

ABSTRACT
Urinary tract infection (UTI) is a major global infectious disease affecting millions of people annually. Human urinary copper (Cu) content is elevated during UTI caused by uropathogenic Escherichia coli (UPEC). UPEC upregulates the expression of Cu efflux genes during clinical UTI in patients as an adaptive response to host-derived Cu. Whether Cu is mobilized to urine as a host response to UTI and its role in protection against UTI remain unresolved. To address these questions, we tested the hypothesis that Cu is a host effector mobilized to urine during UTI to limit bacterial growth. Our results reveal that Cu is mobilized to urine during UTI caused by the major uropathogens Proteus mirabilis and Klebsiella pneumoniae, in addition to UPEC, in humans. Ceruloplasmin, a Cu-containing ferroxidase, is found at higher levels in UTI urine than in healthy control urine and serves as the molecular source of urinary Cu during UTI. Our results demonstrate that ceruloplasmin decreases the bioavailability of iron in urine by a transferrin-dependent mechanism. Experimental UTI with UPEC in nonhuman primates recapitulates the increased urinary Cu content observed during clinical UTI. Furthermore, Cu-deficient mice are highly colonized by UPEC, indicating that Cu is involved in the limiting of bacterial growth within the urinary tract. Collectively, our results indicate that Cu is a host effector that is involved in protection against pathogen colonization of the urinary tract. Because urinary Cu levels are amenable to modulation, augmentation of the Cu-based host defense against UTI represents a novel approach to limiting bacterial colonization during UTI.
KEYWORDS: UTI, urinary tract infection, uropathogenic E. coli, UPEC, copper, ceruloplasmin
INTRODUCTION
Urinary tract infection (UTI) is an extremely common infectious disease in the United States and around the world (1, 2). In the United States alone, UTI leads to 11 million physician visits, 1.7 million emergency room visits, and 470,000 hospitalizations annually, with a direct cost of $3.5 billion (2). Infection of the urinary bladder (cystitis) is the most common form of UTI, whereas kidney infection (pyelonephritis), bacteremia, and sepsis are less common but more serious outcomes of UTI (3, 4). Women are four times as likely to develop UTI as men are because of anatomic differences (4). Children, the elderly, and individuals with catheters, anatomic and physiologic abnormalities of the urinary tract, uroliths, or diabetes mellitus are also highly susceptible to UTI (4).
Uropathogenic Escherichia coli (UPEC) is the etiological agent in ∼85% of the cystitis cases in otherwise healthy people (3). Free-living, adherent, biofilm, and intracellular forms of UPEC are found in the urinary bladder (5, 6). UPEC utilizes multiple virulence factors, including type 1 fimbriae, P fimbriae, flagella, toxins, and iron acquisition systems, to cause UTI (2, 7). In addition to UPEC, Klebsiella pneumoniae and Proteus mirabilis are other common bacterial pathogens that cause UTI (4). Trimethoprim-sulfamethoxazole and ciprofloxacin are widely used in the empirical treatment of acute cystitis (8). In the United States, 11 to 21% and 1 to 6% of UPEC isolates are resistant to trimethoprim-sulfamethoxazole and ciprofloxacin, respectively (9); these drugs are not used when local resistance rates reach 20% and 10%, respectively (8). More troubling is the emergence of multidrug resistance, including resistance to colistin, which is the antibiotic of last resort against Gram-negative bacteria (10). An unabated increase in antibiotic resistance and a lack of new treatments threaten the clinical management of UTI in the future.
We have previously demonstrated that copper (Cu) is mobilized to urine during UTI caused by UPEC in humans (11). A UPEC mutant that lacks the Cus Cu efflux system is attenuated in a mouse model of UTI, compared to the wild-type parental strain during coinfection, indicating that resistance to host-derived Cu is important for in vivo survival (11). Consistent with our results, Chaturvedi et al. have independently reported the detection of Cu-yersiniabactin complexes in UPEC-induced UTI urine (12). The central hypothesis of this study is that Cu is a host effector mobilized to urine during UTI to limit bacterial growth. We tested this hypothesis with human UTI urine samples and nonhuman primate and mouse models of UTI. Our results reveal that Cu mobilization is a conserved host response to UTI in humans and nonhuman primates. We demonstrate that ceruloplasmin, a molecular source of urinary Cu during UTI, is involved in a novel mechanism of iron limitation to UPEC. Furthermore, we demonstrate that Cu is involved in protection against UPEC colonization in the murine urinary tract. In summary, our results highlight an important and novel biological role for Cu in protection against UTI.
RESULTS
Urinary Cu mobilization is a host response to UTI in humans.
First, we tested the hypothesis that Cu mobilization to urine is a host response to UTI. We reasoned that if Cu mobilization is a host response, this phenomenon should be conserved during UTI caused by multiple uropathogens and not just UPEC. Urinary Cu content, determined by inductively coupled plasma mass spectrometry (ICP-MS), was higher during UTI not only with UPEC but also with common Gram-negative uropathogens K. pneumoniae and P. mirabilis (Fig. 1A) than that of healthy controls. To rule out the possibility of differences in the concentration of urine samples between groups, we determined creatinine and specific gravity values in samples from patients with UTI caused by UPEC, K. pneumoniae, and P. mirabilis and in control samples. Urinary creatinine content and specific gravity values were not significantly different between UTI (UPEC, K. pneumoniae, and P. mirabilis) and control samples (Fig. 1B and C), indicating that elevated urinary Cu levels are not due to greater concentration of UTI urine samples than control urine samples. Next, we wanted to identify the source of urinary Cu in UTI urine samples. A possible source of urinary Cu is blood because hematuria is often reported during UTI (3). We reasoned that if blood is the source of Cu, then urinary Cu and hemoglobin levels should be positively correlated. Our results reveal that Cu and hemoglobin levels, however, are not correlated (Pearson's correlation coefficient [r] = 0.2, P = 0.1, Fig. 1D). Indeed, UTI urine with the highest level of Cu contains the lowest level of hemoglobin and vice versa, suggesting that blood is not the source of Cu in UTI urine (Fig. 1D). Our results demonstrate that urinary Cu mobilization is triggered during UTI caused by major Gram-negative bacterial pathogens in humans and urinary Cu mobilization is independent of UTI-induced hematuria.
FIG 1.
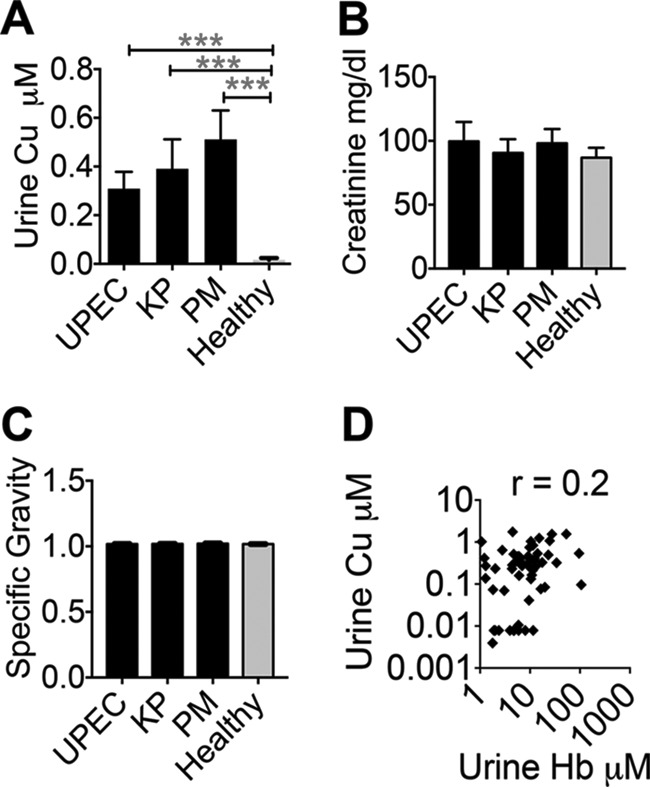
Cu is mobilized to urine during UTI in humans. (A) Urinary Cu content was determined by ICP-MS. ***, P < 0.001; ANOVA with Dunnett's posttest. (B) Creatinine content of urine samples was determined with a colorimetric assay. (C) Specific gravity of urine samples was measured with a refractometer. (D) Hemoglobin (Hb) levels in UTI urine (n = 57) were measured by a colorimetric assay. Correlation to urinary Cu content (n = 57) was determined by Pearson's correlation coefficient (r = 0.2 and P = 0.1). UPEC, n = 30; K. pneumoniae (KP), n = 10; P. mirabilis (PM), n = 17; healthy controls, n = 40. The mean and standard error of the mean are presented.
Cuprous ions are found in human urine.
