although RT027 remains the dominant clone in the United States, Europe has seen a decline in RT027 with a simultaneous increase in other virulent RTs, such as RT017 and RT078 (29).
Using whole-genome sequencing (WGS) and phylogenetic analysis, He et al. (4) identified the presence of two genetically distinct sublineages of RT027 through single-nucleotide polymorphism (SNP) analysis; both had emerged in North America within a relatively short period after acquiring the same fluoroquinolone resistance-conferring mutation containing an alteration in gyrA and a highly related conjugative transposon (4). The two epidemic sublineages showed distinct patterns of global spread, with one lineage spreading more widely and causing health care-associated outbreaks globally (4).
Traditionally, virulent C. difficile strains are characterized and identified in diagnostic laboratories by the presence of two potent toxins, TcdA and TcdB (30). These genes are located on a 19.6-kb pathogenicity locus (PaLoc). There is genetic variation in this region which can be exploited and which has revealed 30 different toxinotypes, including six A− B+ toxinotypes. The most common and clinically relevant is toxinotype VIII, and these isolates belong to RT017 (31). It is well known that the tcdA gene of this type contains a 1.8-kb deletion at the 3′ end and a nonsense mutation at tcdA amino acid 47 that introduces a stop codon leading to a truncated tcdA gene (31). RT017 strains also lack the binary toxin (CDT) found in, for example, pathogenic RT027 strains that produce all three toxins. Despite lacking two toxins, clinically significant C. difficile infection (CDI) has been reported worldwide for the RT017 lineage (32–41).
Historically, these strains were initially identified in CDI outbreaks in Asia and are thought to have spread to Europe and other continents. RT017 strains have been reported in Canada (35, 42), China (34, 43), South Korea (33, 44, 45), Argentina (46), Australia (47, 48), Israel (49), Japan (50), South Africa (51), and throughout Europe (36, 39, 41, 52, 53). These strains have also been isolated from nonhuman sources, including equines, bovines (54), and rabbits (55). We recently performed WGS on 35 human and two hospital environmental isolates of RT017 circulating in London, United Kingdom, and identified three SNP variants (39). One variant was found to be clonal and had persisted in a London hospital ward for at least 5 years (39).
Here, WGS and phylogenetic analysis were used to define the population structure of a collection of 277 RT017 isolates from six continents of human and nonhuman origins with isolation dates between 1990 and 2013. Analyses reveal that RT017 strains have evolved in parallel from at least two independent sources and can readily transmit between continents. Genotypic and phenotypic antimicrobial susceptibilities were also compared.
RESULTS
WGS was performed on a global collection of 277 C. difficile RT017 isolates (Table 1). Collectively, these were isolated from human (n = 251), bovine (n = 9), canine (n = 11), equine (n = 4), and hospital ward environments (n = 2) between 1990 and 2013 (see Information S1 in the supplemental material). All isolates belonged to multilocus sequence type 37. After sequence quality control and mapping to the M68 RT017 reference genome (GenBank accession number FN668375), we identified 1,288 high-quality biallelic SNPs, with 311 present in greater than 1% of samples and greater than 1 bp from an insertion or deletion. Of these non-rare SNPs, 65.6% (n = 204) were nonsynonymous, 17.7% (n = 55) were synonymous, and 16.7% (n = 52) were present in noncoding regions of the genome (nonsynonymous SNPs are shown in Information S2). Twelve SNPs affected stop codons, 11 nonsynonymous and 1 synonymous (Table 1).
TABLE 1.
Stop codon-associated SNPs
| Position in M68 genome | Codona |
Nonsynonymous/synonymous/noncoding | Gene | Predicted function and/or potential impact | No. of isolates with SNP | |
|---|---|---|---|---|---|---|
| M68 reference | Alternative | |||||
| 132573 | TGG | TGA | Nonsynonymous | M68_00168 | Amino acid aminotransferase | 16 |
| 557896 | TTC* | TAA* | Nonsynonymous | feoB3 | Ferrous iron transport protein B | 3 |
| 1204039 | GGA | TGA | Nonsynonymous | M68_01144 | Hydrolase | 36 |
| 1359584 | GGA | TGA | Nonsynonymous | M68_01270 | Extracellular solute-binding protein | 3 |
| 1907433 | TAA | GAA | Nonsynonymous | msrAB | Peptide methionine sulfoxide reductase | 256 |
| 1916756 | AAT* | GAT* | Synonymous | M68_01782 | Unknown | 3 |
| 3304067 | TCA* | GCA* | Nonsynonymous | Sigma-54 | Controls expression of nitrogen-related genes | 29 |
| 3399853 | TTG* | TAA* | Nonsynonymous | M68_03193 | Ca2+/Na+ antiporter | 13 |
| 3402470 | CAA | TAA | Nonsynonymous | plfB | Formate acetyltransferase | 3 |
| 3704987 | CCA* | TGA* | Nonsynonymous | sleB | Spore-cortex-lytic protein | 8 |
| 3784055 | TTC* | TAA* | Nonsynonymous | M68_03513 | Penicillin-binding protein | 3 |
| 4157880 | TTG* | TAA* | Nonsynonymous | M68_03851 | PTS system, IIc component | 6 |
*, Located on the reverse strand.
SNP data revealed 109 haplotypes containing between 0 and 52 SNPs (with respect to the M68 reference), with 76.5% (212/277) of isolates having between 10 and 35 SNPs (Table 2).
TABLE 2.
Summary of details of 277 C. difficile study isolates and their genotypic characteristics
| Sublineage | Total no. of isolates | Country of origin | Isolation date | No. (%) of haplotypes | No. of SNPs | No. (%) of isolates with: |
Resistance inferred according to position, gene, and aa changea [no. (%)] |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Insertion | Deletion | Rifampin |
Fluoroquinolone |
||||||||||
| 34,687, rpoB, R505K | 34,697, rpoB, H502N | 34,747, rpoB, S485F | 112,752, gyrA, T82I | 113,641, gyrB, V426D | 113,642, gyrB, V426I | ||||||||
| 1 | 163 | Argentina, Australia, Bulgaria, Canada, China, Czech Republic, Greece, Hong Kong, Japan, South Korea, Kuwait, Poland, Portugal, Romania, Singapore, Slovenia, South Africa, The Netherlands, UK, USA | 1994 to 2013 | 55 (50.5) | 0–35 | 49 (30.1) | 44 (30) | 73 (44.8) | 79 (48.5) | 0 (0) | 124 (76.1) | 134 (82.2) | 4 (2.5) |
| 2 | 114 | Australia, Hong Kong, Indonesia, Ireland, South Korea, Poland, Singapore, South Africa, Taiwan, The Netherlands, UK, USA | 1990 to 2013 | 54 (49.5) | 17–52 | 65 (57) | 109 (96) | 17 (15) | 13 (11.4) | 3 (2.6) | 55 (48.2) | 114 (100) | 9 (7.9) |
Reference residue/amino acid (aa)/alternative residue.
We generated a maximum-likelihood phylogenetic tree based on the 1,288 SNPs that demonstrated the presence of two genetically diverse sublineages, SL1 and SL2 (Fig. 1 and 2). Of the 1,288 SNPs, 76% (977/1,288) had a minor allele frequency (MAF) of ≤1% and/or were within 1 bp of an insertion or deletion. To control for false-positive identification of SNPs (these SNPs may mask the true phylogeny of RT017), phylogenetic trees with and without these SNPs were generated. The inclusion of 977 SNPs had a minor effect on the overall phylogenetic tree. Four SNPs were found to differentiate the two sublineages, one present in a noncoding region and three nonsynonymous SNPs (Table 3). SL2 is the most distantly related to the reference M68 strain of the two sublineages, and both sublineages are geographically and temporally widespread. All isolates from the previously reported study on London isolates fell into SL2 (39).
FIG 1.

Maximum-likelihood phylogenetic analysis of 277 global RT017 isolates based on core genome SNPs against the M68 reference. We used non-rare (>1% MAF) SNPs that were not in close proximity to insertions or deletions to determine the phylogenic tree. The SL1 and SL2 sublineages were differentiated by four SNPs (Table 3), with the reference strain M68 falling into SL2. The colored nodes indicate the geographical source of isolates.
FIG 2.
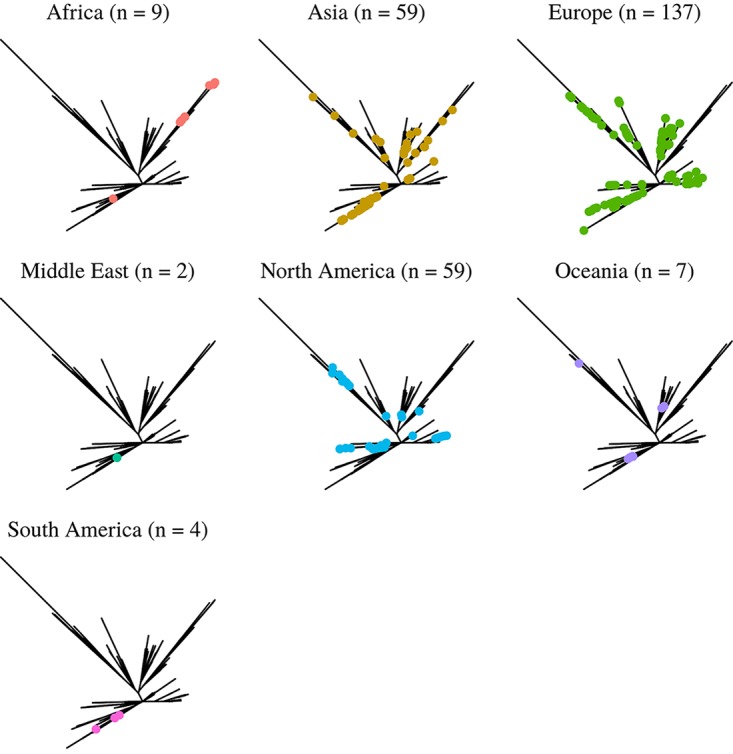
Maximum-likelihood phylogenetic analysis of 277 global RT017 isolates based on core genome SNPs against the M68 reference. The phylogeny is separated into individual panels corresponding to each continent. Data from 5 out of 7 continental designations (Africa, Europe, Asia, Oceania, and North America) include SL1 and SL2 isolates, indicating that both sublineages are global in nature.
TABLE 3.
Lineage-defining SNPs
| Position | Amino acid | Base |
Nonsynonymous/synonymous/noncoding | Gene product | Predicted function and/or potential impact | |
|---|---|---|---|---|---|---|
| Reference | Alternative | |||||
| 650374 | 19 | A | G | Nonsynonymous | MerR | Altered response to environmental stimuli |
| 900866 | C | T | Noncoding | |||
| 2914248 | 257 | A | G | Nonsynonymous | DacF | β-Lactam resistance |
| 3604289 | 329 | C | A | Nonsynonymous | Hypothetical protein | Unknown |
The RT017 strains are documented to have a higher level of antibiotic resistance than other C. difficile RTs (37, 56). Fluoroquinolone resistance in C. difficile has been associated with mutations in codon 82 of the gyrA gene and codon 426 of the gyrB gene. The common SNP found in the gyrA gene is T82I, and those in the gyrB gene are A426V and A426A (57). Remarkably, we found 64.6% (179/277) to have the amino acid substitution found in the gyrA gene (T82I). A substitution in the gyrB gene (V426N) was present in 4.7% of strains (13/277), and an additional 10.1% (28/277), including M68, harbored a valine at position 426 of the predicted gyrB product (Table 2; Information S1). The T82I substitution was globally distributed in both sublineages. Additionally, substitutions in the 81-bp rifampin resistance-determining region of the rpoB gene, R505K, H502N, and S485F, were found in 32.5% (90/277), 33.2% (92/277), and 1.1% (3/277) of isolates, respectively (Table 2; Information S1).