Cu can exist as more bactericidal cuprous (Cu1+) or less bactericidal cupric (Cu2+) ions (13). Redox conditions of human urine, both in health and during UTI, promote the production and maintenance of more bactericidal Cu1+ ions (Fig. 2) from Cu2+ ions. Lysogeny broth, a commonly used medium for culturing E. coli, and phosphate-buffered saline (PBS), however, do not promote the conversion of Cu2+ ions to Cu1+ ions. This result suggests that UPEC is exposed to more bactericidal Cu1+ ions during UTI.
FIG 2.
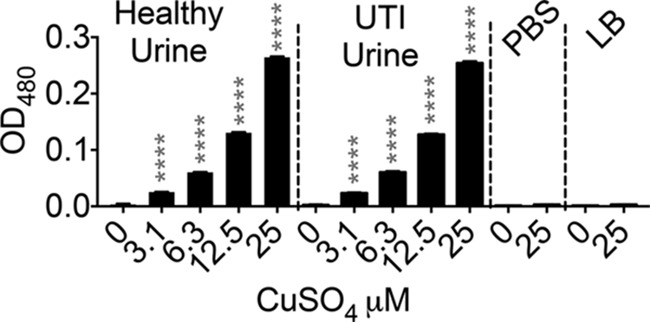
Cu exists as cuprous (Cu1+) ions in human urine. The Cu1+-specific chelator bathocuproine (50 μM) was used to determine the species of Cu ions found in human urine (healthy or UTI) after the addition of cupric (Cu2+) ions. The optical density at 480 nm (OD480) of Cu1+-bathocuproine complexes was measured. Note the absence of Cu1+-bathocuproine complex formation in PBS and lysogeny broth (LB). ****, P < 0.0001; ANOVA with Dunnett's posttest, compared to 0 μM CuSO4. The mean and standard error of the mean are presented.
Ceruloplasmin is a molecular source of Cu in UTI urine.
Ceruloplasmin is a 132-kDa glycoprotein that contains ∼95% of the circulating Cu in mammals and is an acute-phase reactant that is secreted by the liver (14). Urine samples from UTI patients and healthy controls were analyzed by immunoblot assays for the presence of holoceruloplasmin with pure human holoceruloplasmin as a positive control. A representative immunoblot assay, depicted in Fig. 3A, reveals the presence of holoceruloplasmin in UTI but not in healthy urine samples. An enzyme-linked immunosorbent assay (ELISA) was utilized to quantify ceruloplasmin in control and UTI urine samples. Our results demonstrate that ceruloplasmin is found at significantly higher levels in UTI caused by UPEC, K. pneumoniae, and P. mirabilis than in controls (Fig. 3B). Furthermore, urinary Cu content and ceruloplasmin levels are positively correlated (r = 0.58, P < 0.0001; Fig. 3C) and support a model in which ceruloplasmin is the molecular source of Cu in urine during UTI.
FIG 3.
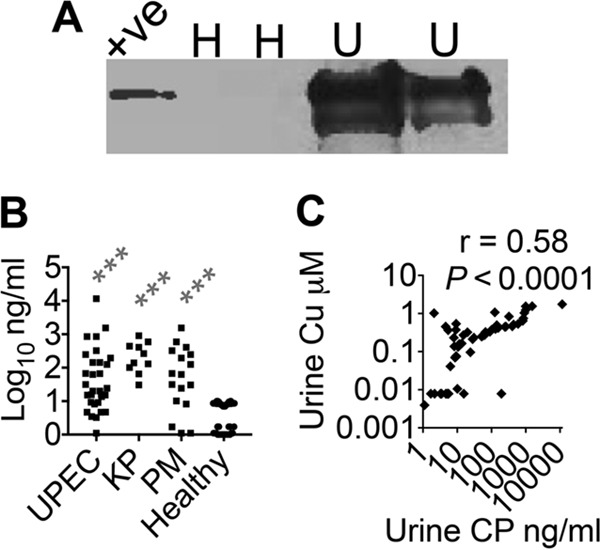
Ceruloplasmin (CP) is a molecular source of urinary Cu during UTI. (A) Representative immunoblot assay with UTI (U; n = 2) and healthy (H; n = 2) human urine samples probed with anti-CP antibody. +ve, human CP (20 ng). (B) Urinary CP levels were measured by ELISA. ***, P < 0.001; Kruskal-Wallis test with Dunn's posttest. UPEC, n = 30; K. pneumoniae (KP), n = 10; P. mirabilis (PM), n = 17; healthy controls, n = 40. (C) Urinary Cu and ceruloplasmin levels are positively correlated in UTI samples. Pearson's correlation coefficient was determined for urinary Cu and ceruloplasmin levels (r = 0.58 and P < 0.0001). UTI urine samples (UPEC, KP, and PM), n = 57.
Ceruloplasmin induces Cu stress in UPEC.
We then tested whether Cu is released from ceruloplasmin to induce Cu stress in UPEC by measuring the transcription of a Cu efflux gene and the growth of a UPEC Cu efflux mutant in the presence of ceruloplasmin in human urine. Because UPEC cus genes are expressed in response to Cu stress (11), we measured the expression of the cusC gene in UPEC cultured in urine with pure human ceruloplasmin by quantitative PCR (qPCR). The expression of the Cu efflux gene cusC, normalized to that of gapA, was induced by ceruloplasmin (Fig. 4A), demonstrating that ceruloplasmin is a source of Cu ions to induce Cu stress in UPEC. The growth of wild-type UPEC (strain CFT073) and a mutant (Δcus) lacking the CusCFBA Cu efflux pump and the CusSR cognate two-component regulatory system was determined in the presence of ceruloplasmin. This Cu efflux pump mutant was previously demonstrated to exhibit a fitness defect during colonization in the mouse model of UTI and was hypersensitive to Cu toxicity (11). Ceruloplasmin did not affect the growth of the wild-type strain but led to a significant decrease in the growth of the Δcus mutant in human urine (Fig. 4B). Together, these two lines of evidence indicate that ceruloplasmin can induce Cu stress in UPEC.
FIG 4.
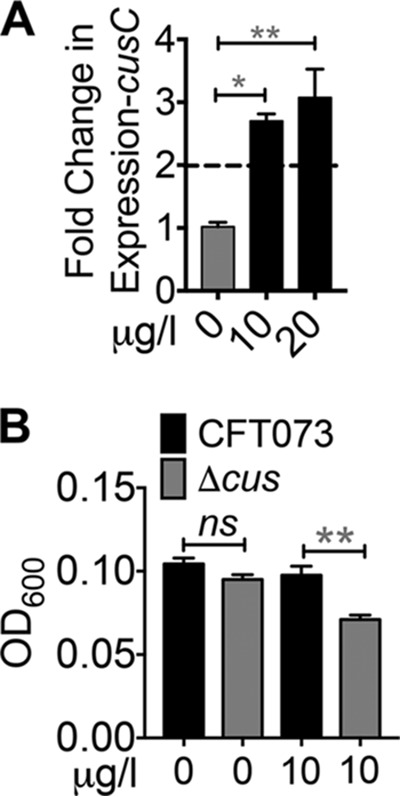
Ceruloplasmin induces Cu stress in UPEC. (A) Expression of cusC in UPEC cultured with or without ceruloplasmin in healthy human urine was determined by qPCR after normalization to gapA. Dotted line, threshold of upregulation. *, P < 0.05; **, P < 0.01; ANOVA with Dunnett's posttest (n = 3, in duplicate). (B) Ceruloplasmin inhibits the growth of a UPEC mutant lacking the Cus Cu efflux system in human urine. The optical density at 600 nm (OD600) of 24-h cultures of wild-type (CFT073) and ΔcusSRCFBA mutant was measured (n = 3, in triplicate). The mean and standard error of the mean are presented. **, P < 0.01; t test.
Ceruloplasmin retains ferroxidase activity in urine during UTI.