To investigate horizontal gene transfer, a key mechanism driving C. difficile evolution, we performed programmatic and visual inspection of the comparisons, which revealed 56 regions of DNA between ∼4 and ∼61.5 kb that were absent from the M68 strain but present in other strains. These had 34 different insertion sites (Table 2 and Fig. 3; Information S1 and S4). Additionally, we found regions of DNA of between ∼8 and ∼29 kb present in the M68 strain at six sites but absent from multiple samples (Table 2; Information S1 and S3). These insertions and deletions were associated with erythromycin, teicoplanin, tetracycline, chloramphenicol, and beta-lactam resistance genes, and their products potentially associated with virulence, such as a two-component response regulator, a SAM protein, an AntA/AntB antirepressor, a cell surface protein, and a sporulation-specific glycosylase (Information S3 and S4). The deletions and insertions were well distributed geographically and temporally, and a 49-kb insertion found only in a clonal cluster of 23/37 London isolates in our previous study (39) was also found to insert at a different site in single isolates from Canada, the United States, and the United Kingdom, with isolation dates of 2006, 2006, and 2011, respectively (Fig. 3). Only one SNP was found in the toxin pathogenicity locus region, which was synonymous and present in the nonfunctioning tcdA gene fragment from five South Korean isolates in SL2 isolated between 2004 and 2008. Visual inspection of the comparisons revealed both tcdA and tcdB genes to be highly conserved; no sequence variations were found.
FIG 3.

Bayesian evolutionary analysis of 277 global RT017 isolates based on core genome SNPs against the M68 reference. Using a geotemporal model, we can orient the evolution of the RT017 isolates though time. The analysis indicates a split from SL1 (lower) into SL2 (upper) c1990, with the M68 reference in SL2. The introduction of resistance-associated SNPs (such as in rpoC) fall within closely related groups in the phylogeny. The continents are colored as described for Fig. 1 and 2. The heat map depicts the sublineage, presence/absence of insertions, and antimicrobial resistance-associated SNPs in relation to the isolates and continent.
MICs were determined for eight C. difficile isolates (including M68 as a control) against the antibiotics chloramphenicol, rifampin, tetracycline, erythromycin, nalidixic acid, gentamicin, teicoplanin, and ampicillin. Their MICs are shown in Table 4. All isolates were resistant to nalidixic acid, gentamicin, and ampicillin, were either resistant or intermediately resistant to tetracycline, and were sensitive to teicoplanin. Out of eight isolates, two were resistant to chloramphenicol, four were resistant to rifampin, and seven were resistant to erythromycin.
TABLE 4.
Antimicrobial susceptibility data and genotypic characteristics
| Parameter | Value(s) for straind: |
|||||||
|---|---|---|---|---|---|---|---|---|
| M68 | S-017.72 | WA 1514 | S-017.92 | S-017.27 | S-017.74 | I6 | 01-116 | |
| Location | Ireland | Walsall, UK | Australia | China | Wrexham, UK | Walsall, UK | Indonesia | South Korea |
| Yr isolated | 2006 | 2011 | 2012 | 2009 | 1996 | 2011 | 2011 | 2001 |
| Insertion | A, B, C | A | D, E | F, G | ||||
| Deletion | H | H, I | J | H, J, K | H, J | |||
| Resistant SNPs | ||||||||
| rpoB (R505K) | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| rpoB (H502N) | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| rpoB (S485F) | ✓ | |||||||
| gyrA (T82I) | ✓ | ✓ | ✓ | ✓ | ||||
| gyrB (V426I) | ✓ | |||||||
| gyrB (V426D) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Antimicrobial agent | ||||||||
| Chloramphenicola | 8 (S) | 8 (S) | 4 (S) | 64 (R) | 8 (S) | 8 (S) | 256 (R) | 8 (S) |
| Rifampina | 0.008 (I) | 2 (I) | 0.004 (S) | >256 (R) | >256 (R) | 0.004 (S) | >256 (R) | >256 (R) |
| Tetracyclineb | 32 (R) | 32 (R) | 0.25 (I) | 32 (R) | 32 (R) | 0.25 (I) | 32 (R) | 32 (R) |
| Erythromycinb | >256 (R) | >256 (R) | >256 (R) | >256 (R) | >256 (R) | <2 (S) | >256 (R) | >256 (R) |
| Nalidixic acidb | 256 (R) | 256 (R) | 256 (R) | 256 (R) | 256 (R) | 256 (R) | 256 (R) | 256 (R) |
| Gentamicinc | >256 (R) | >256 (R) | 256 (R) | >256 (R) | 256 (R) | 256 (R) | >256 (R) | >256 (R) |
| Teicoplaninc | <1 (S) | <1 (S) | <1 (S) | <1 (S) | <1 (S) | <1 (S) | <1 (S) | <1 (S) |
| Ampicillinb | 8 (R) | 8 (R) | 8 (R) | 8 (R) | 8 (R) | 4 (R) | 4 (R) | 8 (R) |
Recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (http://www.eucast.org/clinical_breakpoints/).
No guidance from CLSI or EUCAST. Cutoffs are based on data according to CLSI guideline M100-S23 (interpretative values for Staphylococcus aureus).
S, sensitive; I, intermediate resistance; R, resistant. Insertions: A, putative drug/sodium antiporter and radical SAM protein TetR-family transcriptional regulator; B, transcriptional repressor DicA; C, streptogramin A acetyltransferase and multidrug resistance protein; D, putative beta-lactamase repressor; E, putative drug/sodium antiporter; F, TetR family transcriptional regulator; G, chloramphenicol o-acetyltransferase (M68 has one copy of chloramphenicol); H, dimethyladenosine transferase (ermB); I, putative teicoplanin resistance protein and putative beta-lactamase repressor; J, aminoglycoside 6-adenylyltransferase; K, putative conjugative transposon FtsK_SpoIIIE-related protein.
DISCUSSION
The RT017 lineage, with its unique toxin profile and unusual global prevalence, has been overshadowed by the global outbreak of the RT027 lineage. Reminiscent of the RT027 lineage, two distinct sublineages of C. difficile RT017 that contain multiple independent clonal expansions were revealed in this study. This division demonstrates that toxin variant strains emerged on at least one occasion, suggesting that a full toxin repertoire is not essential for efficient human-to-human transmission.
Based on our gyrA and gyrB SNP data, we predict up to 76.2% (211/277) of isolates are resistant to the fluoroquinolone class of antibiotics. Interestingly, the T82I SNP found in gyrA is the same mutation reported in the global outbreak of RT027 (4). Based on our MIC data, all eight isolates were resistant to nalidixic acid, indicating resistance to the fluoroquinolone class of antimicrobials.
Based on our rifampin SNP data, we predict 34.7% (96/277) of isolates in this study are resistant to the rifampin class of antibiotics. Interestingly, 82% (152/185) of these substitutions were found in SL1. R505K and H502N have previously been associated with rifampin resistance in C. difficile (60); however, based on our MIC data, only two (2/8) isolates were sensitive to rifampin, with one of the isolates containing the R505K and H502N SNP, indicating that these alone do not always lead to phenotypic resistance. Interestingly, S485F was found in three historical isolates from Wrexham, United Kingdom. This resistance-conferring SNP previously has been reported only in Mycobacterium tuberculosis and not in C. difficile (61). All three isolates were phenotypically resistant to rifampin; however, all three isolates also contained the R505K SNP, confirming this SNP's contribution to resistance was not possible.
The multiple haplotypes revealed is similar to those found for the RT027 global study, where >100 distinct genotypes were found in 151 isolates. Despite SNPs and insertions and deletions, there was no variation in susceptibility to ampicillin, teicoplanin, gentamicin, or nalidixic acid. However, there was some variation with chloramphenicol, rifampin, tetracycline, and erythromycin. Whether the insertions carrying chloramphenicol o-acetyltransferase, the TetR-family transcriptional regulator, or the ermB gene played a role in this variation is unknown.
Figure 4 depicts the phylogeny of the isolates by source. Interestingly, the 24 animal strains, which were all isolated from a similar location (Ontario, Canada) over a relatively short time period (2002 and 2005), are distributed among human isolates in SL1 only. This suggests there is transmission between humans and animals.
FIG 4.

Maximum-likelihood phylogenetic analysis of the global RT017 isolates based on core genome SNPs against the M68 reference depicting the 24 animal isolates by colored nodes. Note the three equine isolates are positioned (and masked) by the bovine and canine cluster on the left. The two bovine isolates on the right of the tree have an SNP distance of 17 from the bovine, canine, and equine cluster. All animal isolates are from Ontario, Canada, and were isolated between 2002 and 2005.
The ready global distribution of RT017 suggests determinants independent of toxin B are important in transmission. This could be related to the ready acquisition of antibiotic resistance determinants, efficient germination, and/or spore formation. This study provides the basis to further investigate factors important for the epidemic spread of C. difficile.
The deletions and insertions were well distributed geographically and temporally, suggesting either the rapid dissemination of strains or the multiple independent acquisitions and loss of DNA regions (Fig. 2; Information S1). The insertion of different clusters of genes at the same site suggests hot-spot regions for the uptake of DNA (Information S4), and a 49-kb insertion found only in a clonal cluster of 23/37 London isolates in our previous study (39) was also found to insert at a different site in single isolates from Canada, the United States, and the United Kingdom, with isolation dates of 2006, 2006, and 2011, respectively (Fig. 3). This suggests these isolates have independently acquired this insertion.
Similar to RT027, our analyses support a North American origin for RT017 with multiple, global transmission events, with its earliest movement into Europe in 1986 (Fig. 4 and 5). The North American health system and practices appears to facilitate the ready evolution and epidemic spread of C. difficile for RT027 (4) and now, in this study, for RT017. Our data show that it was Europe that introduced RT017 to Asia and Australia, with subsequent spread from Asia to the Middle East, South America, and South Africa. The analysis indicates over 40 movements back and forth over the span of 30 years, consistent with population movements of a globalized society. Traditionally, it has been considered that RT017 strains emerged from Asia due to the reported high incidence of this RT that could not relate to or depend on toxin A-based assays for diagnosis (40). However, our analysis does not support an “out-of-Asia” hypothesis and supports a North American origin (Fig. 4 and 5).
FIG 5.

Global transmission events inferred from Bayesian evolutionary analysis of RT017. From the geotemporal analyses we can infer the first movements into each continent, with the date and originating continent. The analysis indicates a North American origin with an expansion into Europe in the mid-1980s, followed by a move into Asia and on to Africa and South America through the 1990s and early 2000s. RT017 was not identified in Oceania (Australia) until the late 2000s, via a jump from Europe.
This study investigated the genetic diversity of 277 C. difficile RT017 isolates with temporal, geographical, and source variation. Phylogeographic analysis of the SNPs identified through WGS of the isolates suggests that there are two main sublineages of RT017 that share an ancestry and are globally disseminated. Both sublineages contain isolates from diverse geographical locations and isolation dates, with animal isolates spread among human isolates in SL1. Together with the haplotype diversity and geographically and temporally diverse presence of the transposable elements, these data suggest widespread transcontinental spread and recombination with independent acquisition and loss within different clusters.
MATERIALS AND METHODS
The 277 isolates described in this study are shown in Table 1 and included 37 isolates from a previous study (ENA study accession number ERP009770) (39), with the remaining being new to this study (ENA study accession number PRJEB11868). These were of human (n = 251), environmental/hospital ward (n = 2), equine (n = 4), canine (n = 11), and bovine (n = 9) origin, with isolation dates between 1990 and 2013. These isolates were subjected to genomic DNA extraction as previously described by Stabler et al. (62). WGS data for the isolates was obtained using either the HiSeq 2000 sequencing system or the MiSeq sequencing system (Illumina, California), and libraries were created as previously described (63) or using a Nextera XT kit (Illumina, California), respectively. The sequence data were processed and quality controlled according to a standard pipeline as previously described (64). Briefly, FASTQ-formatted sequencing reads were quality controlled with a minimum quality Phred score of 30 (as a rolling average over 4 bases) using trimmomatic (65). The resulting reads were mapped using the BWA-MEM (66) software against the M68 C. difficile reference strain, and the majority of posttrimmed reads (>92% for all samples passing quality control) were mapped to the reference. SNPs were called using SAMtools/VCFtools (67).