Ceruloplasmin is a ferroxidase that catalyzes the oxidation of Fe2+ to Fe3+ and thereby accelerates iron loading onto apotransferrin because of the higher affinity of apotransferrin for Fe3+ ions than for Fe2+ ions (14). Urine samples from UTI caused by UPEC, K. pneumoniae, and P. mirabilis exhibit significantly greater oxidase activity than healthy samples in the o-dianisidine oxidation assay (Fig. 5A). We tested whether ceruloplasmin can function as an oxidase in human urine in vitro. Our results demonstrate that pure ceruloplasmin exhibits oxidase activity in human urine in a concentration-dependent manner (Fig. 5B). Because ceruloplasmin is a ferroxidase, we tested if ceruloplasmin retains its ferroxidase activity in urine and promotes iron loading onto transferrin. Ferrozine, which binds only Fe2+ ions, was used to determine the Fe2+ ion content in urine in the presence of ceruloplasmin and apotransferrin. Ceruloplasmin oxidizes Fe2+ to Fe3+ in urine, resulting in less production of Fe2+-ferrozine complexes than in controls (Fig. 6A and B). In the presence of apotransferrin, ceruloplasmin-mediated depletion of Fe2+ ions was even higher, suggesting the formation of holotransferrin, which would act as a sink for Fe3+ ions (Fig. 6A and B). Apotransferrin alone does not affect the level of Fe2+ ions in human urine, indicating that ceruloplasmin is necessary for the observed decrease in Fe2+ ions (Fig. 6A). To determine the direct effect of ceruloplasmin on iron bioavailability to UPEC, transcription of the UPEC iron uptake gene iutA, which is involved in the import of the siderophore aerobactin, was determined. Since iron uptake genes are expressed (de-repressed) during growth in a low-iron milieu, the level of expression of iutA serves as an indicator of iron status in UPEC (15). UPEC was grown in urine from healthy volunteers with or without added ceruloplasmin. Addition of ceruloplasmin resulted in a significant increase in iutA transcript expression, indicating that ceruloplasmin induces iron starvation in UPEC (Fig. 6C). Taken together, our data indicate that ceruloplasmin impacts UPEC during UTI by inducing Cu stress and also by limiting the bioavailability of iron, an essential nutrient for UPEC.
FIG 5.
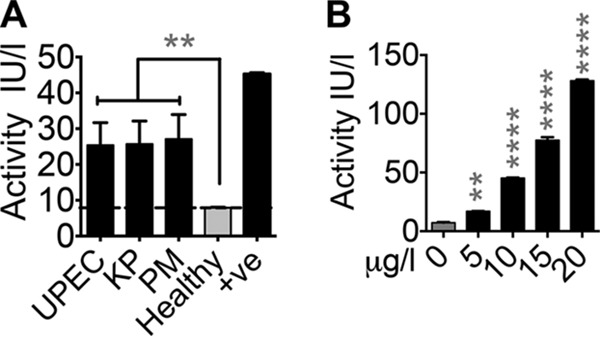
Oxidase activity of ceruloplasmin in urine. (A) Oxidase activity in UTI (UPEC, K. pneumoniae [KP], and P. mirabilis [PM]) and control urine samples was measured with o-dianisidine as the substrate. +ve, 10 μg ceruloplasmin. UPEC, n = 30; KP, n = 10; PM, n = 17; healthy controls, n = 40. **, P < 0.01; Kruskal-Wallis test with Dunn's posttest. Dotted line, limit of detection. (B) Ceruloplasmin functions as an oxidase in human urine, as determined by o-dianisidine oxidation assay. **, P < 0.01; ****, P < 0.0001; ANOVA with Dunnett's posttest. The mean and standard error of the mean are presented.
FIG 6.
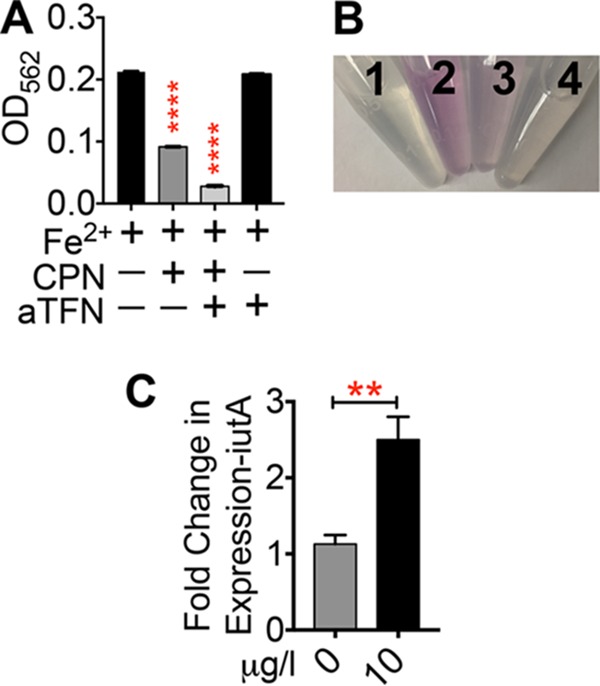
Ferroxidase activity of ceruloplasmin (CP) in urine. (A) The optical density at 562 nm (OD562) of Fe2+-ferrozine complex formation in urine with or without CP and apotransferrin (aTFN) was measured. ****, P < 0.0001; ANOVA with Dunnett's posttest. (B) Representative image of CP- and aTFN-dependent decrease in Fe2+ iron availability, as determined by complex formation with ferrozine. 1, urine alone; 2, FeSO4; 3, FeSO4 and CP; 4, FeSO4, CP, and aTFN. (C) Expression of iutA in UPEC strain CFT073 cultured with or without the indicated concentration of CP in healthy human urine was determined by qPCR after normalization to gapA. **, P < 0.01; t test (n = 3, in duplicate). The mean and standard error of the mean are presented.
Cu is not mobilized to urine during UTI in mice.
Our experiments with human samples demonstrate an association between UTI and elevated urinary Cu content but do not signify a cause-effect relationship. To address whether Cu mobilization to urine is triggered as a host response, we utilized animal models of UTI. We observed a significant difference in the baseline urinary Cu content of humans and nonhuman primates (vervet monkeys) versus that of mice (Fig. 7A). Healthy mouse urine contains much higher levels of Cu than healthy human and nonhuman primate urine does (Fig. 7A). However, urinary Cu levels do not increase during UTI induced by UPEC strain CFT073 in mice, compared to preinfection levels (Fig. 7B). While there is a trend toward decreased urinary Cu content during UTI in mice, compared to the preinfection baseline, this difference is not statistically significant. Infection with UPEC strain HM69, a recent clinical isolate from a woman with cystitis that produces yersiniabactin, a siderophore that binds Cu in addition to Fe3+, unlike CFT073, which does not produce yersiniabactin, also revealed a lack of a UTI-dependent increase in urinary Cu content in mice mirroring the results obtained with strain CFT073 (data not shown). Our results indicate that the Cu mobilization response observed during UTI in patients is not conserved in mice.
FIG 7.
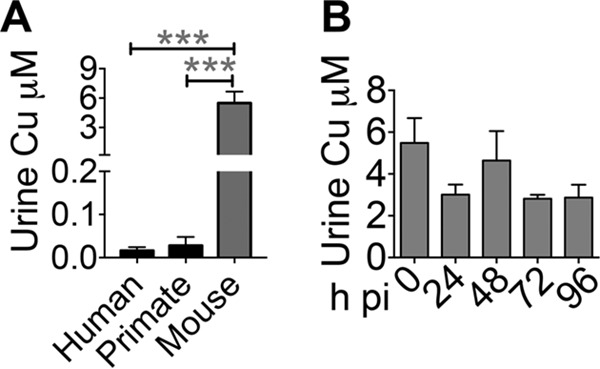
Mice do not mobilize Cu to urine during UTI. (A) Cu contents of healthy human (n = 40), nonhuman primate (adult female vervet monkey, n = 4), and mouse (5-week-old female CBA/J, n = 10) urine samples were determined by ICP-MS. ***, P < 0.001; ANOVA with Dunnett's test. (B) Temporal changes in urinary Cu content of mice infected with UPEC strain CFT073 (n = 10). The mean and standard error of the mean are presented. h pi, hours postinoculation.
A nonhuman primate model recapitulates the increased urinary Cu content observed in human UTI.
We hypothesized that the urinary Cu mobilization response during UTI will be conserved in nonhuman primates because of their closer evolutionary relationship to humans. We adapted a previously described nonhuman primate model of UTI (16) with important differences, including the use of vervet monkeys (Chlorocebus aethiops) and direct instillation of UPEC into the bladder. Adult females (n = 4) were inoculated with UPEC strain CFT073 by cystocentesis. UPEC readily colonized the urinary bladder, which is evident from the high and sustained bacterial load in urine at 2 and 4 days postinfection (Fig. 8A). Enrofloxacin was administered to resolve the UTI, as demonstrated by the absence of detectable UPEC in urine by day 7 (Fig. 8A). As observed in patients, UTI resulted in a massive leukocyte influx into the urine in vervet monkeys (Fig. 8B). Importantly, urinary Cu levels were significantly higher during UTI than at time zero (Fig. 8C), demonstrating that hosts mobilize Cu to urine as part of the innate immune response to UPEC colonization during UTI. Cytologic examination revealed a significant increase in the number of epithelial cells released into urine during UTI (Fig. 8D). The concentration of C-reactive protein, a known acute-phase reactant, was increased in plasma and in urine during UTI in vervet monkeys (Fig. 9A and B). However, after the onset of UTI, C-reactive protein levels were not significantly different from the baseline, possibly because of the high level of intragroup variation. In summary, our nonhuman primate model of UTI faithfully emulates the key features of human UTI and offers direct experimental evidence of urinary Cu mobilization in response to pathogen colonization of the urinary tract.
FIG 8.
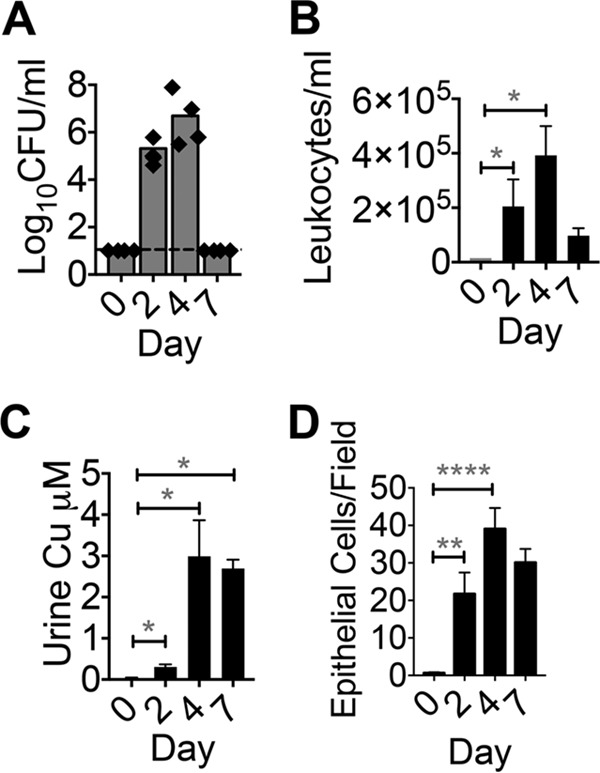
Urinary Cu content in nonhuman primates is elevated during UTI. (A) Urine bacterial loads at time 0 (preinfection), on days 2 and 4 (UTI), and on day 7 (postantibiotic) in vervet monkeys infected with UPEC strain CFT073 (n = 4, adult female). Bars represent median values, and the dotted line denotes the limit of detection. (B) Urine leukocyte counts deduced from leukocyte esterase activity. *, P < 0.05; Kruskal-Wallis test with Dunn's posttest. (C) Urinary Cu content during the course of UTI as determined by ICP-MS. *, P < 0.05; ANOVA with Dunnett's posttest. (D) Number of epithelial cells in urine Cytospin slides (×40 field, five fields per sample, n = 4). **, P < 0.01; ****, P < 0.0001; Kruskal-Wallis test with Dunn's posttest. The mean and standard error of the mean are presented (B to D).
FIG 9.
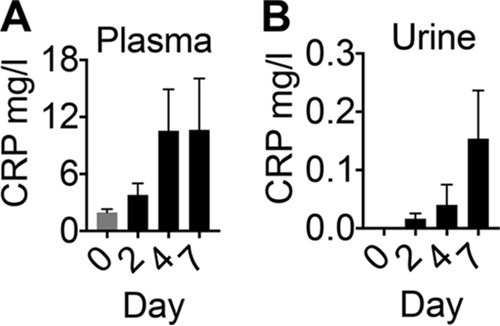
Acute-phase response of vervet monkeys during UTI. C-reactive protein (CRP) levels in plasma (A) and urine (B) during the course of UTI were determined by ELISA. Data from adult female vervet monkeys (n = 4) infected with UPEC strain CFT073 reveal a trend toward increased CRP levels during UTI. P > 0.05; ANOVA with Dunnett's posttest. The mean and standard error of the mean are presented.
Cu deficiency increases UPEC colonization in mice.
To determine the role of Cu in protection against UTI, we tested the hypothesis that Cu deficiency promotes UPEC colonization. The mouse is a relevant model to study the role of Cu in UPEC clearance because of the inherently high levels of urinary Cu regardless of urinary tract health (Fig. 7A and B). Mice were fed a diet lacking Cu for 30 days as described previously (17). Mice in the Cu-adequate control group received Cu in their drinking water. Cu-deficient mice were highly colonized by UPEC in their bladders and kidneys, in contrast to Cu-adequate controls (Fig. 10A). The median bacterial burden in the bladders and kidneys was ∼2 orders of magnitude higher in Cu-deficient mice than in controls, indicating that Cu is involved in protection against UPEC colonization during UTI (Fig. 10A). Cu deficiency status was verified by determining hepatic Cu content because the liver is the primary Cu storage organ. As expected, mice in the Cu-deficient group had a significantly lower hepatic Cu content than controls (Fig. 10B). Cu levels in the urine (Fig. 10C), bladder (Fig. 10D), and kidneys (Fig. 10E) were also lower in the Cu-deficient group than in Cu-adequate controls. Cu deficiency has been reported to induce neutropenia (18), and neutropenic mice are heavily colonized by UPEC during experimental UTI (19). Myeloperoxidase (MPO) is a marker of neutrophil presence during UTI (20). We measured the MPO levels in the bladders and kidneys of control and Cu-deficient mice during UTI by ELISA. Our results reveal that the MPO contents of the bladders (Fig. 11A) and kidneys (Fig. 11B) of control and Cu-deficient mice were not significantly different. Body weight was not different between the control (24.1 ± 0.4 g) and Cu-deficient (24.1 ± 0.6 g) groups. Bladder weight is an indicator of the degree of inflammation because of the edema that follows the host response to pathogen colonization. However, the weights of the urinary bladders of control (19.8 ± 1.4 mg) and Cu-deficient (19.8 ± 1.1 mg) mice were comparable. Because Cu deficiency could lead to anemia, we tested blood hemoglobin levels. The blood hemoglobin levels in the control (14.4 ± 0.3 g/dl) and Cu-deficient (14.5 ± 0.2 g/dl) groups were comparable and within the normal range (10 to 16 g/dl). We also tested blood glucose levels because diabetic mice are highly susceptible to UPEC colonization (21). Blood glucose levels, however, were not significantly different between the control (196.5 ± 12.5 mg/dl) and Cu-deficient (189.4 ± 20.7 mg/dl) groups. In summary, our results demonstrate that Cu deficiency promotes UPEC colonization in the murine urinary tract independent of neutrophil abundance and establishes Cu as an effector involved in protection against UPEC colonization during UTI.
FIG 10.
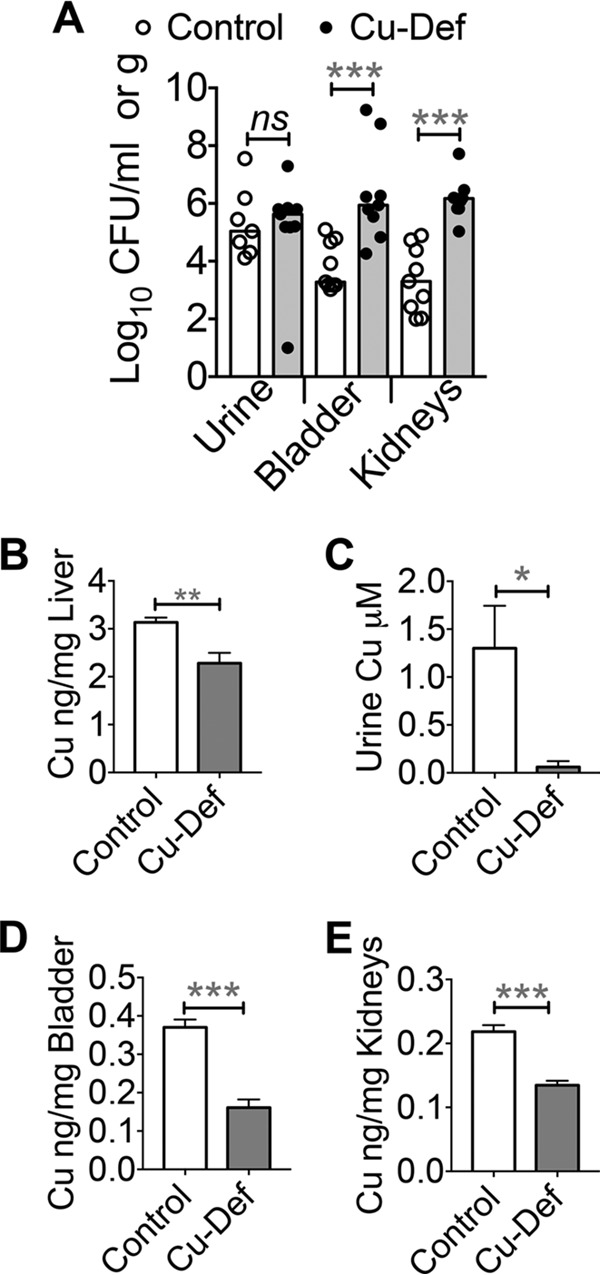
Cu deficiency promotes UPEC colonization in mice. (A) Tissue bacterial loads in control and Cu-deficient (Cu-Def) mice (CBA/J, 8 weeks old, n = 10/group) infected with UPEC strain CFT073 at 48 h postinoculation. Symbols represent bacterial burdens in a mouse, and bars represent median values. ***, P < 0.001; Mann-Whitney test. (B to E) Cu content of the livers (B), urine (C), bladders (D), and kidneys (E) of control and Cu-deficient mice was determined by ICP-MS. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed t test. The mean and standard error of the mean are presented.