Velvet (68) and Velvet Optimizer (http://bioinformatics.net.au/software.velvetoptimiser.shtml) were used to de novo assemble the trimmed reads into contigs, producing 277 assemblies. Optimal k-mers fell between 53 bp and 97 bp, and the mean value for median contig size of genome assembly (n50) was over 928,000 bp. The mean longest contig was 1,067,000 bp, with 71 samples producing contigs that covered over half of the genome (greater than ∼2.15 Mbp), and 16 samples assembled to contigs greater than 4 Mbp (equivalent to greater than 90% of the genome). Pipeline, post-, genetic, phylogenetic, phylogeographic, and cluster analyses were carried out using Perl, R, abacas, prokka, RaXML, Bayesian evolutionary analysis sampling trees (BEAST), and mclust software (69–73). A minor allele frequency (MAF) of less than 1% was used. To remove any SNPs that may be associated with recombination and which would mask the true phylogeny, SNPs within 1 bp of an insertion or deletion site were excluded from further analysis. We used BEAST (72) to produce an SNP phylogeny from the SNPs, as well as geographical and temporal data combined in phylogeographic analysis and mclust software for maximum likelihood cluster analysis.
To determine the MICs of 7/277 isolates, dilutions for the antibiotics chloramphenicol, rifampin, tetracycline, erythromycin, nalidixic acid, gentamicin, teicoplanin, and ampicillin were made as previously described (74). Briefly, 10 ml preequilibrated brain heart infusion broth, supplemented with yeast (Oxoid), l-cysteine (Sigma), and C. difficile supplement (Oxoid) (BHIS), were inoculated with three colonies of 48-h culture on BHIS agar plates. Once the optical density (OD) reached 0.3 nm, 24-well plates containing the antibiotic dilutions were inoculated with 1/100 of the BHIS broths and incubated. The ODs were measured 24 h postinoculation, and MIC data were categorized as susceptible, intermediate, and resistant by following the Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines. The reference strain M68 was used as a control, as were appropriate negative controls.
Accession number(s).
Sequence data were deposited in the European Nucleotide Archive under study accession number PRJEB11868.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Public Health Laboratory, London, United Kingdom, for help with PCR ribotyping. We thank David Harris at the WTSI for assistance with DNA sequencing.
The work was supported by the National Institute for Health Research (NIHR), the Wellcome Trust, and the Medical Research Council (grant reference MR/K000551/1). M.C. is funded by a doctoral research fellowship award from the NIHR. This report is independent research arising from a Chief Scientific Officer (CSO) Healthcare Scientist Award supported by the National Institute for Health Research and the CSO.
The views expressed in this publication are those of the author(s) and not necessarily those of the National Health Service, the NIHR, or the Department of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
M.D.C., R.A.S., and B.W.W. planned the experiments. M.D.C. performed experiments and de novo analysis, and M.D.C., R.A.S., and M.D.P. performed bioinformatics analyses. C.L.H. performed MIC experiments. M.D.C., D.N.G., P.M.H., H.K., H.K., E.J.K., T.D.L., H.P., S.R., T.V.R., K.S., P.J.S., and S.J.W. provided strains. M.D.C. drafted the manuscript, with contributions from R.A.S., M.D.P., and B.W.W., followed by suggestions and comments from all authors.
We have no conflicts of interest to report.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.01296-16.
REFERENCES
- 1.Barbut F, Gariazzo B, Bonne L, Lalande V, Burghoffer B, Luiuz R, Petit JC. 2007. Clinical features of Clostridium difficile-associated infections and molecular characterization of strains: results of a retrospective study, 2000-2004. Infect Control Hosp Epidemiol 28:131–139. doi: 10.1086/511794. [DOI] [PubMed] [Google Scholar]
- 2.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knetsch CW, Terveer EM, Lauber C, Gorbalenya AE, Harmanus C, Kuijper EJ, Corver J, van Leeuwen HC. 2012. Comparative analysis of an expanded Clostridium difficile reference strain collection reveals genetic diversity and evolution through six lineages. Infect Genet Evol 12:1577–1585. doi: 10.1016/j.meegid.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 4.He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, Connor TR, Harris SR, Fairley D, Bamford KB, D'Arc S, Brazier J, Brown D, Coia JE, Douce G, Gerding D, Kim HJ, Koh TH, Kato H, Senoh M, Louie T, Michell S, Butt E, Peacock SJ, Brown NM, Riley T, Songer G, Wilcox M, Pirmohamed M, Kuijper E, Hawkey P, Wren BW, Dougan G, Parkhill J, Lawley TD. 2013. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 45:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camacho-Ortiz A, Lopez-Barrera D, Hernandez-Garcia R, Galvan-De Los Santos AM, Flores-Trevino SM, Llaca-Diaz JM, Garza HJ, Bosques-Padilla FJ, Garza-Gonzalez E. 2015. First report of Clostridium difficile NAP1/027 in a Mexican hospital. PLoS One 10:e0122627. doi: 10.1371/journal.pone.0122627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguayo C, Flores R, Levesque S, Araya P, Ulloa S, Lagos J, Hormazabal JC, Tognarelli J, Ibanez D, Pidal P, Duery O, Olivares B, Fernandez J. 2015. Rapid spread of Clostridium difficile NAP1/027/ST1 in Chile confirms the emergence of the epidemic strain in Latin America. Epidemiol Infect 143:3069–3073. doi: 10.1017/S0950268815000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez-Rocha C, Barra-Carrasco J, Pizarro-Guajardo M, Ibanez P, Bueno SM, Sarker MR, Guzman AM, Alvarez-Lobos M, Paredes-Sabja D. 2012. Epidemic Clostridium difficile ribotype 027 in Chile. Emerg Infect Dis 18:1370–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Zhou Y, Wang Z, Xie S, Zhang T, Lin M, Li R, Tan J, Chen Y, Jiang B. 2014. Identification of Clostridium difficile ribotype 027 for the first time in mainland China. Infect Control Hosp Epidemiol 35:95–98. doi: 10.1086/674405. [DOI] [PubMed] [Google Scholar]