FIG 11.
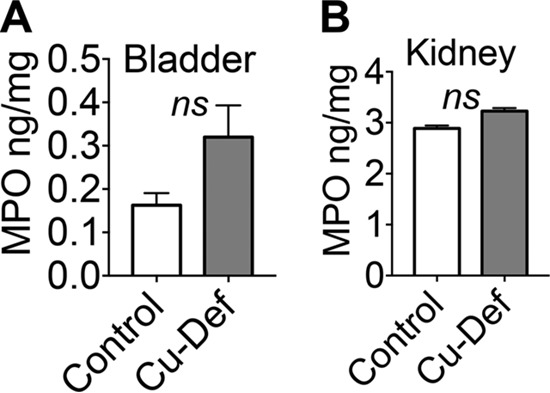
Cu deficiency does not affect myeloperoxidase (MPO) levels. (A, B) The MPO content of control and Cu-deficient (Cu-Def) mice (CBA/J, 8 weeks old, n = 10/group) infected with UPEC-infected mouse bladder (A) and kidney (B) homogenates (48 h postinoculation) was determined by ELISA and normalized to organ weight. The mean and standard error of the mean are presented. ns, not significant; t test.
DISCUSSION
Here, we report that Cu and ceruloplasmin are effectors of the host response to UTI caused by major uropathogens in patients. Our findings are supported by evidence from nonhuman primate and Cu-deficient mouse models of experimental UTI. On the basis of these results, we propose a model in which pathogen colonization triggers Cu mobilization to urine (Fig. 12). Cu is a known bactericidal agent (13), and ceruloplasmin also exhibits bactericidal activity (22). Additionally, the ferroxidase activity of ceruloplasmin in urine decreases iron bioavailability. Cu-deficient mice are highly colonized by UPEC during UTI, and this result supports our hypothesis that Cu deters pathogen colonization in vivo. Recurrent UTI and fatal complications of UTI have been reported in patients with Menkes disease, who are extremely Cu deficient because of a genetic impairment of Cu absorption and trafficking (23, 24). Taken in light of these clinical findings, our results highlight the importance of Cu as a protective effector against UTI.
FIG 12.
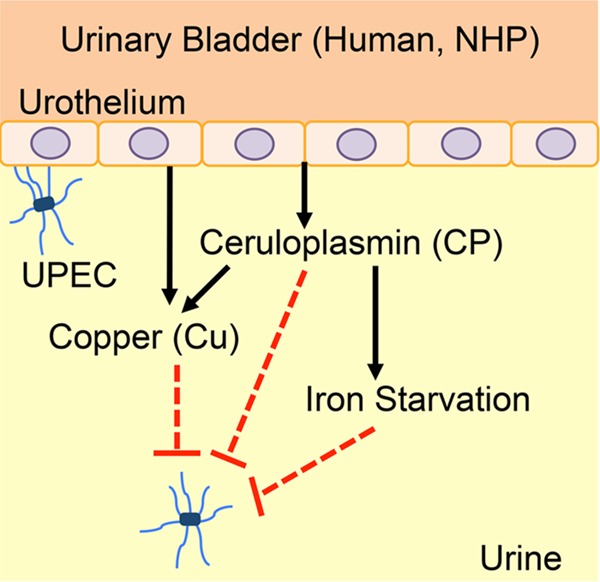
Model of the role of Cu in UTI. In humans and nonhuman primates (NHP), Cu is mobilized to urine during UTI. Ceruloplasmin is the molecular source of Cu in urine. Cu and ceruloplasmin exert direct and indirect effects to limit the growth of UPEC. Ceruloplasmin promotes the loading of iron onto apotransferrin to generate holotransferrin. Holotransferrin is then internalized by the urothelial cells during UTI, resulting in iron starvation of UPEC. Mobilization of Cu to urine during UTI limits bacterial growth by direct toxicity and also by hampering the bioavailability of iron, an essential nutrient.
During the acute-phase reaction triggered by infection and inflammation in humans, the circulating Cu content increases because of increased secretion of ceruloplasmin from the liver (14, 25). While our work demonstrates urinary Cu mobilization via ceruloplasmin during UTI with Gram-negative pathogens, whether the Cu mobilization response is conserved during UTI with Gram-positive pathogens remains to be examined. Mutants lacking Cu detoxification systems in diverse pathogens, including Cryptococcus neoformans (a fungus), K. pneumoniae, Mycobacterium tuberculosis, Salmonella enterica serovar Typhimurium, Streptococcus pneumoniae, and UPEC, are attenuated in animal models of infection (11, 26–31). Our results presented here elucidate the protective role of Cu in the pathogenesis of UTI. Taken together, Cu emerges as a critical host effector involved in protection against multiple infectious diseases, including UTI.
E. coli utilizes Cop and Cus Cu efflux pumps to overcome Cu toxicity (32). In addition, yersiniabactin, a siderophore that is typically used for iron acquisition in pathogenic but not commensal strains of E. coli, also binds Cu to limit Cu toxicity during UTI (12). Cu-yersiniabactin complexes have also been reported to exhibit superoxide dismutase-like activity to confer protection from reactive oxygen species (33). These findings and our results highlight the role of Cu in the mammalian host-UPEC interaction, where the host mobilizes Cu to limit pathogen colonization and UPEC co-opts a molecule involved in iron acquisition to resist Cu toxicity. Additionally, vaccination with the yersiniabactin receptor FyuA decreases UPEC colonization in the mouse model of UTI, further strengthening the role of yersiniabactin in the virulence of UPEC (34). Further studies are required to define the extent to which yersiniabactin-mediated sequestration of Cu, independent of its role in iron acquisition, is critical for bacterial survival during UTI. Such studies are critical to evaluate the effectiveness of augmentation of the host Cu response as a therapeutic strategy to limit UPEC colonization during UTI.
In addition to reactive oxygen species, lysozyme, and other lytic effectors, macrophages utilize Cu for bacterial killing. Mycobacterium avium-containing phagosomes in mouse macrophages exposed to gamma interferon (IFN-γ) contain higher Cu levels than controls (35). RAW 264.7 murine macrophages exposed to proinflammatory molecules (IFN-γ and lipopolysaccharide) import higher levels of Cu via the CTR1 Cu transporter than controls (36). Cu is then trafficked into the phagolysosome by the ATP7A Cu transporter to kill a laboratory strain of E. coli in a Cu-dependent mechanism (36). Here, we report significantly greater bacterial loads in the bladders and kidneys of Cu-deficient mice than in those of controls. However, there is no difference in the pathogen load in urine between these two groups, although the urine, bladder, and kidney Cu levels are significantly lower in Cu-deficient mice than in controls. The well-described role of Cu in bacterial killing within macrophages raises the important question of whether Cu kills UPEC extracellularly and/or within phagocytes in the urine, bladder, and kidneys during UTI.
Iron acquisition is vital for the survival and virulence of UPEC during UTI (37, 38). Nephrons secrete lipocalin into urine to hamper enterobactin-dependent iron uptake by UPEC during UTI (39–41). By utilizing lipocalin-resistant siderophores such as yersiniabactin and salmochelin, UPEC continues to acquire iron within the urinary tract. Similar to Cu, the human urinary iron content during UTI is higher than that of healthy urine (42). Paradoxically, UPEC iron uptake genes, whose expression is induced by iron starvation, are highly expressed during UTI (11, 43), indicating lower bioavailability of iron. The presence of transferrin in human urine has been reported (44), and this observation further supports our model, where ceruloplasmin and transferrin are involved in the limiting of iron bioavailability to UPEC. Our work has revealed a novel mechanism of iron limitation during UTI where ceruloplasmin catalyzes the production of Fe3+ ions that are then loaded onto transferrin in urine. During UPEC colonization, urothelial cells upregulate the expression of transferrin receptors that then rapidly import holotransferrin to deplete iron from the extracellular milieu (45). Therefore, we posit that Cu mobilization to urine exerts distinct yet convergent effects to limit bacterial survival by direct bactericidal action, by bactericidal action via delivery of Cu, and also indirectly by inducing iron starvation (Fig. 12). Our results indicate that ceruloplasmin is an effector of nutritional immunity, a part of the innate immune response that impedes pathogen growth by depriving the pathogen of essential nutrients such as iron (46).