- 9.Kato H, Ito Y, van den Berg RJ, Kuijper EJ, Arakawa Y. 2007. First isolation of Clostridium difficile 027 in Japan. Euro Surveill 12:E070111–E070113. [DOI] [PubMed] [Google Scholar]
- 10.Cheng VC, Yam WC, Chan JF, To KK, Ho PL, Yuen KY. 2009. Clostridium difficile ribotype 027 arrives in Hong Kong. Int J Antimicrob Agents 34:492–493. doi: 10.1016/j.ijantimicag.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Kim H, Lee Y, Moon HW, Lim CS, Lee K, Chong Y. 2011. Emergence of Clostridium difficile ribotype 027 in Korea. Korean J Lab Med 31:191–196. doi: 10.3343/kjlm.2011.31.3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tae CH, Jung SA, Song HJ, Kim SE, Choi HJ, Lee M, Hwang Y, Kim H, Lee K. 2009. The first case of antibiotic-associated colitis by Clostridium difficile PCR ribotype 027 in Korea. J Korean Med Sci 24:520–524. doi: 10.3346/jkms.2009.24.3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai MJ, Chiueh TS, Huang ZY, Lin JC. 2015. The first Clostridium difficile ribotype 027 strain isolated in Taiwan. J Formos Med Assoc 115:210–212. [DOI] [PubMed] [Google Scholar]
- 14.Lim PL, Ling ML, Lee HY, Koh TH, Tan AL, Kuijper EJ, Goh SS, Low BS, Ang LP, Harmanus C, Lin RT, Krishnan P, James L, Lee CE. 2011. Isolation of the first three cases of Clostridium difficile polymerase chain reaction ribotype 027 in Singapore. Singapore Med J 52:361–364. [PubMed] [Google Scholar]
- 15.Riley TV, Thean S, Hool G, Golledge CL. 2009. First Australian isolation of epidemic Clostridium difficile PCR ribotype 027. Med J Aust 190:706–708. [DOI] [PubMed] [Google Scholar]
- 16.Richards M, Knox J, Elliott B, Mackin K, Lyras D, Waring LJ, Riley TV. 2011. Severe infection with Clostridium difficile PCR ribotype 027 acquired in Melbourne, Australia. Med J Aust 194:369–371. [DOI] [PubMed] [Google Scholar]
- 17.Alzahrani N, Johani SA. 2013. Emergence of a highly resistant Clostridium difficile strain (NAP/BI/027) in a tertiary care center in Saudi Arabia. Ann Saud Med 33:198–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiener-Well Y, Ben-Chetrit E, Abed-Eldaim M, Assous MV, Miller-Roll T, Adler A. 2014. Clinical and molecular characteristics of an outbreak caused by the pandemic (BI/NAP1/027) Clostridium difficile clone in a single center in Israel. Infect Control Hosp Epidemiol 35:1306–1308. doi: 10.1086/678070. [DOI] [PubMed] [Google Scholar]
- 19.Bacci S, St-Martin G, Olesen B, Bruun B, Olsen KE, Nielsen EM, Molbak K. 2009. Outbreak of Clostridium difficile 027 in North Zealand, Denmark, 2008-2009. Euro Surveill 14:19183. [PubMed] [Google Scholar]
- 20.Lachowicz D, Szulencka G, Obuch-Woszczatynski P, van Belkum A, Pituch H. 2015. First Polish outbreak of Clostridium difficile ribotype 027 infections among dialysis patients. Eur J Clin Microbiol Infect Dis 34:63–67. doi: 10.1007/s10096-014-2204-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orsi GB, Conti C, Mancini C, Giordano A. 2014. Clostridium difficile 027 increasing detection in a teaching hospital in Rome, Italy. Infection 42:941–942. doi: 10.1007/s15010-014-0652-y. [DOI] [PubMed] [Google Scholar]
- 22.Arvand M, Vollandt D, Bettge-Weller G, Harmanus C, Kuijper EJ, Clostridium difficile study group Hesse. 2014. Increased incidence of Clostridium difficile PCR ribotype 027 in Hesse, Germany, 2011 to 2013. Euro Surveill 19:20732. doi: 10.2807/1560-7917.ES2014.19.10.20732. [DOI] [PubMed] [Google Scholar]
- 23.Lagier JC, Dubourg G, Cassir N, Fournier PE, Colson P, Richet H, Brouqui P, Raoult D. 2013. Clostridium difficile 027 emerging outbreak in Marseille, France. Infect Control Hosp Epidemiol 34:1339–1341. doi: 10.1086/673995. [DOI] [PubMed] [Google Scholar]
- 24.Di Bella S, Paglia MG, Johnson E, Petrosillo N. 2012. Clostridium difficile 027 infection in central Italy. BMC Infect Dis 12:370. doi: 10.1186/1471-2334-12-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Indra A, Huhulescu S, Fiedler A, Kernbichler S, Blaschitz M, Allerberger F. 2009. Outbreak of Clostridium difficile 027 infection in Vienna, Austria 2008-2009. Euro Surveill 14:19186. [PubMed] [Google Scholar]
- 26.Long S, Fenelon L, Fitzgerald S, Nolan N, Burns K, Hannan M, Kyne L, Fanning S, Drudy D. 2007. First isolation and report of clusters of Clostridium difficile PCR 027 cases in Ireland. Euro Surveill 12:E070423–E070426. [DOI] [PubMed] [Google Scholar]
- 27.Drudy D, Goorhuis B, Bakker D, Kyne L, van den Berg R, Fenelon L, Fanning S, Kuijper EJ. 2008. Clindamycin-resistant clone of Clostridium difficile PCR Ribotype 027, Europe. Emerg Infect Dis 14:1485–1487. doi: 10.3201/eid1409.071346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuijper EJ, Coignard B, Tull P, ESCMID Study Group for Clostridium difficile, EU Member States, European Centre for Disease Prevention and Control. 2006. Emergence of Clostridium difficile-associated disease in North America and Europe. Clinical Microbiol Infect 12(Suppl 6):S2–S18. [DOI] [PubMed] [Google Scholar]
- 29.Public Health England. 2016. Clostridium difficile ribotyping network (CDRN) for England and Northern Ireland, 2013 to 2015. PHE Publication Report. Public Health England, London, United Kingdom. [Google Scholar]
- 30.Dupuy B, Govind R, Antunes A, Matamouros S. 2008. Clostridium difficile toxin synthesis is negatively regulated by TcdC. J Med Microbiol 57:685–689. doi: 10.1099/jmm.0.47775-0. [DOI] [PubMed] [Google Scholar]
- 31.Rupnik M, Janezic S. 2016. An update on Clostridium difficile toxinotyping. J Clin Microbiol 54:13–18. doi: 10.1128/JCM.02083-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin BM, Kuak EY, Yoo SJ, Shin WC, Yoo HM. 2008. Emerging toxin A-B+ variant strain of Clostridium difficile responsible for pseudomembranous colitis at a tertiary care hospital in Korea. Diagn Microbiol Infect Dis 60:333–337. doi: 10.1016/j.diagmicrobio.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 33.Kim SJ, Kim H, Seo Y, Yong D, Jeong SH, Chong Y, Lee K. 2010. Molecular characterization of toxin A-negative, toxin B-positive variant strains of Clostridium difficile isolated in Korea. Diagn Microbiol Infect Dis 67:198–201. doi: 10.1016/j.diagmicrobio.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Weintraub A, Fang H, Wu S, Zhang Y, Nord CE. 2010. Antimicrobial susceptibility and heteroresistance in Chinese Clostridium difficile strains. Anaerobe 16:633–635. doi: 10.1016/j.anaerobe.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 35.al-Barrak A, Embil J, Dyck B, Olekson K, Nicoll D, Alfa M, Kabani A. 1999. An outbreak of toxin A negative, toxin B positive Clostridium difficile-associated diarrhea in a Canadian tertiary-care hospital. Can Commun Dis Rep 25:65–69. [PubMed] [Google Scholar]
- 36.Kuijper EJ, de Weerdt J, Kato H, Kato N, van Dam AP, van der Vorm ER, Weel J, van Rheenen C, Dankert J. 2001. Nosocomial outbreak of Clostridium difficile-associated diarrhoea due to a clindamycin-resistant enterotoxin A-negative strain. Eur J Clin Microbiol Infect Dis 20:528–534. doi: 10.1007/s100960100550. [DOI] [PubMed] [Google Scholar]
- 37.Drudy D, Harnedy N, Fanning S, Hannan M, Kyne L. 2007. Emergence and control of fluoroquinolone-resistant, toxin A-negative, toxin B-positive Clostridium difficile. Infect Control Hosp Epidemiol 28:932–940. doi: 10.1086/519181. [DOI] [PubMed] [Google Scholar]
- 38.Arvand M, Hauri AM, Zaiss NH, Witte W, Bettge-Weller G. 2009. Clostridium difficile ribotypes 001, 017, and 027 are associated with lethal C. difficile infection in Hesse, Germany. Euro Surveill 14:19403. [DOI] [PubMed] [Google Scholar]
- 39.Cairns MD, Preston MD, Lawley TD, Clark TG, Stabler RA, Wren BW. 2015. Genomic epidemiology of a protracted hospital outbreak caused by a toxin A negative, Clostridium difficile sublineage PCR Ribotype 017 strain in London, England. J Clin Microbiol 53:3141–3147. doi: 10.1128/JCM.00648-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins DA, Hawkey PM, Riley TV. 2013. Epidemiology of Clostridium difficile infection in Asia. Antimicrob Resist Infect Control 2:21. doi: 10.1186/2047-2994-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pituch H, Brazier JS, Obuch-Woszczatynski P, Wultanska D, Meisel-Mikolajczyk F, Luczak M. 2006. Prevalence and association of PCR ribotypes of Clostridium difficile isolated from symptomatic patients from Warsaw with macrolide-lincosamide-streptogramin B (MLSB) type resistance. J Med Microbiol 55:207–213. doi: 10.1099/jmm.0.46213-0. [DOI] [PubMed] [Google Scholar]
- 42.Alfa MJ, Kabani A, Lyerly D, Moncrief S, Neville LM, Al-Barrak A, Harding GK, Dyck B, Olekson K, Embil JM. 2000. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile responsible for a nosocomial outbreak of Clostridium difficile-associated diarrhea. J Clin Microbiol 38:2706–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hawkey PM, Marriott C, Liu WE, Jian ZJ, Gao Q, Ling TK, Chow V, So E, Chan R, Hardy K, Xu L, Manzoor S. 2013. Molecular epidemiology of Clostridium difficile infection in a major chinese hospital: an underrecognized problem in Asia? J Clin Microbiol 51:3308–3313. doi: 10.1128/JCM.00587-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H, Riley TV, Kim M, Kim CK, Yong D, Lee K, Chong Y, Park JW. 2008. Increasing prevalence of toxin A-negative, toxin B-positive isolates of Clostridium difficile in Korea: impact on laboratory diagnosis. J Clin Microbiol 46:1116–1117. doi: 10.1128/JCM.01188-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim H, Jeong SH, Roh KH, Hong SG, Kim JW, Shin MG, Kim MN, Shin HB, Uh Y, Lee H, Lee K. 2010. Investigation of toxin gene diversity, molecular epidemiology, and antimicrobial resistance of Clostridium difficile isolated from 12 hospitals in South Korea. Korean J Lab Med 30:491–497. doi: 10.3343/kjlm.2010.30.5.491. [DOI] [PubMed] [Google Scholar]
- 46.Goorhuis A, Legaria MC, van den Berg RJ, Harmanus C, Klaassen CH, Brazier JS, Lumelsky G, Kuijper EJ. 2009. Application of multiple-locus variable-number tandem-repeat analysis to determine clonal spread of toxin A-negative Clostridium difficile in a general hospital in Buenos Aires, Argentina. Clin Microbiol Infect 15:1080–1086. doi: 10.1111/j.1469-0691.2009.02759.x. [DOI] [PubMed] [Google Scholar]