Our results reveal that, unlike humans and nonhuman primates, mice lack the Cu mobilization response during UTI. The urinary Cu content of mice has been reported to decrease during nephritis subsequent to systemic infection caused by Candida albicans (47). Taken together, the results show that Cu mobilization to urine during UTI is lacking in mice during UTI of systemic origin (C. albicans) and also during ascending UTI (UPEC). We demonstrate that the basal levels of urinary Cu in mice are higher than those in humans and nonhuman primates, raising the possibility that a further increase in Cu content might not affect Cu-dependent killing of UPEC within the murine urinary tract. Our results reveal a dichotomy in the urinary Cu mobilization response between humans and mice and highlight the importance of nonhuman primates as physiologically relevant models for investigation of the host-pathogen interaction during UTI. Mouse models, although incredibly useful for the investigation of human diseases, have well-recognized limitations in faithfully emulating the pathophysiological changes encountered in humans, especially during inflammation (48). In humans and nonhuman primates, ceruloplasmin is an abundant acute-phase reactant (25, 49, 50). However, ceruloplasmin is a minor acute-phase reactant in mice (51). Because ceruloplasmin contains ∼95% of the circulating Cu (14), this is a plausible reason that humans and nonhuman primates, but not mice, mobilize Cu to urine during UTI. Here, we adapted the nonhuman primate model of UTI (16) to vervet monkeys to garner experimental evidence for UTI-induced mobilization of Cu into urine. This result is significant because it provides direct experimental evidence that UTI is the cause of elevated urinary Cu content.
In summary, our results demonstrate that Cu is a host effector involved in protection against UTI. Further studies on the mechanism(s) of Cu-dependent impairment of bacterial survival within the urinary tract will advance our understanding of an innate defense effector against UTI, a ubiquitous infectious disease. A potential translational application of our study is to develop Cu-based therapeutics against UTI because urinary Cu levels are amenable to modulation by dietary and pharmacological intervention.
MATERIALS AND METHODS
Human urine samples.
UTI and healthy urine samples were collected in accordance with protocol IRB00035856, approved by the Wake Forest Baptist Medical Center. Urine samples were cultured on MacConkey agar. Lactose-positive colonies were tested for the presence of E. coli gadA/B and uidA by PCR as described previously (52). Lactose fermentation, motility, colony phenotype (mucoid), and urease production phenotypes were used to differentiate K. pneumoniae (lactose positive, mucoid, nonmotile) from P. mirabilis (lactose negative, urease strong positive, nonmucoid, swimming and swarming motility positive) (53). Pooled, filter-sterilized urine from six healthy volunteers was used for in vitro culture of UPEC in urine.
ICP-MS.
Cu content of urine samples was determined as described in reference 11. Briefly, 100-μl human urine samples were digested with trace metal-grade nitric acid (100 μl) for 1 h at 100°C and diluted with deionized water. Cu content was determined in a blinded fashion by ICP-MS (8800 Triple Quadrupole ICP-MS; Agilent) with a Cu detection limit, calculated according to IUPAC recommendations (3σ, n = 15), of 0.31 nM.
Urinalyses.
Creatinine levels were measured with a colorimetric assay according to the manufacturer's instructions (Cayman Chemicals). Specific gravity was determined with a refractometer according to the manufacturer's instructions (Sper Scientific). Hemoglobin levels were measured with a colorimetric assay according to the manufacturer's instructions (Sigma). All of these assays were conducted in duplicate.
Cu speciation assay.
The indicated concentrations of CuSO4 were incubated in healthy or UTI human urine samples (two from UPEC; two from P. mirabilis; and two from K. pneumoniae), PBS, or lysogeny broth (1% tryptone, 0.5% yeast extract, 1% sodium chloride) at room temperature for 30 min. The Cu1+-specific chelator bathocuproine disodium disulfonate (Sigma) was added (50 μM final concentration), and absorbance at 480 nm was measured (54). The experiment was repeated three times independently.
Immunoblotting.
UTI and control urine samples (10 μl) were resolved by 10% SDS-PAGE, transferred to polyvinylidene difluoride membrane, and blocked with 5% milk in Tris-buffered saline with Tween 20. The protein content of urine samples was measured with Bradford's assay (Thermo Scientific) according to the manufacturer's instructions. Dilute urine samples were concentrated with columns (3-kDa molecular mass cutoff) to achieve comparable concentration of protein. Blots were probed with rabbit anticeruloplasmin antibody (Thermo Scientific). Goat anti-rabbit IgG-horseradish peroxidase conjugate (Sigma) was used for detection by chemiluminescence (ECL Prime; GE Healthcare Life Sciences). Pure human ceruloplasmin (Athens Research and Technology) was used as a positive control. All samples were analyzed by immunoblotting, and a representative blot, with the layout described in the figure legend, is depicted in Fig. 3A. Autoradiography films were scanned, and entire images were converted from color scale to gray scale. The image was then cropped to display the signal corresponding to holoceruloplasmin (132 kDa).
Ceruloplasmin assays.
Urine samples were assayed in duplicate to determine ceruloplasmin content by ELISA according to the manufacturer's instructions (Molecular Innovations). The specificity of this assay was experimentally verified by preincubation of a ceruloplasmin standard or representative UTI urine samples (ceruloplasmin positive by immunoblotting) with rabbit anticeruloplasmin antibody (Thermo Scientific) prior to ELISA. Preincubation ablated the signal to background levels. Oxidase activity in UTI and healthy urine samples and also of pure ceruloplasmin in healthy urine samples was measured with o-dianisidine in acetate buffer as the substrate in a colorimetric assay as described previously (55). According to the original publication (55), this method has little to no interference from reducing or colored components. However, the activity of oxidases in addition to or other than ceruloplasmin in UTI urine might also generate a signal in this assay. These experiments were repeated three times independently.
Effect of ceruloplasmin on UPEC growth.
Wild-type (CFT073) (56) and ΔcusSRCFBA mutant (11) UPEC strains were cultured in healthy human urine collected as described earlier. Overnight cultures in lysogeny broth were diluted 1:100 in urine with or without ceruloplasmin (10 μg/liter), dispensed into 96-well plates (200 μl/well), and incubated statically at 37°C with 5% CO2. Absorbance was measured at 24 h in a SpectraMax M2 multimode plate reader (Molecular Devices). This experiment was repeated three times independently and in triplicate.
qPCR.
RNA was extracted from UPEC strain CFT073 (56) cultured in healthy human urine with or without the indicated amounts of ceruloplasmin for 2 h at 37°C (SurePrep RNA extraction kit; Fisher). Contaminating DNA was removed by treatment with TURBO DNase (Ambion), and cDNA was synthesized with the Superscript II system (Invitrogen). Expression of cusC and iutA, normalized to gapA, was determined by qPCR by SYBR green chemistry and a relative quantitation method (7500 real-time PCR system; Applied Biosystems). The primers used (5′ to 3′; described in reference 11) were cusC Forward (AATGTCGCGCAAAGCTATTT), cusC Reverse (CGACAAACGCATATGACTGC), iutA Forward (AAAGAGCTGAAAGACGCACTGG), iutA Reverse (TGTCGGAACGTGAAGAGTTGAG), gapA Forward (AAGTTGGTGTTGACGTTGTCGC), and gapA Reverse (AGCGCCTTTAACGAACATCG). The experiment was repeated three times in duplicate.
Ferroxidase activity assay.
The selective binding property of ferrozine to Fe2+ ions was utilized for the ferroxidase activity assay (57). Pure human ceruloplasmin (5 μg) and/or apotransferrin (10 μg; Sigma) was added to 500 μl of healthy human urine with or without ferrous sulfate (1 μM; Sigma). After 10 min of incubation, an equal volume of ferrozine reagent (1 mM in 50 mM HEPES; Acros Organics) was added and absorbance at 562 nm was measured. The experiment was repeated three times independently and in triplicate with pooled urine samples from six healthy adult volunteers.
Nonhuman primate model of UTI.
This study was conducted at the Wake Forest Primate Center according to protocol A16-070, approved by the Wake Forest Baptist Medical Center. We have adapted a nonhuman primate model of UTI (16) with the major modification of direct instillation of UPEC into the bladders of vervet monkeys. Adult female vervet monkeys (C. aethiops, n = 4) were anesthetized with ketamine and dexmedetomidine and inoculated with 107 CFU of UPEC strain CFT073 in the urinary bladder with ultrasound guidance. UPEC, cultured in lysogeny broth overnight at 37°C and shaking at 200 rpm, was pelleted and resuspended in PBS. Urine samples were collected by cystocentesis at time zero prior to inoculation, on days 2 and 4 during the course of experimental UTI, and on day 7 after UTI resolution. When ultrasonography revealed an empty bladder, furosemide and/or 0.9% sodium chloride infusion was used to promote diuresis. Animals were administered enrofloxacin after cystocentesis on day 4. UPEC load in urine was determined by culture on MacConkey agar. Urine leukocyte counts were deduced from leukocyte esterase activity with dipsticks (Fisher). Urinary Cu content was determined by ICP-MS as described above. Urine samples (100 μl) were spun onto Cytospin slides and stained with Kwik-Diff (Fisher). A board-certified veterinary pathologist determined the number of epithelial cells per field under ×40 magnification (five fields per sample per time point) in a blinded manner. Plasma and urine samples taken from vervet monkeys during the course of UTI were assayed in duplicate to determine C-reactive protein levels by ELISA (Abnova) according to the manufacturer's instructions.
Mouse model of UTI.
Experiments were conducted according to protocol A15-178, approved by the Wake Forest Baptist Medical Center. Adult female CBA/J mice from Jackson Laboratories were used (n = 10/group). Mice were inoculated in the urinary bladder with 108 CFU of UPEC strain CFT073, cultured as described above, in 50 μl of PBS (58). Briefly, a catheter was introduced into the bladder of a tribromoethanol-anesthetized mouse and the inoculum was deposited slowly (100 μl/min) to avoid vesicoureteral reflux with a syringe pump (Braintree Scientific). The Cu content of urine samples collected before and after infection was determined by ICP-MS as described earlier. Mice were fed a diet lacking Cu prepared at the Wake Forest Diet Lab for 30 days prior to infection as described previously (17). Cu-adequate controls received Cu (20 mg/liter) in their drinking water. Mice were euthanized at 48 h postinoculation to collect their bladders, kidneys, and livers. Urine and homogenates of bladders and kidneys were plated to determine bacterial loads. Liver tissue, urine, and kidney and bladder homogenates were digested with nitric acid. Cu content was determined by ICP-MS as described earlier and was normalized to the weight of the organ. The MPO content of bladder and kidney homogenates was determined in duplicate by ELISA (Thermo Scientific) according to the manufacturer's instructions and normalized to the weight of the organ. Blood samples were collected immediately after euthanasia to measure hemoglobin levels with a HemoCue (Hb201) analyzer and glucose levels with a glucometer (Bayer).
Statistical analyses.
Data were analyzed by t test, analysis of variance (ANOVA), or nonparametric equivalents (Mann-Whitney and Kruskal-Wallis tests) with GraphPad Prism version 7. P < 0.05 was considered a statistically significant difference.
ACKNOWLEDGMENTS
We thank Matthew Jorgensen, Elizabeth Palavecino, Russell Talley, Katherine Turnbull, Keith Ballentine, Kevin Zhou, and Stephen Richardson for their help.
Author contributions are as follows: conceptualization, S.S.; investigation, A.N.H., K.K., N.D.K., G.L.D., and S.S.; writing, S.S.; funding acquisition, S.S. and G.L.D.; supervision, S.S.
This work was supported by funds from the Wake Forest School of Medicine, the Errett Fisher Foundation, and the Wake Forest Clinical and Translational Science Institute to S.S.; NIH awards P40 OD010965 to the Wake Forest Primate Center and UL1TR001420 to the Wake Forest Clinical and Translational Science Institute; and NSF MRI grant CHE-1531698 to G.L.D. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Stamm WE, Norrby SR. 2001. Urinary tract infections: disease panorama and challenges. J Infect Dis 183(Suppl 1):S1–S4. doi: 10.1086/318850. [DOI] [PubMed] [Google Scholar]
- 2.Subashchandrabose S, Mobley HL. 2015. Virulence and fitness determinants of uropathogenic Escherichia coli. Microbiol Spectr 3. doi: 10.1128/microbiolspec.UTI-0015-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooton TM. 2012. Clinical practice. Uncomplicated urinary tract infection. N Engl J Med 366:1028–1037. doi: 10.1056/NEJMcp1104429. [DOI] [PubMed] [Google Scholar]
- 4.Foxman B. 2010. The epidemiology of urinary tract infection. Nat Rev Urol 7:653–660. doi: 10.1038/nrurol.2010.190. [DOI] [PubMed] [Google Scholar]
- 5.Mulvey MA, Schilling JD, Hultgren SJ. 2001. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun 69:4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med 4:e329. doi: 10.1371/journal.pmed.0040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. 2015. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13:269–284. doi: 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, Moran GJ, Nicolle LE, Raz R, Schaeffer AJ, Soper DE, Infectious Diseases Society of America, European Society for Microbiology and Infectious Diseases . 2011. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis 52:e103-120. doi: 10.1093/cid/ciq257. [DOI] [PubMed] [Google Scholar]
- 9.Karlowsky JA, Kelly LJ, Thornsberry C, Jones ME, Sahm DF. 2002. Trends in antimicrobial resistance among urinary tract infection isolates of Escherichia coli from female outpatients in the United States. Antimicrob Agents Chemother 46:2540–2545. doi: 10.1128/AAC.46.8.2540-2545.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGann P, Snesrud E, Maybank R, Corey B, Ong AC, Clifford R, Hinkle M, Whitman T, Lesho E, Schaecher KE. 2016. Escherichia coli harboring mcr-1 and blaCTX-M on a novel IncF plasmid: first report of mcr-1 in the United States. Antimicrob Agents Chemother 60:4420–4421. doi: 10.1128/AAC.01103-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subashchandrabose S, Hazen TH, Brumbaugh AR, Himpsl SD, Smith SN, Ernst RD, Rasko DA, Mobley HL. 2014. Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc Natl Acad Sci U S A 111:18327–18332. doi: 10.1073/pnas.1415959112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, Henderson JP. 2012. The siderophore yersiniabactin binds copper to protect pathogens during infection. Nat Chem Biol 8:731–736. doi: 10.1038/nchembio.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ladomersky E, Petris MJ. 2015. Copper tolerance and virulence in bacteria. Metallomics 7:957–964. doi: 10.1039/C4MT00327F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellman NE, Gitlin JD. 2002. Ceruloplasmin metabolism and function. Annu Rev Nutr 22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- 15.Alteri CJ, Mobley HL. 2007. Quantitative profile of the uropathogenic Escherichia coli outer membrane proteome during growth in human urine. Infect Immun 75:2679–2688. doi: 10.1128/IAI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Söderhäll M, Normark S, Ishikawa K, Karlsson K, Teneberg S, Winberg J, Mollby R. 1997. Induction of protective immunity after Escherichia coli bladder infection in primates. Dependence of the globoside-specific P-fimbrial tip adhesin and its cognate receptor. J Clin Invest 100:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, Counter CM. 2014. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509:492–496. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Percival SS. 1995. Neutropenia caused by copper deficiency: possible mechanisms of action. Nutr Rev 53:59–66. [DOI] [PubMed] [Google Scholar]
- 19.Haraoka M, Hang L, Frendeus B, Godaly G, Burdick M, Strieter R, Svanborg C. 1999. Neutrophil recruitment and resistance to urinary tract infection. J Infect Dis 180:1220–1229. doi: 10.1086/315006. [DOI] [PubMed] [Google Scholar]
- 20.Ozer A, Altuntas CZ, Bicer F, Izgi K, Hultgren SJ, Liu G, Daneshgari F. 2015. Impaired cytokine expression, neutrophil infiltration and bacterial clearance in response to urinary tract infection in diabetic mice. Pathog Dis 73:ftv002. doi: 10.1093/femspd/ftv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosen DA, Hung CS, Kline KA, Hultgren SJ. 2008. Streptozocin-induced diabetic mouse model of urinary tract infection. Infect Immun 76:4290–4298. doi: 10.1128/IAI.00255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klebanoff SJ. 1992. Bactericidal effect of Fe2+, ceruloplasmin, and phosphate. Arch Biochem Biophys 295:302–308. doi: 10.1016/0003-9861(92)90522-X. [DOI] [PubMed] [Google Scholar]