- 47.Elliott B, Squire MM, Thean S, Chang BJ, Brazier JS, Rupnik M, Riley TV. 2011. New types of toxin A-negative, toxin B-positive strains among clinical isolates of Clostridium difficile in Australia. J Med Microbiol 60:1108–1111. doi: 10.1099/jmm.0.031062-0. [DOI] [PubMed] [Google Scholar]
- 48.Elliott B, Reed R, Chang BJ, Riley TV. 2009. Bacteremia with a large clostridial toxin-negative, binary toxin-positive strain of Clostridium difficile. Anaerobe 15:249–251. doi: 10.1016/j.anaerobe.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Samra Z, Talmor S, Bahar J. 2002. High prevalence of toxin A-negative toxin B-positive Clostridium difficile in hospitalized patients with gastrointestinal disease. Diagn Microbiol Infect Dis 43:189–192. doi: 10.1016/S0732-8893(02)00400-5. [DOI] [PubMed] [Google Scholar]
- 50.Komatsu M, Kato H, Aihara M, Shimakawa K, Iwasaki M, Nagasaka Y, Fukuda S, Matsuo S, Arakawa Y, Watanabe M, Iwatani Y. 2003. High frequency of antibiotic-associated diarrhea due to toxin A-negative, toxin B-positive Clostridium difficile in a hospital in Japan and risk factors for infection. Eur J Clin Microbiol Infect Dis 22:525–529. doi: 10.1007/s10096-003-0992-5. [DOI] [PubMed] [Google Scholar]
- 51.Rajabally N, Kullin B, Ebrahim K, Brock T, Weintraub A, Whitelaw A, Bamford C, Watermeyer G, Thomson S, Abratt V, Reid S. 9 February 2016. A comparison of Clostridium difficile diagnostic methods for identification of local strains in a South African centre. J Med Microbiol doi: 10.1099/jmm.0.000231. [DOI] [PubMed] [Google Scholar]
- 52.Drudy D, Harnedy N, Fanning S, O'Mahony R, Kyne L. 2007. Isolation and characterisation of toxin A-negative, toxin B-positive Clostridium difficile in Dublin, Ireland. Clin Microbiol Infect 13:298–304. doi: 10.1111/j.1469-0691.2006.01634.x. [DOI] [PubMed] [Google Scholar]
- 53.Pituch H, van den Braak N, van Leeuwen W, van Belkum A, Martirosian G, Obuch-Woszczatynski P, Luczak M, Meisel-Mikolajczyk F. 2001. Clonal dissemination of a toxin-A-negative/toxin-B-positive Clostridium difficile strain from patients with antibiotic-associated diarrhea in Poland. Clin Microbiol Infect 7:442–446. doi: 10.1046/j.1198-743x.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez-Palacios A, Stampfli HR, Duffield T, Peregrine AS, Trotz-Williams LA, Arroyo LG, Brazier JS, Weese JS. 2006. Clostridium difficile PCR ribotypes in calves, Canada. Emerg Infect Dis 12:1730–1736. doi: 10.3201/eid1211.051581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drigo I, Mazzolini E, Bacchin C, Tonon E, Puiatti C, Bano L, Spigaglia P, Barbanti F, Agnoletti F. 2015. Molecular characterization and antimicrobial susceptibility of Clostridium difficile isolated from rabbits raised for meat production. Vet Microbiol 181:303–317. doi: 10.1016/j.vetmic.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Lee JH, Lee Y, Lee K, Riley TV, Kim H. 2014. The changes of PCR ribotype and antimicrobial resistance of Clostridium difficile in a tertiary care hospital over 10 years. J Med Microbiol 63:819–823. doi: 10.1099/jmm.0.072082-0. [DOI] [PubMed] [Google Scholar]
- 57.Drudy D, Quinn T, O'Mahony R, Kyne L, O'Gaora P, Fanning S. 2006. High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J Antimicrob Chemother 58:1264–1267. doi: 10.1093/jac/dkl398. [DOI] [PubMed] [Google Scholar]
- 58.Clinical and Laboratory Standards Institute. 2012. Methods for antimicrobial susceptibility testing of anaerobic bacteria, 8th ed M11-A8, approved standard. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 59.Clinical and Laboratory Standards Institute. 2013. Performance standards for antimicrobial susceptibility testing, 23rd informational supplement. M100-S23. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 60.Curry SR, Marsh JW, Shutt KA, Muto CA, O'Leary MM, Saul MI, Pasculle AW, Harrison LH. 2009. High frequency of rifampin resistance identified in an epidemic Clostridium difficile clone from a large teaching hospital. Clin Infect Dis 48:425–429. doi: 10.1086/596315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bahrmand AR, Titov LP, Tasbiti AH, Yari S, Graviss EA. 2009. High-level rifampin resistance correlates with multiple mutations in the rpoB gene of pulmonary tuberculosis isolates from the Afghanistan border of Iran. J Clin Microbiol 47:2744–2750. doi: 10.1128/JCM.r00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Preston MD, Assefa SA, Ocholla H, Sutherland CJ, Borrmann S, Nzila A, Michon P, Hien TT, Bousema T, Drakeley CJ, Zongo I, Ouedraogo JB, Djimde AA, Doumbo OK, Nosten F, Fairhurst RM, Conway DJ, Roper C, Clark TG. 2014. PlasmoView: a web-based resource to visualise global Plasmodium falciparum genomic variation. J Infect Dis 209:1808–1815. doi: 10.1093/infdis/jit812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zerbino DR. 2010. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics Chapter 11:Unit 11.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. 2009. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25:1968–1969. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 71.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fraley C, Raftery AE. 2002. Model-based clustering, discriminant analysis and density estimation. J Am Stat Assoc 97:611. doi: 10.1198/016214502760047131. [DOI] [Google Scholar]
- 74.Andrews JM. 2001. Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48(Suppl 1):S5–S16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.