- 23.Tümer Z, Moller LB. 2010. Menkes disease. Eur J Hum Genet 18:511–518. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler EM, Roberts PF. 1976. Menkes's steely hair syndrome. Arch Dis Child 51:269–274. doi: 10.1136/adc.51.4.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markowitz H, Gubler CJ, Mahoney JP, Cartwright GE, Wintrobe MM. 1955. Studies on copper metabolism. XIV. Copper, ceruloplasmin and oxidase activity in sera of normal human subjects, pregnant women, and patients with infection, hepatolenticular degeneration and the nephrotic syndrome. J Clin Invest 34:1498–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding C, Festa RA, Chen YL, Espart A, Palacios O, Espin J, Capdevila M, Atrian S, Heitman J, Thiele DJ. 2013. Cryptococcus neoformans copper detoxification machinery is critical for fungal virulence. Cell Host Microbe 13:265–276. doi: 10.1016/j.chom.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachman MA, Breen P, Deornellas V, Mu Q, Zhao L, Wu W, Cavalcoli JD, Mobley HL. 2015. Genome-wide identification of Klebsiella pneumoniae fitness genes during lung infection. mBio 6:e00775-15. doi: 10.1128/mBio.00775-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 108:1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achard ME, Tree JJ, Holden JA, Simpfendorfer KR, Wijburg OL, Strugnell RA, Schembri MA, Sweet MJ, Jennings MP, McEwan AG. 2010. The multi-copper-ion oxidase CueO of Salmonella enterica serovar Typhimurium is required for systemic virulence. Infect Immun 78:2312–2319. doi: 10.1128/IAI.01208-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shafeeq S, Yesilkaya H, Kloosterman TG, Narayanan G, Wandel M, Andrew PW, Kuipers OP, Morrissey JA. 2011. The cop operon is required for copper homeostasis and contributes to virulence in Streptococcus pneumoniae. Mol Microbiol 81:1255–1270. doi: 10.1111/j.1365-2958.2011.07758.x. [DOI] [PubMed] [Google Scholar]
- 31.Johnson MD, Kehl-Fie TE, Klein R, Kelly J, Burnham C, Mann B, Rosch JW. 2015. Role of copper efflux in pneumococcal pathogenesis and resistance to macrophage-mediated immune clearance. Infect Immun 83:1684–1694. doi: 10.1128/IAI.03015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Outten FW, Huffman DL, Hale JA, O'Halloran TV. 2001. The independent cue and cus systems confer copper tolerance during aerobic and anaerobic growth in Escherichia coli. J Biol Chem 276:30670–30677. doi: 10.1074/jbc.M104122200. [DOI] [PubMed] [Google Scholar]
- 33.Chaturvedi KS, Hung CS, Giblin DE, Urushidani S, Austin AM, Dinauer MC, Henderson JP. 2014. Cupric yersiniabactin is a virulence-associated superoxide dismutase mimic. ACS Chem Biol 9:551–561. doi: 10.1021/cb400658k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brumbaugh AR, Smith SN, Subashchandrabose S, Himpsl SD, Hazen TH, Rasko DA, Mobley HL. 2015. Blocking yersiniabactin import attenuates extraintestinal pathogenic Escherichia coli in cystitis and pyelonephritis and represents a novel target to prevent urinary tract infection. Infect Immun 83:1443–1450. doi: 10.1128/IAI.02904-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner D, Maser J, Lai B, Cai Z, Barry CE III, Honer Zu Bentrup K, Russell DG, Bermudez LE. 2005. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J Immunol 174:1491–1500. doi: 10.4049/jimmunol.174.3.1491. [DOI] [PubMed] [Google Scholar]
- 36.White C, Lee J, Kambe T, Fritsche K, Petris MJ. 2009. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem 284:33949–33956. doi: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres AG, Redford P, Welch RA, Payne SM. 2001. TonB-dependent systems of uropathogenic Escherichia coli: aerobactin and heme transport and TonB are required for virulence in the mouse. Infect Immun 69:6179–6185. doi: 10.1128/IAI.69.10.6179-6185.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alteri CJ, Hagan EC, Sivick KE, Smith SN, Mobley HL. 2009. Mucosal immunization with iron receptor antigens protects against urinary tract infection. PLoS Pathog 5:e1000586. doi: 10.1371/journal.ppat.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paragas N, Kulkarni R, Werth M, Schmidt-Ott KM, Forster C, Deng R, Zhang Q, Singer E, Klose AD, Shen TH, Francis KP, Ray S, Vijayakumar S, Seward S, Bovino ME, Xu K, Takabe Y, Amaral FE, Mohan S, Wax R, Corbin K, Sanna-Cherchi S, Mori K, Johnson L, Nickolas T, D'Agati V, Lin CS, Qiu A, Al-Awqati Q, Ratner AJ, Barasch J. 2014. α-Intercalated cells defend the urinary system from bacterial infection. J Clin Invest 124:2963–2976. doi: 10.1172/JCI71630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steigedal M, Marstad A, Haug M, Damas JK, Strong RK, Roberts PL, Himpsl SD, Stapleton A, Hooton TM, Mobley HL, Hawn TR, Flo TH. 2014. Lipocalin 2 imparts selective pressure on bacterial growth in the bladder and is elevated in women with urinary tract infection. J Immunol 193:6081–6089. doi: 10.4049/jimmunol.1401528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shields-Cutler RR, Crowley JR, Miller CD, Stapleton AE, Cui W, Henderson JP. 2016. Human metabolome-derived cofactors are required for the antibacterial activity of siderocalin in urine. J Biol Chem 291:25901–25910. doi: 10.1074/jbc.M116.759183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subashchandrabose S, Mobley HL. 2015. Back to the metal age: battle for metals at the host-pathogen interface during urinary tract infection. Metallomics 7:935–942. doi: 10.1039/C4MT00329B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagan EC, Lloyd AL, Rasko DA, Faerber GJ, Mobley HL. 2010. Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog 6:e1001187. doi: 10.1371/journal.ppat.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poortmans J, Jeanloz RW. 1968. Quantitative immunological determination of 12 plasma proteins excreted in human urine collected before and after exercise. J Clin Invest 47:386–393. doi: 10.1172/JCI105735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dikshit N, Bist P, Fenlon SN, Pulloor NK, Chua CE, Scidmore MA, Carlyon JA, Tang BL, Chen SL, Sukumaran B. 2015. Intracellular uropathogenic E. coli exploits host Rab35 for iron acquisition and survival within urinary bladder cells. PLoS Pathog 11:e1005083. doi: 10.1371/journal.ppat.1005083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cassat JE, Skaar EP. 2013. Iron in infection and immunity. Cell Host Microbe 13:509–519. doi: 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li CX, Gleason JE, Zhang SX, Bruno VM, Cormack BP, Culotta VC. 2015. Candida albicans adapts to host copper during infection by swapping metal cofactors for superoxide dismutase. Proc Natl Acad Sci U S A 112:E5336–E5342. doi: 10.1073/pnas.1513447112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation Host Response to Injury, Large Scale Collaborative Research Program . 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabay C, Kushner I. 1999. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 50.Berendt RF, Knutsen GL, Powanda MC. 1978. Nonhuman primate model for the study of respiratory Klebsiella pneumoniae infection. Infect Immun 22:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wait R, Chiesa G, Parolini C, Miller I, Begum S, Brambilla D, Galluccio L, Ballerio R, Eberini I, Gianazza E. 2005. Reference maps of mouse serum acute-phase proteins: changes with LPS-induced inflammation and apolipoprotein A-I and A-II transgenes. Proteomics 5:4245–4253. doi: 10.1002/pmic.200401292. [DOI] [PubMed] [Google Scholar]
- 52.McDaniels AE, Rice EW, Reyes AL, Johnson CH, Haugland RA, Stelma GN Jr. 1996. Confirmational identification of Escherichia coli, a comparison of genotypic and phenotypic assays for glutamate decarboxylase and beta-d-glucuronidase. Appl Environ Microbiol 62:3350–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guentzel MN. 1997. Escherichia, Klebsiella, Enterobacter, Serratia, Citrobacter, and Proteus, chapter 26. In Baron S, Albrecht T, Castro G, Couch RB, Davis CP, Dianzani F, McGinnis MR, Niesel DW, Olson LJ, Woods GL (ed), Medical microbiology, 4th ed University of Texas Medical Branch at Galveston, Galveston, TX. [PubMed] [Google Scholar]
- 54.Zak B, Ressler N. 1958. Serum copper and iron on a single sample. Clin Chem 4:43–48. [PubMed] [Google Scholar]
- 55.Schosinsky KH, Lehmann HP, Beeler MF. 1974. Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin Chem 20:1556–1563. [PubMed] [Google Scholar]
- 56.Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun 58:1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carter P. 1971. Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine). Anal Biochem 40:450–458. doi: 10.1016/0003-2697(71)90405-2. [DOI] [PubMed] [Google Scholar]
- 58.Hagberg L, Engberg I, Freter R, Lam J, Olling S, Svanborg Eden C. 1983. Ascending, unobstructed urinary tract infection in mice caused by pyelonephritogenic Escherichia coli of human origin. Infect Immun 40:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]